LDSC_enrichment
Last updated: 2021-09-01
Checks: 7 0
Knit directory: funcFinemapping/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it's best to always run the code in an empty environment.
The command set.seed(20210404) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 4db9eb3. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .ipynb_checkpoints/
Ignored: analysis/ldsc_results.nb.html
Ignored: analysis/results.nb.html
Ignored: analysis/snp_finemapping_results.nb.html
Ignored: analysis/splicing.nb.html
Untracked files:
Untracked: SNPs_categories,png
Untracked: SNPs_categories.png
Untracked: analysis/enhancer_gene_feature.Rmd
Untracked: analysis/feedback.Rmd
Untracked: analysis/gene_finemapping_results.Rmd
Untracked: analysis/learn_susie.Rmd
Untracked: analysis/notes.Rmd
Untracked: analysis/snp_finemapping_results.Rmd
Untracked: analysis/splicing.Rmd
Untracked: code/.ipynb_checkpoints/
Untracked: code/ldsc_regression.sh
Untracked: code/make_plots.R
Untracked: code/run_ldsc.sh
Untracked: code/run_ldsc_with_bed.sh
Untracked: code/run_torus.sh
Untracked: code/split_vcf.sh
Untracked: data/num_overlaps_finemapped_SNPs_and_ctcf.txt
Untracked: data/scz_2018
Untracked: data/torus_enrichment_novel_annot.est
Untracked: data/torus_joint_enrichment.est
Untracked: data/torus_joint_refined_enrichment.est
Untracked: enhancer_gene_feature.rmd
Untracked: fig1_panels.pdf
Untracked: fig2.pdf
Untracked: fig_panel2.pdf
Untracked: gene_mapping.pdf
Untracked: output/joint_history/
Untracked: output/joint_torus_conservation_enrichment.est
Untracked: output/proposal_fig1.pdf
Untracked: output/proposal_fig2.pdf
Untracked: output/proposal_gene_mapping_figure.pdf
Untracked: output/scz_bmi_joint_enrichment.png
Untracked: output/scz_immune_joint_enrichment.png
Untracked: output/scz_lipid_joint_enrichment.png
Untracked: output/torus_enrichment_CDTS_vs_OCR.est
Untracked: output/torus_enrichment_DMR.est
Untracked: output/torus_enrichment_constraints.est
Untracked: output/torus_enrichment_ctcf.est
Untracked: output/torus_enrichment_m6A.est
Untracked: output/torus_joint_refined_enrichment.est
Untracked: output/torus_marginal_enrich_DMR.est
Untracked: output/torus_marginal_enrich_ctcf.est
Untracked: output/torus_marginal_enrich_m6A.est
Untracked: panel_figure2.pdf
Unstaged changes:
Modified: analysis/biology_bkg.Rmd
Modified: analysis/method_bkg.Rmd
Deleted: analysis/results.Rmd
Deleted: data/joint_torus_conservation_enrichment.est
Deleted: data/torus_enrichment.est
Deleted: data/torus_enrichment_DMR.est
Modified: data/torus_enrichment_ambigousSNPs.est
Deleted: data/torus_enrichment_m6A.est
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/ldsc_results.Rmd) and HTML (docs/ldsc_results.html) files. If you've configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 4db9eb3 | Jing Gu | 2021-09-01 | compute standardized effect sizes |
| html | 05f73e9 | Jing Gu | 2021-09-01 | Build site. |
| Rmd | bed838e | Jing Gu | 2021-09-01 | update evaluations on slicing |
Partitioned-LDSC
Technical details
HapMap3 SNPs (~1.2M): used as a proxy for well imputed SNPs and the regression SNPs in LDSC. They were generated by the international HapMap project on a collection of 1301 samples from a variaty of human populations.[https://www.sanger.ac.uk/resources/downloads/human/hapmap3.html]
Reference panel SNPs (~9M): the set of 1000G SNPs with MAF > 5% in ~500 Euproean samples
LDSC inputs/outputs: * .M/.l2.M_5_50: The .M file contains the total number of SNPs; the .l2.M_5_50 file contains the number of SNPs with minor allele frequency above 5%. By default, ldsc uses common SNPs (MAF > 5%) to estimate per SNP heritability, which is different for rare variants.
