Finemapping using Mapgen with UKBB reference LD matrices
Kaixuan Luo
Last updated: 2023-11-06
Checks: 7 0
Knit directory: analysis_pipelines/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200524) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version b117f93. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Untracked files:
Untracked: analysis/test_sldsc_splicingAnnot.Rmd
Untracked: code/compute_ldscore_generic_annot.sbatch
Untracked: code/extract_baselineLD_generic_annot.R
Untracked: code/ldsc_make_binary_annot_compute_ldscores_bedfiles.sbatch
Untracked: code/make_ldsc_binary_annots_from_bedfiles.R
Untracked: code/sldsc_annot_generic_baselineLD_separate.sbatch
Untracked: scripts/tmp.R
Unstaged changes:
Modified: analysis/mapgen_torus_enrichment_heart_atlas.Rmd
Modified: analysis/sldsc_example_GTEx_QTLs.Rmd
Modified: analysis/sldsc_pipeline.Rmd
Modified: code/extract_baselineLDv2.2_generic_annot.R
Modified: code/mapgen_trackplots.R
Modified: scripts/run_finemapping.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown
(analysis/mapgen_finemapping_diagnosis_ukbb_ld.Rmd) and
HTML (docs/mapgen_finemapping_diagnosis_ukbb_ld.html)
files. If you’ve configured a remote Git repository (see
?wflow_git_remote), click on the hyperlinks in the table
below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | b117f93 | kevinlkx | 2023-11-06 | wflow_rename("analysis/mapgen_finemapping_ukbb_ld.Rmd", "analysis/mapgen_finemapping_diagnosis_ukbb_ld.Rmd") |
| html | b117f93 | kevinlkx | 2023-11-06 | wflow_rename("analysis/mapgen_finemapping_ukbb_ld.Rmd", "analysis/mapgen_finemapping_diagnosis_ukbb_ld.Rmd") |
library(mapgen)
library(tidyverse)
library(susieR)
library(ggplot2)Load an example asthma GWAS summary statistics
gwas.file <- '/project2/xinhe/shared_data/mapgen/example_data/GWAS/aoa_v3_gwas_ukbsnps_susie_input.txt.gz'
gwas.sumstats <- vroom::vroom(gwas.file, col_names = TRUE, show_col_types = FALSE)
head(gwas.sumstats)# A tibble: 6 × 10
chr pos beta se a0 a1 snp pval zscore locus
<dbl> <dbl> <dbl> <dbl> <chr> <chr> <chr> <dbl> <dbl> <dbl>
1 1 693731 0.00277 0.0156 A G rs12238997 0.859 0.178 1
2 1 707522 0.00337 0.0169 G C rs371890604 0.841 0.200 1
3 1 717587 -0.0538 0.0429 G A rs144155419 0.210 -1.25 1
4 1 723329 0.00182 0.128 A T rs189787166 0.989 0.0143 1
5 1 729679 0.00577 0.0142 C G rs4951859 0.684 0.407 1
6 1 730087 -0.00465 0.0220 T C rs148120343 0.832 -0.212 1n = 336210LD blocks
LD_blocks <- readRDS(system.file('extdata', 'LD.blocks.EUR.hg19.rds', package='mapgen'))
head(LD_blocks, 3) chr start end locus
1 1 10583 1892607 1
2 1 1892607 3582736 2
3 1 3582736 4380811 3Process GWAS summary statistics
gwas.sumstats <- process_gwas_sumstats(gwas.sumstats,
chr='chr',
pos='pos',
beta='beta',
se='se',
a0='a0',
a1='a1',
snp='snp',
pval='pval',
LD_Blocks=LD_blocks)Cleaning summary statistics...
Assigning GWAS SNPs to LD blocks...Select GWAS significant loci with -log10(pval) < 5e-8
if(max(gwas.sumstats$pval) <= 1){
gwas.sumstats <- gwas.sumstats %>% dplyr::mutate(pval = -log10(pval))
}
sig.loci <- gwas.sumstats %>% dplyr::filter(pval > -log10(5e-8)) %>% dplyr::pull(locus) %>% unique()
sumstats.sigloci <- gwas.sumstats[gwas.sumstats$locus %in% sig.loci, ]
cat(length(sig.loci), "significant loci.\n")19 significant loci.Load LD matrix, match variants between GWAS and LD matrix
locus <- sig.loci[2]
LDREF <- load_UKBB_LDREF(LD_blocks, locus = locus,
LDREF.dir = "/project2/mstephens/wcrouse/UKB_LDR_0.1_b37", prefix = "ukb_b37_0.1")
matched.sumstat.LD.res <- match_gwas_LDREF(gwas.sumstats, LDREF$R, LDREF$var_info)
sumstats.locus <- matched.sumstat.LD.res$sumstats
R.locus <- matched.sumstat.LD.res$ROriginal data
Run SuSiE with LD matrices
LD_matrices <- list(R.locus)
names(LD_matrices) <- locus
susie.locus.res <- run_finemapping(sumstats.locus, LD_matrices = LD_matrices, priortype = 'uniform', n = n, L = 10)Finemapping locus 193...
