cTWAS_analysis_for_LDL
Jing Gu
2023-08-16
Last updated: 2023-08-16
Checks: 6 1
Knit directory: m6A_in_disease_genetics/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230331) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| ~/projects/m6A_in_disease_genetics/code/ctwas/ctwas_config_b37.R | code/ctwas/ctwas_config_b37.R |
| ~/projects/m6A_in_disease_genetics/code/ctwas/qiansheng/locus_plot.R | code/ctwas/qiansheng/locus_plot.R |
| ~/projects/m6A_in_disease_genetics/data/zhao_silver_genes.csv | data/zhao_silver_genes.csv |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 08eaf44. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .ipynb_checkpoints/
Ignored: analysis/m6A_switch_to_disease_h2g.nb.html
Ignored: data/plots/
Untracked files:
Untracked: HMGCR_locus_gene_tracks.pdf
Untracked: Rplots.pdf
Untracked: analysis/.ipynb_checkpoints/
Untracked: analysis/IBD_E_S_m6A.Rmd
Untracked: analysis/IBD_E_S_m6A_output.Rmd
Untracked: analysis/LDL_E_S_m6A.Rmd
Untracked: analysis/LDL_m6A_output.Rmd
Untracked: analysis/RA_m6A_output.Rmd
Untracked: analysis/WhiteBlood_WholeBlood_E_M.Rmd
Untracked: analysis/identify_m6A_mechanisms_with_finemapping.Rmd
Untracked: analysis/lymph_m6A_output.Rmd
Untracked: analysis/pre_weights_m6AQTL.txt
Untracked: analysis/rbc_E_S_m6A_output.Rmd
Untracked: analysis/rbc_m6A_output.Rmd
Untracked: analysis/rbc_m6A_output_hg19.Rmd
Untracked: analysis/wbc_E_S_m6A_output.Rmd
Untracked: code/.ipynb_checkpoints/
Untracked: code/all_m6a_sites_with_paired_cisNATs_summary.csv
Untracked: code/check_double_strand.ipynb
Untracked: code/check_double_strand_v2.ipynb
Untracked: code/ctwas/
Untracked: code/figure/
Untracked: code/learn_gviz.Rmd
Untracked: code/learn_gviz.html
Untracked: code/learn_gviz.nb.html
Untracked: code/m6AQTL_finemapping.Rmd
Untracked: code/summary_TWAS_coloc_m6A_2023.Rmd
Untracked: code/test_gviz.ipynb
Untracked: code/twas_genes_PP4_0.3_immune_traits_trackplots.pdf
Untracked: data/.ipynb_checkpoints/
Untracked: data/ADCY7_gwas_input.tsv
Untracked: data/ADCY7_qtl_input.tsv
Untracked: data/Allergy_full_coloc.txt
Untracked: data/Asthma_full_coloc.txt
Untracked: data/CAD_full_coloc.txt
Untracked: data/Eosinophil_count_full_coloc.txt
Untracked: data/GSE125377_jointPeakReadCount.txt
Untracked: data/HMGCR_ctwas_dat.Rd
