Accounting for technical batch effects: Response to infection with Mycobacterium tuberculosis
John Blischak
2018-08-09
Last updated: 2018-08-20
workflowr checks: (Click a bullet for more information)-
✔ R Markdown file: up-to-date
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
-
✔ Environment: empty
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
-
✔ Seed:
set.seed(12345)The command
set.seed(12345)was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible. -
✔ Session information: recorded
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
-
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.✔ Repository version: fcc513c
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can usewflow_publishorwflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.Ignored files: Ignored: .Rhistory Ignored: .Rproj.user/ Untracked files: Untracked: code/counts_per_sample.txt Untracked: code/table-s1.txt
Expand here to see past versions:
An example of diagnosing and correcting batch effects from one of my own studies on the response to infection with Mycobacterium tuberculosis (paper, code, data).
Setup
library(dplyr)
library(limma)
library(edgeR)
# Have to load Biobase after dplyr so that exprs function works
library(Biobase)Download data.
file_url <- "https://bitbucket.org/jdblischak/tb-data/raw/bc0f64292eb8c42a372b3a2d50e3d871c70c202e/counts_per_sample.txt"
full <- read.delim(file_url, stringsAsFactors = FALSE)Convert to ExpressionSet.
dim(full)[1] 156 19419full <- full[order(full$dir), ]
rownames(full) <- paste(full$ind, full$bact, full$time, sep = ".")
x <- t(full[, grep("ENSG", colnames(full))])
p <- full %>% select(ind, bact, time, extr, rin)
stopifnot(colnames(x) == rownames(p))
eset <- ExpressionSet(assayData = x,
phenoData = AnnotatedDataFrame(p))Filter lowly expressed genes.
keep <- rowSums(cpm(exprs(eset)) > 1) > 6
sum(keep)[1] 12728eset <- eset[keep, ]
dim(eset)Features Samples
12728 156 Normalize with TMM.
norm_factors <- calcNormFactors(exprs(eset))
exprs(eset) <- cpm(exprs(eset), lib.size = colSums(exprs(eset)) * norm_factors,
log = TRUE)
plotDensities(eset, legend = FALSE)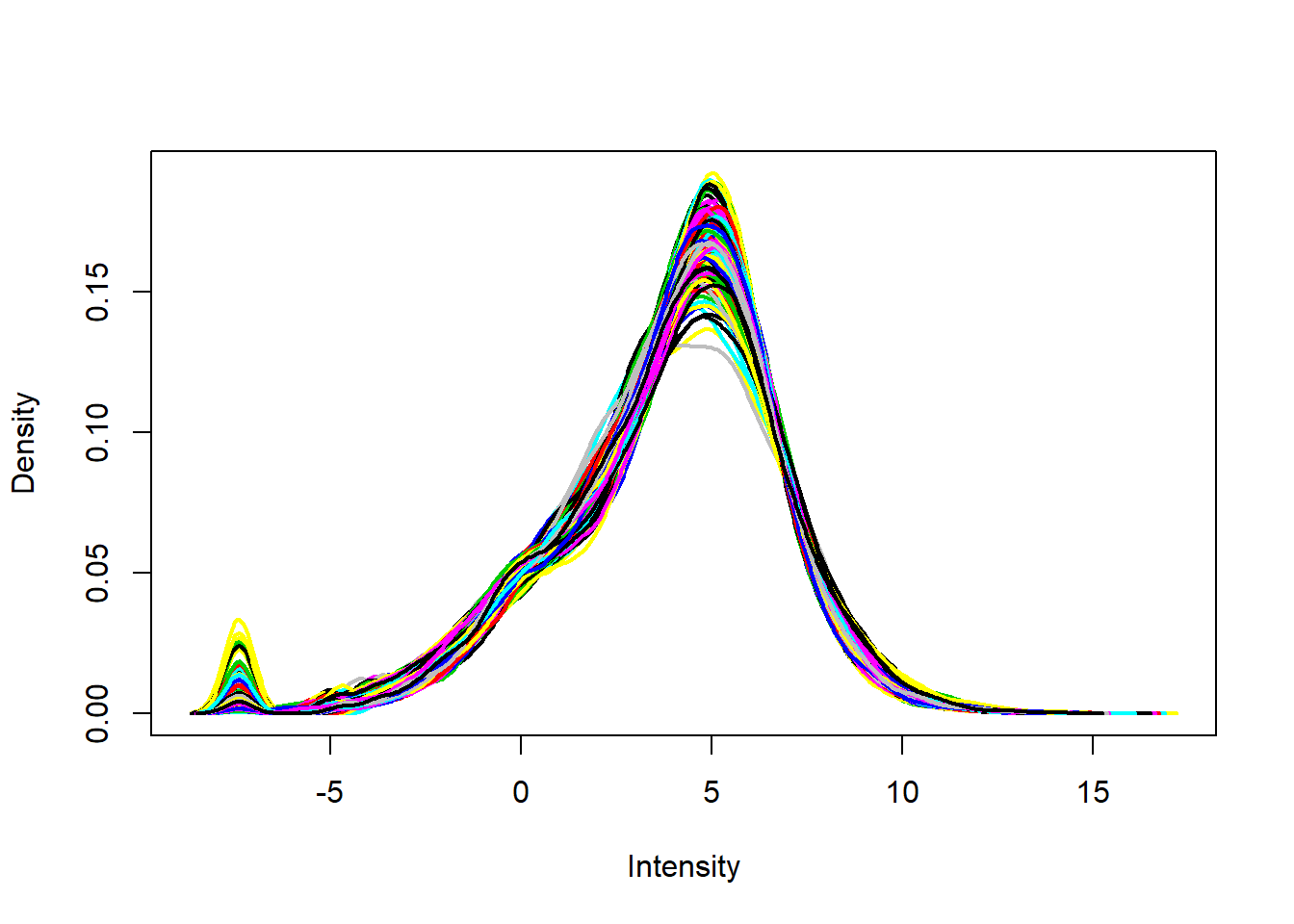
Expand here to see past versions of normalize-1.png:
| Version | Author | Date |
|---|---|---|
| f2c0198 | John Blischak | 2018-08-09 |
Clean up phenotype data frame to focus on early versus late timepoint for this example.