Run susie_rss...susie.locus.res[[1]]$sets$cs
$cs$L2
[1] 464 485 498 499 508 517 530 564 574 593 597 615 628 630 636
[16] 644 649 653 654 668 672 682 684 689 690 726 729 731 732 733
[31] 738 747 771 773 778 789 804 835 841 852 856 859 873 881 882
[46] 889 893 903 905 909 921 923 924 926 929 947 956 960 965 984
[61] 987 1010 1014 1018 1034 1062 1067 1146
$cs$L1
[1] 602 632 637 660 706 711 714 734 735 739 740 749 751 759 763 766 768 769 775
[20] 779 782 787 790 791 794 797 812 813 814 854 857 865 894 895 896 899 916 932
$purity
min.abs.corr mean.abs.corr median.abs.corr
L2 0.7923272 0.9172110 0.9318115
L1 0.5835378 0.8666369 0.8125560
$cs_index
[1] 2 1
$coverage
[1] 0.9517707 0.9512414
$requested_coverage
[1] 0.95susie_plot(susie.locus.res[[1]], y='PIP')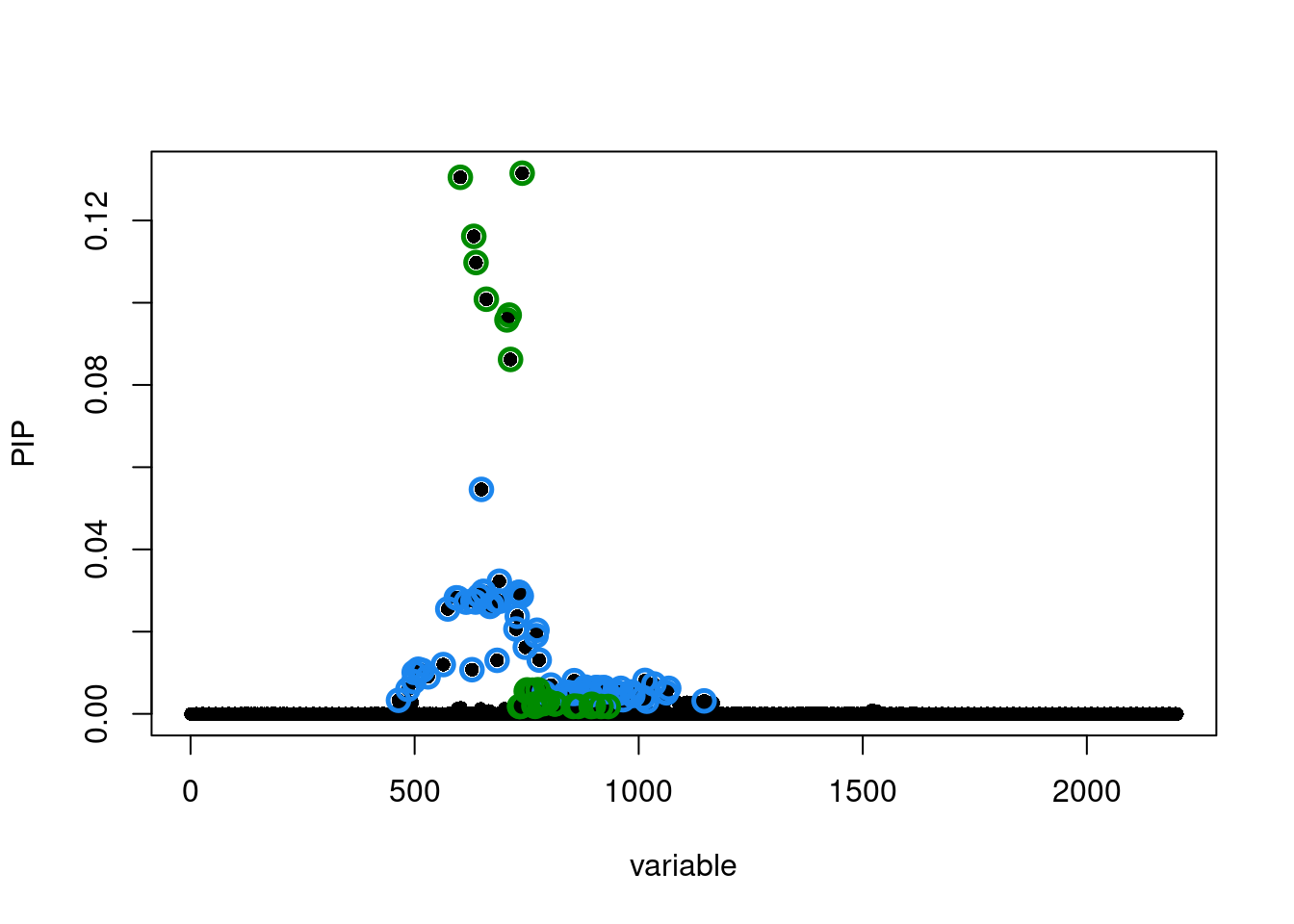
susie.locus.sumstats <- merge_susie_sumstats(susie.locus.res, sumstats.locus)condz <- LD_diagnosis_rss(sumstats.locus$zscore, R = R.locus, n = n)Estimate consistency between the z-scores and LD matrix in susie_rss model using regularized LD ...