Untracked: data/IBD_full_coloc.txt
Untracked: data/JointPeaks.bed
Untracked: data/Li2022_dsRNAs.xlsx
Untracked: data/Lupus_full_coloc.txt
Untracked: data/RA_full_coloc.txt
Untracked: data/TABLE1_hg19.txt
Untracked: data/TABLE1_hg19.txt.zip
Untracked: data/__MACOSX/
Untracked: data/coloc_blood_traits.csv
Untracked: data/crohns_disease_full_coloc.txt
Untracked: data/edit_sites_and_GE_neg_correlated.txt
Untracked: data/edit_sites_and_GE_pos_correlated.txt
Untracked: data/features
Untracked: data/human_EERs.csv
Untracked: data/human_EERs.txt
Untracked: data/lymph_full_coloc.txt
Untracked: data/m6A_TWAS_results.csv
Untracked: data/m6a_TWAS_genes.txt
Untracked: data/m6a_joint_calling_peaks.csv
Untracked: data/nasser_2021_ABC_IBD_genes.txt
Untracked: data/nat_sense_pairs.csv
Untracked: data/plt_full_coloc.txt
Untracked: data/rbc_full_coloc.txt
Untracked: data/rdw_full_coloc.txt
Untracked: data/reported_AS_targets_S1.txt
Untracked: data/reported_AS_wanowska.txt
Untracked: data/sig_coloc_results/
Untracked: data/test_locuscomparer.pdf
Untracked: data/ulcerative_colitis_full_coloc.txt
Untracked: data/wbc_full_coloc.txt
Untracked: data/zhao_silver_genes.csv
Untracked: output/.ipynb_checkpoints/
Untracked: output/HMGCR_gene_track_plot.pdf
Untracked: output/HMGCR_locus_plot.pdf
Untracked: output/all_m6a_sites_with_cisNATs.csv
Untracked: output/all_m6a_sites_with_paired_cisNATs_summary.csv
Untracked: output/all_m6a_sites_with_paired_cisNATs_summary_PP40.3.csv
Untracked: output/all_m6a_sites_with_paired_cisNATs_summary_PP40.5.csv
Untracked: output/all_m6a_sites_with_paired_cis_NATs.csv
Untracked: output/fine_mapped_m6AQTLs_TWAS_genes_highPP4.rds
Untracked: output/gene_summary.csv
Untracked: output/immune_related_m6A_targets.csv
Untracked: output/m6aQTL_dsRNAs_PPP2R3C_PRORP.pdf
Untracked: output/m6a_peaks_nearby_dsRNAs.csv
Untracked: output/m6a_sites_near_all_dsRNAs_twas.csv
Untracked: output/m6a_sites_near_dsRNAs_coloc.csv
Untracked: output/m6a_sites_near_dsRNAs_twas.csv
Untracked: output/m6a_sites_near_dsRNAs_twas_summary.csv
Untracked: output/m6a_sites_overlapping_NAT_twas.csv
Untracked: output/m6a_sites_overlapping_dsRNAs_coloc.csv
Untracked: output/m6a_sites_overlapping_dsRNAs_twas.csv
Untracked: output/m6a_sites_overlapping_dsRegions.csv