pData(eset)[, "infection"] <- ifelse(pData(eset)[, "bact"] == "none",
"con", "inf")
pData(eset)[, "time"] <- ifelse(pData(eset)[, "time"] == 4,
"early", "late")
pData(eset)[, "batch"] <- sprintf("b%02d", pData(eset)[, "extr"])
table(pData(eset)[, c("time", "batch")]) batch
time b01 b02 b03 b04 b05 b06 b07 b08 b09 b10 b11 b12 b13
early 4 4 4 4 4 4 4 4 4 4 4 4 6
late 8 8 8 8 8 8 8 8 8 8 8 8 6Remove batch effect
Visualize principal components 1 and 2 for the original data.
plotMDS(eset, labels = pData(eset)[, "time"], gene.selection = "common")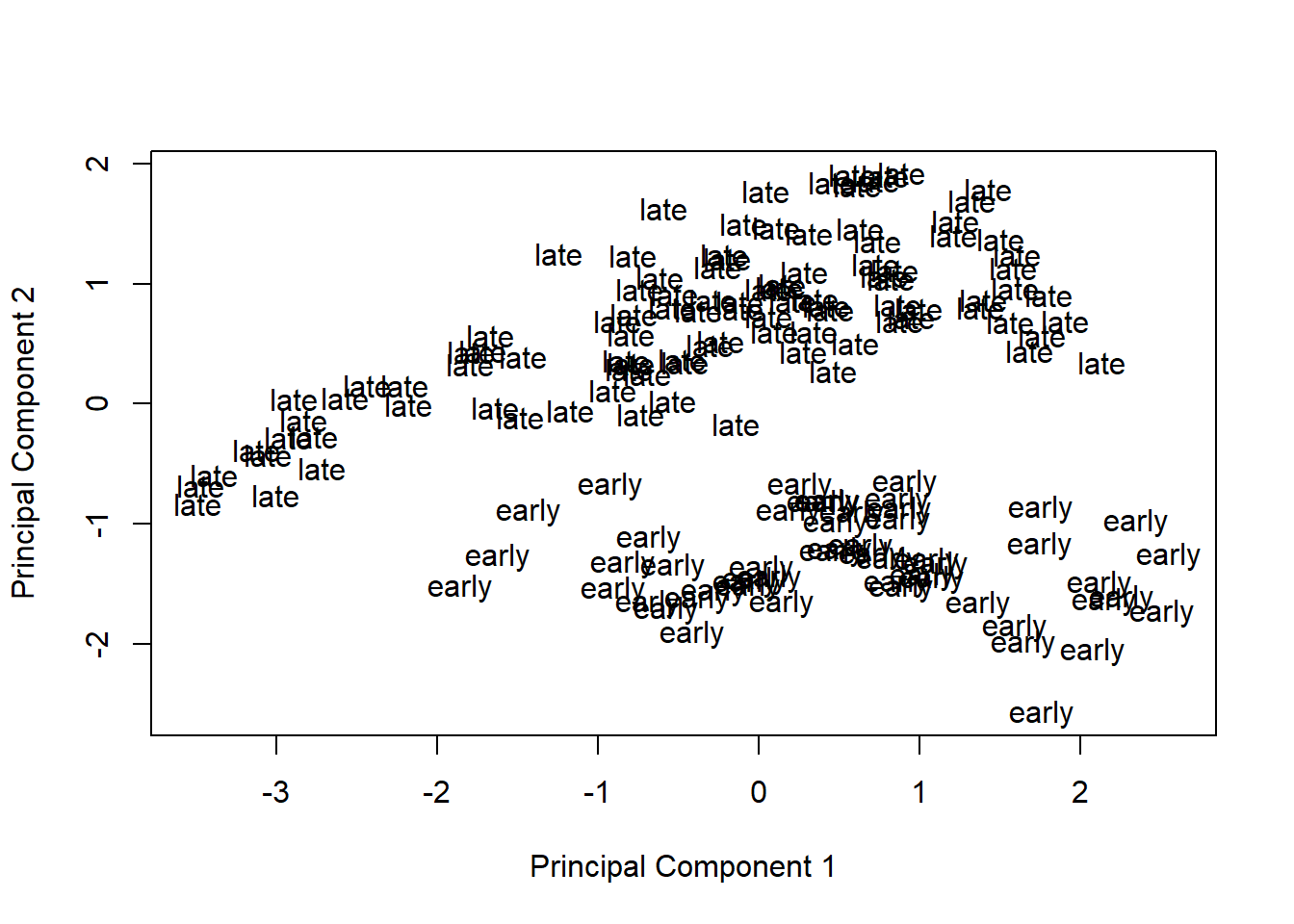
Expand here to see past versions of pre-1.png:
| Version | Author | Date |
|---|---|---|
| f2c0198 | John Blischak | 2018-08-09 |
Remove the effect of the technical variables: batch (discrete) and RIN (continuous; a measure of RNA quality).
exprs(eset) <- removeBatchEffect(eset, batch = pData(eset)[, "batch"],
covariates = pData(eset)[, "rin"])Visualize principal components 1 and 2 for the corrected data.
plotMDS(eset, labels = pData(eset)[, "time"], gene.selection = "common")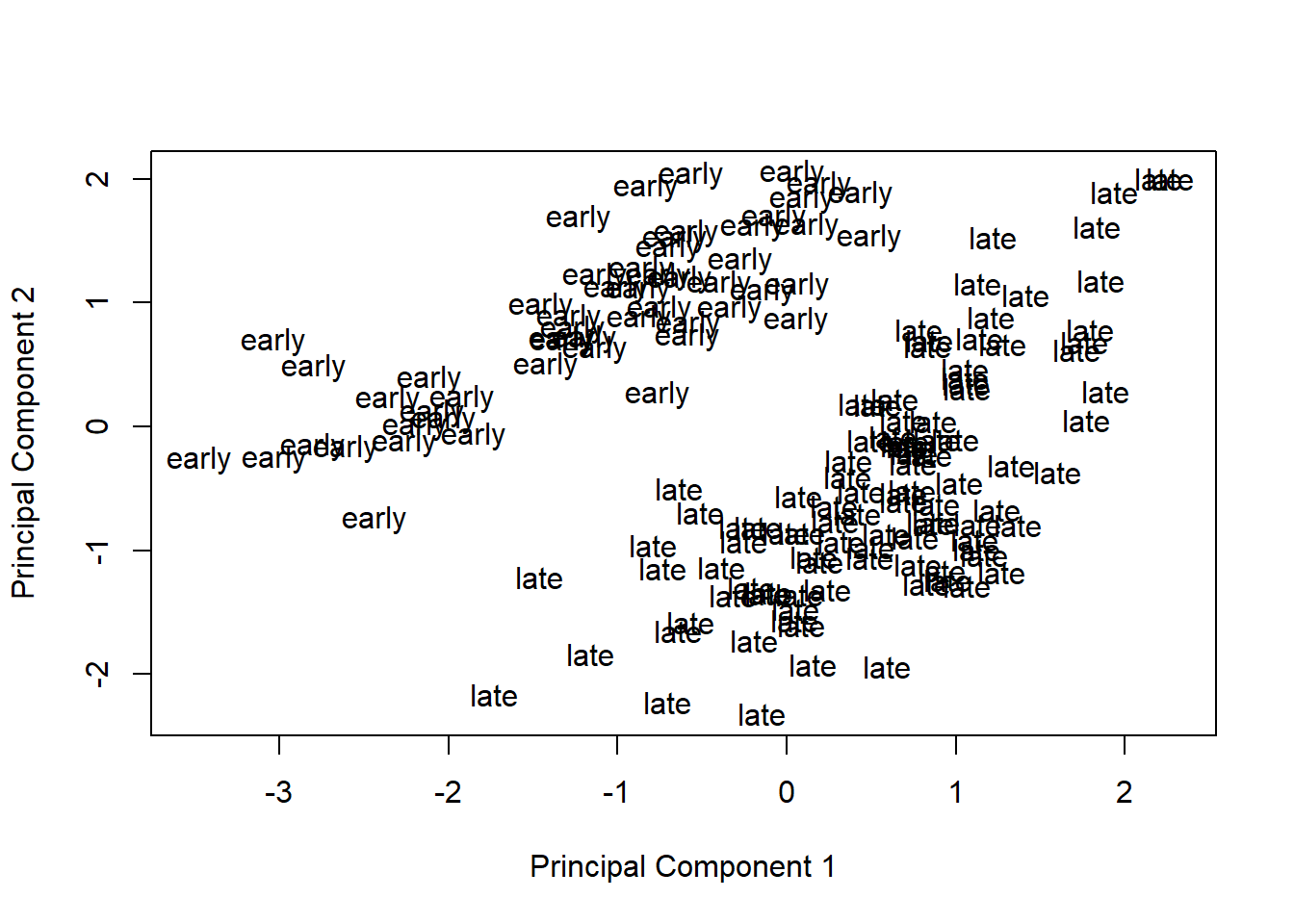
Expand here to see past versions of post-1.png:
| Version | Author | Date |
|---|---|---|
