Genetic data quality control and processing
Last updated: 2021-03-03
Checks: 6 1
Knit directory: PSYMETAB/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20191126) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| /data/sgg2/jenny/projects/PSYMETAB | . |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: ._docs
Ignored: .drake/
Ignored: analysis/.Rhistory
Ignored: analysis/._GWAS.Rmd
Ignored: analysis/._data_processing_in_genomestudio.Rmd
Ignored: analysis/._quality_control.Rmd
Ignored: analysis/GWAS/
Ignored: analysis/GWAS_results.knit.md
Ignored: analysis/GWAS_results.utf8.md
Ignored: analysis/PRS/
Ignored: analysis/QC/
Ignored: analysis/Rlogo2.png
Ignored: analysis/figure/
Ignored: analysis/rplot.jpg
Ignored: data/processed/
Ignored: data/raw/
Ignored: packrat/lib-R/
Ignored: packrat/lib-ext/
Ignored: packrat/lib/
Ignored: process_init_10_clustermq.out
Ignored: process_init_11_clustermq.out
Ignored: process_init_12_clustermq.out
Ignored: process_init_13_clustermq.out
Ignored: process_init_14_clustermq.out
Ignored: process_init_15_clustermq.out
Ignored: process_init_16_clustermq.out
Ignored: process_init_17_clustermq.out
Ignored: process_init_18_clustermq.out
Ignored: process_init_19_clustermq.out
Ignored: process_init_1_clustermq.out
Ignored: process_init_20_clustermq.out
Ignored: process_init_21_clustermq.out
Ignored: process_init_22_clustermq.out
Ignored: process_init_2_clustermq.out
Ignored: process_init_3_clustermq.out
Ignored: process_init_4_clustermq.out
Ignored: process_init_5_clustermq.out
Ignored: process_init_6_clustermq.out
Ignored: process_init_7_clustermq.out
Ignored: process_init_8_clustermq.out
Ignored: process_init_9_clustermq.out
Ignored: prs_1_clustermq.out
Ignored: prs_2_clustermq.out
Ignored: prs_3_clustermq.out
Ignored: prs_4_clustermq.out
Ignored: prs_5_clustermq.out
Ignored: prs_6_clustermq.out
Ignored: prs_7_clustermq.out
Ignored: prs_8_clustermq.out
Ignored: ukbb_analysis_10_clustermq.out
Ignored: ukbb_analysis_11_clustermq.out
Ignored: ukbb_analysis_12_clustermq.out
Ignored: ukbb_analysis_13_clustermq.out
Ignored: ukbb_analysis_14_clustermq.out
Ignored: ukbb_analysis_15_clustermq.out
Ignored: ukbb_analysis_16_clustermq.out
Ignored: ukbb_analysis_17_clustermq.out
Ignored: ukbb_analysis_18_clustermq.out
Ignored: ukbb_analysis_19_clustermq.out
Ignored: ukbb_analysis_1_clustermq.out
Ignored: ukbb_analysis_20_clustermq.out
Ignored: ukbb_analysis_21_clustermq.out
Ignored: ukbb_analysis_22_clustermq.out
Ignored: ukbb_analysis_2_clustermq.out
Ignored: ukbb_analysis_3_clustermq.out
Ignored: ukbb_analysis_4_clustermq.out
Ignored: ukbb_analysis_5_clustermq.out
Ignored: ukbb_analysis_6_clustermq.out
Ignored: ukbb_analysis_7_clustermq.out
Ignored: ukbb_analysis_8_clustermq.out
Ignored: ukbb_analysis_9_clustermq.out
Untracked files:
Untracked: Rlogo.png
Untracked: Rlogo2.png
Untracked: analysis_prep.log
Untracked: download_impute.log
Untracked: extract_sig.log
Untracked: grs.log
Untracked: init_analysis.log
Untracked: output/PSYMETAB_GWAS_UKBB_comparison.csv
Untracked: output/PSYMETAB_GWAS_UKBB_comparison2.csv
Untracked: output/PSYMETAB_GWAS_baseline_CEU_result.csv
Untracked: output/PSYMETAB_GWAS_subgroup_CEU_result.csv
Untracked: output/coffee_consumed_Neale_UKBB_analysis.csv
Untracked: process_init.log
Untracked: prs.log
Untracked: rplot.jpg
Untracked: test
Untracked: ukbb_analysis.log
Unstaged changes:
Modified: analysis/plans.Rmd
Modified: cache_log.csv
Modified: post_impute.log
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | cf00a10 | Jenny Sjaarda | 2021-03-03 | Testing genetic QC Rmd |
| html | 6d2a07e | Jenny Sjaarda | 2021-03-03 | Build site. |
| Rmd | 105ad4f | Jenny Sjaarda | 2021-03-03 | Testing genetic QC Rmd |
| Rmd | 551bdb5 | Jenny Sjaarda | 2021-03-03 | testing genetics_qc |
| html | 843aad9 | Jenny Sjaarda | 2021-03-03 | Build site. |
| Rmd | e98e3a0 | Jenny Sjaarda | 2021-03-03 | testing genetics_qc |
| Rmd | 941b66d | Jenny Sjaarda | 2021-03-02 | add new Rmd files and respective html files |
| html | 941b66d | Jenny Sjaarda | 2021-03-02 | add new Rmd files and respective html files |
The following document outlines and summarizes the genetic quality control and processing procedure that was followed to create a clean, imputed dataset.
