Circle fit to intensities
Joyce Hsiao
Last updated: 2018-02-23
Code version: f434aa3
Overview/Results
Here we estimate a circle fit on the two-dimensional intensity distriubtion of GFP and RFP.
Data and packages
Packages
library(circular)
library(conicfit)
library(Biobase)
library(dplyr)
library(matrixStats)
library(CorShrink)
source("../code/circle.intensity.fit.R")Load data
df <- readRDS(file="../data/eset-filtered.rds")
pdata <- pData(df)
fdata <- fData(df)
# select endogeneous genes
counts <- exprs(df)[grep("ENSG", rownames(df)), ]
# log2cpm <- readRDS("../output/seqdata-batch-correction.Rmd/log2cpm.rds")
# log2cpm.adjust <- readRDS("../output/seqdata-batch-correction.Rmd/log2cpm.adjust.rds")
# import corrected intensities
pdata.adj <- readRDS("../output/images-normalize-anova.Rmd/pdata.adj.rds")Circle fitting
Based on all data.
source("../code/circle.intensity.fit.R")
#sample_names <- rownames(pdata.adj)
pdata.adj <- pdata.adj %>% group_by(chip_id) %>%
mutate(rfp.z=scale(rfp.median.log10sum.adjust.ash),
gfp.z=scale(gfp.median.log10sum.adjust.ash),
dapi.z=scale(dapi.median.log10sum.adjust.ash))
pdata.adj <- data.frame(pdata.adj)
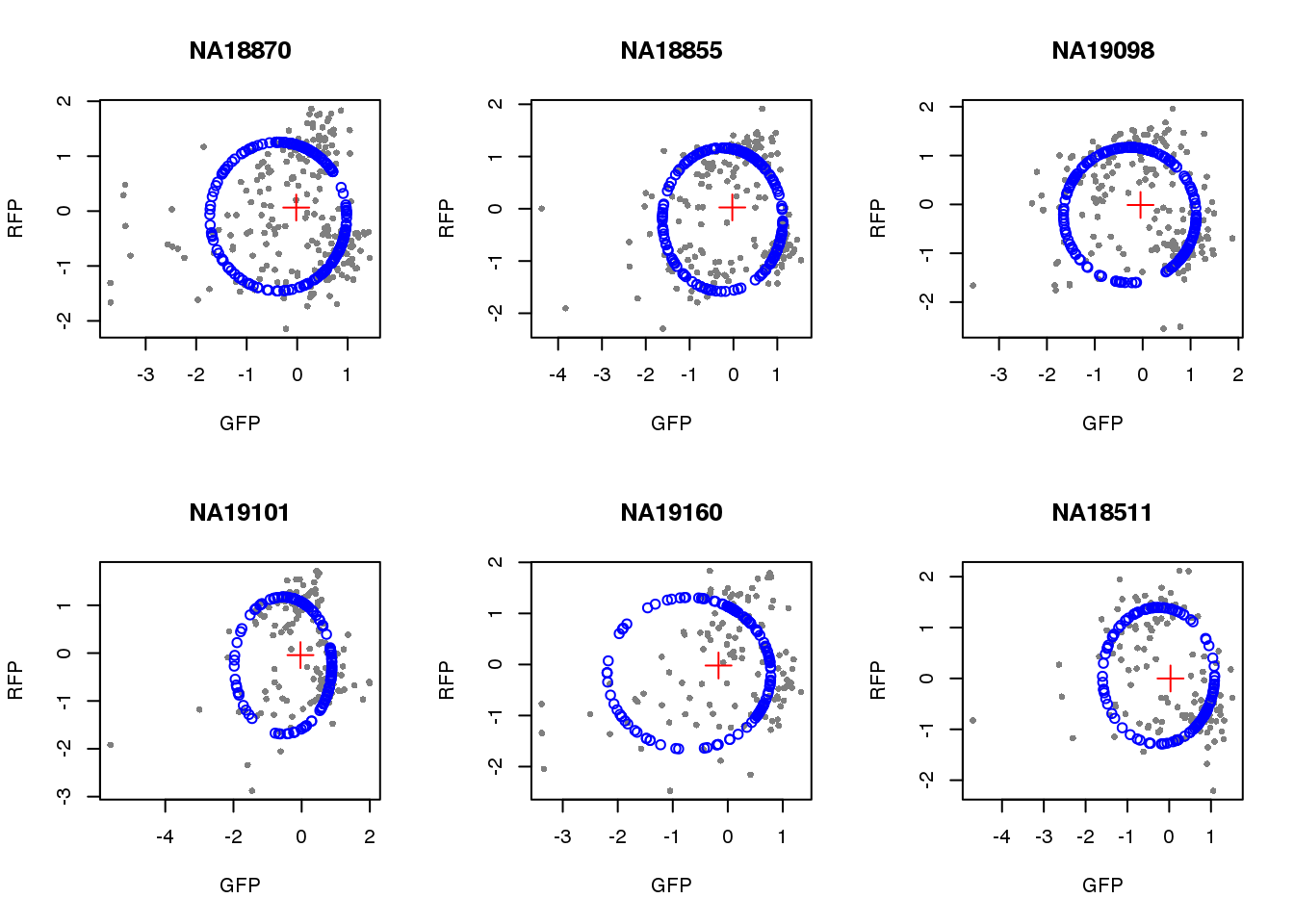
par(mfrow=c(2,3))
for(i in 1:length(unique(pdata.adj$chip_id))) {
id <- unique(as.character(pdata.adj$chip_id))[i]
df_sub <- subset(pdata.adj, chip_id == id, select=c(gfp.z, rfp.z))
cpred <- circle.fit(df_sub)
xlims <- range(df_sub[,1])
ylims <- range(df_sub[,2])
plot(df_sub, pch=16, col="gray50", xlim=xlims, ylim=ylims, cex=.7,
main = id, xlab="GFP", ylab="RFP")
points(cpred[,1], cpred[,2], col="blue", type = "p")
points(mean(cpred[,1]), mean(cpred[,2]), col="red", pch=3, cex=2)
}
Consider deleted residuals.
resids.del <- lapply(1:length(unique(pdata.adj$chip_id)), function(i) {
id <- unique(as.character(pdata.adj$chip_id))[i]
df_sub <- subset(pdata.adj, chip_id == id, select=c(gfp.z, rfp.z))
resids <- circle.fit.resid.delete(df_sub)
scale(resids)
})
names(resids.del) <- unique(pdata.adj$chip_id)
par(mfrow=c(2,3))
for(i in 1:length(unique(pdata.adj$chip_id))) {
# id <- unique(as.character(pdata.adj$chip_id))[i]
# df_sub <- subset(pdata.adj, chip_id == id, select=c(gfp.z, rfp.z))
hist(resids.del[[i]], main = unique(pdata.adj$chip_id)[i])
}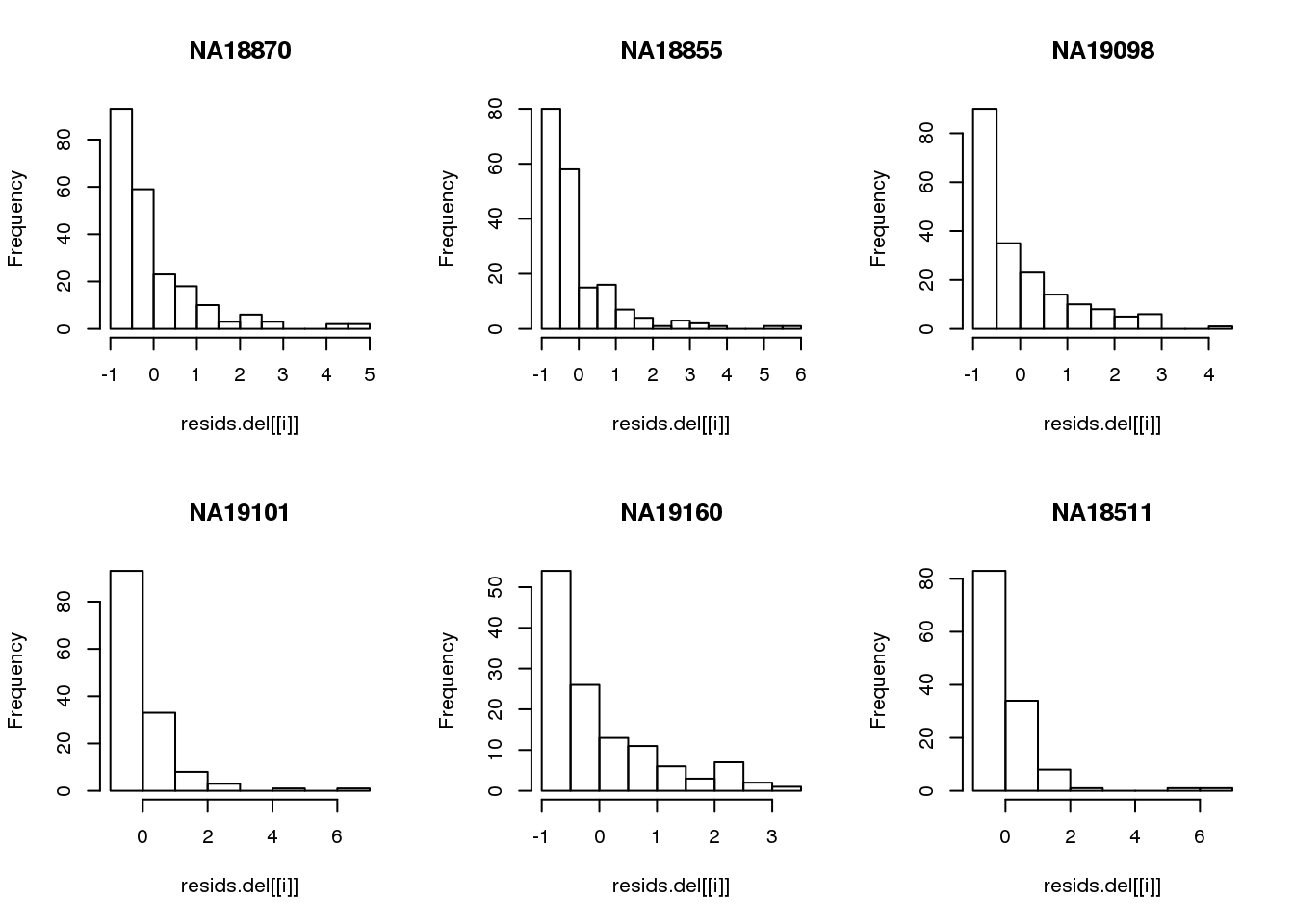
Remove samples with standardized residuals greater than 3.
