cTWAS_analysis_for_neutrophill_counts
Jing Gu
2023-08-22
Last updated: 2023-08-22
Checks: 6 1
Knit directory: m6A_in_disease_genetics/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230331) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| ~/projects/m6A_in_disease_genetics/code/ctwas/ctwas_config_b37.R | code/ctwas/ctwas_config_b37.R |
| ~/projects/m6A_in_disease_genetics/code/ctwas/qiansheng/locus_plot.R | code/ctwas/qiansheng/locus_plot.R |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 0560ec9. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .ipynb_checkpoints/
Ignored: analysis/figure/
Ignored: analysis/m6A_switch_to_disease_h2g.nb.html
Ignored: data/plots/
Untracked files:
Untracked: HMGCR_locus_gene_tracks.pdf
Untracked: Rplots.pdf
Untracked: analysis/.ipynb_checkpoints/
Untracked: analysis/IBD_E_S_m6A.Rmd
Untracked: analysis/IBD_E_S_m6A_output.Rmd
Untracked: analysis/LDL_E_S_m6A.Rmd
Untracked: analysis/LDL_m6A_output.Rmd
Untracked: analysis/RA_m6A_output.Rmd
Untracked: analysis/WhiteBlood_WholeBlood_E_M.Rmd
Untracked: analysis/identify_m6A_mechanisms_with_finemapping.Rmd
Untracked: analysis/lymph_m6A_output.Rmd
Untracked: analysis/pre_weights_m6AQTL.txt
Untracked: analysis/rbc_E_S_m6A_output.Rmd
Untracked: analysis/rbc_m6A_output.Rmd
Untracked: analysis/summarize_ctwas_m6A_results.Rmd
Untracked: analysis/wbc_E_S_m6A_output.Rmd
Untracked: code/.ipynb_checkpoints/
Untracked: code/all_m6a_sites_with_paired_cisNATs_summary.csv
Untracked: code/annotating_fine-mapped_m6A_QTLs.Rmd
Untracked: code/check_double_strand.ipynb
Untracked: code/check_double_strand_v2.ipynb
Untracked: code/ctwas/
Untracked: code/figure/
Untracked: code/learn_gviz.Rmd
Untracked: code/learn_gviz.html
Untracked: code/learn_gviz.nb.html
Untracked: code/m6AQTL_finemapping.Rmd
Untracked: code/plot_genomic_tracks_gviz.ipynb
Untracked: code/summary_TWAS_coloc_m6A_2023.Rmd
Untracked: code/test_gviz.ipynb
Untracked: code/twas_genes_PP4_0.3_immune_traits_trackplots.pdf
Untracked: data/.ipynb_checkpoints/
Untracked: data/ADCY7_gwas_input.tsv
Untracked: data/ADCY7_qtl_input.tsv
Untracked: data/Allergy_full_coloc.txt
Untracked: data/Asthma_full_coloc.txt
Untracked: data/CAD_full_coloc.txt
Untracked: data/Eosinophil_count_full_coloc.txt
Untracked: data/GSE125377_jointPeakReadCount.txt
Untracked: data/G_list.Rd
Untracked: data/HMGCR_ctwas_dat.Rd
Untracked: data/IBD_full_coloc.txt
Untracked: data/JointPeaks.bed
Untracked: data/Li2022_dsRNAs.xlsx
Untracked: data/Lupus_full_coloc.txt
Untracked: data/RA_full_coloc.txt
Untracked: data/TABLE1_hg19.txt
Untracked: data/TABLE1_hg19.txt.zip
Untracked: data/__MACOSX/
Untracked: data/coloc_blood_traits.csv
Untracked: data/crohns_disease_full_coloc.txt
Untracked: data/ctwas_m6a_joint_top_PIP.txt
Untracked: data/edit_sites_and_GE_neg_correlated.txt
Untracked: data/edit_sites_and_GE_pos_correlated.txt
Untracked: data/features
Untracked: data/human_EERs.csv
Untracked: data/human_EERs.txt
Untracked: data/lymph_full_coloc.txt
Untracked: data/m6A_TWAS_results.csv
Untracked: data/m6a_TWAS_genes.txt
Untracked: data/m6a_joint_calling_peaks.csv
Untracked: data/nasser_2021_ABC_IBD_genes.txt
Untracked: data/nat_sense_pairs.csv
Untracked: data/plt_full_coloc.txt
Untracked: data/rbc_full_coloc.txt
Untracked: data/rdw_full_coloc.txt
Untracked: data/reported_AS_targets_S1.txt
Untracked: data/reported_AS_wanowska.txt
Untracked: data/sig_coloc_results/
Untracked: data/test_locuscomparer.pdf
Untracked: data/ulcerative_colitis_full_coloc.txt
Untracked: data/wbc_full_coloc.txt
Untracked: data/zhao_silver_genes.csv