Step 1: Prepare and cluster genomestudio files.
Step 1 was performed entirely on CHUV computer
Part A: Randomize IDs.
- Genetic sampleIDs were recoded according to GPCR algorithm to ensure genetic participants are not identifiable.
code/radomize_IDs.rwas run on CHUV computer before building GenomeStudio project.- Creates a new csv file which was used to create a GenomeStudio project with data provided by lab in Geneva.
- Requires manual addition of header before uploading to GenomeStudio.
[Header],,,,,,,,,,,,,
Investigator Name,,,,,,,,,,,,,
Project Name,,,,,,,,,,,,,
Experiment Name,,,,,,,,,,,,,
Date,,,,,,,,,,,,,
[Manifests],,,,,,,,,,,,,
A,GSA_UPPC_20023490X357589_A1,,,,,,,,,,,,
[Data],,,,,,,,,,,,,- Some samples were found to be duplicates (i.e. 2 samples at 2 different time points were analyzed for the same individual) and they were recoded to have ID as:
${ID}002.
Part B: Create GenomeStudio files.
- Instructions can be found here.
- Required files:
- Sample sheet: as csv file (created above).
- Data repository: as idat files.
- Manifest file: as bpm file.
- Cluster file: as egt file.
- Data provided from Mylene Docquier, copied from sftp and saved here:
L:\PCN\UBPC\ANALYSES_RECHERCHE\Jenny\PSYMETAB_GWAS\data. - Create new IDs based on GPCR randomization (see
code/randomize_IDs.r), and save to above folder as:Eap0819_1t26_27to29corrected_7b9b_randomizedID.csv. - Note that original IDs can be found in the same folder at the file:
Eap0819_1t26_27to29corrected_7b9.csv, if needed. - Create empty folder here:
L:\PCN\UBPC\ANALYSES_RECHERCHE\Jenny\PSYMETAB_GWAS, named:GS_project_26092019(data of creation). - Using new IDs, create genome studio project as follows:
- Open GenomeStudio.
- Select: File > New Genotyping Project.
- Select
L:\PCN\UBPC\ANALYSES_RECHERCHE\Jenny\PSYMETAB_GWASas project repository. - Under project name: use “GS_project_26092019” and click “Next”.
- Select “Use sample sheet to load intensities” and click “Next”.
- Select sample, data and manifests as specified below and click “Next”:
- Sample sheet:
L:\PCN\UBPC\ANALYSES_RECHERCHE\Jenny\PSYMETAB_GWAS\data\Eap0819_1t26_27to29corrected_7b9b_randomizedID.csv, - Data repository:
L:\PCN\UBPC\ANALYSES_RECHERCHE\Jenny\PSYMETAB_GWAS\data, - Manifest repository:
L:\PCN\UBPC\ANALYSES_RECHERCHE\Jenny\PSYMETAB_GWAS\data.
- Sample sheet:
- Select “Import cluster positions from cluster file” and choose cluster file located here:
L:\PCN\UBPC\ANALYSES_RECHERCHE\Jenny\PSYMETAB_GWAS\data\GSPMA24v1_0-A_4349HNR_Samples.egtand click “Finish”.
- Genome studio files were created using the following required files:
- Sample sheet: as csv file (created above).
- Data repository: as idat files (provided from Mylene Docquier).
- Manifest file: as bpm file (provided from Mylene Docquier).
- Cluster file: as egt file (provided from Mylene Docquier).
Part C: Clustering and PLINK conversion.
- Data saved on CHUV servers at the following location:
L:\PCN\UBPC\ANALYSES_RECHERCHE\Jenny\PSYMETAB_GWAS\GS_project_26092019. GS_project_26092019.bscwas opened (requires Genome Studio) and used for clustering.- Clustering was performed according to the guidelines in the webinar (see notes on creating custom cluster files) with the following procedure:
- Cluster: cluster all SNPs, evaluate samples.