resids.del.remove <- lapply(1:length(unique(pdata.adj$chip_id)), function(i) {
which(resids.del[[i]] > 3)
})
names(resids.del.remove) <- unique(pdata.adj$chip_id)
pdata.adj.filt <- do.call(rbind, lapply(1:length(unique(pdata.adj$chip_id)), function(i) {
id <- unique(as.character(pdata.adj$chip_id))[i]
df_sub <- pdata.adj[which(pdata.adj$chip_id == id),]
ii.remove <- resids.del.remove[[i]]
df_sub_return <- df_sub[-ii.remove,]
rownames(df_sub_return) <- (rownames(pdata.adj)[which(pdata.adj$chip_id == id)])[-ii.remove]
data.frame(df_sub_return)
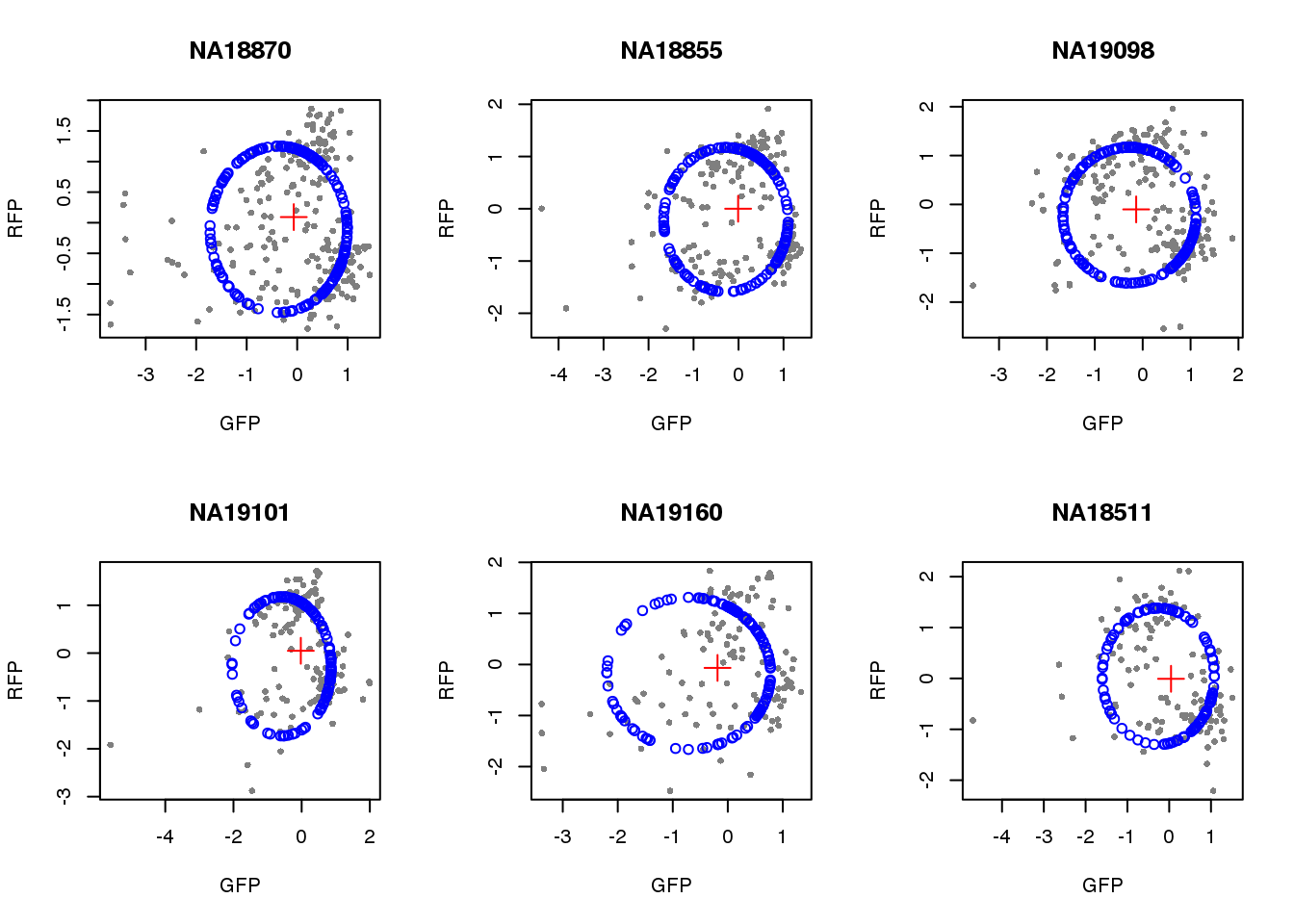
}) )Visualize fit after removing outliers.
par(mfrow=c(2,3))
for(i in 1:length(unique(pdata.adj.filt$chip_id))) {
id <- unique(as.character(pdata.adj.filt$chip_id))[i]
df_sub <- subset(pdata.adj.filt, chip_id == id, select=c(gfp.z, rfp.z))
cpred <- circle.fit(df_sub)
xlims <- range(df_sub[,1])
ylims <- range(df_sub[,2])
plot(df_sub, pch=16, col="gray50", xlim=xlims, ylim=ylims, cex=.7,
main = id, xlab="GFP", ylab="RFP")
points(cpred[,1], cpred[,2], col="blue", type = "p")
points(mean(cpred[,1]), mean(cpred[,2]), col="red", pch=3, cex=2)
}
saveRDS(pdata.adj.filt,
file = "../output/images-circle-ordering.Rmd/pdata.adj.filt.rds")Project positions
pdata.adj.filt <- readRDS("../output/images-circle-ordering.Rmd/pdata.adj.filt.rds")
proj.res <- vector("list", length=length(unique((pdata.adj$chip_id))))
for(i in 1:length(unique((pdata.adj$chip_id)))) {
proj.res[[i]] <- vector("list",2)
id <- unique(as.character(pdata.adj.filt$chip_id))[i]
df_sub <- subset(pdata.adj.filt,
chip_id == id, select=c(gfp.z, rfp.z))
# sample_ids <-
cpred <- circle.fit(df_sub)
proj.res[[i]][[1]] <- data.frame(cpred, df_sub)
colnames(proj.res[[i]][[1]]) <- c("pos.pred.x", "pos.pred.y", "gfp.z", "rfp.z")
# convert projected coordinates to radians
# modulo 2*pi
proj.res[[i]][[1]]$rads <- coord2rad(cbind(proj.res[[i]][[1]]$pos.pred.x,
proj.res[[i]][[1]]$pos.pred.y))
rownames(proj.res[[i]][[1]]) <- rownames(df_sub)
# compute centers
centers <- LMcircleFit(as.matrix(df_sub), ParIni=colMeans(as.matrix(df_sub)), IterMAX=50)
proj.res[[i]][[2]] <- data.frame(x.center=centers[1], y.center=centers[2])
}
names(proj.res) <- unique(pdata.adj.filt$chip_id)Save output
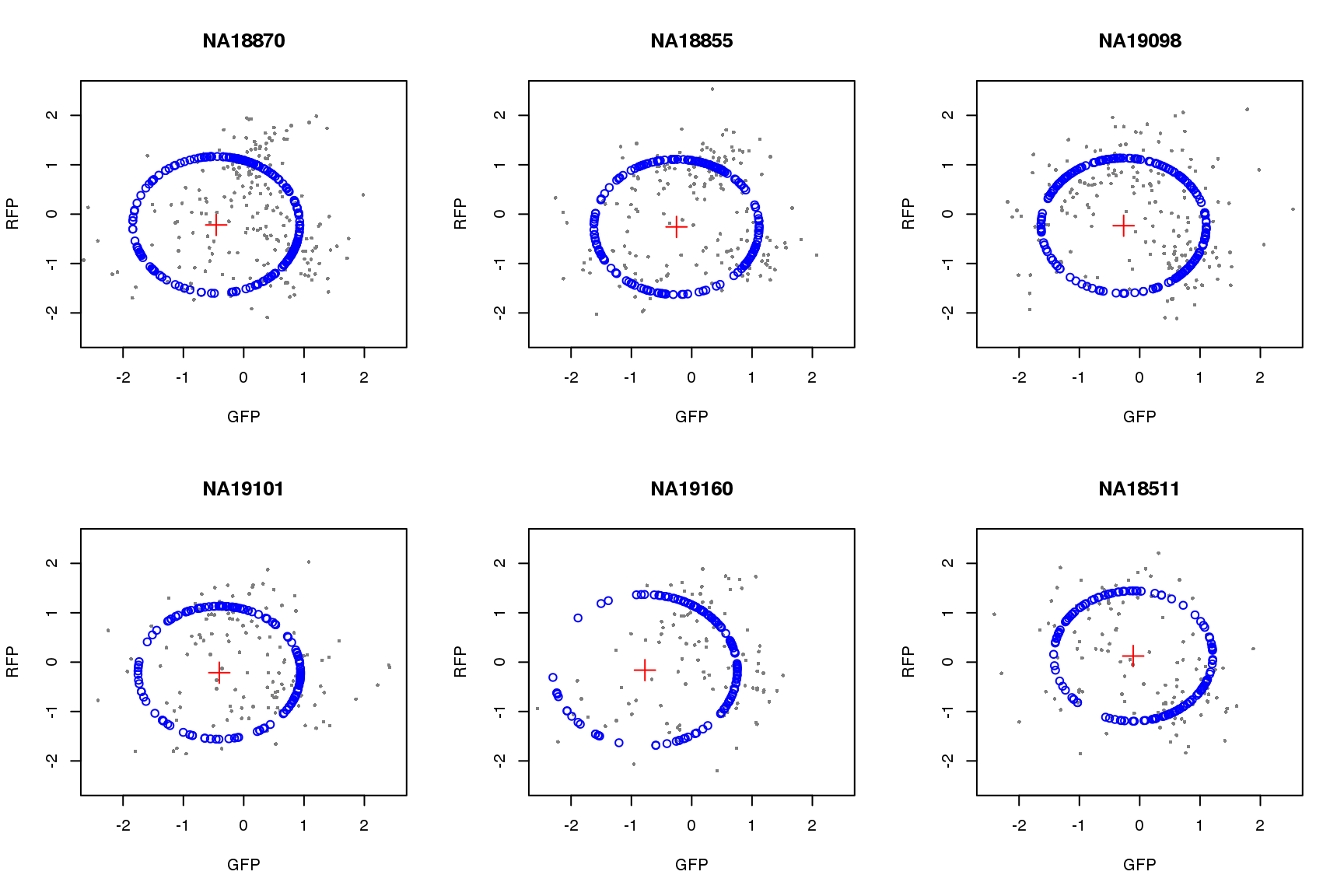
saveRDS(proj.res, file = "../output/images-circle-ordering.Rmd/proj.res.rds")Plot circle fit.