Untracked: output/.ipynb_checkpoints/
Untracked: output/HMGCR_gene_track_plot.pdf
Untracked: output/HMGCR_locus_plot.pdf
Untracked: output/IBD_DHX38_plot.pdf
Untracked: output/IBD_DHX38_plot_genetrack.pdf
Untracked: output/all_m6a_sites_with_cisNATs.csv
Untracked: output/all_m6a_sites_with_paired_cisNATs_summary.csv
Untracked: output/all_m6a_sites_with_paired_cisNATs_summary_PP40.3.csv
Untracked: output/all_m6a_sites_with_paired_cisNATs_summary_PP40.5.csv
Untracked: output/all_m6a_sites_with_paired_cis_NATs.csv
Untracked: output/fine_mapped_m6AQTLs_TWAS_genes_highPP4.rds
Untracked: output/gene_summary.csv
Untracked: output/immune_related_m6A_targets.csv
Untracked: output/lupus_MIR210HG_plot.pdf
Untracked: output/lupus_MIR210HG_plot_genetrack.pdf
Untracked: output/m6aQTL_dsRNAs_PPP2R3C_PRORP.pdf
Untracked: output/m6a_QTL_genes.csv
Untracked: output/m6a_genes_PIP_0.6_blood_immune.csv
Untracked: output/m6a_genes_PIP_0.6_blood_immune.txt
Untracked: output/m6a_peaks_nearby_dsRNAs.csv
Untracked: output/m6a_sites_near_all_dsRNAs_twas.csv
Untracked: output/m6a_sites_near_dsRNAs_coloc.csv
Untracked: output/m6a_sites_near_dsRNAs_twas.csv
Untracked: output/m6a_sites_near_dsRNAs_twas_summary.csv
Untracked: output/m6a_sites_overlapping_NAT_twas.csv
Untracked: output/m6a_sites_overlapping_dsRNAs_coloc.csv
Untracked: output/m6a_sites_overlapping_dsRNAs_twas.csv
Untracked: output/m6a_sites_overlapping_dsRegions.csv
Untracked: output/m6a_sites_overlapping_dsRegions_coloc.csv
Untracked: output/negatively_correlated_genes.txt
Untracked: output/postively_correlated_genes.txt
Untracked: output/rs1806261_RABEP1-NUP88_focused_locusview.pdf
Untracked: output/rs1806261_RABEP1-NUP88_locusview.pdf
Untracked: output/rs3177647_MAPKAPK5-AS1-MAPKAPK5_locusview.pdf
Untracked: output/rs3204541_DDX55-EIF2B1_locusview.pdf
Untracked: output/rs7184802_ADCY7-BRD7_locusview.pdf
Untracked: output/rs7184802_ADCY7_locuscompare.pdf
Untracked: output/twas_genes_PP4_0.3_immune_traits_trackplots.pdf
Untracked: output/twas_genes_PP4_0.5_blood_traits_trackplots.pdf
Untracked: output/twas_m6a_sites_with_all_cisNATs.RDS
Untracked: output/twas_m6a_sites_with_cisNATs_range.RDS
Untracked: output/twas_m6a_sites_with_the_nearest_cisNAT.RDS
Untracked: twas_genes_PP4_0.3_immune_traits_trackplots.pdf
Unstaged changes:
Deleted: analysis/learn_ctwas.Rmd
Modified: analysis/lymph_m6A_output_hg19.Rmd
Modified: analysis/m6A_switch_to_disease_h2g.Rmd
Modified: analysis/rbc_m6A_output_hg19.Rmd
Modified: analysis/wbc_m6A_output.Rmd
Modified: analysis/wbc_m6A_output_hg19.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown
(analysis/neutroph_m6A_output_hg19.Rmd) and HTML
(docs/neutroph_m6A_output_hg19.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 0560ec9 | Jing Gu | 2023-08-22 | analyzed neutrph |
Load ctwas results
# top 1 method
res <- impute_expr_z(z_snp, weight = weight, ld_R_dir = ld_R_dir,
method = NULL, outputdir = outputdir, outname = outname.e,
harmonize_z = T, harmonize_wgt = T, scale_by_ld_variance=F,
strand_ambig_action_z = "recover",
recover_strand_ambig_wgt = T
# lasso/elastic-net method
res <- impute_expr_z(z_snp, weight = weight, ld_R_dir = ld_R_dir,
method = NULL, outputdir = outputdir, outname = outname.e,
harmonize_z = T, harmonize_wgt = T, scale_by_ld_variance=F,
strand_ambig_action_z = "none",
recover_strand_ambig_wgt = FGWAS: UK Biobank GWAS summary statistics - European individuals
Weights: FUSION weights using top1, lasso, or elastic-net models were converted into PredictDB format and were not needed to do scaling when running ctwas.
