OutOfAfrica_3G09 [fix shared factor]
Joseph Marcus
2020-05-18
Last updated: 2020-05-18
Checks: 6 1
Knit directory: drift-workflow/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190211) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version efb1fba. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .snakemake/
Ignored: data/datasets/
Ignored: data/raw/
Ignored: data/simulations/
Ignored: nb-log-1570452.err
Ignored: nb-log-1570452.out
Ignored: notebooks/.ipynb_checkpoints/
Ignored: output/
Ignored: sandbox/.ipynb_checkpoints/
Untracked files:
Untracked: analysis/OutOfAfrica_3G09_fix.Rmd
Unstaged changes:
Modified: analysis/OutOfAfrica_3G09.Rmd
Modified: analysis/index.Rmd
Modified: code/structure_plot.R
Modified: start-rstudio.sbatch
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with wflow_publish() to start tracking its development.
Here I visualize population structure with simulated data from the OutOfAfrica_3G09 scenario. See Figure 2. from Gutenkunst et al. 2009.
Below, I show a number of EBMF solutions and in each of them I don’t display the first shared factor which is prefixed to the one-vector and scale the loadings by the prior variance. I only describe the loadings that remain after the shared factor.
Imports
Import the required libraries and scripts:
suppressMessages({
library(lfa)
library(flashier)
library(drift.alpha)
library(ggplot2)
library(RColorBrewer)
library(reshape2)
library(tidyverse)
library(alstructure)
source("../code/structure_plot.R")
})Data
data_path <- "../output/simulations/OutOfAfrica_3G09/rep1.txt"
Y <- t(as.matrix(read.table(data_path, sep=" ")))
colnames(Y) <- NULL
rownames(Y) <- NULL
n <- nrow(Y)
daf <- colSums(Y) / (2 * n)
colors <- brewer.pal(8, "Set2")
# filter out too rare and too common SNPs
Y <- Y[,((daf>=.05) & (daf <=.95))]
p <- ncol(Y)
print(n)[1] 120print(p)[1] 29815# sub-population labels from stdpop
labs <- rep(c("YRI", "CEU", "HAN"), each=40)we end up with 120 individuals and ~30000 SNPs. View fitted the sample covariance matrix:
plot_cov((1.0 / p) * Y %*% t(Y), as.is=T)
flash [greedy]
Run the greedy algorithm:
ones <- matrix(1, nrow = n, ncol = 1)
ls.soln <- t(solve(crossprod(ones), crossprod(ones, Y)))
fl <- flash.init(Y) %>%
flash.init.factors(EF = list(ones, ls.soln),
prior.family=c(prior.bimodal(), prior.normal())) %>%
flash.fix.loadings(kset = 1, mode = 1L) %>%
flash.backfit() %>%
flash.add.greedy(Kmax=6, prior.family=c(prior.bimodal(), prior.normal()))Backfitting 1 factors (tolerance: 5.33e-02)...
Difference between iterations is within 1.0e-01...
Wrapping up...
Done.
Adding factor 2 to flash object...
Adding factor 3 to flash object...
Adding factor 4 to flash object...
Adding factor 5 to flash object...
Factor doesn't significantly increase objective and won't be added.
Wrapping up...