- Once data was opened, all SNPs were clustered by clicking “Cluster all SNPs button” in the top panel (icon with 3 red, purple, blue ovals). SNP statistics and heritability estimate updates were ignored.
- In the samples table (bottom left panel) call rate was recalculated by clicking “Calculate” (calculator icon) to recalculate SNP call rates with new cluster positions. SNP statistics and heritability estimate updates were ignored.
- The sample table was then sorted by the “Call Rate” column and samples with call rate <95% were selected and removed from downstream processing (right click, select “Exclude Selected Samples”), 8 samples fell below this cut-off. Updates were ignored. In “SNP Graph” right click and deselect “Show Excluded Samples”.
- Recluster: cluster sex chromosomes, cluster autosomes.
- “SNP Table” was filtered by (“Chr” = Y) using filter icon.
- Using the 3rd and 7th SNP (index 3010 and 3014, 3 samples had missing values for 3010 and sex was determined to be female using 3014), males were selected as those with high intensities (females have no Y chromosome) and set to have an aux value of 101 by right clicking the selected samples in the “Samples Table”. Similarly, females were selected and set to have an aux value of 102.
- “Samples Table” was filtered to have (“Aux” > 100).
- “Samples Table” was sorted on “Aux” column and females were selected (value 102) and excluded. Updates were ignored.
- In “SNP Table”, all SNPs were selected and Y-snps were clustered (right click and “Cluster Selected SNPs”) on only male samples. Updates were ignored.
- Females were re-added to the project in the “Samples Table”.
- “SNP Table” was filtered by (“Chr” = X) using filter icon.
- In “Samples Table”, males were selected (Aux value 101) and excluded. Updates were ignored.
- In “SNP Table”, all SNPs were selected and X-SNPs were clustered (right click and “Cluster Selected SNPs”) on only female samples. Updates were ignored.
- Males were re-added to the project in the “Samples Table”.
- “SNP Table” was filtered by [ !(“Chr” = X ) AND !(“Chr” = Y ) ], using filter icon.
- In “SNP Table”, all SNPs were selected and autosomal SNPs were clustered (right click and “Cluster Selected SNPs”) on all good quality samples. Update SNP statistics.
- In the samples table (bottom left panel) call rate was recalculated by clicking “Calculate” (calculator icon) to recalculate SNP call rates with new cluster positions. SNP statistics and heritability estimate updates were ignored.
- SNP statistics were updated (“Analysis” > “Update SNP statistics”).
- Review and edit: use filters and scores to evaluate SNPs, correct or zero SNPs as needed. No manual editing was performed.
- New genome studio was exported to the following location:
L:\PCN\UBPC\ANALYSES_RECHERCHE\Jenny\PSYMETAB_GWAS, and named:GS_project_26092019_cluster. - Remove filtered individuals:
- In samples table, remove filter for “Aux” > 100.
- Recalculate SNP statistics for these 8 samples only (to save time).
- Note that sample call rates have increased with new clustering positions.
- Project was exported as PLINK file to the following location:
L:\PCN\UBPC\ANALYSES_RECHERCHE\Jenny\PSYMETAB_GWAS, and named:PLINK_091019_0920. - Individual PLINK files within above folder were named according to parent directory as: PSYMETAB_GWAS.ped and PSYMETAB_GWAS.map.
- Ultimately, the final ped/map files reside here:
L:\PCN\UBPC\ANALYSES_RECHERCHE\Jenny\PSYMETAB_GWAS\PLINK_091019_0920.
Notes on GenomeStudio.
- Processing data within GS software is slow in comparison to the rest of the pipeline.
- There is no command line version, only a licensed, GUI version available.
- It is impossible to fully automate and/or parallelize.
Data updates and releases.
- Initial data was received on April 8, 2019:
- Final two plates were received on May 17, 2019.
- Processing began with initial files.
- July 18, 2019 update:
- It came to our attention that 15 participants were genotyped that did not consent.
- This list was sent to Mylene to be removed.
- Plates 27 to 29 were re-provided on 08/08/2019 without these 15 individuals (list below, and provided by Severine in email).
013CB
017CB
074CB
095CB
150CRV
192CRV
193CRV
156CSM
181CSM
191CSM
224UAS
234GL
058GP
246GP
089PP- Until new file was provided, these participants were removed using PLINK to avoid any further analysis of these individuals.
- The new genomestudio file was copied to:
L:\PCN\UBPC\ANALYSES_RECHERCHE\Jenny\PSYMETAB_GWAS\PSYMETAB_GS2\Plates27to29_0819. - The same process above was followed (data opened in GS, cluster positions imported, and data saved to
Plates27to29_0819_cluster, andPLINK_270819_0457). - Old files were deleted to remove all data containing these individuals.