proj.res <- readRDS(file = "../output/images-circle-ordering.Rmd/proj.res.rds")
par(mfrow=c(2,3))
for (i in 1:length(proj.res)) {
# xlims <- range(proj.res[[i]]$gfp.z)
# ylims <- range(proj.res[[i]]$rfp.z)
xlims <- c(-2.5, 2.5)
ylims <- c(-2.5, 2.5)
plot(subset(proj.res[[i]][[1]], select=c(gfp.z, rfp.z)),
pch=16, col="gray50", xlim=xlims, ylim=ylims, cex=.5,
main = names(proj.res)[i],
xlab = "GFP", ylab = "RFP")
points(proj.res[[i]][[1]]$pos.pred.x, proj.res[[i]][[1]]$pos.pred.y,
col="blue", pch=1)
points(proj.res[[i]][[2]]$x.center, proj.res[[i]][[2]]$y.center,
col="red", pch=3, cex=2)
}
par(mfrow=c(2,3))
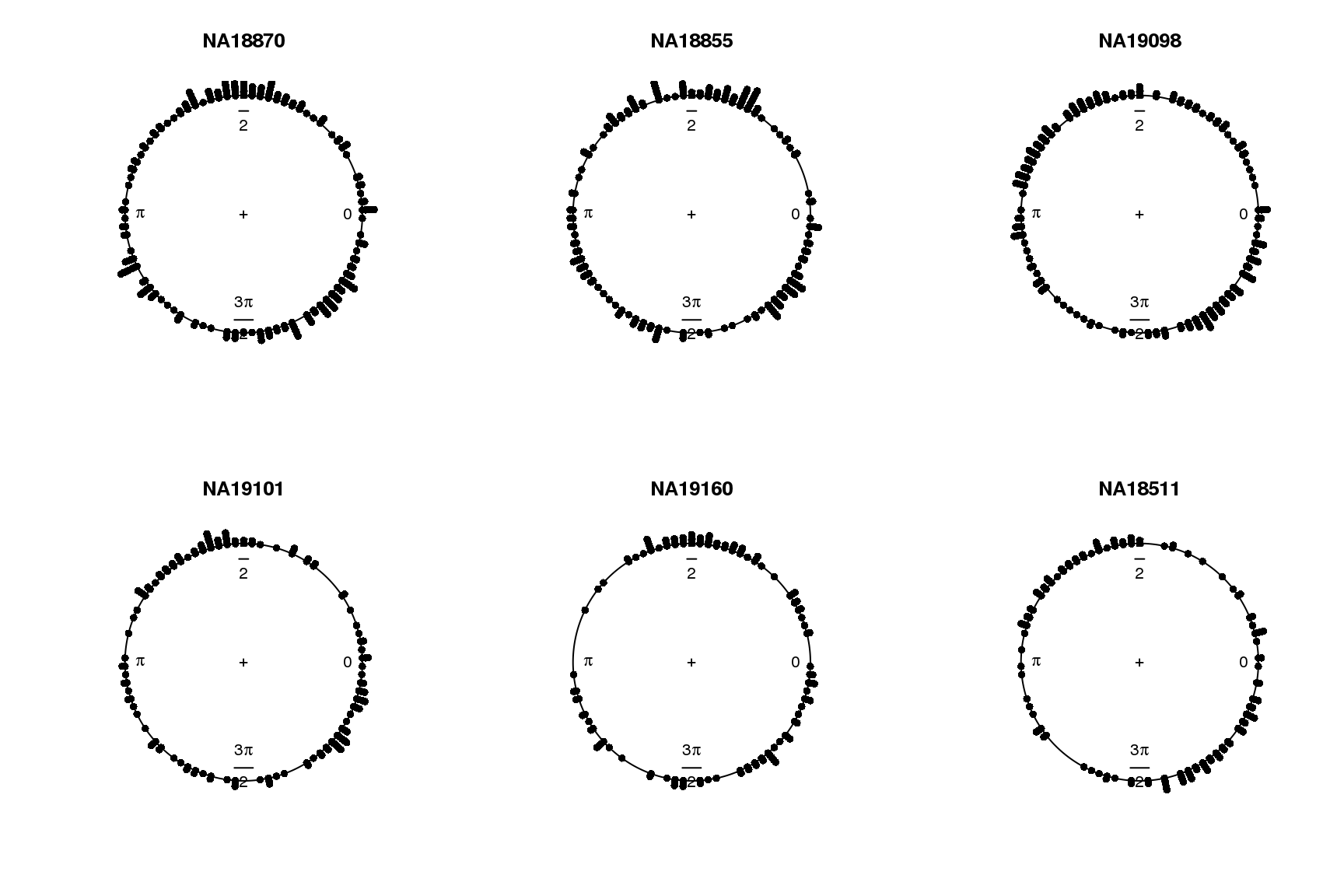
for (i in 1:length(proj.res)) {
plot(proj.res[[i]][[1]]$rads, stack=TRUE, bins=90,
main = names(proj.res)[i])
}
Property of the circle fit
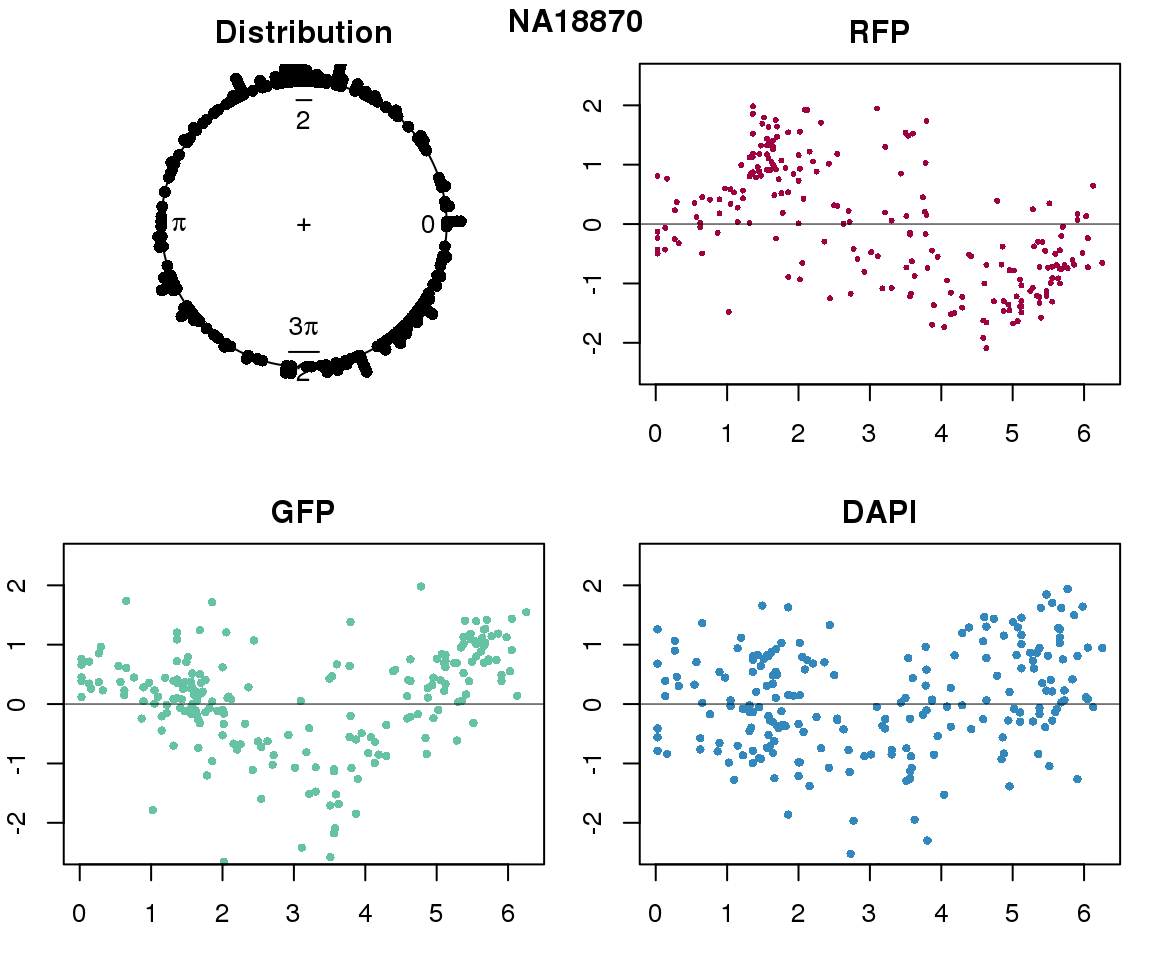
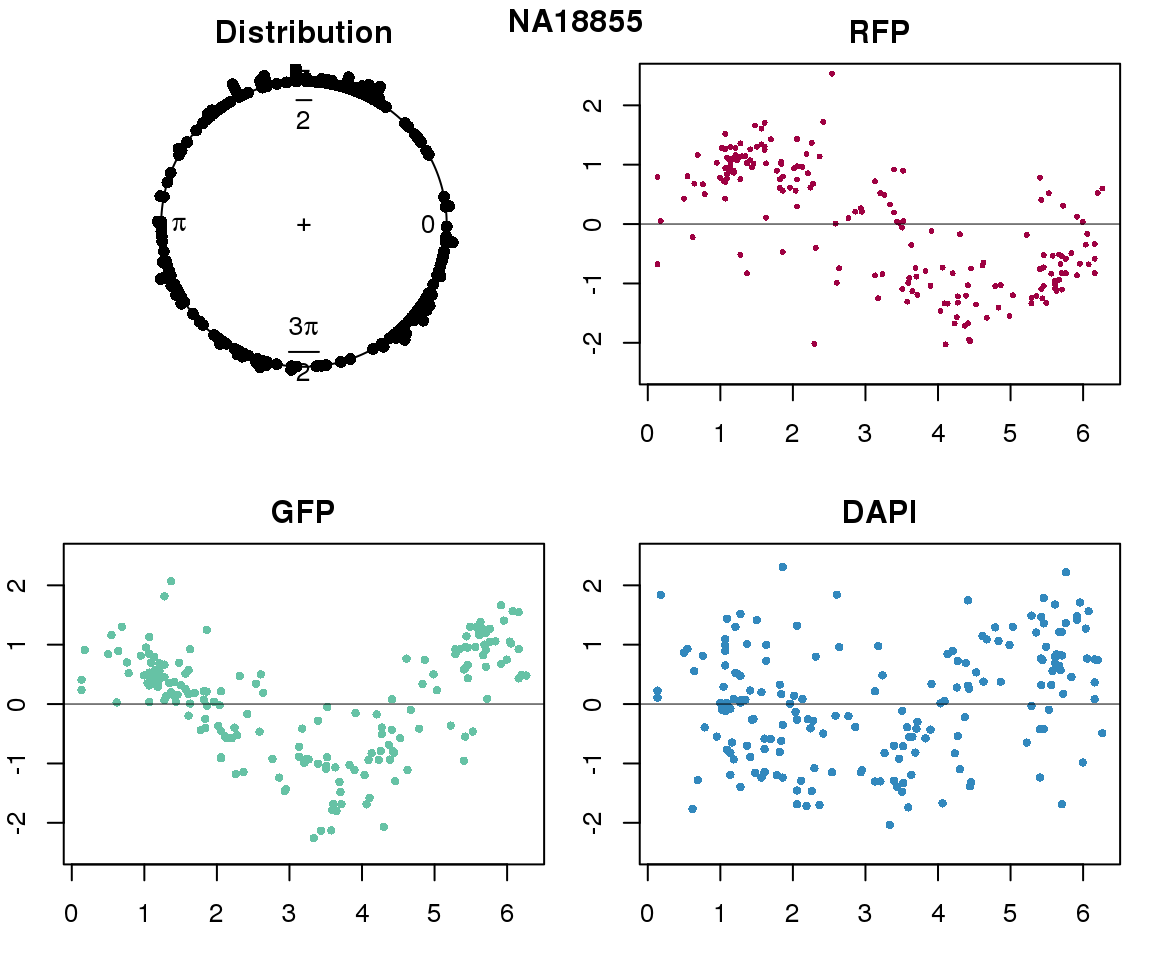
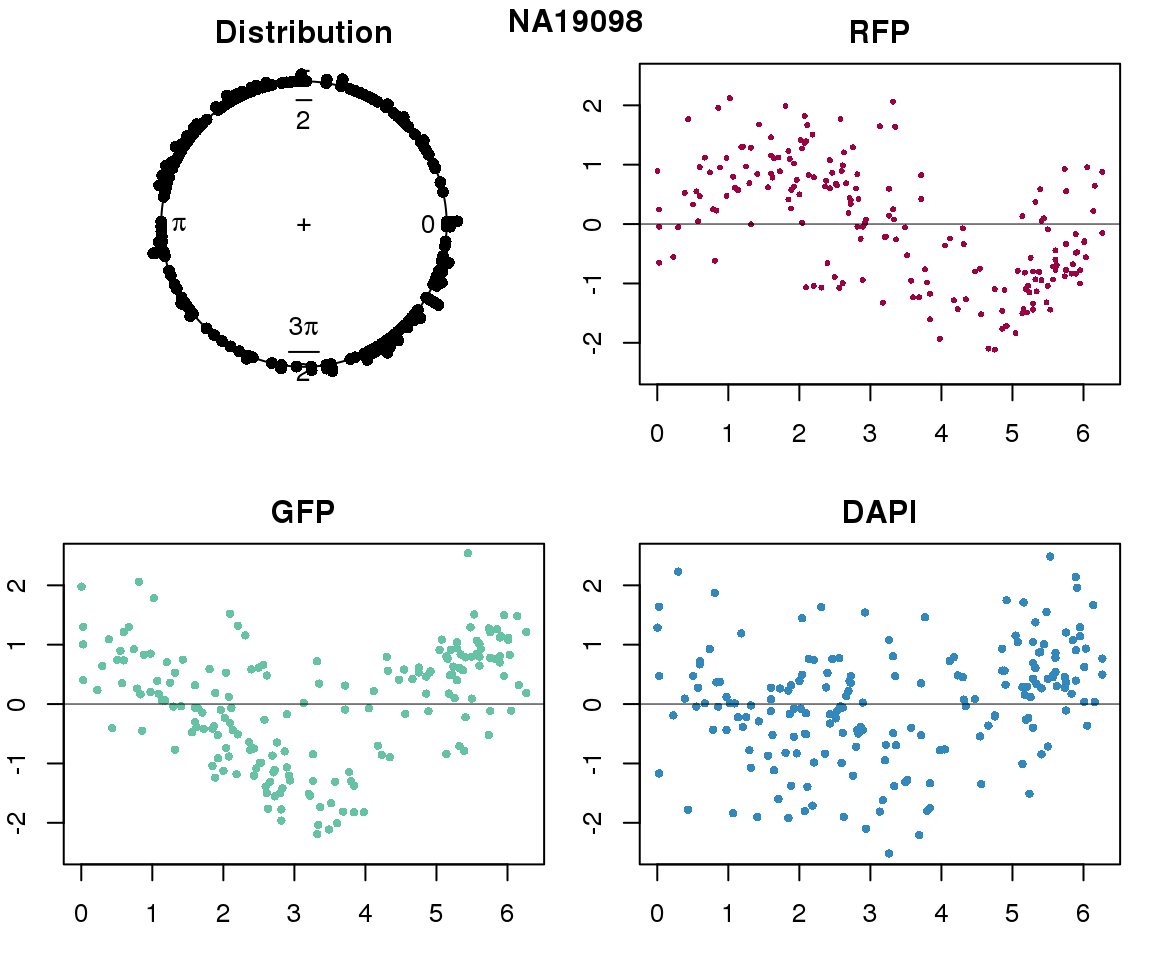
Intensity values by circle fit
pdata.adj.filtered <- readRDS("../output/images-circle-ordering.Rmd/pdata.adj.filt.rds")
proj.res <- readRDS("../output/images-circle-ordering.Rmd/proj.res.rds")
for (i in 1:length(unique(pdata.adj.filt$chip_id))) {
par(mfrow=c(2,2), mar = c(3,2,2,1))
ids <- unique(as.character(pdata.adj.filt$chip_id))
p_sub <- subset(pdata.adj.filt, chip_id == ids[i])
#all.equal(rownames(p_sub), rownames(proj.res$NA18870[[1]]))
plot(proj.res[[i]][[1]]$rads, stack=T, bins=180, main = "Distribution")