| f2c0198 | John Blischak | 2018-08-09 |
Session information
sessionInfo()R version 3.5.0 (2018-04-23)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 17134)
Matrix products: default
locale:
[1] LC_COLLATE=English_United States.1252
[2] LC_CTYPE=English_United States.1252
[3] LC_MONETARY=English_United States.1252
[4] LC_NUMERIC=C
[5] LC_TIME=English_United States.1252
attached base packages:
[1] parallel stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] Biobase_2.40.0 BiocGenerics_0.26.0 edgeR_3.22.2
[4] limma_3.36.1 dplyr_0.7.5
loaded via a namespace (and not attached):
[1] Rcpp_0.12.17 knitr_1.20 bindr_0.1.1
[4] whisker_0.3-2 magrittr_1.5 workflowr_1.1.1
[7] tidyselect_0.2.4 lattice_0.20-35 R6_2.2.2
[10] rlang_0.2.1 stringr_1.3.1 tools_3.5.0
[13] grid_3.5.0 R.oo_1.22.0 git2r_0.21.0
[16] htmltools_0.3.6 yaml_2.1.19 rprojroot_1.3-2
[19] digest_0.6.15 assertthat_0.2.0 tibble_1.4.2
[22] bindrcpp_0.2.2 purrr_0.2.5 R.utils_2.6.0
[25] glue_1.2.0 evaluate_0.10.1 rmarkdown_1.10
[28] stringi_1.2.3 pillar_1.2.3 compiler_3.5.0
[31] backports_1.1.2 R.methodsS3_1.7.1 locfit_1.5-9.1
[34] pkgconfig_2.0.1 This reproducible R Markdown analysis was created with workflowr 1.1.1