Check convergence of parameters
cTWAS analysis on m6A alone
[1] "Check convergence for the top1 model:"
[1] "Table of group size:"
SNP gene
8713250 888
SNP gene
estimated_group_prior 2.387e-04 1.829e-02
estimated_group_prior_var 1.683e+01 1.718e+01
estimated_group_pve 1.000e-01 7.976e-04
attributable_group_pve 9.921e-01 7.909e-03$top1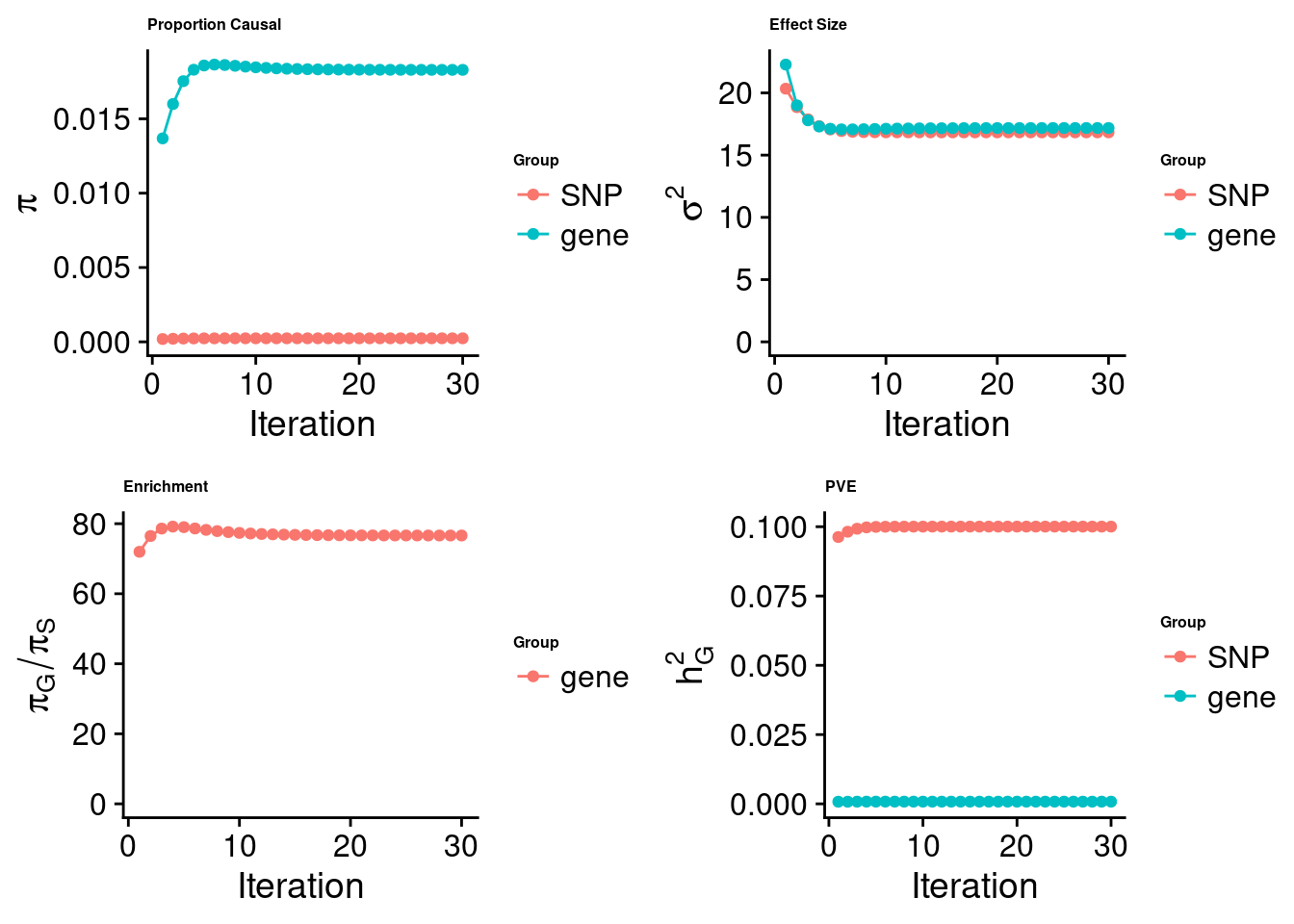
Joint analysis of expression, splicing and m6A
[1] "Check convergence for the top1 model when jointly analyzing expression, splicing and m6A:"
[1] "Table of group size before/after matching with UKBB SNPs:"
SNP eQTL sQTL m6AQTL
prior_group_size 9.324e+06 2005.0000 2191.000 918.0000
group_size 8.713e+06 1928.0000 2123.000 888.0000
percent_of_overlaps 9.345e-01 0.9616 0.969 0.9673
SNP eQTL sQTL m6AQTL
estimated_group_prior 0.000234 0.015405 0.008797 7.876e-04
estimated_group_prior_var 15.762762 16.480563 25.106901 1.010e+01
estimated_group_pve 0.091849 0.001399 0.001340 2.020e-05
attributable_group_pve 0.970832 0.014788 0.014167 2.135e-04
[1] "Check convergence for the lasso model when jointly analyzing expression, splicing and m6A:"
[1] "Table of group size before/after matching with UKBB SNPs:"
SNP eQTL sQTL m6AQTL
prior_group_size 9.324e+06 2005.0000 2191.000 918.0000
group_size 8.713e+06 1998.0000 2180.000 912.0000
percent_of_overlaps 9.345e-01 0.9965 0.995 0.9935
SNP eQTL sQTL m6AQTL
estimated_group_prior 2.067e-04 0.01459 1.479e-03 3.526e-04
estimated_group_prior_var 1.838e+01 13.68762 3.091e+01 1.039e+01