library(RColorBrewer)
color <- colorRampPalette(brewer.pal(11,"Spectral"))(11)
plot(x=as.numeric(proj.res[[i]][[1]]$rads),
y=p_sub$rfp.z, pch=16, cex=.5, col=color[1], ylim=c(-2.5, 2.5),
xlab = "Position on the circle",
ylab = "RFP", main = "RFP")
abline(h=0, lwd=.5)
plot(x=as.numeric(proj.res[[i]][[1]]$rads),
y=p_sub$gfp.z, pch=16, cex=.7, col=color[9], ylim=c(-2.5, 2.5),
xlab = "Position on the circle",
ylab = "GFP", main = "GFP")
abline(h=0, lwd=.5)
plot(x=as.numeric(proj.res[[i]][[1]]$rads),
y=p_sub$dapi.z, pch=16, cex=.7, col=color[10], ylim=c(-2.5, 2.5),
xlab = "Position on the circle",
ylab = "DAPI", main = "DAPI")
abline(h=0, lwd=.5)
title(names(proj.res)[i], outer=TRUE, line =-1)
}
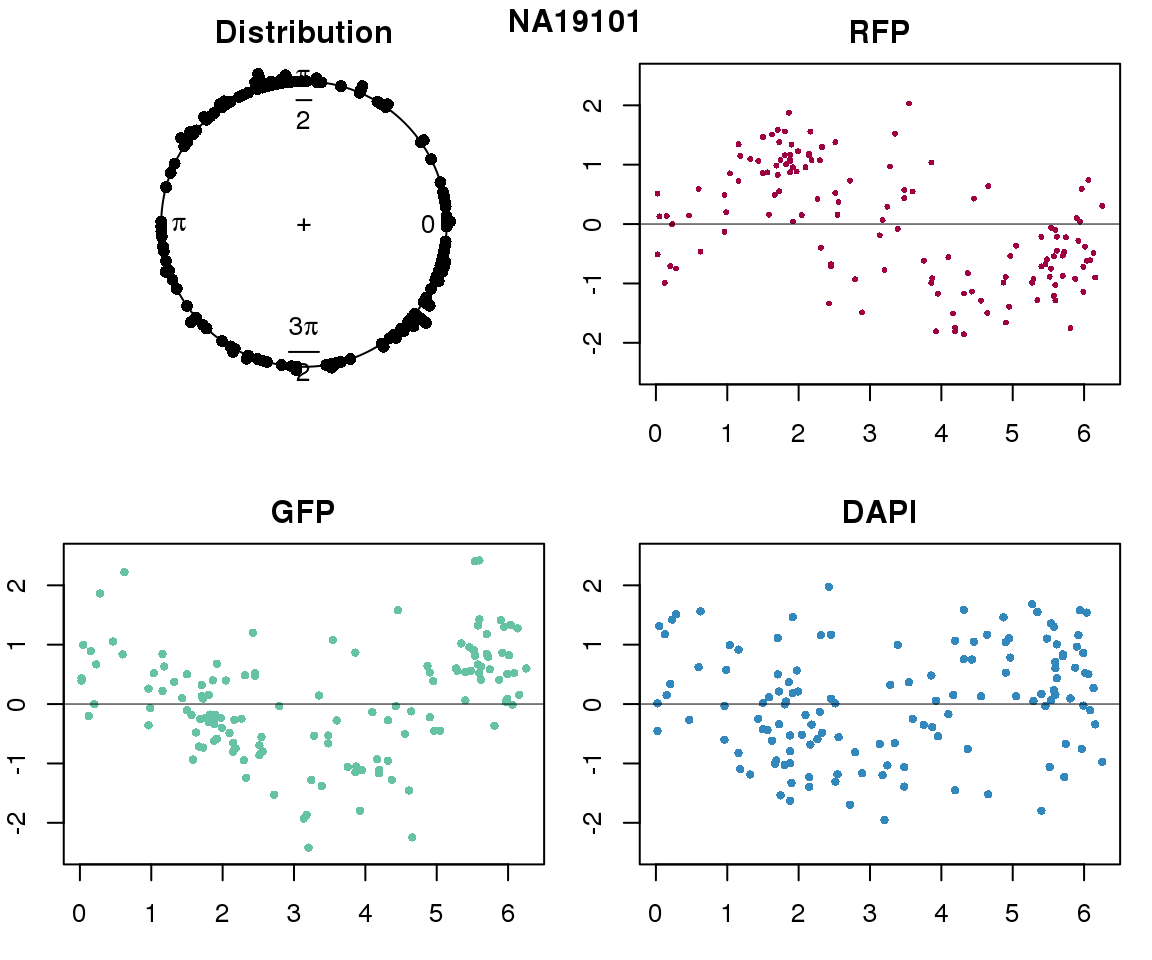
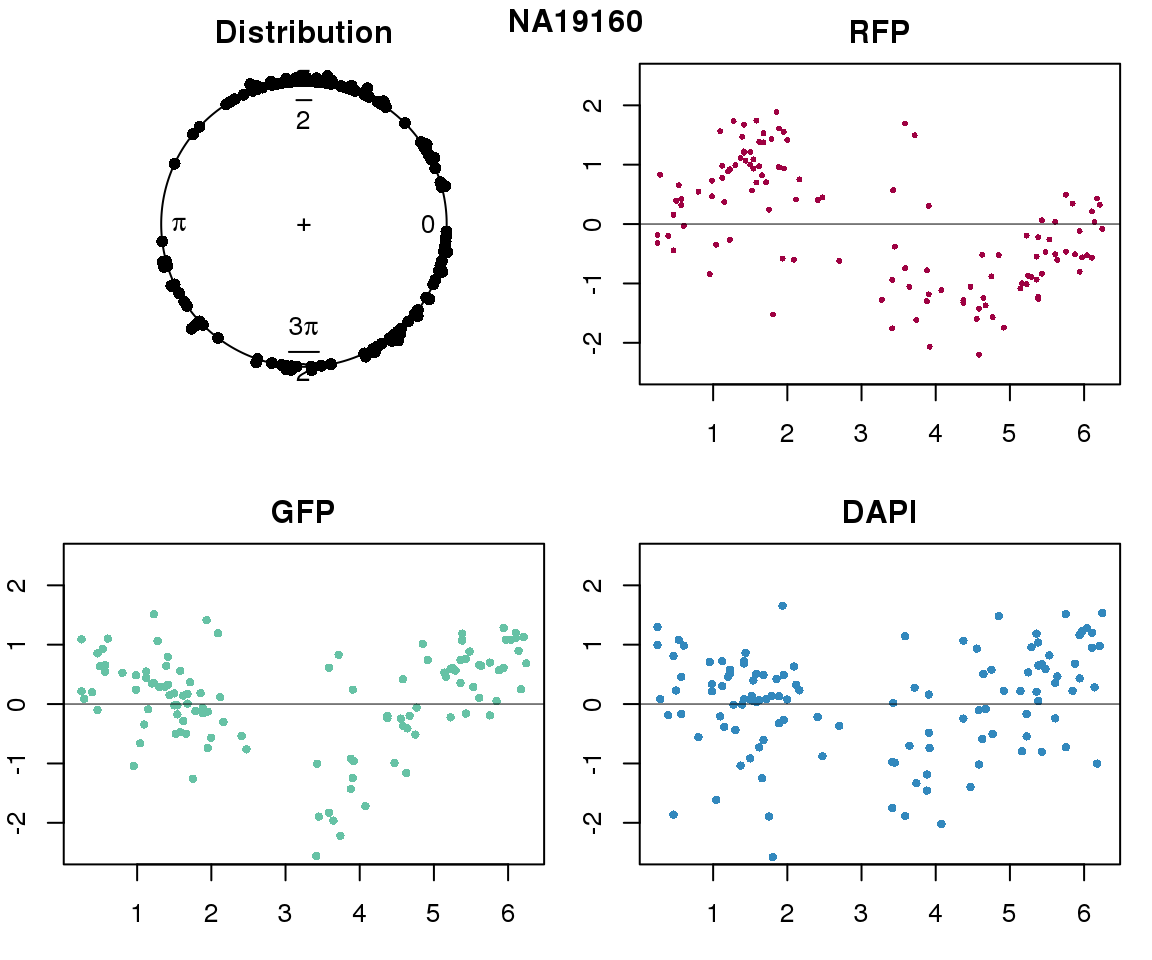
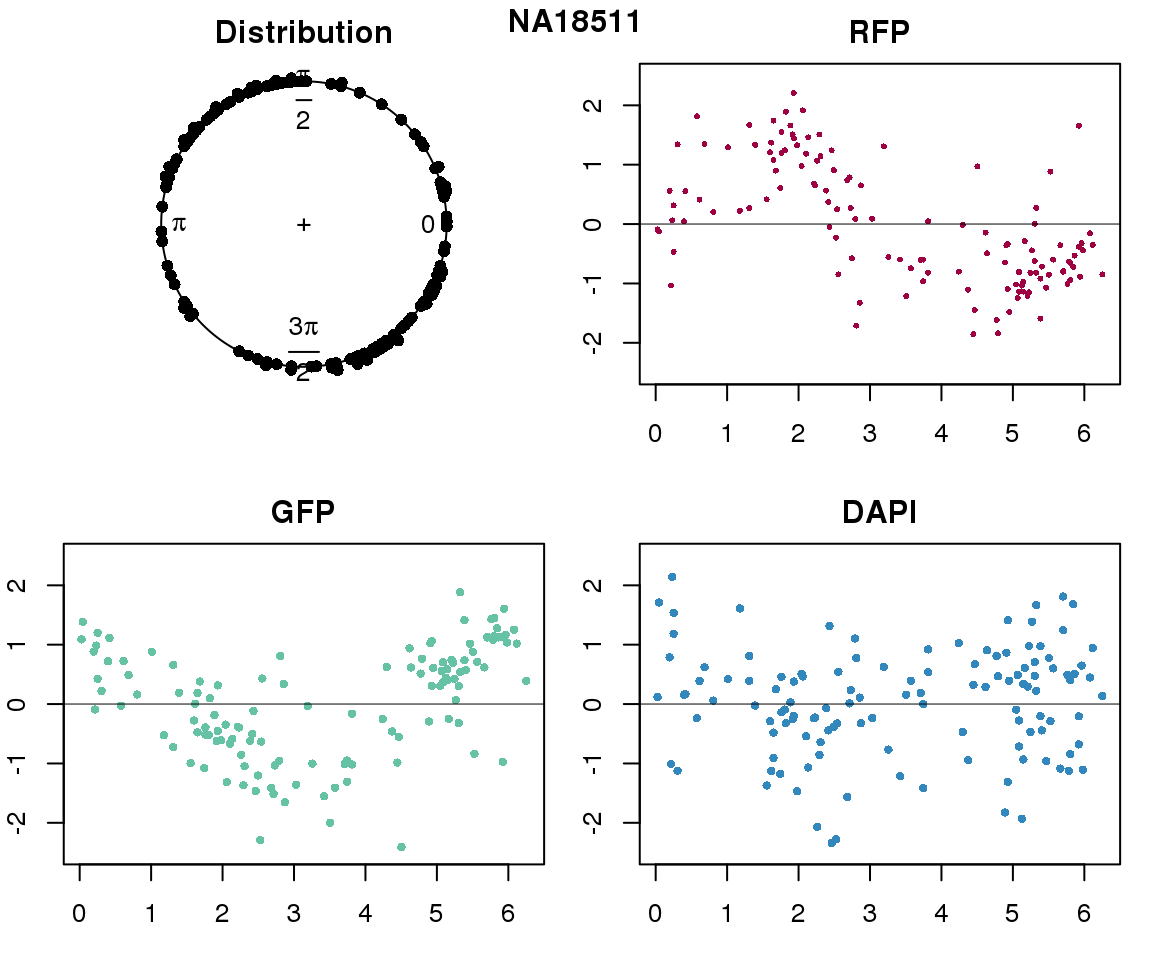
Expression variation by cell time
# load cell cycle genes
genes.cycle <- readRDS("../output/seqdata-select-cellcyclegenes.Rmd/genes.cycle.detect.rds")
# log2cpm
log2cpm <- readRDS("../output/seqdata-batch-correction.Rmd/log2cpm.rds")
log2cpm.adjust <- readRDS("../output/seqdata-batch-correction.Rmd/log2cpm.adjust.rds")
counts.cycle <- counts[rownames(counts) %in% genes.cycle, ]
log2cpm.cycle <- log2cpm[rownames(log2cpm) %in% genes.cycle, ]
log2cpm.adjust.cycle <- log2cpm.adjust[rownames(log2cpm.adjust) %in% genes.cycle, ]Pearson correlation