- Updated files were then copied to PSYMETAB_GS1.
- August 28, 2019 update:
- Mylene provided one single genome studio file with all samples (excluding the list from Severine).
- These files were copied to UPPC folders and custom clustering was re-performed.
- These changes are reflected in the above description.
- All old files were subsequently deleted to ensure the data from these participants is completely removed from all databases.
- As of September 3, 2019, all clustering was complete and final PLINK files (
PLINK_030919_0149) were copied to SGG directory (names of plink files according to parent directory:DATA). - September 6, 2019 update:
- It was decided that all IDs part of PSYMETAB should be randomized to ensure they are not identifiable.
- We had a meeting to discuss (Celine, Fred, Chin, Nermine, and Claire), and decided to use a CHUV program (GPCR) for the randomization process.
- We requested with Mylene to create a new project with the new IDs, but she suggested to create our own GS project.
- She provided all relevant data to create our own GS project.
- The description above reflects these changes.
- As of October 11, all GS file were created, clustered and exported as PLINK files and subsequently moved to the sgg server.
Part D: Copy files to SGG server.
- PLINK files were copied to SGG servers using FileZilla
- Host name:
je4649@hpc1.chuv.ch - Password:
<chuv-password> - Port:
22 - Output saved to:
/data/sgg2/jenny/projects/PSYMETAB_GWAS/data/raw.
All subsequent steps were performed on the sgg server and run using drake plan
Step 2: Pre-quality control data prep.
See qc_prep drake plan in code/plans.sh.
- Processed sex and ethnicity files to be used in QC scripts.
- Sex file was created according to the input specified on plink man page (FID, IID, sex [M/F]).
- Ethnicity input file to be used in R script for comparison to genetically derived ethnic groups (by snpweights).
- Recodes ethnic groups as follows:
- Changes French codes to English.
- Changes missing to unknown.
- Groups small ethnic groups to missing.
- A1 rsid conversion file was updated to remove all SNPs labeled with a [.] (see data sources).
- Create duplicates file
Step 3: Pre-imputation quality control.
Results of Step 3-6 are saved to analysis/QC. The majority of analyses were performed using PLINK (either version 2.0 or 1.9) Each sub-spet (i.e. 0-15) corresponds to one folder within analysis/QC
Source code for Step 3 can be found at: code/pre_imputation_qc.sh.
0. Preprocessing.
- Create binary PLINK files (if necessary):
- 761641 variants initially.
- 2767 individuals initially.
- Exclude Y and MT variants:
- 11641 Y / MT variants removed.
- 750000 variants remaining.
- Remove duplicate variants:
- 7945 duplicates removed.
- 742055 variants remaining.
- Update sex:
- Using the file located at:
data/processed/phenotype_data/PSYMETAB_GWAS_sex.txt(created above).
F M 1298 1469 - Using the file located at:
- Remove duplicate individuals that were identified previously (i.e. two different IDs for the same participant):
- 2 duplicates identified and removed
- 2752 indviduals remaining.
- Sanity check: ensure that duplicates have the same genetic info"
- From the PLINK webpage: Note that KING kinship coefficients are scaled such that duplicate samples have kinship 0.5, not 1. First-degree relations (parent-child, full siblings) correspond to ~0.25, second-degree relations correspond to ~0.125, etc.
- If all duplicate ID samples have KINSHIP ~0.5, then indeed they are genetic duplicates.
#FID1 ID1 FID2 ID2 NSNP HETHET IBS0 KINSHIP
1 2071 BEEEDIGO002 224 BEEEDIGO 703110 0.178137 0.00000000000 0.499539
2 1873 CQLIXEZP002 64 CQLIXEZP 703045 0.153413 0.00000142238 0.499504
3 1965 EFWKQOIK002 1433 EFWKQOIK 697403 0.151525 0.00000860335 0.496680
4 1886 HFNWJHCI002 1448 HFNWJHCI 702845 0.153089 0.00000426837 0.499547
5 2075 HROOJNCI002 553 HROOJNCI 702167 0.155970 0.00000284833 0.499257
6 1974 IOAWLZGK002 549 IOAWLZGK 704278 0.153028 0.00000000000 0.499847
7 2314 KLFEBCIE002 1916 KLFEBCIE 700799 0.153949 0.00000570777 0.499007
8 2073 LWCGLSDP002 317 LWCGLSDP 702226 0.150114 0.00000427213 0.499363
9 2379 PBAIFEMQ002 2070 PBAIFEMQ 700642 0.154083 0.00000285452 0.498820
10 2009 PNWDYVRH002 494 PNWDYVRH 703993 0.153736 0.00000284094 0.499806
11 2068 QHNUPGWK002 318 QHNUPGWK 702891 0.154500 0.00000569078 0.499363
12 1928 QZAUHIPY002 559 QZAUHIPY 702896 0.144711 0.00000142269 0.499826
13 2067 SSITXXAY002 283 SSITXXAY 702409 0.152603 0.00000284734 0.499418
14 1947 WKBFDWJF002 566 WKBFDWJF 703642 0.153506 0.00000284235 0.499783
15 1657 XABRILAR002 1385 XABRILAR 698282 0.154672 0.00000000000 0.4973151. Strand alignment (so all SNPs are on positive strand).