estimated_group_pve 9.463e-02 0.00114 2.848e-04 9.552e-06
attributable_group_pve 9.851e-01 0.01187 2.965e-03 9.943e-05$top1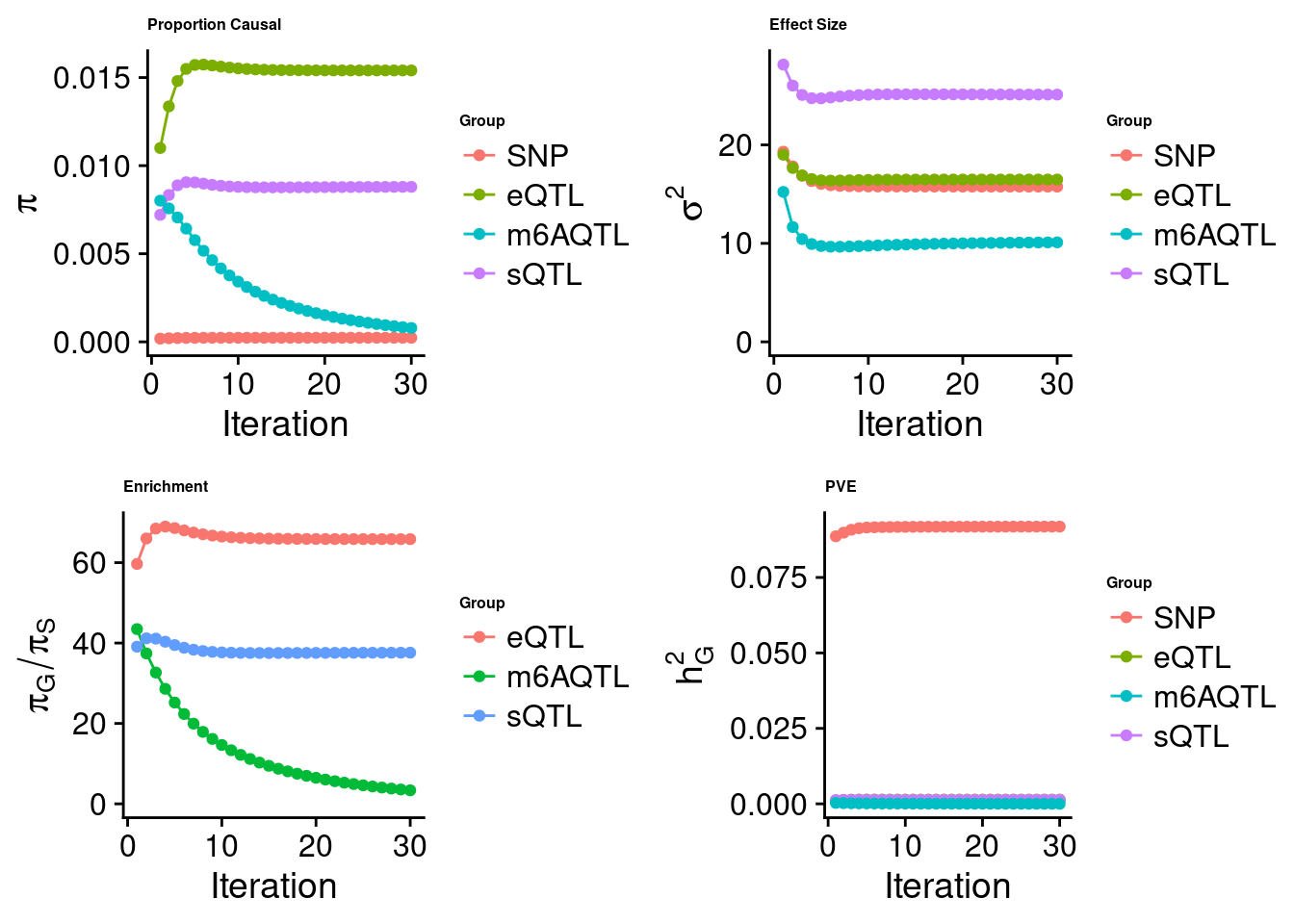
$lasso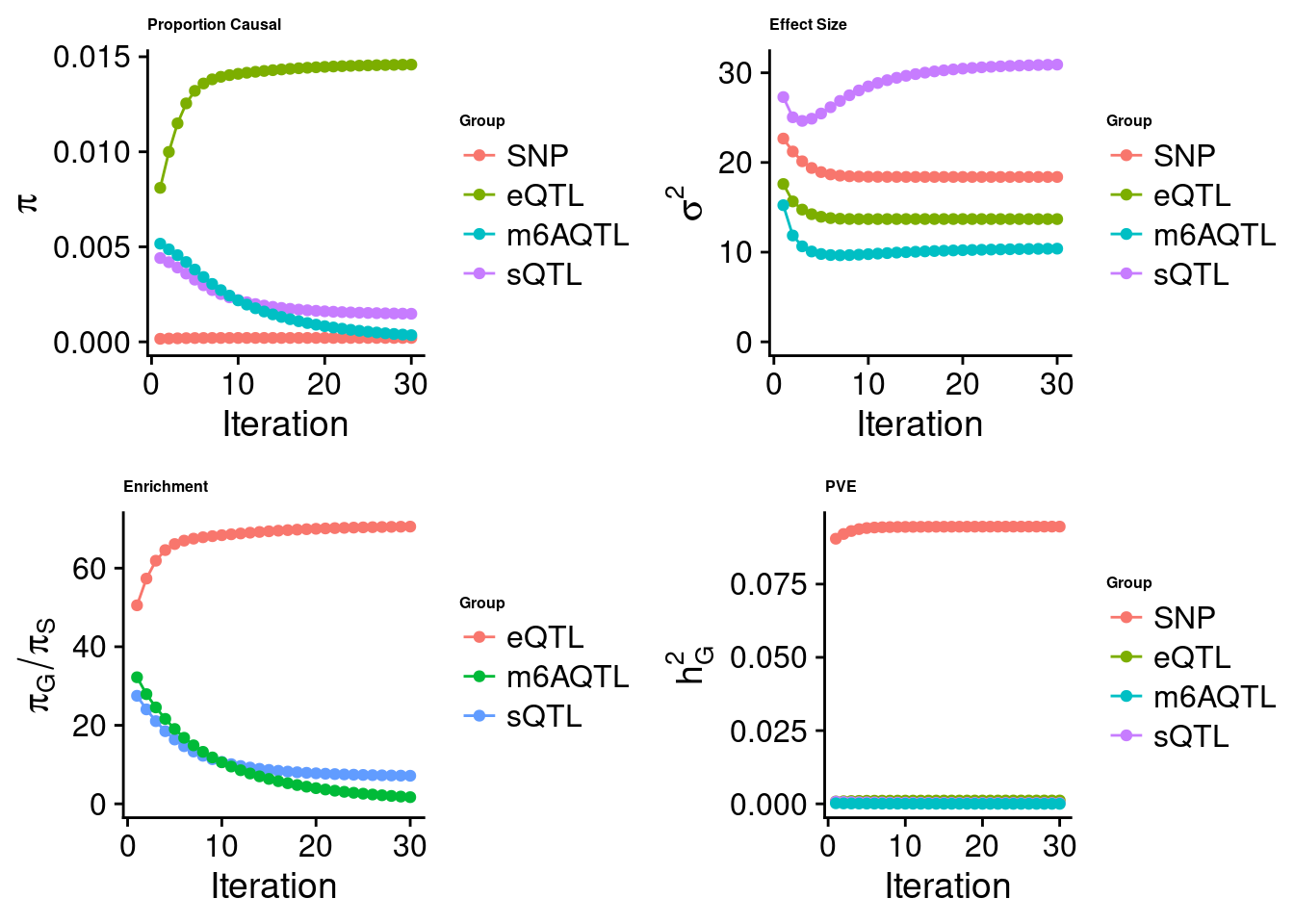
cTWAS results for individual analysis with m6A
top1 model
genename region_tag susie_pip z
1 SLC9A3R1 17_42 0.9664 -7.384
2 LETMD1 12_31 0.8989 -5.107
3 HMGN4 6_20 0.7958 3.861
4 ASCC1 10_48 0.7928 3.989
5 HNRNPK 9_41 0.7313 7.884
6 BANF1 11_36 0.7053 4.664
7 SQSTM1 5_108 0.6327 -5.743
8 THEMIS2 1_19 0.6237 3.913Summing up PIPs for m6A peaks located in the same gene
Top m6A PIPs by genes
# A tibble: 9 × 2
genename total_susie_pip
<chr> <dbl>
1 SLC9A3R1 0.966
2 LETMD1 0.899
3 HMGN4 0.796
4 ASCC1 0.793
5 HNRNPK 0.731
6 BANF1 0.705
7 SH2D3C 0.634
8 SQSTM1 0.633
9 THEMIS2 0.624cTWAS results for joint analysis using a lasso model
Top m6A modification pip
Top expression/splicing/m6A units
For m6A or splicing QTLs, they are assigned to the nearest genes (m6A needs to be confirmed with Kevin).
Top SNPs or genes with PIP > 0.6
$eQTL
genename susie_pip group region_tag
1956 CSNK1G1 0.9995 eQTL 15_29
1933 ZMIZ1 0.9909 eQTL 10_51
1993 TTLL12 0.9888 eQTL 22_18
1916 NDUFS2 0.9267 eQTL 1_81
1971 CCDC9 0.8776 eQTL 19_34
1923 MXD3 0.8298 eQTL 5_106
256 KYNU 0.7896 eQTL 2_85
1959 RAPGEFL1 0.7579 eQTL 17_23
970 BORCS7 0.7408 eQTL 10_66
782 ENSG00000255310 0.7163 eQTL 8_14
68 ENSG00000229431 0.6882 eQTL 1_27
1905 ZNF593 0.6366 eQTL 1_18
$m6AQTL
[1] genename susie_pip group region_tag