corrs <- lapply(1:length(unique(pdata.adj.filt$chip_id)), function(i) {
id <- unique(pdata.adj.filt$chip_id)[i]
log2cpm_sub <- log2cpm.adjust.cycle[, match(rownames(proj.res[[i]][[1]]), colnames(log2cpm.adjust.cycle))]
counts_sub <- counts.cycle[, match(rownames(proj.res[[i]][[1]]), colnames(counts.cycle))]
corrs <- do.call(rbind, lapply(1:nrow(counts_sub), function(g) {
vec <- cbind(as.numeric(proj.res[[i]][[1]]$rads),
log2cpm_sub[g,])
filt <- counts_sub[g,] > 1
nsamp <- sum(filt)
if (nsamp > ncol(counts_sub)/2) {
vec <- vec[filt,]
corr <- cor(vec[,1], vec[,2])
nsam <- nrow(vec)
data.frame(corr=corr, nsam=nsam)
} else {
data.frame(corr=NA, nsam=nrow(vec))
}
}))
rownames(corrs) <- rownames(counts_sub)
return(corrs)
})
names(corrs) <- unique(pdata.adj.filt$chip_id)par(mfrow=c(2,3))
for (i in 1:length(corrs)) {
hist(corrs[[i]]$corr, main = names(corrs)[i])
}
title(main = "Pearson correlation", outer = TRUE, line = -1)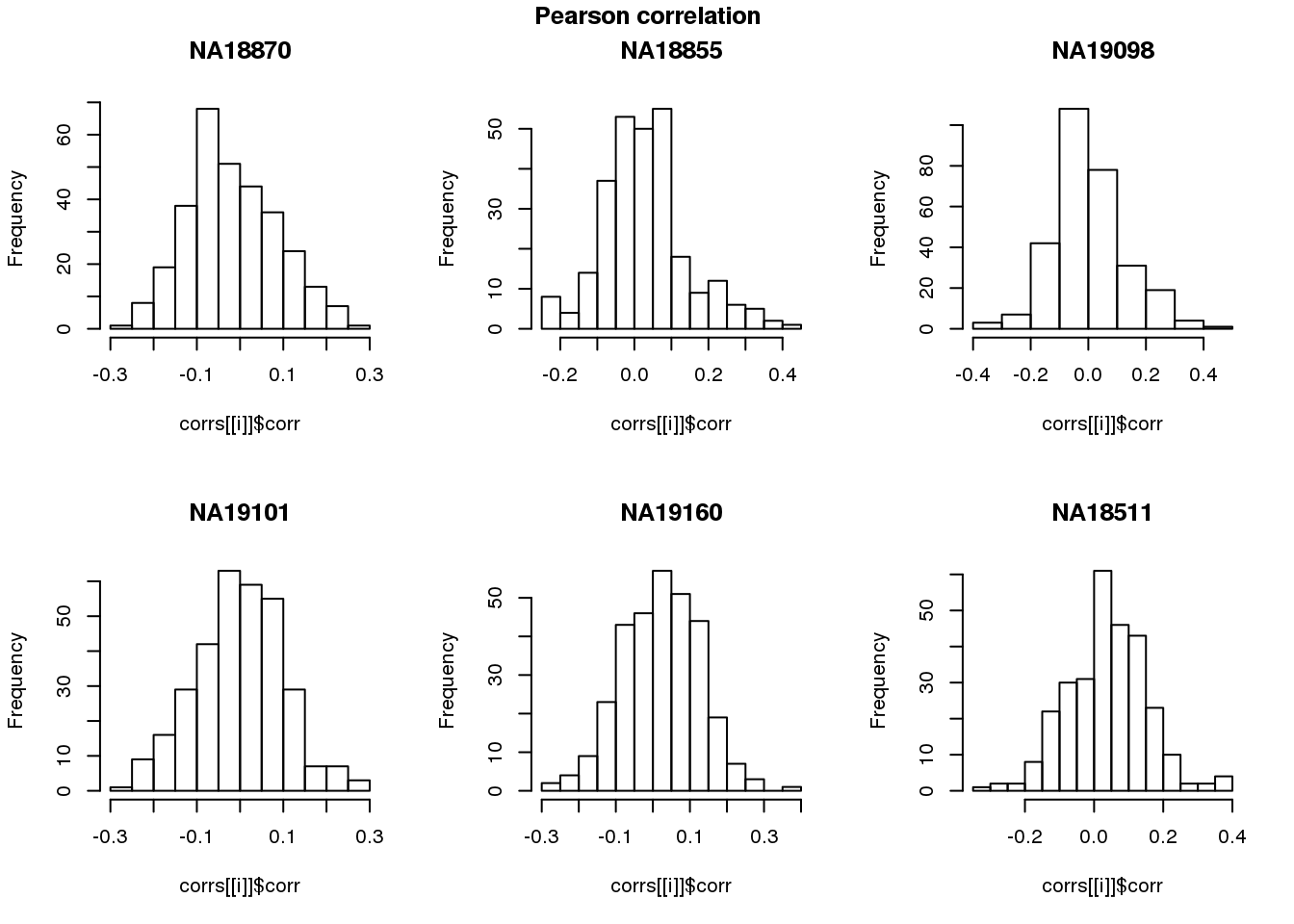
Apply CorShrink
par(mfrow=c(2,3))
for (i in 1:length(corrs)) {
corrs_sub <- corrs[[i]]
corr.shrink <- CorShrinkVector(corrs_sub$corr, nsamp_vec = corrs_sub$nsam,
optmethod = "mixEM", report_model = TRUE)
names(corr.shrink$estimate) <- rownames(corrs_sub)
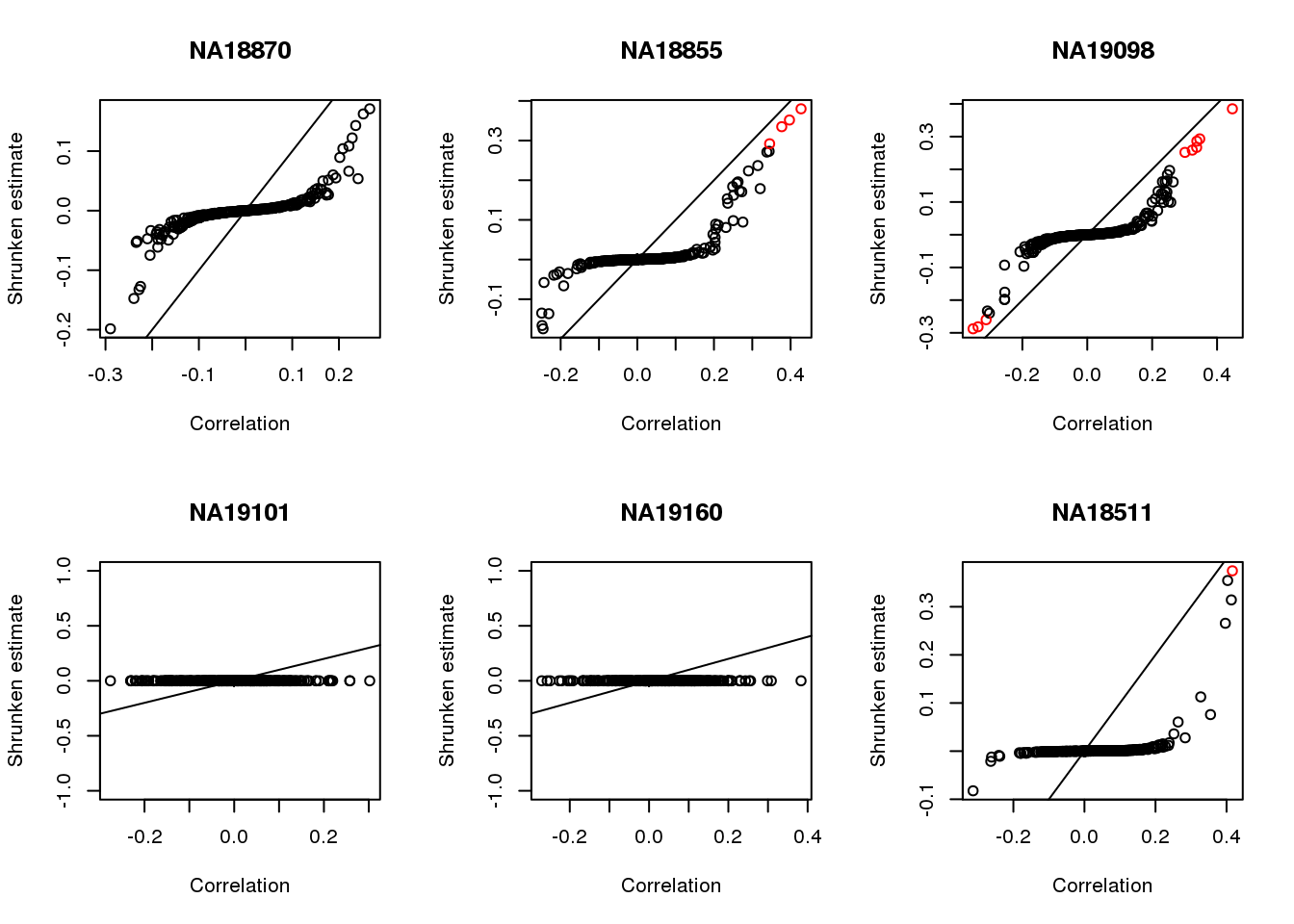
plot(corr.shrink$model$result$betahat,
corr.shrink$model$result$PosteriorMean,
col = 1+as.numeric(corr.shrink$model$result$svalue < .01),
xlab = "Correlation", ylab = "Shrunken estimate")