Untracked: output/m6a_sites_overlapping_dsRegions_coloc.csv
Untracked: output/negatively_correlated_genes.txt
Untracked: output/postively_correlated_genes.txt
Untracked: output/rs1806261_RABEP1-NUP88_focused_locusview.pdf
Untracked: output/rs1806261_RABEP1-NUP88_locusview.pdf
Untracked: output/rs3177647_MAPKAPK5-AS1-MAPKAPK5_locusview.pdf
Untracked: output/rs3204541_DDX55-EIF2B1_locusview.pdf
Untracked: output/rs7184802_ADCY7-BRD7_locusview.pdf
Untracked: output/rs7184802_ADCY7_locuscompare.pdf
Untracked: output/twas_genes_PP4_0.3_immune_traits_trackplots.pdf
Untracked: output/twas_genes_PP4_0.5_blood_traits_trackplots.pdf
Untracked: output/twas_m6a_sites_with_all_cisNATs.RDS
Untracked: output/twas_m6a_sites_with_cisNATs_range.RDS
Untracked: output/twas_m6a_sites_with_the_nearest_cisNAT.RDS
Untracked: twas_genes_PP4_0.3_immune_traits_trackplots.pdf
Unstaged changes:
Modified: analysis/m6A_switch_to_disease_h2g.Rmd
Modified: analysis/wbc_m6A_output.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/LDL_m6A_output_hg19.Rmd)
and HTML (docs/LDL_m6A_output_hg19.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 08eaf44 | Jing Gu | 2023-08-16 | run ctwas for multiple traits |
Load ctwas results
# top 1 method
res <- impute_expr_z(z_snp, weight = weight, ld_R_dir = ld_R_dir,
method = NULL, outputdir = outputdir, outname = outname.e,
harmonize_z = T, harmonize_wgt = T, scale_by_ld_variance=F,
strand_ambig_action_z = "recover",
recover_strand_ambig_wgt = T
# lasso/elastic-net method
res <- impute_expr_z(z_snp, weight = weight, ld_R_dir = ld_R_dir,
method = NULL, outputdir = outputdir, outname = outname.e,
harmonize_z = T, harmonize_wgt = T, scale_by_ld_variance=F,
strand_ambig_action_z = "none",
recover_strand_ambig_wgt = FGWAS: UK Biobank GWAS summary statistics - European individuals
Weights: FUSION weights using top1, lasso, or elastic-net models were converted into PredictDB format and were not needed to do scaling when running ctwas.
Check convergence of parameters
cTWAS analysis on m6A alone
[1] "Check convergence for the top1 model:"
[1] "Table of group size:"
SNP gene
8713250 888
SNP gene
estimated_group_prior 7.333e-05 0.050687
estimated_group_prior_var 5.562e+01 7.859621
estimated_group_pve 1.034e-01 0.001030