<0 rows> (or 0-length row.names)
$sQTL
genename susie_pip group region_tag
4108 MYO1G 0.9619 sQTL 7_33
3558 ETFA 0.7622 sQTL 15_36
2456 GSK3B 0.7306 sQTL 3_74Top m6A modification pip
genename region_tag susie_pip z
1 TAPBP 6_28 0.48279 -8.315
2 TGOLN2 2_54 0.33394 -7.771
3 SLC9A3R1 17_42 0.08616 -7.384
4 LETMD1 12_31 0.06916 -4.766
5 BANF1 11_36 0.04132 4.670
6 THEMIS2 1_19 0.02911 3.876
7 C2CD2L 11_71 0.02885 3.482
8 PPP2R5C 14_54 0.02779 -3.812
9 TRIT1 1_25 0.02766 3.964
10 ASCC1 10_49 0.02600 4.016Summing up PIPs for m6A peaks located in the same gene
Top 10 m6A PIPs by genes
# A tibble: 819 × 2
genename total_susie_pip
<chr> <dbl>
1 TAPBP 0.507
2 TGOLN2 0.334
3 SLC9A3R1 0.0862
4 LETMD1 0.0692
5 BANF1 0.0413
6 THEMIS2 0.0291
7 C2CD2L 0.0289
8 PPP2R5C 0.0278
9 TRIT1 0.0277
10 ASCC1 0.0260
# ℹ 809 more rowsTop splicing PIPs
peak_id genename pos region_tag susie_pip z
1 chr7:45009474-45009639 MYO1G 44925489 7_33 0.9619 -8.315
2 chr15:76588078-76602273 ETFA 76496232 15_36 0.7622 -5.564
3 chr3:119582452-119624602 GSK3B 119503971 3_74 0.7306 6.695
4 chr2:85823772-85824227 RNF181 85818886 2_54 0.3726 3.817
5 chr10:97007123-97023621 PDLIM1 97001124 10_61 0.2946 -7.031
6 chr9:86593367-86595418 HNRNPK 86592026 9_41 0.2547 7.912
7 chr7:56120178-56123317 CCT6A 56033141 7_40 0.2220 -4.773
8 chr19:1036561-1037624 CNN2 1038445 19_2 0.1991 3.367
9 chr19:1036999-1037624 CNN2 1038445 19_2 0.1991 -3.367
10 chr1:207940540-207943666 CD46 207923081 1_107 0.1979 -9.808Summing up PIPs for spliced introns located in the same gene