Done.sd <- unlist(lapply(fl$fitted.g[[2]], '[[', 3))
L <- fl$flash.fit$EF[[1]]
LDsqrt <- L %*% diag(sd)
K <- ncol(LDsqrt)
plot_loadings(LDsqrt[,2:K], labs) + scale_color_brewer(palette="Set2")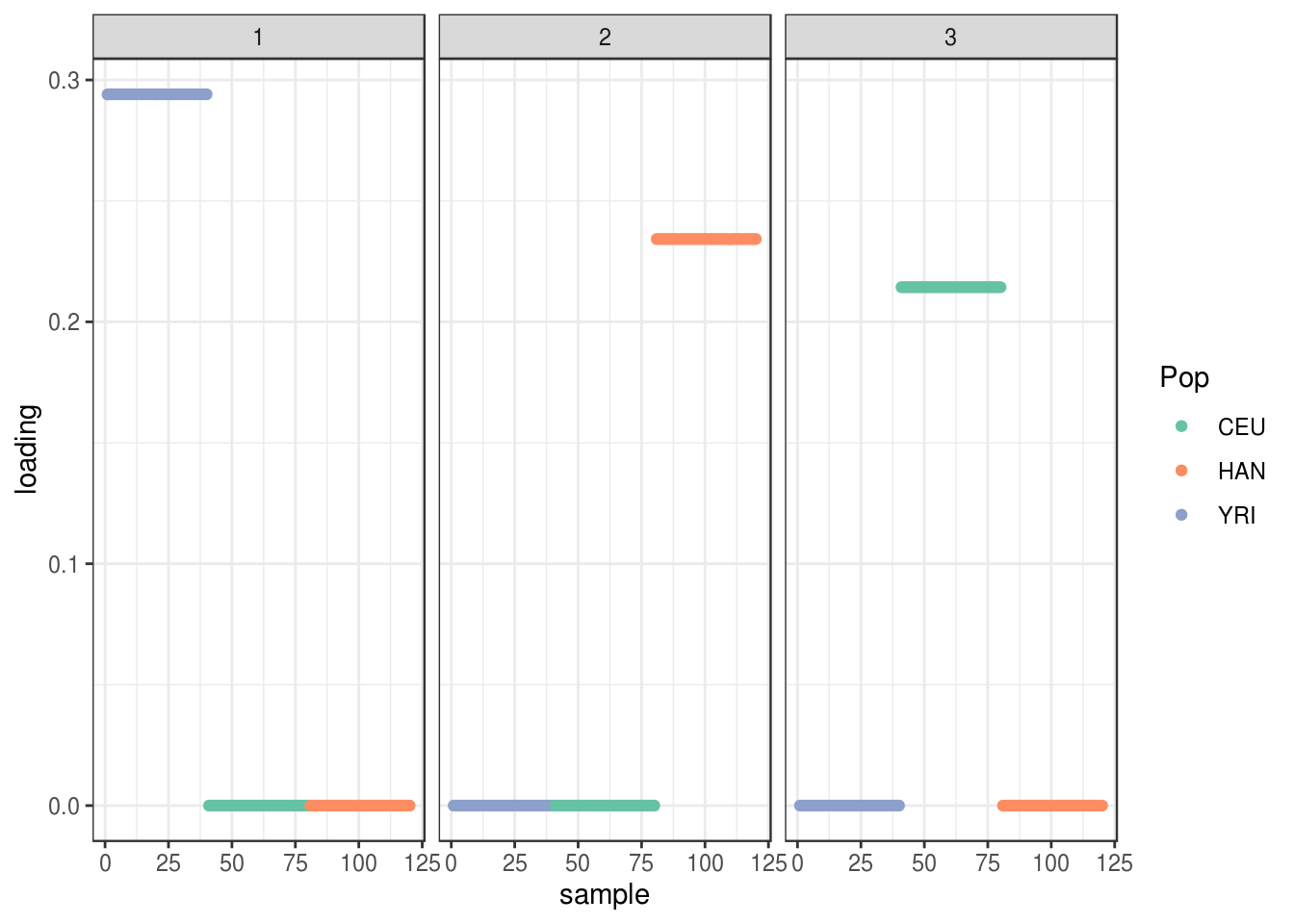
view structure plot:
create_structure_plot(L=LDsqrt[,2:K], labels=labs, colors=colors)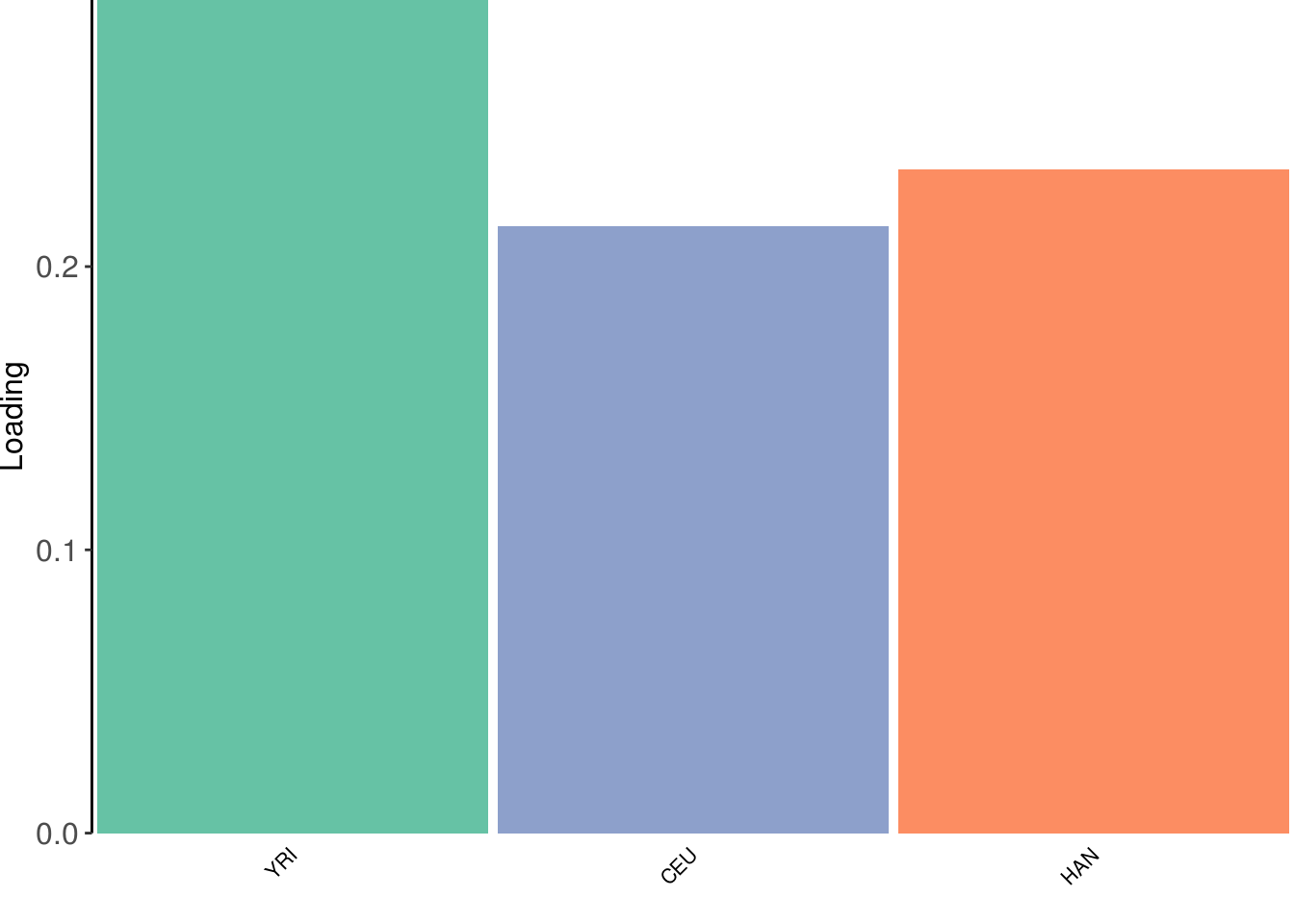
view fitted covariance matrix:
plot_cov(LDsqrt %*% t(LDsqrt), as.is=T)
the greedy algorithm finds 3 population specific factors.
flash [backfit]
Run flash [backfit] initializing from the greedy solution:
flbf <- fl %>%
flash.backfit() %>%
flash.nullcheck(remove=TRUE)Backfitting 4 factors (tolerance: 5.33e-02)...
Difference between iterations is within 1.0e+02...
Difference between iterations is within 1.0e+01...
Difference between iterations is within 1.0e+00...