- Update chromosome
- Update position
- Flip alleles on negative strand
- Extract SNPs that are not in strand file
- Remove any non autosomal / X chromosomal variants (might have changed due to the pos and chr update):
- 11 duplicates removed.
- 741937 variants remaining.
- Update rsids using
data/processed/reference_files/rsid_conversion.txt - Remove duplicate variants:
- 6723 duplicates removed.
- 734328 variants remaining.
2. Removal of SNPs that have MAF zero.
- Calculate frequency of all SNPs
- Remove MAF 0 SNPs:
- 93578 variants with
MAF = 0. - 640750 variants remaining.
- 93578 variants with
3. Missingness.
- Exclude variants with >10% missingness (using
geno --0.1):- 6480 variants removed.
- 634270 variants remaining.
- Exclude individuals with >10% missingness (using
mind --0.1):- 3 individuals removed.
- 2749 individuals remaining.
- Exclude variants with >5% missingness (using
geno --0.05):- 8256 variants removed.
- 626014 variants remaining.
- Exclude individuals with >5% missingness (using
mind --0.05):- 3 individuals removed.
- 2746 individuals remaining.
- Exclude variants with >1% missingness (using
geno --0.01):- 35957 variants removed.
- 590057 variants remaining.
- Exclude individuals with >1% missingness (using
mind --0.01):- 5 individuals removed.
- 2741 individuals remaining.
*Total removed: 50693 variants (7.91%) and 11 individuals (0.40%).**
4. Sex check.
- Perform sex check.
- Remove unambiguous sex violations.
- 26 individuals removed.
- 2715 individuals remaining.
5. Imputation preparation.
- Write frequency of final QC’d file (from #4) to file (using
-- freq). - Using McCarthy Group Tools, QC’d files were prepared for imputation using the script
HRC-1000G-check-bim-NoReadKey.pl(download link).- This script checks: Strand, alleles, position, Ref/Alt assignments and frequency differences.
- Produces: A set of plink commands to update or remove SNPs based on the checks as well as a file (FreqPlot) of cohort allele frequency vs reference panel allele frequency.
- Updates: Strand, position, ref/alt assignment.
- Removes: A/T & G/C SNPs if MAF > 0.4, SNPs with differing alleles, SNPs with > 0.2 allele frequency difference (can be removed/changed in V4.2.2), SNPs not in reference panel
- Replace underscores in fam file since vcf conversion uses understcores between
FIDandIID. - Run the generated
Run-plink.shscript from #2. - Sort outputed
vcffiles. - Download zipped files to personl or CHUV files to copy to Michigan Imputation Server.
Step 4: Imputation.
Source code for Step 4 can be found at: code/download_imputation.sh and code/check_imputation.sh.