Top 10 splicing PIPs by genes
# A tibble: 10 × 2
genename total_susie_pip
<chr> <dbl>
1 MYO1G 0.962
2 ETFA 0.765
3 GSK3B 0.731
4 CD46 0.463
5 CNN2 0.399
6 RNF181 0.397
7 HNRNPK 0.356
8 PDLIM1 0.295
9 AC253536.7 0.236
10 CCT6A 0.224Top genes by combined PIP
genename combined_pip expression_pip splicing_pip m6A_pip
624 CSNK1G1 1.001 0.99947 0.000000 0.001205
3320 ZMIZ1 0.991 0.99091 0.000000 0.000000
3141 TTLL12 0.989 0.98885 0.000000 0.000000
1957 MYO1G 0.962 0.00000 0.961931 0.000000
2012 NDUFS2 0.927 0.92667 0.000000 0.000000
437 CCDC9 0.878 0.87764 0.000000 0.000000
1947 MXD3 0.830 0.82980 0.000000 0.000000
1663 KYNU 0.791 0.78963 0.001790 0.000000
1265 ETFA 0.776 0.01132 0.764910 0.000000
2438 RAPGEFL1 0.758 0.75791 0.000000 0.000000
317 BORCS7 0.741 0.74077 0.000000 0.000000
1439 GSK3B 0.731 0.00000 0.730646 0.000000
1085 ENSG00000255310 0.716 0.71625 0.000000 0.000000
967 ENSG00000229431 0.688 0.68816 0.000000 0.000000
3371 ZNF593 0.637 0.63657 0.000000 0.000000
3230 VPS16 0.576 0.57565 0.000000 0.000000
360 C19orf54 0.538 0.53245 0.004023 0.001566
1264 ESYT2 0.537 0.53457 0.002218 0.000000
1626 KDELR2 0.520 0.52024 0.000000 0.000000
1195 ENSG00000270081 0.510 0.51042 0.000000 0.000000
region_tag
624 15_29
3320 10_51
3141 22_18
1957 7_33
2012 1_81
437 19_34
1947 5_106
1663 2_85
1265 15_36
2438 17_23
317 10_66
1439 3_74
1085 8_14
967 1_27
3371 1_18
3230 20_3
360 19_28
1264 7_99
1626 7_9