attributable_group_pve 9.901e-01 0.009857$top1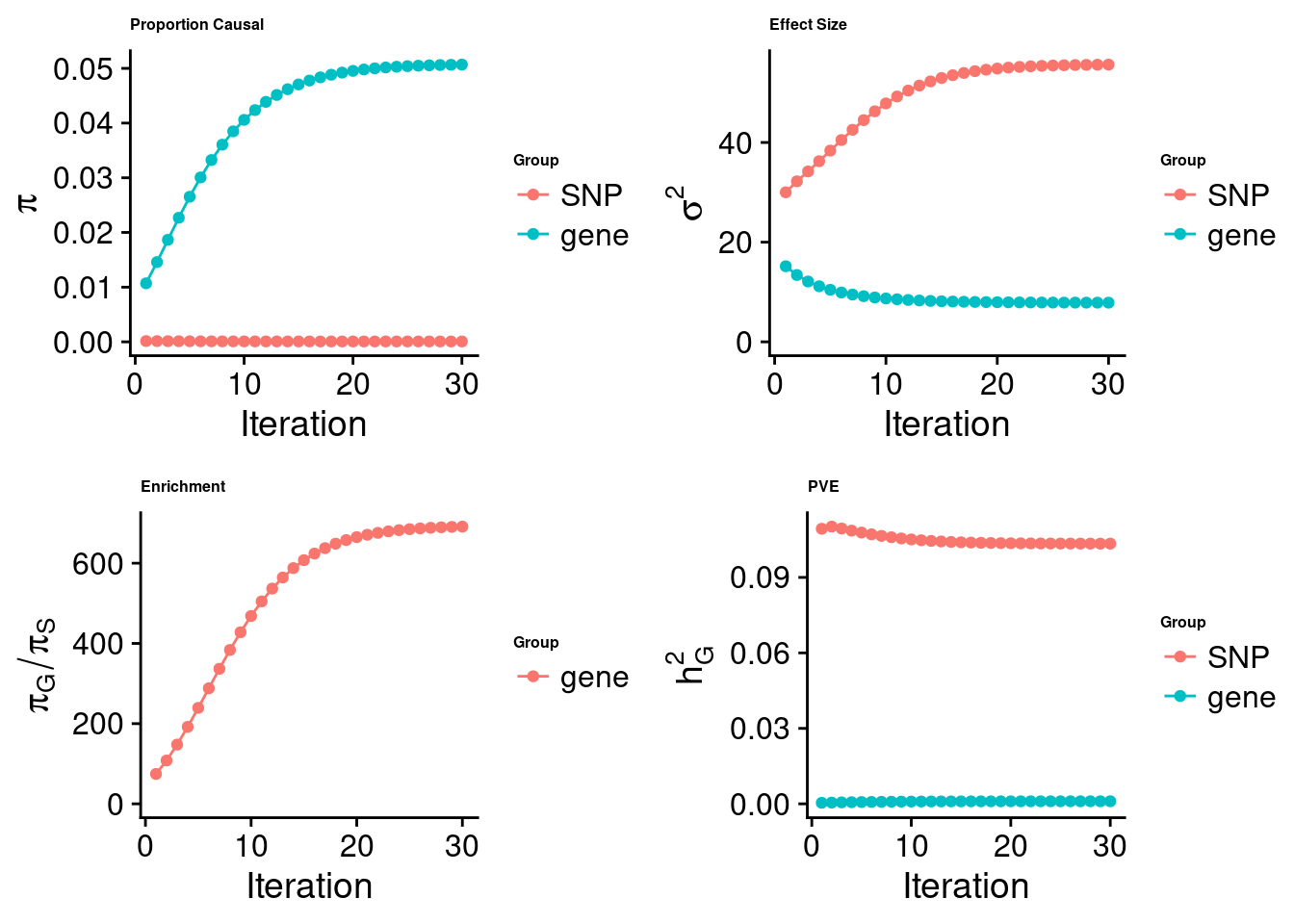
Joint analysis of expression, splicing and m6A
[1] "Check convergence for the top1 model when jointly analyzing expression, splicing and m6A:"
[1] "Table of group size before/after matching with UKBB SNPs:"
SNP eQTL sQTL m6AQTL
prior_group_size 9.324e+06 2005.0000 2191.000 918.0000
group_size 8.713e+06 1928.0000 2123.000 888.0000
percent_of_overlaps 9.345e-01 0.9616 0.969 0.9673
SNP eQTL sQTL m6AQTL
estimated_group_prior 5.872e-05 0.021347 0.03260 0.0290148
estimated_group_prior_var 6.634e+01 9.304196 6.55138 9.1182195
estimated_group_pve 9.878e-02 0.001114 0.00132 0.0006837
attributable_group_pve 9.694e-01 0.010937 0.01295 0.0067097
[1] "Check convergence for the lasso model when jointly analyzing expression, splicing and m6A:"
[1] "Table of group size before/after matching with UKBB SNPs:"
SNP eQTL sQTL m6AQTL
prior_group_size 9.324e+06 2005.0000 2191.000 918.0000
group_size 8.713e+06 1998.0000 2180.000 912.0000
percent_of_overlaps 9.345e-01 0.9965 0.995 0.9935
SNP eQTL sQTL m6AQTL
estimated_group_prior 9.786e-05 0.0116409 0.0239019 1.470e-02
estimated_group_prior_var 2.182e+01 7.4765802 5.1365664 1.663e+01
estimated_group_pve 5.414e-02 0.0005061 0.0007789 6.489e-04
attributable_group_pve 9.655e-01 0.0090243 0.0138897 1.157e-02$top1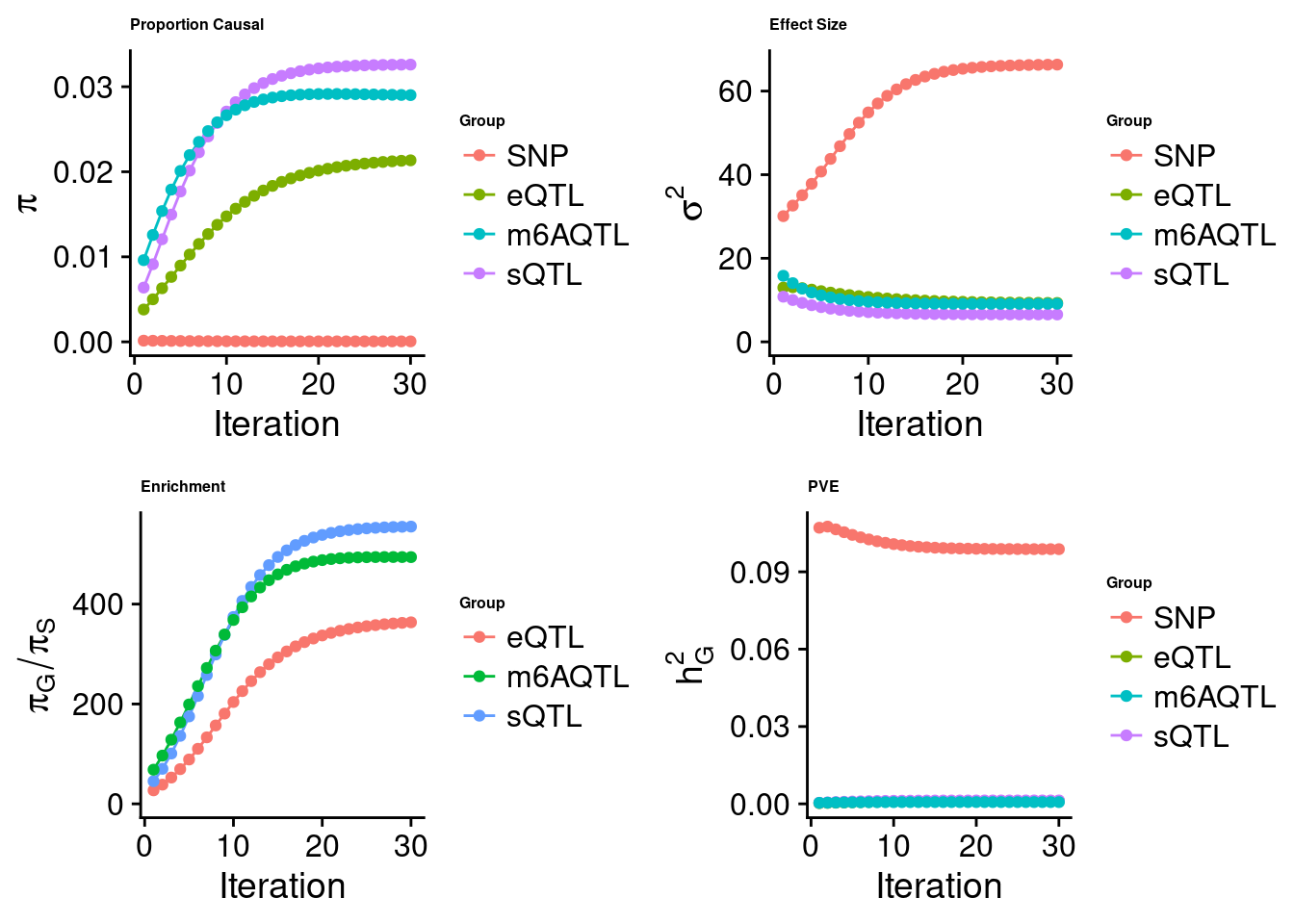
$lasso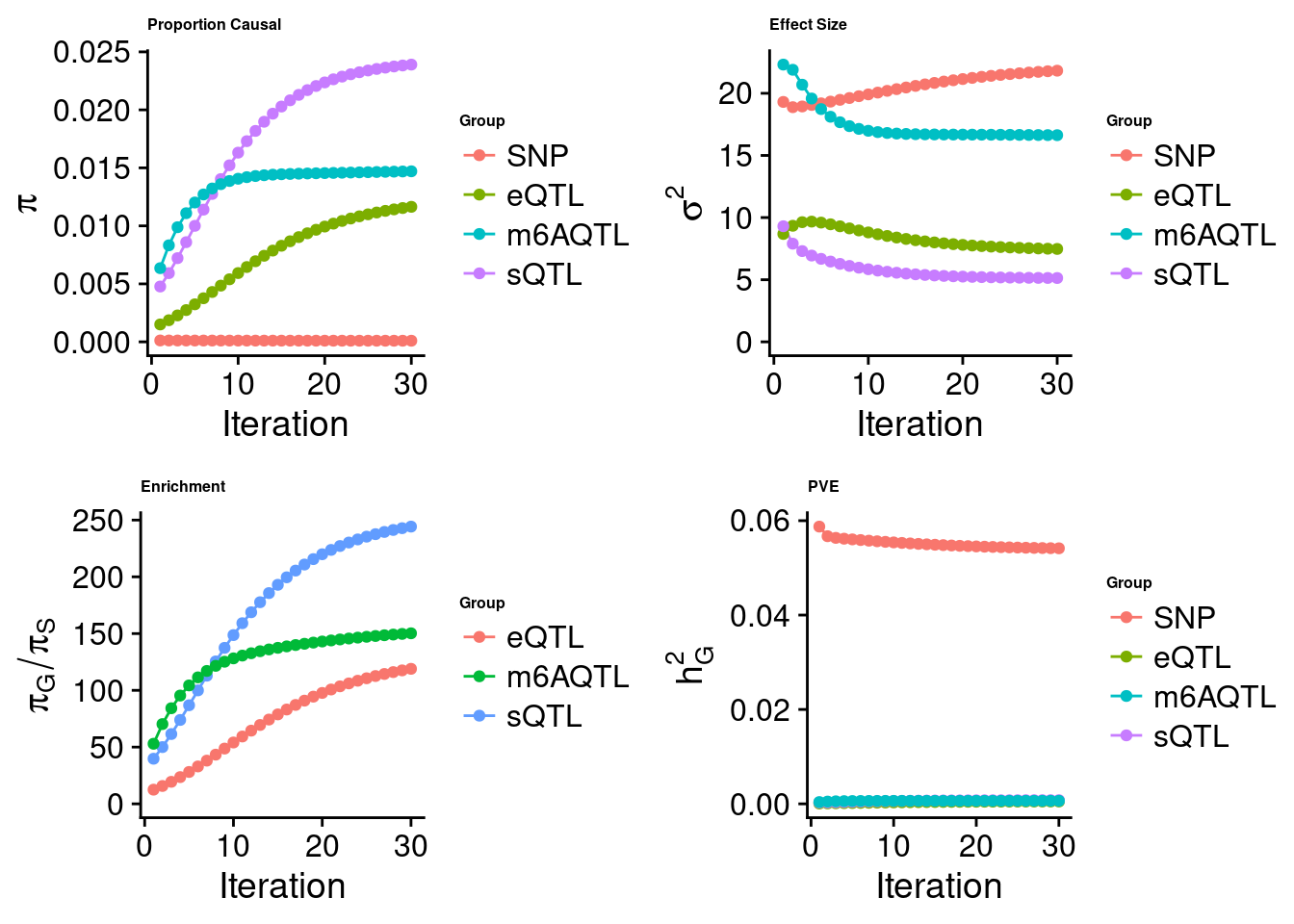
cTWAS results for individual analysis with m6A
top1 model
genename region_tag susie_pip z
1 PMPCA 9_73 0.9816 6.364
2 PGAM5 12_82 0.9385 4.457
3 TAF6L 11_35 0.9366 4.016
4 C7orf50 7_2 0.9165 -4.260
5 RABEP1 17_5 0.8514 -3.553
6 TMEM199 17_17 0.8416 -6.098
7 MYL12B 18_3 0.8288 3.524
8 HERC1 15_29 0.7916 -2.865
9 TCTN3 10_61 0.7764 3.980
10 MVK 12_66 0.7549 -3.789
11 ICOSLG 21_22 0.7456 -3.467
12 VKORC1 16_24 0.7376 -3.587
13 POLD4 11_37 0.7330 -3.379
14 DIABLO 12_75 0.6965 3.143
15 HMGCR 5_44 0.6849 -19.989Summing up PIPs for m6A peaks located in the same gene
Top m6A PIPs by genes
# A tibble: 22 × 2
genename total_susie_pip
<chr> <dbl>
1 ICOSLG 1.06
2 PMPCA 0.982
3 RABEP1 0.969
4 PGAM5 0.938
5 TAF6L 0.937
6 C7orf50 0.916
7 TMEM199 0.842
8 MYL12B 0.829
9 HERC1 0.792
10 TCTN3 0.776
# ℹ 12 more rowscTWAS results for joint analysis using a lasso model
Top m6A modification pip