6. Run and download imputation.
- Upload downloaded QC’d
vcf.gzfiles to Michigan Imputation Server as follows:- Select
Run,Genotype Imputation (Minimac4). - Reference panel:
HRC r1.1 2016 (GRCh37/hg19). - Array build:
GRCh37/hg19. - rsq Filter:
off. - Phasing:
Eagle v2.4 (phased output). - Population:
EUR. - Mode:
Quality Control & Imputation.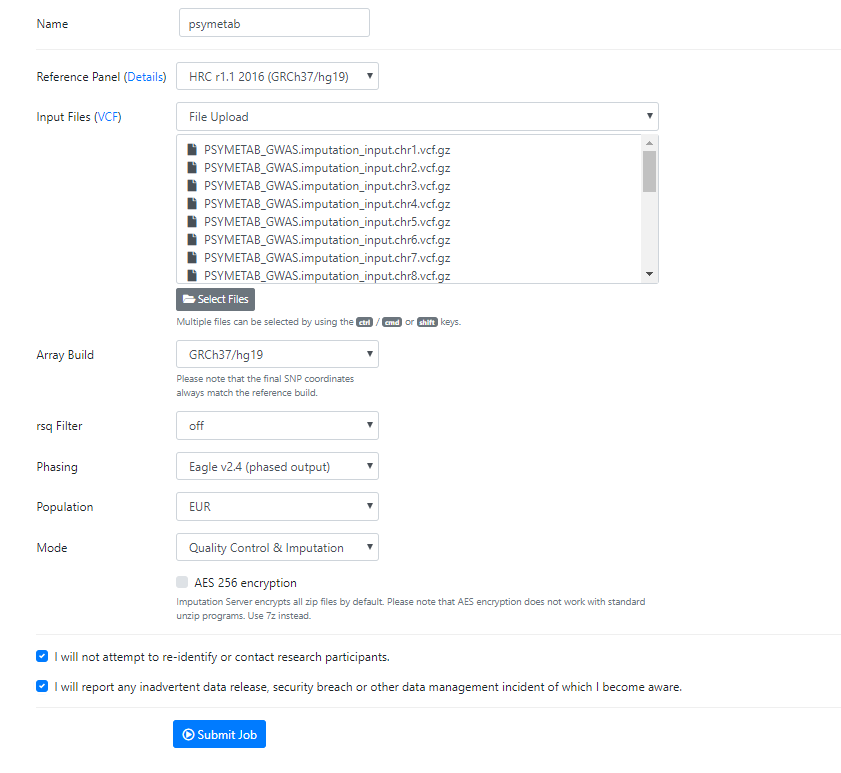
- Select
2. Download imputation, using password from email retrieve the following files: - QC report. - QC stats. - Logs. - Imputation results.
[1] "archive" "chr1.info.gz" "chr10.info.gz"
[4] "chr11.info.gz" "chr12.info.gz" "chr13.info.gz"
[7] "chr14.info.gz" "chr15.info.gz" "chr16.info.gz"
[10] "chr17.info.gz" "chr18.info.gz" "chr19.info.gz"
[13] "chr2.info.gz" "chr20.info.gz" "chr21.info.gz"
[16] "chr22.info.gz" "chr3.info.gz" "chr4.info.gz"
[19] "chr5.info.gz" "chr6.info.gz" "chr7.info.gz"
[22] "chr8.info.gz" "chr9.info.gz" "qcreport.html"
[25] "snps-excluded.txt" Chr Num imputed variants
1 1 3069931
2 2 3392237
3 3 2821894
4 4 2787581
5 5 2588168
6 6 2460111
7 7 2289305
8 8 2242705
9 9 1686471
10 10 1927503
11 11 1936990
12 12 1848117
13 13 1385433
14 14 1270436
15 15 1139215
16 16 1281297
17 17 1090072
18 18 1104755
19 19 868554
20 20 884983
21 21 531276
22 22 524544
23 all 391315787. Check imputation.
- Using Will Rayner’s post imputation tool, produce a visual summary of the imputated results from the Michigan Imputation Server:
Step 5: Post imputation quality control.
8. PLINK conversion.
Source code for Step 5 can be found at: code/post_imputation_qc.sh.
- Remove SNPs with
info < 0.30
Chr Num imputed variants R2 filtered
1 1 3069931 2223452
2 2 3392237 2454557
3 3 2821894 2059285
4 4 2787581 2043343
5 5 2588168 1887050
6 6 2460111 1813223
7 7 2289305 1659374
8 8 2242705 1632513
9 9 1686471 1218207
10 10 1927503 1411585
11 11 1936990 1421749
12 12 1848117 1352003
13 13 1385433 1019900
14 14 1270436 920528
15 15 1139215 816986
16 16 1281297 894859
17 17 1090072 771058
18 18 1104755 798012
19 19 868554 607422
20 20 884983 631946
21 21 531276 374376
22 22 524544 367153
23 all 39131578 28378581- Update
bimfile to include rsIDs instead ofchr:bpconvention:- Output of Michigan Impuation server is in format
chr:bp:ref:alt. - Convenient to have SNPs in regular rsIDs for extraction, etc.
- If there is no known rsID, SNP name is left as
chr:bp:ref:alt. - Reference file for determining rsIDs can be found here:
/data/sgg2/jenny/data/dbSNP/dbSNP_SNP_list_chr${chr}.txt, which was processed according to description injenny/SGG_generic/scripts/public_data.sh.
- Output of Michigan Impuation server is in format
9. Extract typed SNPs.
- Write a list of only genotypd data to run in snpweights software (using
require=info "TYPED"flag).