abline(0,1)
title(names(corrs)[i])
}
Ciruclar correlation
source("../code/corr.cl.R")
corrs.cl <- lapply(1:length(unique(pdata.adj.filt$chip_id)), function(i) {
id <- unique(pdata.adj.filt$chip_id)[i]
log2cpm_sub <- log2cpm.adjust.cycle[, match(rownames(proj.res[[i]][[1]]), colnames(log2cpm.adjust.cycle))]
counts_sub <- counts.cycle[, match(rownames(proj.res[[i]][[1]]), colnames(counts.cycle))]
corrs <- do.call(rbind, lapply(1:nrow(counts_sub), function(g) {
vec <- cbind(as.numeric(proj.res[[i]][[1]]$rads),
log2cpm_sub[g,])
filt <- counts_sub[g,] > 1
nsamp <- sum(filt)
if (nsamp > ncol(counts_sub)/2) {
vec <- vec[filt,]
corr <- R2xtCorrCoeff(lvar=vec[,2], cvar=vec[,1])
nsam <- nrow(vec)
data.frame(corr=corr, nsam=nsam)
} else {
data.frame(corr=NA, nsam=nrow(vec))
}
}))
rownames(corrs) <- rownames(counts_sub)
return(corrs)
})
names(corrs.cl) <- unique(pdata.adj.filt$chip_id)par(mfrow=c(2,3))
for (i in 1:length(corrs)) {
hist(corrs.cl[[i]]$corr, main = names(corrs)[i])
}
title(main = "Circular-linear correlation", outer = TRUE, line = -1)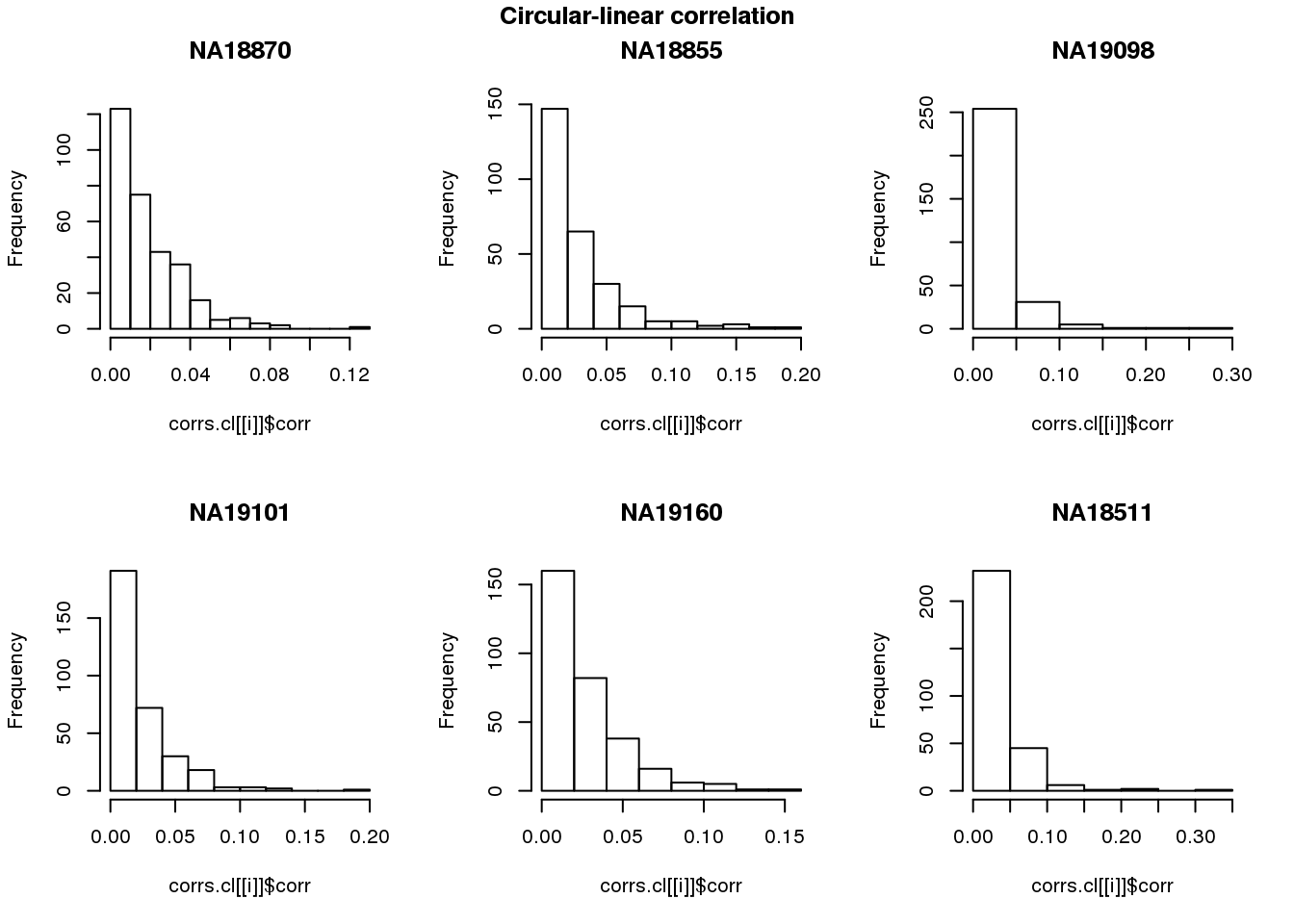
Compute significance value
corrs.cl.sig <- lapply(1:length(unique(pdata.adj.filt$chip_id)), function(i) {
id <- unique(pdata.adj.filt$chip_id)[i]
log2cpm_sub <- log2cpm.adjust.cycle[, match(rownames(proj.res[[i]][[1]]), colnames(log2cpm.adjust.cycle))]
counts_sub <- counts.cycle[, match(rownames(proj.res[[i]][[1]]), colnames(counts.cycle))]