Difference between iterations is within 1.0e-01...
Difference between iterations is within 1.0e-02...
Wrapping up...
Done.
Nullchecking 4 factors...
Wrapping up...
Done.sd <- unlist(lapply(flbf$fitted.g[[2]], '[[', 3))
L <- flbf$flash.fit$EF[[1]]
LDsqrt <- L %*% diag(sd)
K <- ncol(LDsqrt)
plot_loadings(LDsqrt[,2:K], labs) + scale_color_brewer(palette="Set2")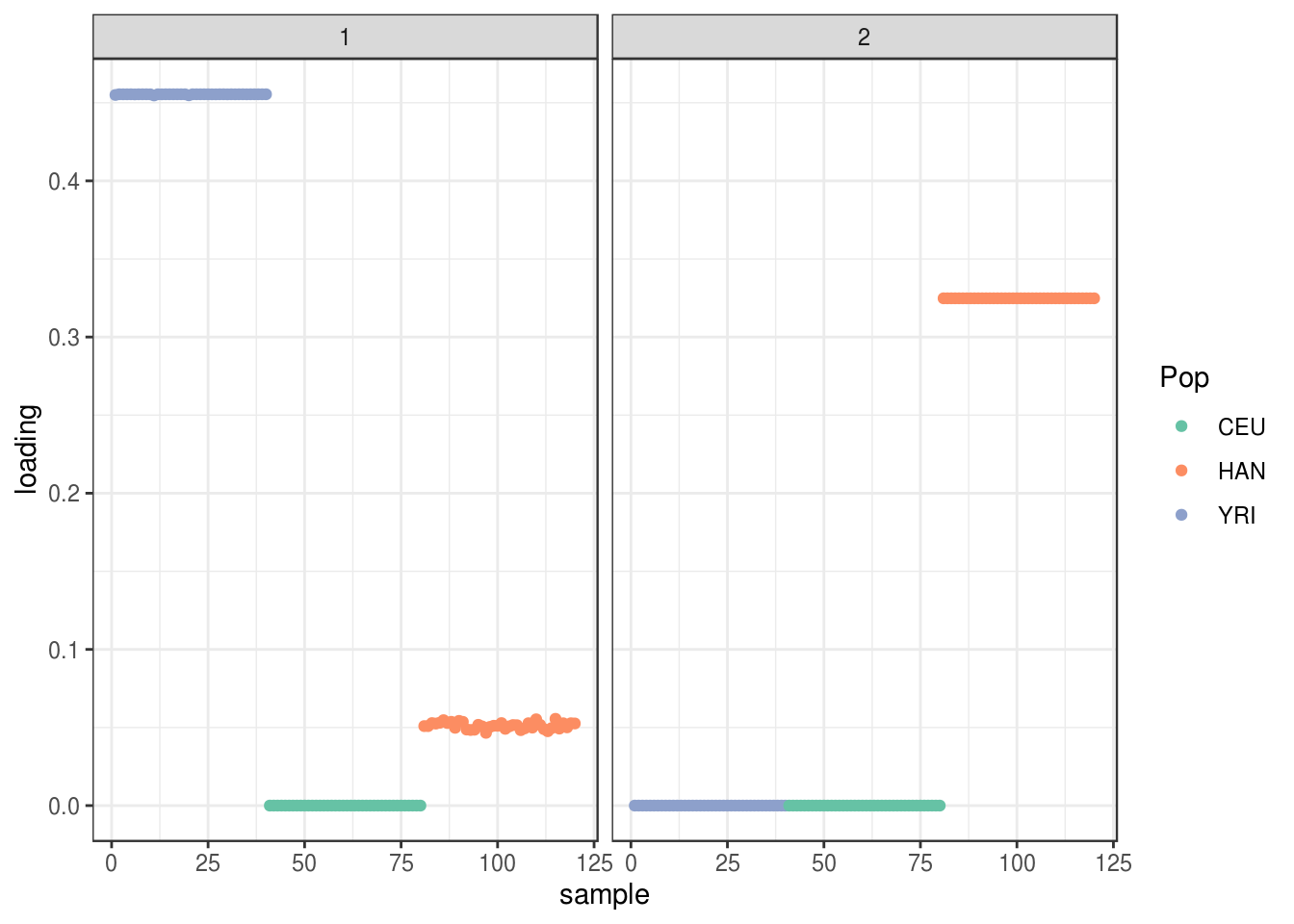
view structure plot:
create_structure_plot(L=LDsqrt[,2:K], labels=labs, colors=colors)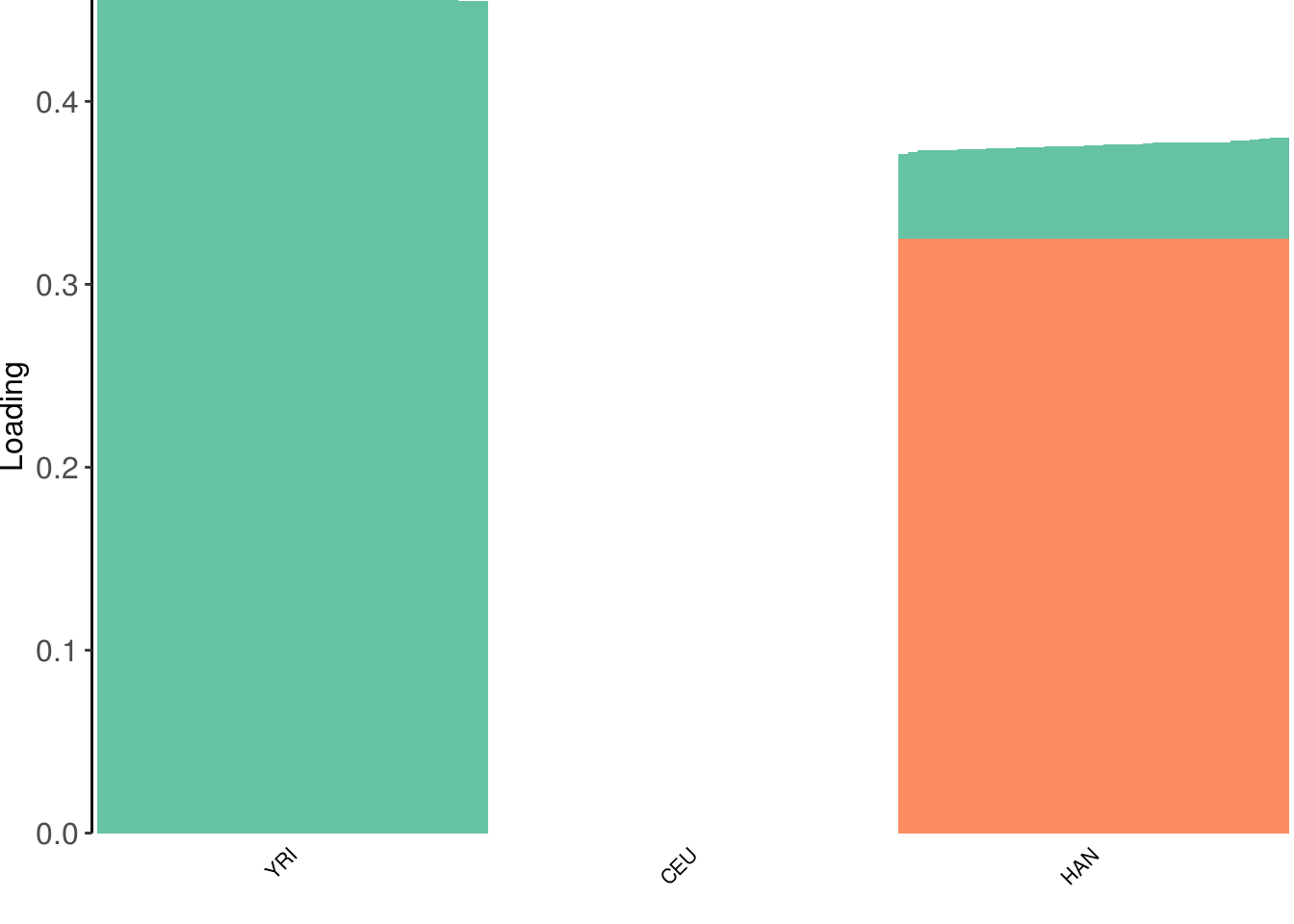
view fitted covariance matrix:
plot_cov(LDsqrt %*% t(LDsqrt), as.is=T)