Chr Num imputed variants R2 filtered Typed SNPs
1 1 3069931 2223452 44252
2 2 3392237 2454557 45514
3 3 2821894 2059285 37277
4 4 2787581 2043343 34139
5 5 2588168 1887050 32223
6 6 2460111 1813223 38957
7 7 2289305 1659374 30649
8 8 2242705 1632513 28611
9 9 1686471 1218207 23815
10 10 1927503 1411585 27714
11 11 1936990 1421749 27722
12 12 1848117 1352003 26603
13 13 1385433 1019900 19346
14 14 1270436 920528 17920
15 15 1139215 816986 17002
16 16 1281297 894859 18447
17 17 1090072 771058 16646
18 18 1104755 798012 15625
19 19 868554 607422 12997
20 20 884983 631946 13557
21 21 531276 374376 7582
22 22 524544 367153 8104
23 all 39131578 28378581 544702- Hard call back to genotypes (
bim,bed, andfamfiles) using the--hard-call-thresholdflag set at0.1. - Merge by chromosome genotyped data into single file.
10. Merge imputed SNPs.
- Convert fileset from step #8 back to
vcfas there is no merge function in plink (using the flag--recode vcf id-paste=iid vcf-dosage=HDS). - Merge using
bcftools concat. - Convert back to pgen using
plink2 --vcf <output-name> dosage=HDS.
12. Ethnicty check and admixture estimation.
- Clean typed data (from #9).
- Remove duplicate samples (from #11).
- Remove genotypes with missingness > 10% (using
--geno 0.1). - Remove genotypes with MAF < 5% (using
--maf 0.05). - Remove genotypes with HWE < 5e-4 (using
--hwe 5e-4). - After filters, 249634 SNPs remaining.
- After filters, 2697 individuals remaining.
- Run
snpweightssoftware from Alkes Price software page using SNP weights for European, West African, East Asian and Native American ancestral populations (downloaded here).- SNPweights is a software package for inferring genome-wide genetic ancestry using SNP weights precomputed from large external reference panels (Chen et al. 2013 Bioinformatics).
- The software relies on external databases to infer ancestry weights, so related individuals can safely be included.
- This software creates a file with the name
${output_name}.NA.predpc. - The 10 columns in the
${output_name}.NA.predpcoutput file are:sample ID,population label,number of SNPs used for inference,predicted PC1,predicted PC2,predicted PC3,% YRI ancestry,% CEU ancestry,% East Asian ancestry, and% Native American ancestry.
- Filter to Europeans.
- Using threshold
% CEU ancestry>0.8as done in other papers. - 543 non-European individuals removed.
- 2154 European individuals remaining.
- Using threshold
- Run
snpweightsin Europeans using SNP weights for NW, SE and AJ ancestral populations of European Americans (downloaded here).- The 8 columns in the
${output_name}.CEU80.EA.predpcoutput file are:sample ID,population label,number of SNPs used for inference,predicted PC1,predicted PC2,% Northwest European ancestry,% Southeast European ancestry,% Ashkenazi Jewish ancestry. - Note that nothing was done with these computed snpweights.
- The 8 columns in the
- Using data from step 1, create pruned set of SNPs.
- Remove related individuals (idenified in #11).
- PLINK parameters set as:
--indep-pairwise 50 5 0.2. - After pruning, 152879 SNPs removed
- Could consider excluding regions suggested by flaspca by adding the following line:
--exclude range $flashpca/exclusion_regions_hg19.txt.
- Create an unrelated dataset for calculating PCs using snps from step 5.
- After pruning, 96755 SNPs remaining.
- 50 related samples removed.
- 2647 unrelated samples remaining.
- Create a full set for projecting PCs with same set of pruned SNPs.
- As above, 96755 SNPs remaining.
- No samples removed, 2697 samples in total.
- Perform PCA using
flashpcain unrelated set (from step 6). - Perform PCA projections onto related sample set (from 12.7)
- Split data into seperate ethnic groups as determined in 12.2.
- For all ethnic groups except
YRI(African), use a threshold of% ancestry>0.8. - For African, use a threshold of
% ancestry>0.7. - If a participant has no ethnic group that meets this criteria (note that it is impossible to follow into multiple categories), they are classified as
MIXED, in this way 5 ethnic groups remain:CEU(European),EA(East Asian),NA(Native American),YRI(West African), andMIXED.