1195 2_75Loading required package: gridWarning: replacing previous import 'utils::download.file' by
'restfulr::download.file' when loading 'rtracklayer'Locus plots for specific examples
R version 4.2.0 (2022-04-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.3.13-el7-x86_64/lib/libopenblas_haswellp-r0.3.13.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=C
[4] LC_COLLATE=C LC_MONETARY=C LC_MESSAGES=C
[7] LC_PAPER=C LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=C LC_IDENTIFICATION=C
attached base packages:
[1] grid stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] biomaRt_2.52.0 Gviz_1.40.1 cowplot_1.1.1
[4] ggplot2_3.4.3 GenomicRanges_1.48.0 GenomeInfoDb_1.32.2
[7] IRanges_2.30.1 S4Vectors_0.34.0 BiocGenerics_0.42.0
[10] ctwas_0.1.38 dplyr_1.1.2 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] colorspace_2.1-0 deldir_1.0-6
[3] rjson_0.2.21 rprojroot_2.0.3
[5] biovizBase_1.44.0 htmlTable_2.4.0
[7] XVector_0.36.0 base64enc_0.1-3
[9] fs_1.6.3 dichromat_2.0-0.1
[11] rstudioapi_0.15.0 farver_2.1.1
[13] bit64_4.0.5 AnnotationDbi_1.58.0
[15] fansi_1.0.4 xml2_1.3.3
[17] codetools_0.2-18 logging_0.10-108
[19] cachem_1.0.8 knitr_1.39
[21] Formula_1.2-4 jsonlite_1.8.7
[23] Rsamtools_2.12.0 cluster_2.1.3
[25] dbplyr_2.3.3 png_0.1-7
[27] compiler_4.2.0 httr_1.4.7
[29] backports_1.4.1 lazyeval_0.2.2
[31] Matrix_1.6-1 fastmap_1.1.1
[33] cli_3.6.1 later_1.3.0
[35] htmltools_0.5.2 prettyunits_1.1.1
[37] tools_4.2.0 gtable_0.3.3
[39] glue_1.6.2 GenomeInfoDbData_1.2.8
[41] rappdirs_0.3.3 Rcpp_1.0.11