the backfitting algorithm represents the data with a sparser solution and finds a factor represented by YRI and a small loading from Han and
drift
Run drift initializing from the greedy solution:
init <- init_from_flash(fl)
dr <- drift(init, miniter=2,
maxiter=1000,
tol=0.01,
verbose=FALSE,
update_order=c(0, 2:ncol(init$EL), -1))
sd <- sqrt(dr$prior_s2)
L <- dr$EL
LDsqrt <- L %*% diag(sd)
K <- ncol(LDsqrt)
plot_loadings(LDsqrt[,2:K], labs) + scale_color_brewer(palette="Set2")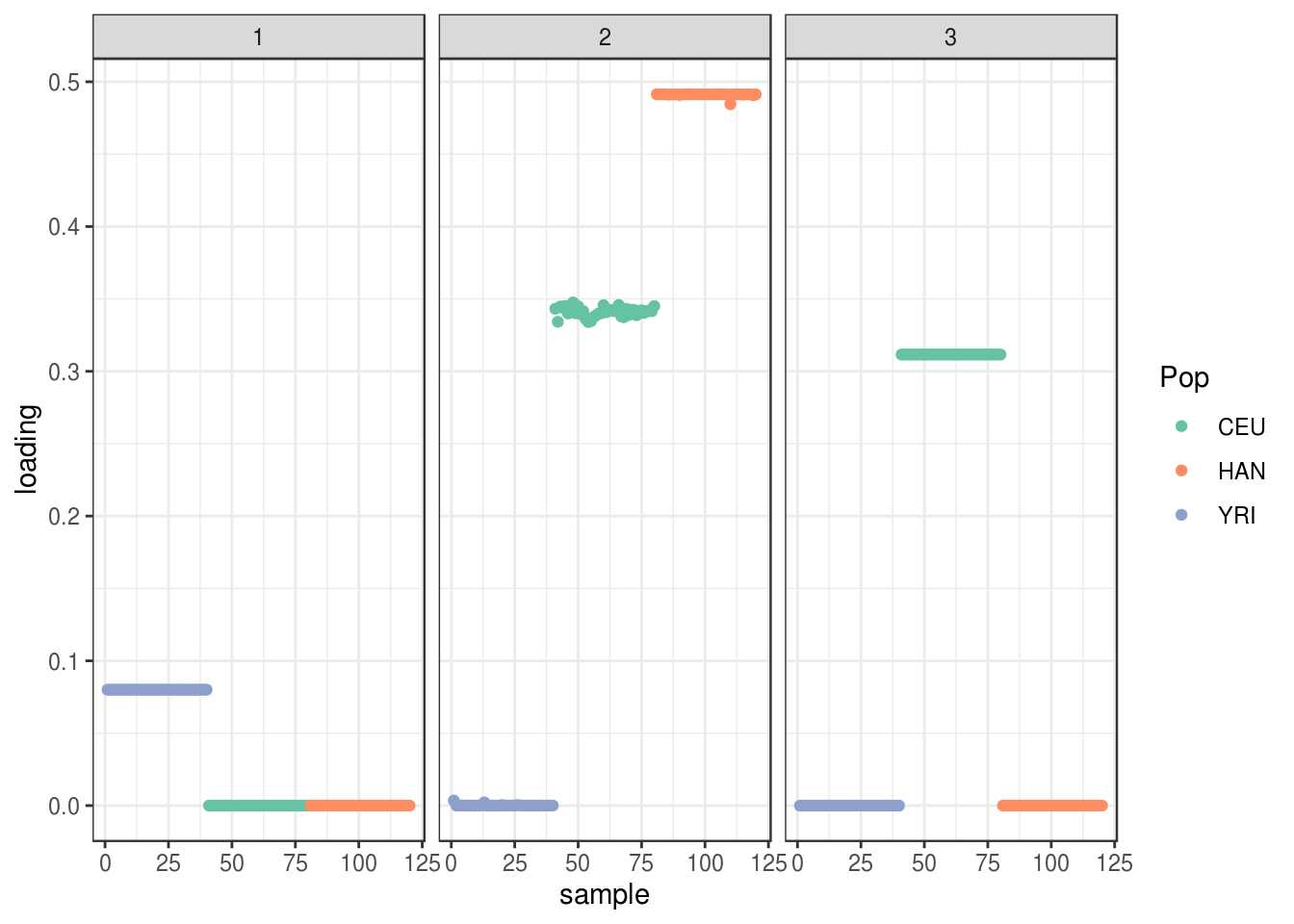
view structure plot:
create_structure_plot(L=LDsqrt[,2:K], labels=labs, colors=colors)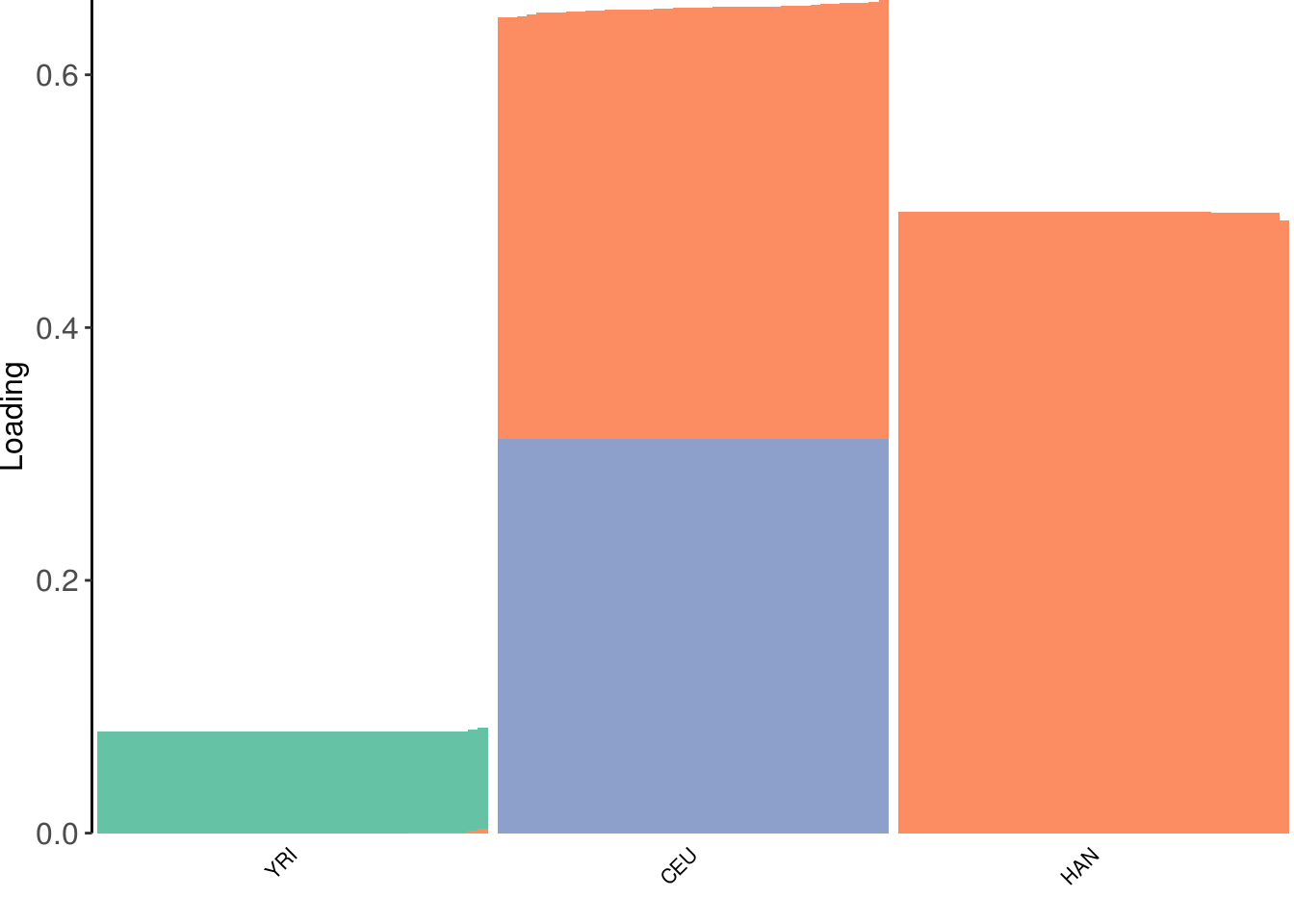