- For all ethnic groups except
AFRICAN EAST_ASIAN EUROPEAN LATIN OTHER SOUTH_ASIAN UNKNOWN Total
CEU 0 0 1540 0 72 1 541 2154
EA 0 17 0 0 2 3 7 29
MIXED 54 4 79 0 99 55 135 426
NA 0 0 0 1 3 0 1 5
YRI 47 4 1 1 3 0 27 83

sessionInfo()R version 3.5.3 (2019-03-11)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS: /data/sgg2/jenny/bin/R-3.5.3/lib64/R/lib/libRblas.so
LAPACK: /data/sgg2/jenny/bin/R-3.5.3/lib64/R/lib/libRlapack.so
locale:
[1] en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] tidyselect_0.2.5 rbgen_0.1 ukbtools_0.11.3
[4] hrbrthemes_0.8.0 OpenImageR_1.1.6 fuzzyjoin_0.1.5
[7] kableExtra_1.1.0 R.utils_2.9.2 R.oo_1.23.0
[10] R.methodsS3_1.7.1 TwoSampleMR_0.4.25 reader_1.0.6
[13] NCmisc_1.1.6 optparse_1.6.4 readxl_1.3.1
[16] ggthemes_4.2.0 tryCatchLog_1.1.6 futile.logger_1.4.3
[19] DataExplorer_0.8.0 taRifx_1.0.6.1 qqman_0.1.4
[22] MASS_7.3-51.5 bit64_0.9-7 bit_1.1-14
[25] rslurm_0.5.0 rmeta_3.0 devtools_2.2.1
[28] usethis_1.5.1 data.table_1.12.8 clustermq_0.8.8.1
[31] future.batchtools_0.8.1 future_1.15.1 rlang_0.4.5
[34] knitr_1.26 drake_7.12.0.9000 forcats_0.4.0
[37] stringr_1.4.0 dplyr_0.8.3 purrr_0.3.3
[40] readr_1.3.1 tidyr_1.0.3 tibble_2.1.3
[43] ggplot2_3.3.2 tidyverse_1.3.0 pacman_0.5.1
[46] processx_3.4.1 workflowr_1.6.0
loaded via a namespace (and not attached):
[1] backports_1.1.6 systemfonts_0.2.3 plyr_1.8.5
[4] igraph_1.2.5 storr_1.2.1 listenv_0.8.0
[7] digest_0.6.25 foreach_1.4.7 htmltools_0.4.0
[10] tiff_0.1-5 fansi_0.4.1 magrittr_1.5
[13] checkmate_1.9.4 memoise_1.1.0 base64url_1.4
[16] doParallel_1.0.15 remotes_2.1.0 globals_0.12.5
[19] extrafont_0.17 modelr_0.1.5 extrafontdb_1.0
[22] prettyunits_1.1.0 jpeg_0.1-8.1 colorspace_1.4-1
[25] rvest_0.3.5 rappdirs_0.3.1 haven_2.2.0
[28] xfun_0.11 callr_3.4.0 crayon_1.3.4
[31] jsonlite_1.6 iterators_1.0.12 brew_1.0-6
[34] glue_1.4.0 gtable_0.3.0 webshot_0.5.2
[37] pkgbuild_1.0.6 Rttf2pt1_1.3.8 scales_1.1.0
[40] futile.options_1.0.1 DBI_1.1.0 Rcpp_1.0.3
[43] xtable_1.8-4 viridisLite_0.3.0 progress_1.2.2
[46] txtq_0.2.0 htmlwidgets_1.5.1 httr_1.4.1
[49] getopt_1.20.3 calibrate_1.7.5 ellipsis_0.3.0
[52] farver_2.0.1 XML_3.98-1.20 pkgconfig_2.0.3
[55] dbplyr_1.4.2 labeling_0.3 reshape2_1.4.3
[58] later_1.0.0 munsell_0.5.0 cellranger_1.1.0
[61] tools_3.5.3 cli_2.0.1 generics_0.0.2
[64] broom_0.5.3 fastmap_1.0.1 evaluate_0.14
[67] yaml_2.2.0 fs_1.3.1 packrat_0.5.0
[70] nlme_3.1-143 mime_0.8 whisker_0.4
[73] formatR_1.7 proftools_0.99-2 xml2_1.2.2
[76] compiler_3.5.3 rstudioapi_0.10 png_0.1-7
[79] filelock_1.0.2 testthat_2.3.1 reprex_0.3.0
[82] stringi_1.4.5 highr_0.8 ps_1.3.0
[85] desc_1.2.0 gdtools_0.2.2 lattice_0.20-38
[88] vctrs_0.2.4 pillar_1.4.3 lifecycle_0.1.0
[91] networkD3_0.4 httpuv_1.5.2 R6_2.4.1
[94] promises_1.1.0 gridExtra_2.3 sessioninfo_1.1.1
[97] codetools_0.2-16 lambda.r_1.2.4 assertthat_0.2.1
[100] pkgload_1.0.2 rprojroot_1.3-2 withr_2.1.2
[103] batchtools_0.9.12 parallel_3.5.3 hms_0.5.3
[106] grid_3.5.3 rmarkdown_1.18 git2r_0.26.1
[109] shiny_1.4.0 lubridate_1.7.4