corrs <- do.call(rbind, lapply(1:nrow(counts_sub), function(g) {
vec <- cbind(as.numeric(proj.res[[i]][[1]]$rads),
log2cpm_sub[g,])
filt <- counts_sub[g,] > 1
nsamp <- sum(filt)
if (nsamp > ncol(counts_sub)/2) {
vec <- vec[filt,]
corr <- R2xtIndTestRand(lvar=vec[,2], cvar=vec[,1], NR=100)
nsam <- nrow(vec)
return(corr)
} else {
return(data.frame(corr=NA, pval=NA))
}
}))
rownames(corrs) <- rownames(counts_sub)
return(corrs)
})
names(corrs.cl.sig) <- unique(pdata.adj.filt$chip_id)par(mfrow=c(2,3))
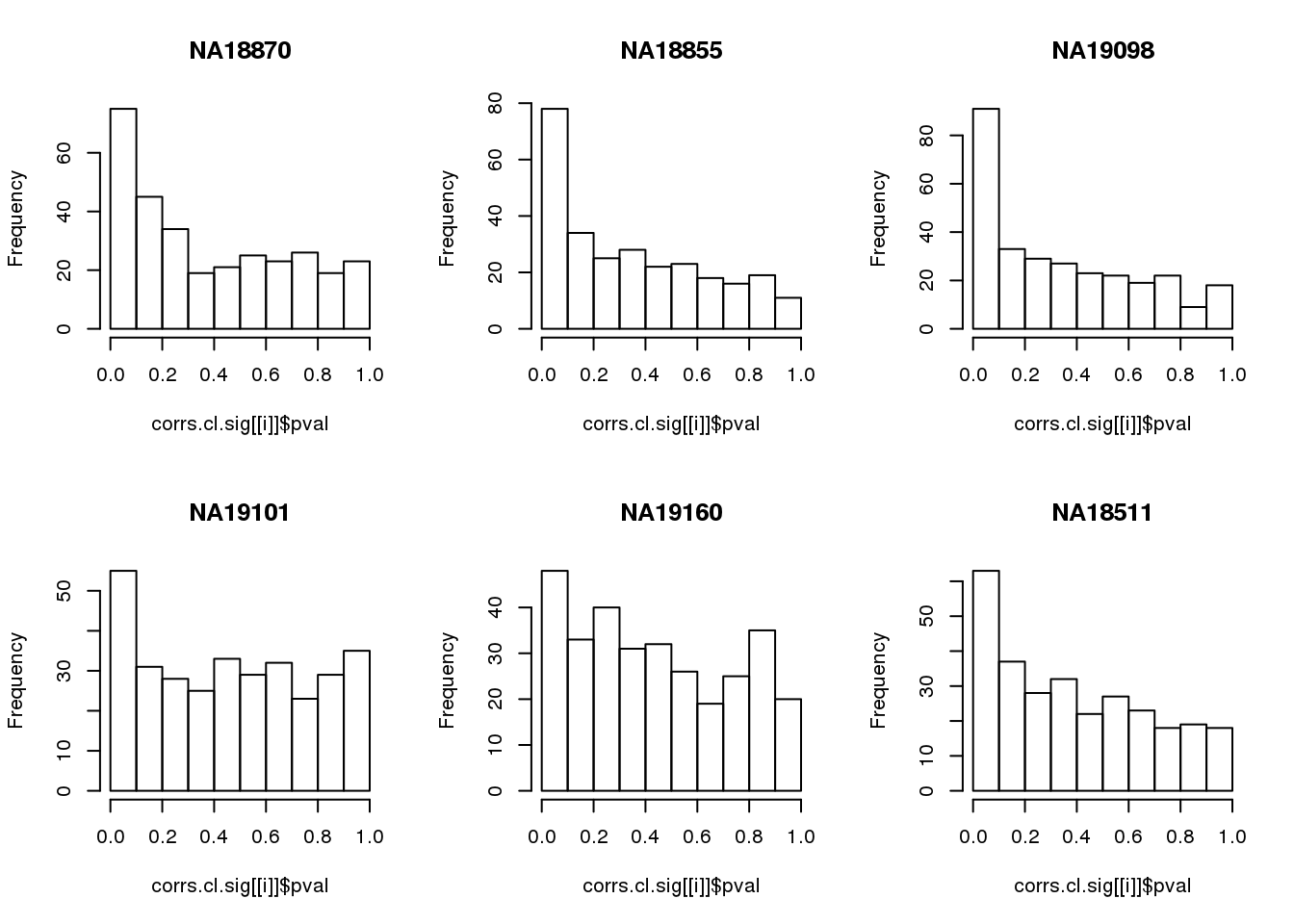
for(i in 1:length(corrs.cl.sig)) {
hist(corrs.cl.sig[[i]]$pval,
main = names(corrs.cl.sig)[i])
}
Consider significant ones
for (i in 1:length(corrs.cl.sig)) {
ii.sig <- corrs.cl.sig[[i]]$pval < .01
print(sum(ii.sig, na.rm=TRUE))
}[1] 12
[1] 21
[1] 23
[1] 11
[1] 4
[1] 10Print some genes
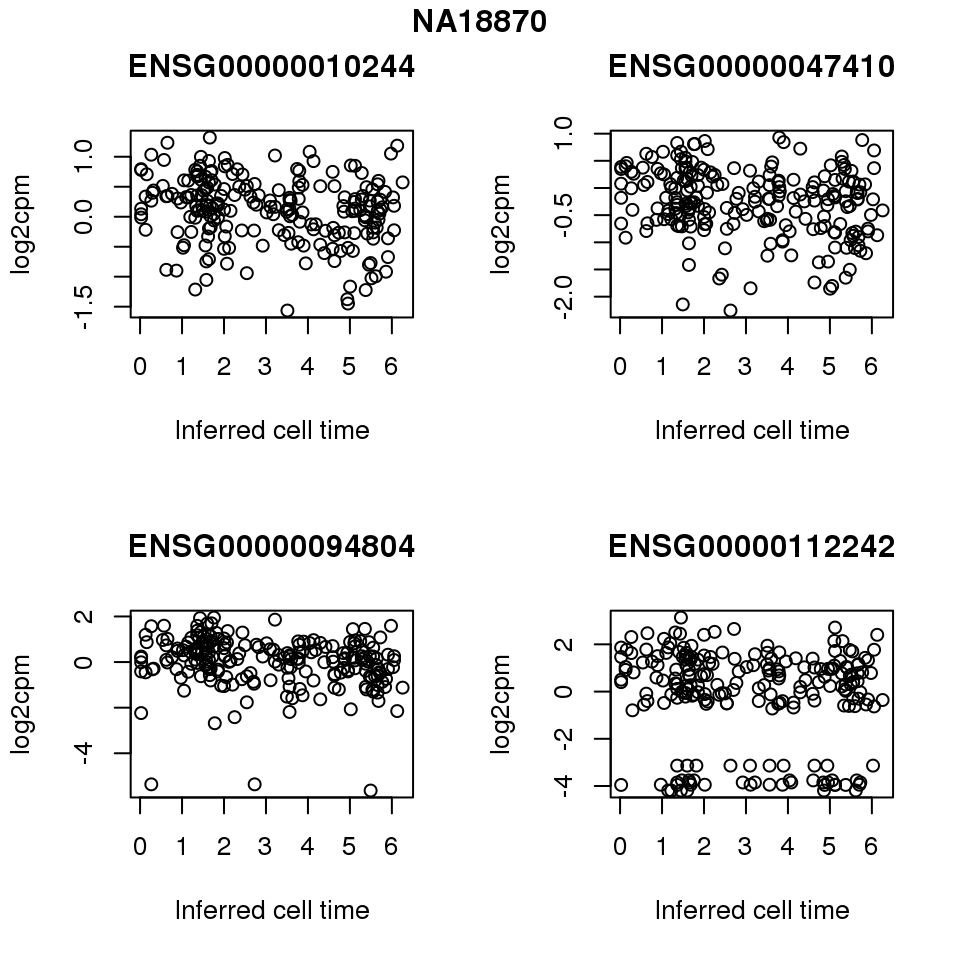
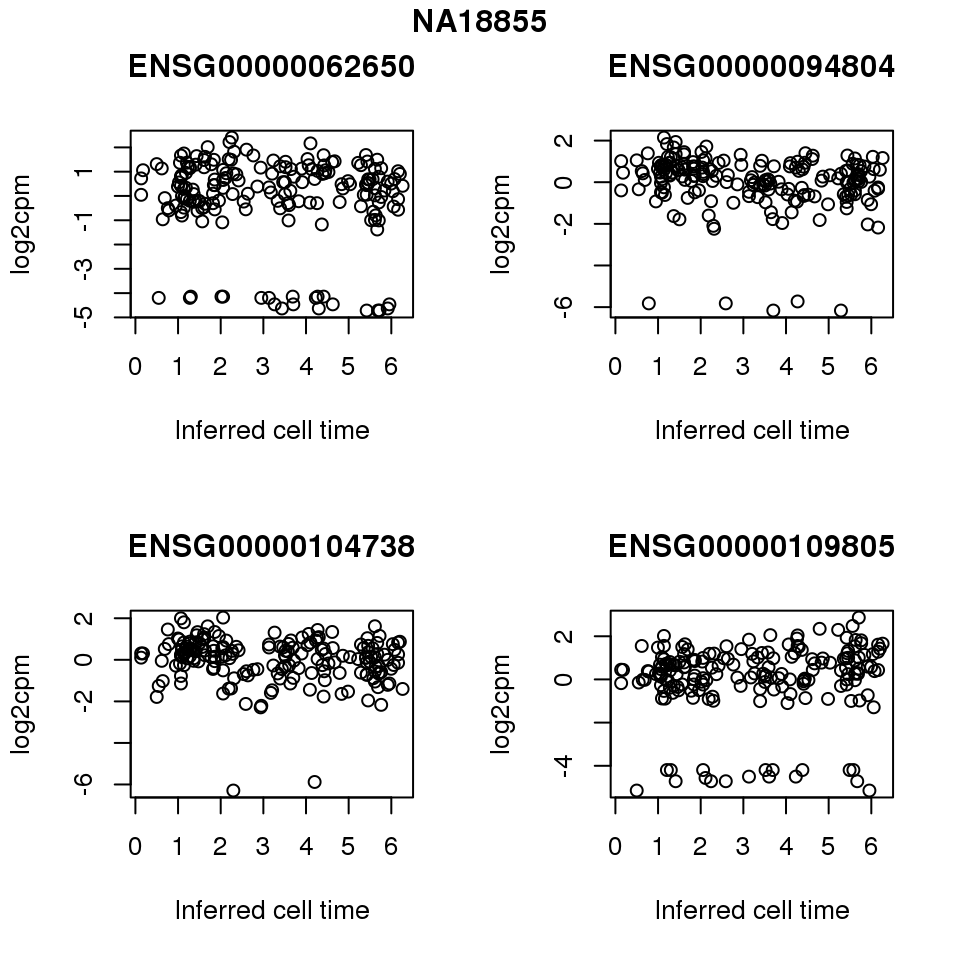
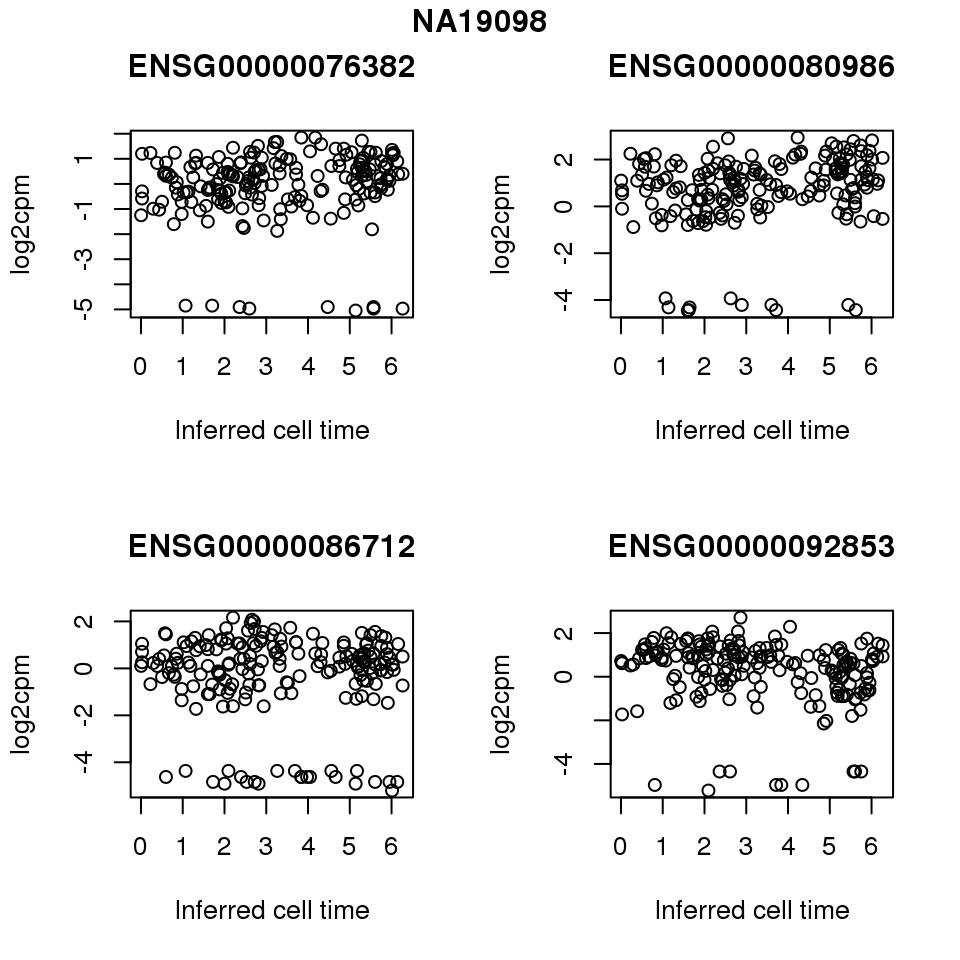
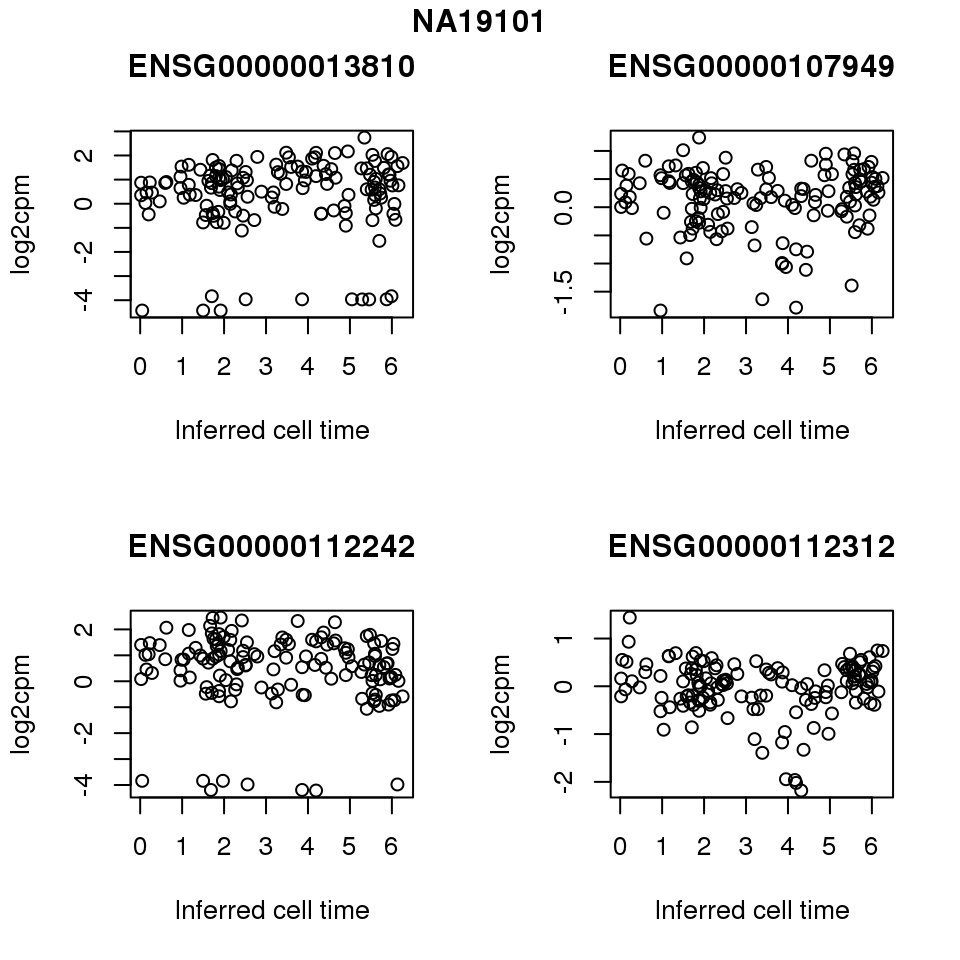
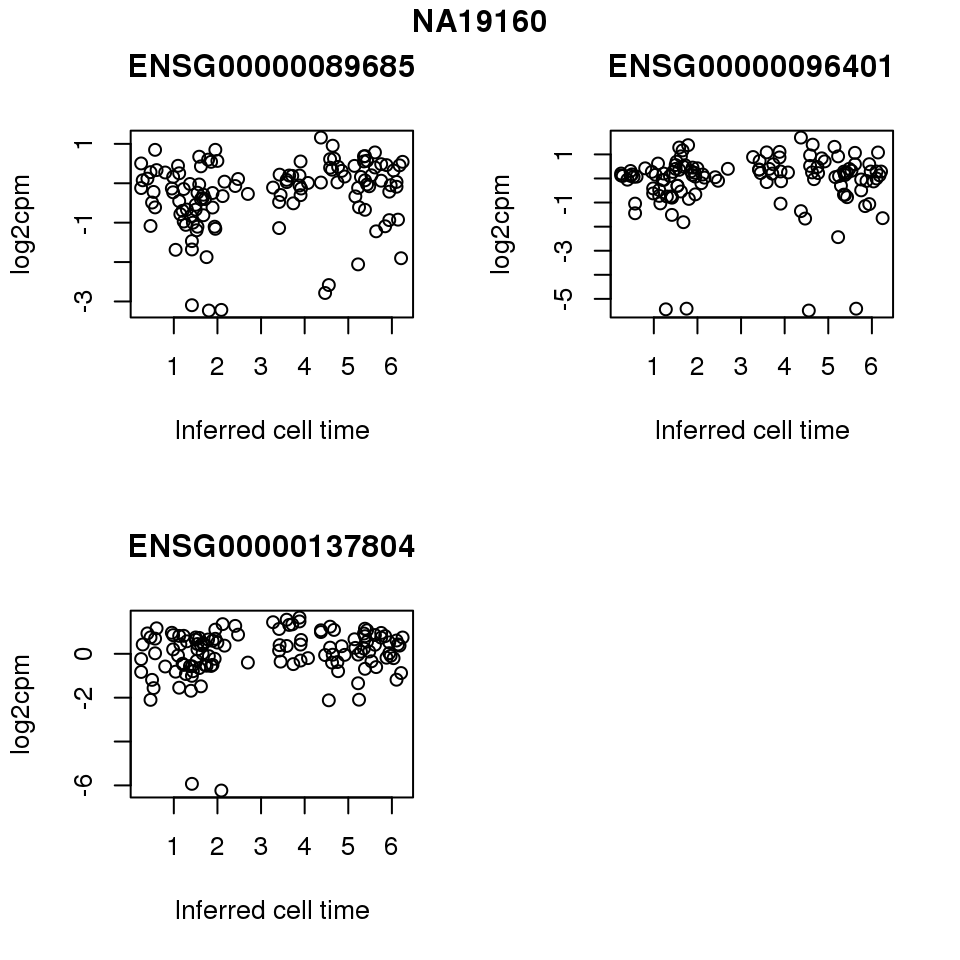
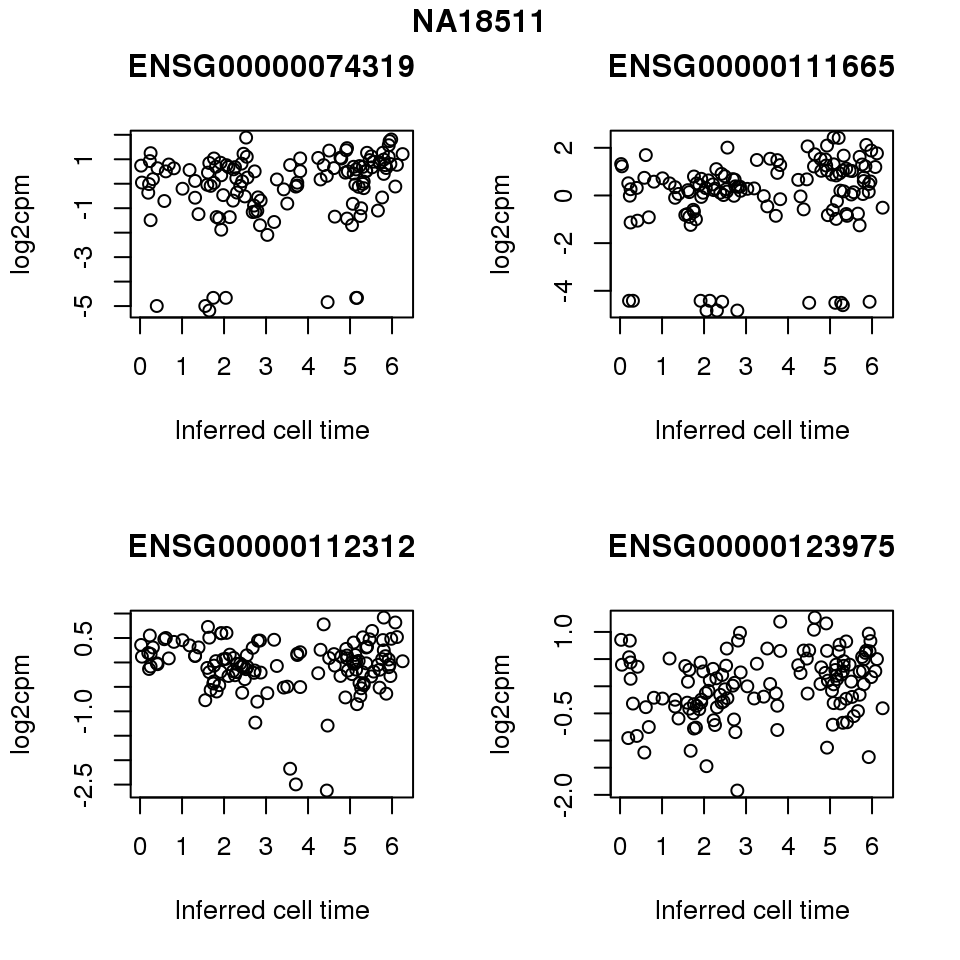
for (i in 1:length(corrs.cl.sig)) {
ii.sig <- corrs.cl.sig[[i]]$pval < .01
id <- unique(pdata.adj.filt$chip_id)[i]
log2cpm_sub <- log2cpm.adjust.cycle[, match(rownames(proj.res[[i]][[1]]), colnames(log2cpm.adjust.cycle))]
genes <- rownames(corrs.cl.sig[[i]])
if (i == 5) {numgene <- 3} else {numgene <- 4}