- freqfile/w-ld-chr: computed on European of Phase 3 of 1kg Genomes
Regression weights: used to correct for non-independence and heteroskedasticity among the \(\chi^2\) statistics.
Regression coefficients: Quoted from the website that "They measure the additional contribution of one annotation to the model and are interpretable for both binary and continuous annotations"
GC correction:
Baseline annotations:
DHS, H3K4me1, H3K4me3, and H3K9ac - peaks w/o flanking regions Coding, Conserved, CTCF, DGF
FANTOM5-Enhancer, Enhancer, Fetal_DHS, H3K27ac
Intron, PromoterFlanking, Promoter, Repressed, Super-enhancer, TFBS, Transcribed TSS
3-prime UTR, 5-prime UTR, Weak Enhancer
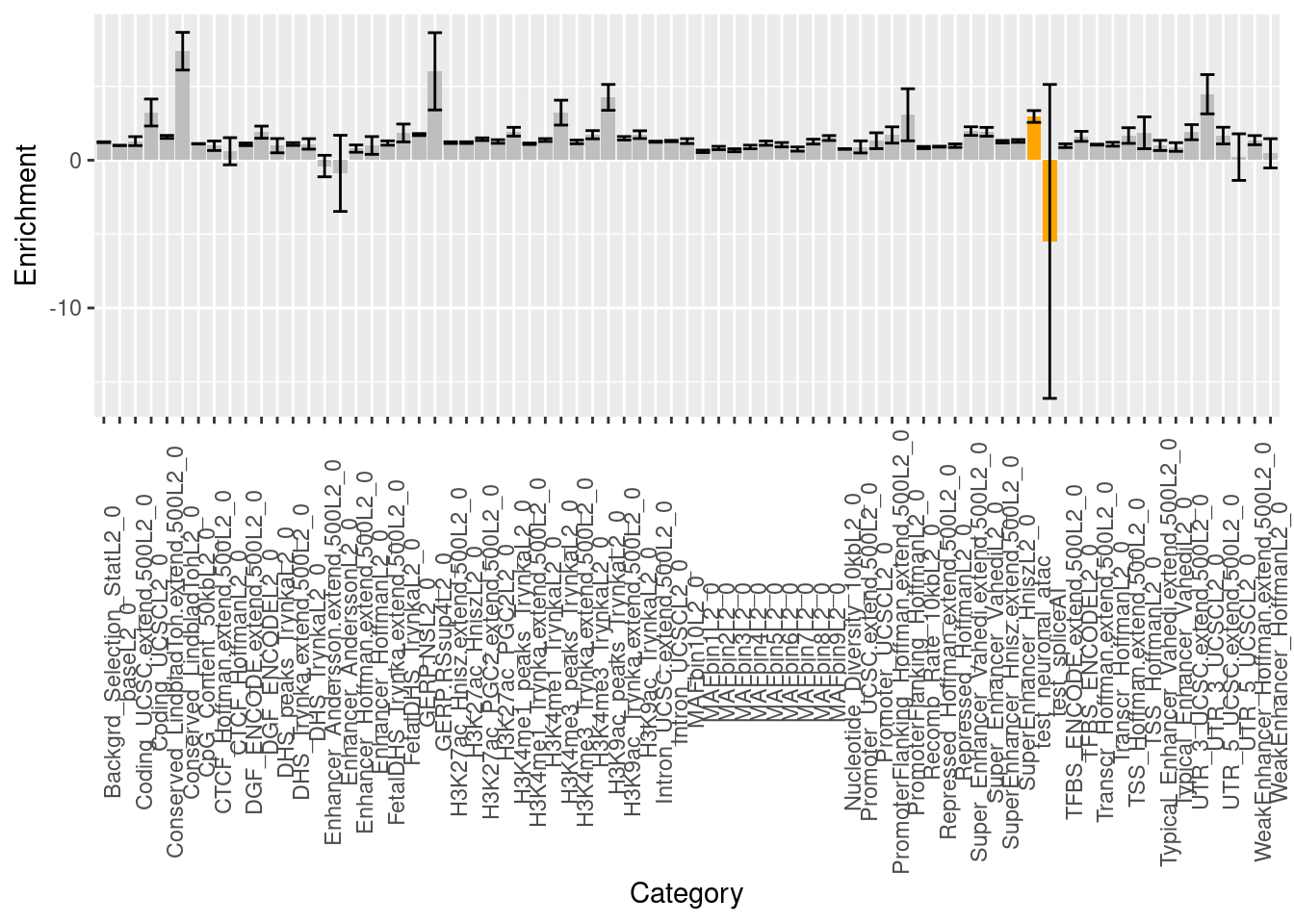
Across traits