[43] Biobase_2.56.0 jquerylib_0.1.4
[45] vctrs_0.6.3 Biostrings_2.64.0
[47] rtracklayer_1.56.0 iterators_1.0.14
[49] xfun_0.30 stringr_1.5.0
[51] ps_1.7.0 lifecycle_1.0.3
[53] ensembldb_2.20.2 restfulr_0.0.14
[55] XML_3.99-0.14 getPass_0.2-2
[57] zlibbioc_1.42.0 scales_1.2.1
[59] BSgenome_1.64.0 VariantAnnotation_1.42.1
[61] ProtGenerics_1.28.0 hms_1.1.3
[63] promises_1.2.0.1 MatrixGenerics_1.8.0
[65] parallel_4.2.0 SummarizedExperiment_1.26.1
[67] AnnotationFilter_1.20.0 RColorBrewer_1.1-3
[69] yaml_2.3.5 curl_5.0.2
[71] memoise_2.0.1 gridExtra_2.3
[73] sass_0.4.1 rpart_4.1.16
[75] latticeExtra_0.6-30 stringi_1.7.12
[77] RSQLite_2.3.1 highr_0.9
[79] BiocIO_1.6.0 foreach_1.5.2
[81] checkmate_2.1.0 GenomicFeatures_1.48.4
[83] filelock_1.0.2 BiocParallel_1.30.3
[85] rlang_1.1.1 pkgconfig_2.0.3
[87] matrixStats_0.62.0 bitops_1.0-7
[89] evaluate_0.15 lattice_0.20-45
[91] htmlwidgets_1.5.4 GenomicAlignments_1.32.0
[93] labeling_0.4.2 bit_4.0.5
[95] processx_3.8.0 tidyselect_1.2.0
[97] magrittr_2.0.3 R6_2.5.1
[99] generics_0.1.3 Hmisc_5.1-0
[101] DelayedArray_0.22.0 DBI_1.1.3
[103] pgenlibr_0.3.6 pillar_1.9.0
[105] whisker_0.4 foreign_0.8-82
[107] withr_2.5.0 KEGGREST_1.36.2
[109] RCurl_1.98-1.7 nnet_7.3-17
[111] tibble_3.2.1 crayon_1.5.2
[113] interp_1.1-4 utf8_1.2.3
[115] BiocFileCache_2.4.0 rmarkdown_2.14
[117] jpeg_0.1-10 progress_1.2.2
[119] data.table_1.14.8 blob_1.2.4
[121] callr_3.7.3 git2r_0.30.1
[123] digest_0.6.33 httpuv_1.6.5
[125] munsell_0.5.0 bslib_0.3.1