par(mfrow=c(2,2))
for (g in 1:numgene) {
gene <- genes[which(ii.sig)[g]]
plot(x=as.numeric(proj.res[[i]][[1]]$rads),
y = log2cpm_sub[rownames(log2cpm_sub) == gene,] ,
xlab = "Inferred cell time",
ylab = "log2cpm",
main = gene)
}
title(names(proj.res)[i], outer = TRUE, line = -1)
}
Session information
R version 3.4.1 (2017-06-30)
Platform: x86_64-redhat-linux-gnu (64-bit)
Running under: Scientific Linux 7.2 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /usr/lib64/R/lib/libRblas.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] RColorBrewer_1.1-2 bindrcpp_0.2 CorShrink_0.1.1
[4] matrixStats_0.53.1 dplyr_0.7.4 Biobase_2.38.0
[7] BiocGenerics_0.24.0 conicfit_1.0.4 geigen_2.1
[10] pracma_2.1.4 circular_0.4-93
loaded via a namespace (and not attached):
[1] Rcpp_0.12.15 plyr_1.8.4 compiler_3.4.1
[4] pillar_1.1.0 git2r_0.21.0 bindr_0.1
[7] iterators_1.0.9 tools_3.4.1 boot_1.3-19
[10] digest_0.6.15 evaluate_0.10.1 tibble_1.4.2
[13] lattice_0.20-35 pkgconfig_2.0.1 rlang_0.2.0
[16] foreach_1.4.4 Matrix_1.2-10 yaml_2.1.16
[19] mvtnorm_1.0-7 stringr_1.3.0 knitr_1.20
[22] rprojroot_1.3-2 grid_3.4.1 glue_1.2.0
[25] R6_2.2.2 rmarkdown_1.8 reshape2_1.4.3
[28] ashr_2.2-4 magrittr_1.5 MASS_7.3-47
[31] codetools_0.2-15 backports_1.1.2 htmltools_0.3.6
[34] assertthat_0.2.0 stringi_1.1.6 pscl_1.5.2
[37] doParallel_1.0.11 truncnorm_1.0-7 SQUAREM_2017.10-1This R Markdown site was created with workflowr