per-standardized-annotation effect sizes
<ggproto object: Class ScaleDiscrete, Scale, gg>
aesthetics: fill
axis_order: function
break_info: function
break_positions: function
breaks: waiver
call: call
clone: function
dimension: function
drop: TRUE
expand: waiver
get_breaks: function
get_breaks_minor: function
get_labels: function
get_limits: function
guide: legend
is_discrete: function
is_empty: function
labels: cutoff0.01 cutoff0.03 cutoff0.05 cutoff0.07 cutoff0.1
limits: NULL
make_sec_title: function
make_title: function
map: function
map_df: function
n.breaks.cache: NULL
na.translate: TRUE
na.value: grey50
name: Annotations
palette: function
palette.cache: NULL
position: left
range: <ggproto object: Class RangeDiscrete, Range, gg>
range: NULL
reset: function
train: function
super: <ggproto object: Class RangeDiscrete, Range, gg>
rescale: function
reset: function
scale_name: hue
train: function
train_df: function
transform: function
transform_df: function
super: <ggproto object: Class ScaleDiscrete, Scale, gg>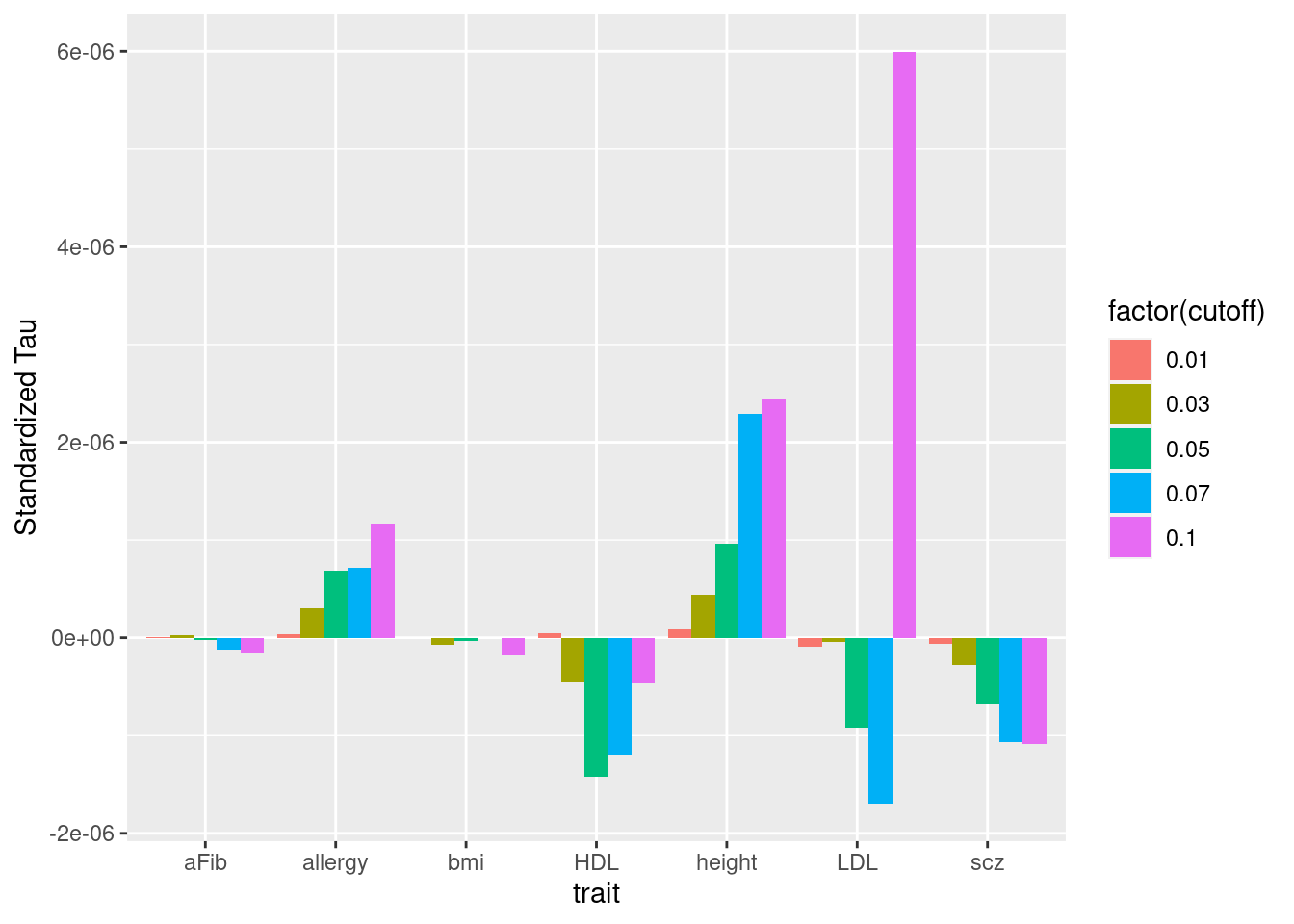
| Version | Author | Date |
|---|---|---|
| 05f73e9 | Jing Gu | 2021-09-01 |
Heritability Enrichment across traits and annotations 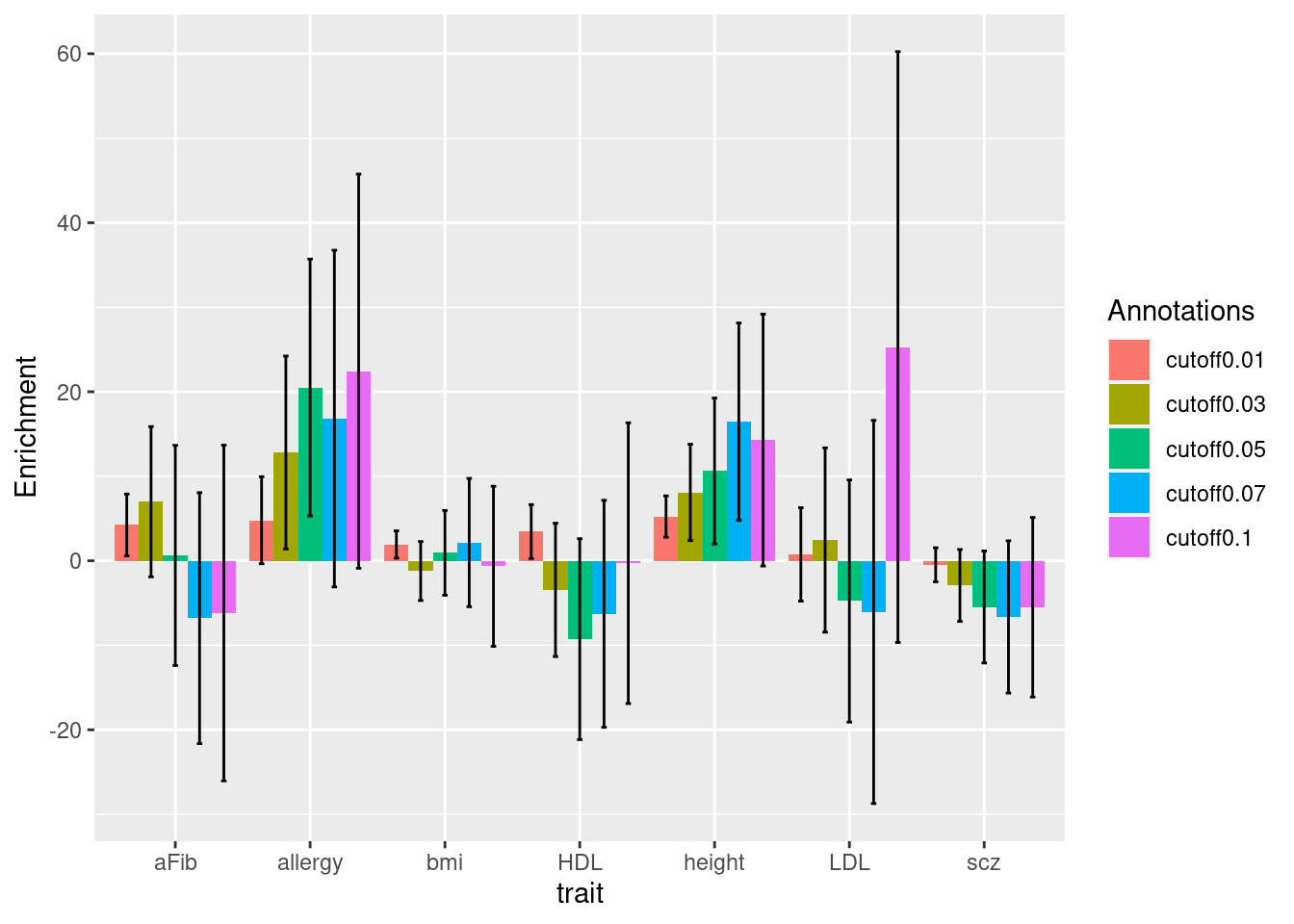
sessionInfo()R version 4.0.4 (2021-02-15)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.3.13-el7-x86_64/lib/libopenblas_haswellp-r0.3.13.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] ggplot2_3.3.3 workflowr_1.6.2
loaded via a namespace (and not attached):