Top expression/splicing/m6A units
For m6A or splicing QTLs, they are assigned to the nearest genes (m6A needs to be confirmed with Kevin).
Top SNPs or genes with PIP > 0.6
$eQTL
genename susie_pip group region_tag
1987 ABHD8 0.9777 eQTL 19_14
1966 TRIM5 0.9565 eQTL 11_4
1960 SPRED2 0.9248 eQTL 2_42
1832 PCMTD2 0.6698 eQTL 20_38
911 CDK9 0.6393 eQTL 9_66
$m6AQTL
genename susie_pip group region_tag
5082 TMEM199 0.9698 m6AQTL 17_18
5078 PMPCA 0.9584 m6AQTL 9_73
5073 TAP2 0.7896 m6AQTL 6_27
4702 PGAM5 0.7499 m6AQTL 12_82
4905 MYL12B 0.7133 m6AQTL 18_3
$sQTL
genename susie_pip group region_tag
4176 ABHD12 0.9996 sQTL 20_19
4135 SCAMP3 0.8896 sQTL 1_79
2453 GSK3B 0.7947 sQTL 3_74
4152 RAF1 0.7858 sQTL 3_9
3109 ENTPD1 0.6742 sQTL 10_61
2932 BLK 0.6438 sQTL 8_15
3082 ANAPC16 0.6368 sQTL 10_49Top m6A modification pip
genename region_tag susie_pip z
1 TMEM199 17_18 0.9698 -6.649
2 PMPCA 9_73 0.9584 6.350
3 TAP2 6_27 0.7896 -8.346
4 PGAM5 12_82 0.7499 4.457
5 MYL12B 18_3 0.7133 3.653
6 POLD4 11_37 0.5925 -4.179
7 C7orf50 7_2 0.5123 -4.212
8 HERC1 15_29 0.5029 -3.818
9 TAF6L 11_35 0.4885 4.021
10 FGD3 9_47 0.2726 -3.213Summing up PIPs for m6A peaks located in the same gene
Top 10 m6A PIPs by genes
# A tibble: 819 × 2
genename total_susie_pip
<chr> <dbl>
1 TMEM199 0.970
2 PMPCA 0.958
3 TAP2 0.790
4 PGAM5 0.750
5 MYL12B 0.713
6 POLD4 0.593
7 C7orf50 0.512
8 HERC1 0.503
9 TAF6L 0.488
10 DIDO1 0.283
# ℹ 809 more rowsTop splicing PIPs
peak_id genename pos region_tag susie_pip z
1 chr20:25275666-25282855 ABHD12 25260931 20_19 0.9996 6.644
2 chr1:155230450-155231448 SCAMP3 155149718 1_79 0.8896 4.305
3 chr3:119582452-119624602 GSK3B 119503971 3_74 0.7947 -5.789
4 chr3:12650834-12660014 RAF1 12574512 3_9 0.7858 -5.628
5 chr10:97602251-97602973 ENTPD1 97507473 10_61 0.6742 -4.590
6 chr8:11397080-11400733 BLK 11368731 8_15 0.6438 4.373
7 chr10:73980137-73983646 ANAPC16 73949708 10_49 0.6368 4.415
8 chr2:85823772-85824227 RNF181 85818886 2_54 0.5906 3.595
9 chr6:29691304-29691460 HLA-F 29644502 6_23 0.5565 5.293
10 chr12:53856351-53859716 PCBP2 53770941 12_33 0.5476 4.063Summing up PIPs for spliced introns located in the same gene
Top 10 splicing PIPs by genes
# A tibble: 10 × 2
genename total_susie_pip
<chr> <dbl>