view fitted covariance matrix:
plot_cov(LDsqrt %*% t(LDsqrt), as.is=T)
the drift algorithm finds two population specific factors and a shared factor between HAN CEU. Also notably the population specific factor for Africa has lower total magnitude then for OOA populations which makes senses as the prior variance for OOA should be higher (i.e. more drift). Note the solution here is a bit of hack in that we don’t update the loadings or factors for the first factor. This will be fixed in the drift.alpha package soon.
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] alstructure_0.1.0 forcats_0.5.0 stringr_1.4.0
[4] dplyr_0.8.5 purrr_0.3.4 readr_1.3.1
[7] tidyr_1.0.2 tibble_3.0.1 tidyverse_1.3.0
[10] reshape2_1.4.3 RColorBrewer_1.1-2 ggplot2_3.3.0
[13] drift.alpha_0.0.9 flashier_0.2.4 lfa_1.9.0
loaded via a namespace (and not attached):
[1] httr_1.4.1 jsonlite_1.6 modelr_0.1.6 assertthat_0.2.1
[5] mixsqp_0.3-17 cellranger_1.1.0 yaml_2.2.0 ebnm_0.1-24
[9] pillar_1.4.3 backports_1.1.6 lattice_0.20-38 glue_1.4.0
[13] digest_0.6.25 promises_1.0.1 rvest_0.3.5 colorspace_1.4-1
[17] htmltools_0.3.6 httpuv_1.4.5 Matrix_1.2-15 plyr_1.8.4
[21] pkgconfig_2.0.3 invgamma_1.1 broom_0.5.6 haven_2.2.0
[25] corpcor_1.6.9 scales_1.1.0 later_0.7.5 git2r_0.26.1
[29] farver_2.0.3 generics_0.0.2 ellipsis_0.3.0 withr_2.2.0
[33] ashr_2.2-50 cli_2.0.2 magrittr_1.5 crayon_1.3.4
[37] readxl_1.3.1 evaluate_0.14 fs_1.3.1 fansi_0.4.1
[41] nlme_3.1-137 xml2_1.3.2 truncnorm_1.0-8 tools_3.5.1
[45] hms_0.5.3 lifecycle_0.2.0 munsell_0.5.0 reprex_0.3.0
[49] irlba_2.3.3 compiler_3.5.1 rlang_0.4.5 grid_3.5.1
[53] rstudioapi_0.11 labeling_0.3 rmarkdown_1.10 gtable_0.3.0
[57] DBI_1.0.0 R6_2.4.1 lubridate_1.7.4 knitr_1.20
[61] workflowr_1.6.1 rprojroot_1.3-2 stringi_1.4.6 parallel_3.5.1
[65] SQUAREM_2020.2 Rcpp_1.0.4.6 vctrs_0.2.4 dbplyr_1.4.3
[69] tidyselect_1.0.0