[1] Rcpp_1.0.6 highr_0.8 pillar_1.5.0 compiler_4.0.4
[5] bslib_0.2.4 later_1.1.0.1 jquerylib_0.1.3 git2r_0.28.0
[9] tools_4.0.4 digest_0.6.27 jsonlite_1.7.2 evaluate_0.14
[13] lifecycle_1.0.0 tibble_3.0.6 gtable_0.3.0 pkgconfig_2.0.3
[17] rlang_0.4.11 DBI_1.1.1 yaml_2.2.1 xfun_0.21
[21] withr_2.4.1 dplyr_1.0.4 stringr_1.4.0 knitr_1.31
[25] generics_0.1.0 fs_1.5.0 vctrs_0.3.8 sass_0.3.1
[29] tidyselect_1.1.1 rprojroot_2.0.2 grid_4.0.4 glue_1.4.2
[33] R6_2.5.0 fansi_0.4.2 rmarkdown_2.7 farver_2.0.3
[37] purrr_0.3.4 magrittr_2.0.1 whisker_0.4 scales_1.1.1
[41] promises_1.2.0.1 ellipsis_0.3.2 htmltools_0.5.1.1 assertthat_0.2.1
[45] colorspace_2.0-0 httpuv_1.5.5 labeling_0.4.2 utf8_1.1.4
[49] stringi_1.5.3 munsell_0.5.0 crayon_1.4.1