1 RMDN1 1.31
2 LBP 1.16
3 WARS1 1.13
4 SCAMP3 1.10
5 ANAPC16 1.07
6 ERGIC3 1.07
7 HLA-F 1.05
8 IFI44L 1.02
9 CCT7 1.01
10 ABHD12 1.00Top genes by combined PIP
genename combined_pip expression_pip splicing_pip m6A_pip region_tag
2503 RMDN1 1.314 0.00000 1.31366 0.00000 8_62
1505 HLA-F 1.201 0.05645 1.05500 0.08978 6_23
1680 LBP 1.162 0.00000 1.16237 0.00000 20_23
3241 WARS1 1.160 0.03006 1.13042 0.00000 14_52
2623 SCAMP3 1.129 0.00000 1.10088 0.02771 1_79
149 ANAPC16 1.072 0.00000 1.07160 0.00000 10_49
1251 ERGIC3 1.066 0.00000 1.06575 0.00000 20_21
1552 IFI44L 1.053 0.00000 1.01540 0.03795 1_48
3095 TRIM5 1.043 0.95648 0.08641 0.00000 11_4
454 CCT7 1.010 0.00000 1.00965 0.00000 2_48
14 ABHD12 1.000 0.00000 0.99960 0.00000 20_19
18 ABHD8 0.978 0.97771 0.00000 0.00000 19_14
3010 TMEM199 0.970 0.00000 0.00000 0.96979 17_18
2259 PMPCA 0.958 0.00000 0.00000 0.95843 9_73
1572 IMMP1L 0.940 0.00000 0.94015 0.00000 11_21
1790 MCOLN2 0.933 0.03269 0.90027 0.00000 1_52
2815 SPRED2 0.925 0.92476 0.00000 0.00000 2_42
1928 MTERF4 0.909 0.03773 0.67259 0.19843 2_144
2809 SPG7 0.889 0.00000 0.81819 0.07106 16_54
1233 ENTPD1 0.886 0.02109 0.86474 0.00000 10_61
638 CTSH 0.858 0.18503 0.60610 0.06653 15_37
1862 MMAB 0.857 0.01508 0.84150 0.00000 12_67
1968 NADSYN1 0.855 0.13755 0.71720 0.00000 11_40
2942 TDP1 0.846 0.06640 0.77954 0.00000 14_45
310 BLK 0.825 0.00000 0.82520 0.00000 8_15
508 CENPU 0.795 0.09120 0.70348 0.00000 4_119
1439 GSK3B 0.795 0.00000 0.79472 0.00000 3_74
2910 TAP2 0.790 0.00000 0.00000 0.78964 6_27
2428 RAF1 0.786 0.00000 0.78581 0.00000 3_9
2166 PCBP2 0.776 0.00000 0.77585 0.00000 12_33
2801 SP140 0.774 0.00000 0.77396 0.00000 2_135
1689 LGALS8 0.763 0.00000 0.68077 0.08214 1_124
2560 RPL8 0.763 0.04376 0.71959 0.00000 8_94
3260 WDR91 0.756 0.02902 0.55332 0.17384 7_82
2201 PGAM5 0.750 0.00000 0.00000 0.74988 12_82
3073 TRAF1 0.733 0.00000 0.73315 0.00000 9_63
2889 SYNCRIP 0.716 0.00000 0.71582 0.00000 6_58
1953 MYL12B 0.713 0.00000 0.00000 0.71328 18_3
207 ARIH2 0.700 0.00000 0.63860 0.06122 3_35
471 CD46 0.700 0.00000 0.70025 0.00000 1_107Loading required package: gridWarning: replacing previous import 'utils::download.file' by
'restfulr::download.file' when loading 'rtracklayer'Compared with the results from Zhao et al.
[1] "Table of combined PIPs for LCL silver standard genes:" genename combined_pip expression_pip splicing_pip m6A_pip region_tag
1 HMGCR 0.216 0.00000 0.0000 0.21569 5_44
2 VDAC1 0.174 0.00000 0.1738 0.00000 5_80
3 CETP 0.145 0.14520 0.0000 0.00000 16_31
4 DHCR7 0.129 0.00000 0.0000 0.12937 11_40
5 VAPA 0.087 0.00000 0.0868 0.00000 18_7
6 PLTP 0.082 0.08167 0.0000 0.00000 20_28
7 VAPB 0.061 0.06068 0.0000 0.00000 20_34
8 STARD3 0.054 0.05385 0.0000 0.00000 17_23
9 TNKS 0.035 0.02106 0.0000 0.01417 8_12
10 LIPA 0.030 0.02955 0.0000 0.00000 10_57
11 EPHX2 0.029 0.02887 0.0000 0.00000 8_27
12 ITIH4 0.027 0.02655 0.0000 0.00000 3_36
13 LDLR 0.000 0.00000 0.0000 0.00000 19_10
annotation
1 known
2 known
3 known
4 known
5 known
6 known
7 known
8 known
9 known
10 known
11 known
12 known
13 known[1] "Table of combined PIPs for LCL bystander genes:" genename combined_pip expression_pip splicing_pip m6A_pip region_tag
1 NADSYN1 0.855 0.13755 0.7172 0.00000 11_40
2 RAF1 0.786 0.00000 0.7858 0.00000 3_9
3 ACOT8 0.493 0.01311 0.4801 0.00000 20_28
4 ITGB3BP 0.469 0.03102 0.4381 0.00000 1_40
5 GNL3 0.435 0.00000 0.3376 0.09772 3_36
6 SMUG1 0.420 0.00000 0.2614 0.15811 12_33
7 NT5DC2 0.407 0.02316 0.3837 0.00000 3_36
8 YWHAB 0.327 0.07149 0.2553 0.00000 20_28
9 DNPEP 0.313 0.00000 0.3126 0.00000 2_129
10 NUMA1 0.306 0.00000 0.3062 0.00000 11_40
annotation
1 bystander
2 bystander
3 bystander
4 bystander
5 bystander
6 bystander
7 bystander
8 bystander
9 bystander
10 bystander[1] "Overlaps with previously identified high PIP genes that are either silver standard or bystander genes:" genename combined_pip expression_pip splicing_pip m6A_pip region_tag
1 USP1 0.094 0.02297 0.0712 0.00000 1_39
2 PLTP 0.082 0.08167 0.0000 0.00000 20_28
3 TNKS 0.035 0.02106 0.0000 0.01417 8_12
annotation
1 bystander
2 known
3 knownLocus plots for specific examples
genename combined_pip expression_pip splicing_pip m6A_pip region_tag
412 HMGCR 0.216 0 0 0.2157 5_44
annotation
412 known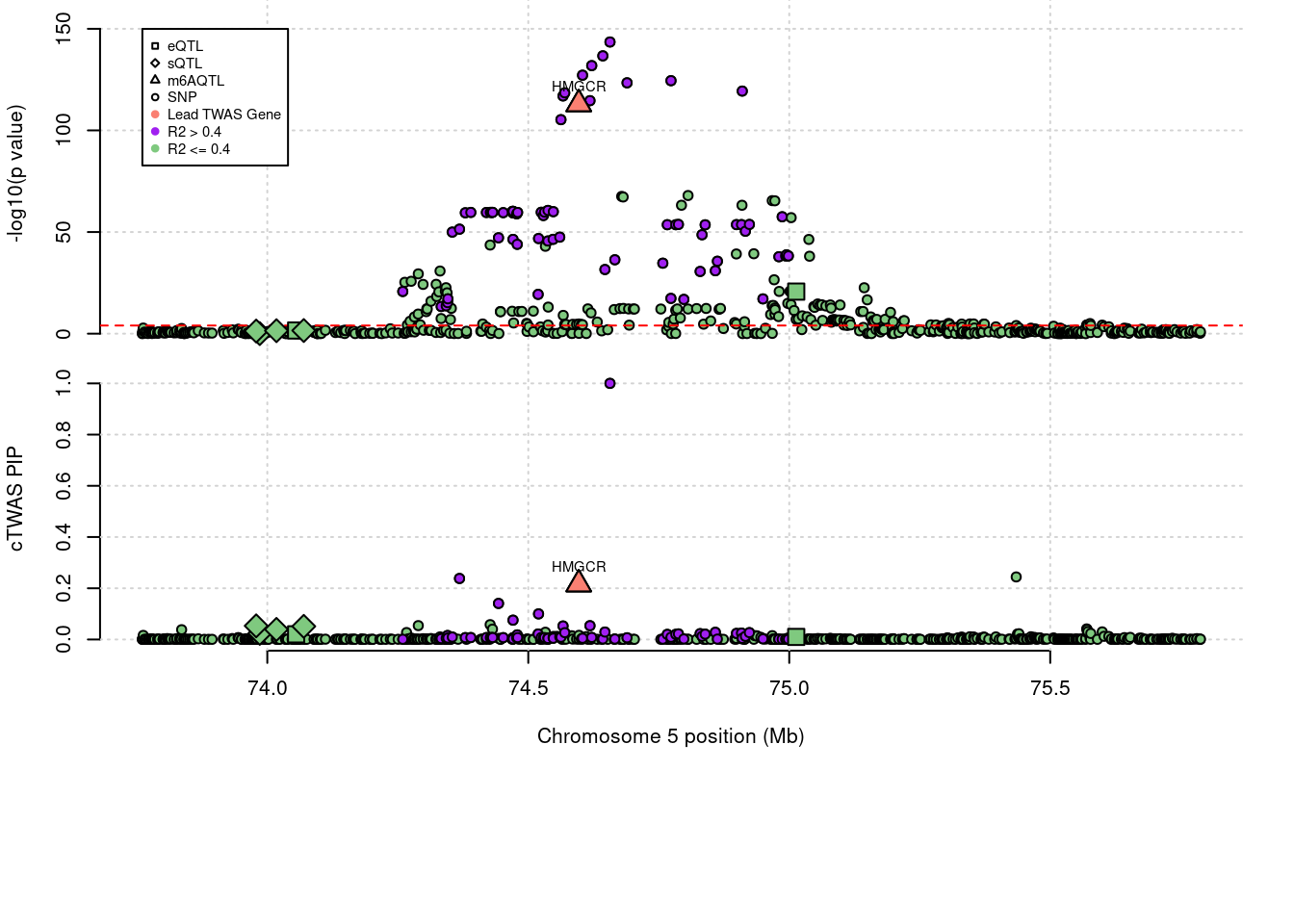
R version 4.2.0 (2022-04-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.3.13-el7-x86_64/lib/libopenblas_haswellp-r0.3.13.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=C
[4] LC_COLLATE=C LC_MONETARY=C LC_MESSAGES=C
[7] LC_PAPER=C LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=C LC_IDENTIFICATION=C
attached base packages:
[1] grid stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] biomaRt_2.52.0 Gviz_1.40.1 cowplot_1.1.1
[4] ggplot2_3.4.3 GenomicRanges_1.48.0 GenomeInfoDb_1.32.2
[7] IRanges_2.30.1 S4Vectors_0.34.0 BiocGenerics_0.42.0
[10] ctwas_0.1.38 dplyr_1.1.2 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] colorspace_2.1-0 deldir_1.0-6
[3] rjson_0.2.21 rprojroot_2.0.3
[5] biovizBase_1.44.0 htmlTable_2.4.0
[7] XVector_0.36.0 base64enc_0.1-3
[9] fs_1.6.3 dichromat_2.0-0.1
[11] rstudioapi_0.15.0 farver_2.1.1
[13] bit64_4.0.5 AnnotationDbi_1.58.0
[15] fansi_1.0.4 xml2_1.3.3
[17] codetools_0.2-18 logging_0.10-108
[19] cachem_1.0.8 knitr_1.39
[21] Formula_1.2-4 jsonlite_1.8.7
[23] Rsamtools_2.12.0 cluster_2.1.3
[25] dbplyr_2.3.3 png_0.1-7
[27] compiler_4.2.0 httr_1.4.6
[29] backports_1.4.1 lazyeval_0.2.2
[31] Matrix_1.6-1 fastmap_1.1.1
[33] cli_3.6.1 later_1.3.0
[35] htmltools_0.5.2 prettyunits_1.1.1
[37] tools_4.2.0 gtable_0.3.3
[39] glue_1.6.2 GenomeInfoDbData_1.2.8
[41] rappdirs_0.3.3 Rcpp_1.0.11
[43] Biobase_2.56.0 jquerylib_0.1.4
[45] vctrs_0.6.3 Biostrings_2.64.0
[47] rtracklayer_1.56.0 iterators_1.0.14
[49] xfun_0.30 stringr_1.5.0
[51] ps_1.7.0 lifecycle_1.0.3
[53] ensembldb_2.20.2 restfulr_0.0.14
[55] XML_3.99-0.14 getPass_0.2-2
[57] zlibbioc_1.42.0 scales_1.2.1
[59] BSgenome_1.64.0 VariantAnnotation_1.42.1
[61] ProtGenerics_1.28.0 hms_1.1.3
[63] promises_1.2.0.1 MatrixGenerics_1.8.0
[65] parallel_4.2.0 SummarizedExperiment_1.26.1
[67] AnnotationFilter_1.20.0 RColorBrewer_1.1-3
[69] yaml_2.3.5 curl_5.0.2
[71] memoise_2.0.1 gridExtra_2.3
[73] sass_0.4.1 rpart_4.1.16
[75] latticeExtra_0.6-30 stringi_1.7.12
[77] RSQLite_2.3.1 highr_0.9
[79] BiocIO_1.6.0 foreach_1.5.2
[81] checkmate_2.1.0 GenomicFeatures_1.48.4
[83] filelock_1.0.2 BiocParallel_1.30.3
[85] rlang_1.1.1 pkgconfig_2.0.3
[87] matrixStats_0.62.0 bitops_1.0-7
[89] evaluate_0.15 lattice_0.20-45
[91] htmlwidgets_1.5.4 GenomicAlignments_1.32.0
[93] labeling_0.4.2 bit_4.0.5
[95] processx_3.8.0 tidyselect_1.2.0
[97] magrittr_2.0.3 R6_2.5.1
[99] generics_0.1.3 Hmisc_5.1-0
[101] DelayedArray_0.22.0 DBI_1.1.3
[103] pgenlibr_0.3.6 pillar_1.9.0
[105] whisker_0.4 foreign_0.8-82
[107] withr_2.5.0 KEGGREST_1.36.2
[109] RCurl_1.98-1.7 nnet_7.3-17
[111] tibble_3.2.1 crayon_1.5.2
[113] interp_1.1-4 utf8_1.2.3
[115] BiocFileCache_2.4.0 rmarkdown_2.14
[117] jpeg_0.1-10 progress_1.2.2
[119] data.table_1.14.8 blob_1.2.4
[121] callr_3.7.3 git2r_0.30.1
[123] digest_0.6.33 httpuv_1.6.5
[125] munsell_0.5.0 bslib_0.3.1