Estimated lambda = 7.436622e-05
Compute expected z-scores based on conditional distribution of other z-scores ...condz$plot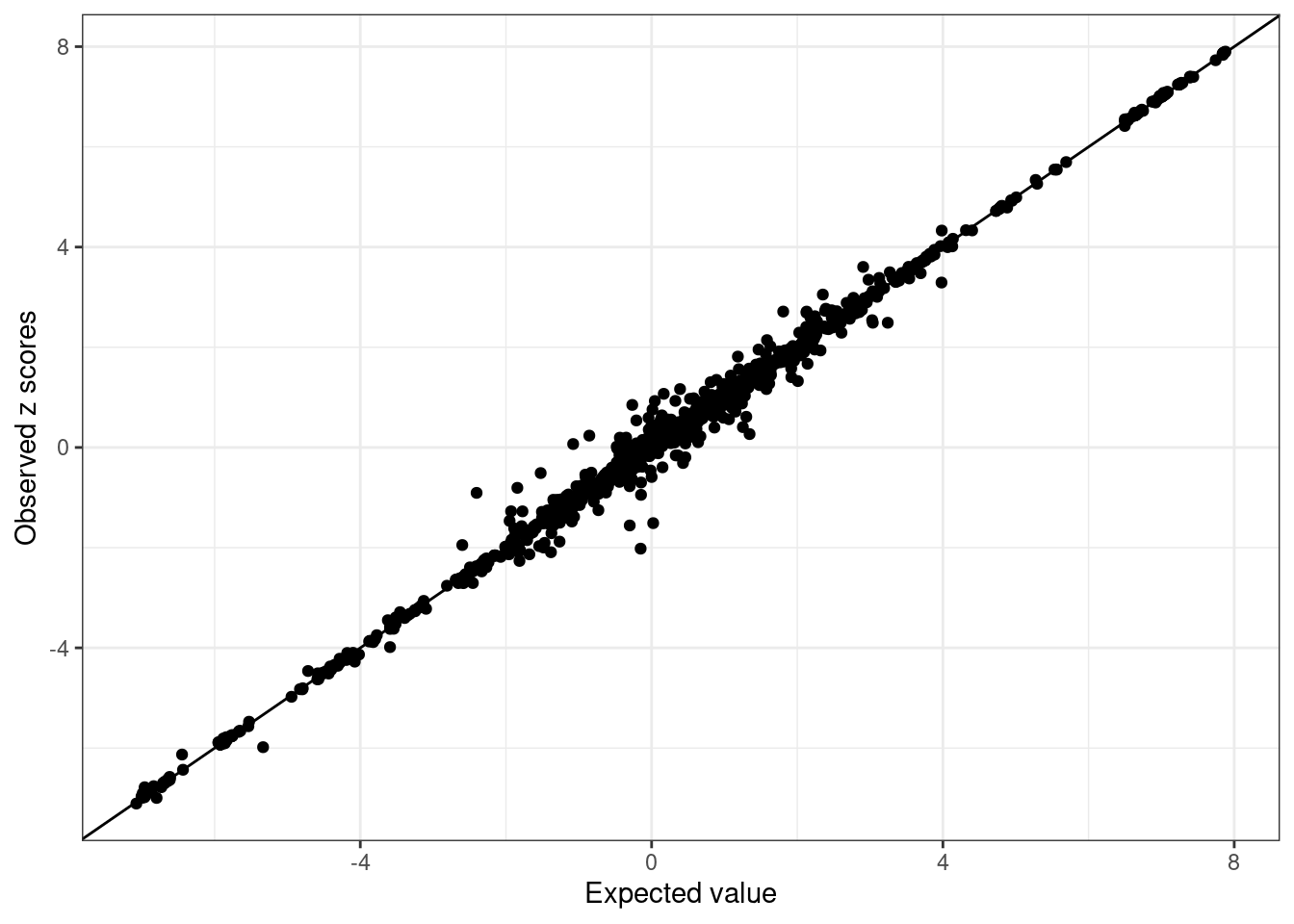
| Version | Author | Date |
|---|---|---|
| b117f93 | kevinlkx | 2023-11-06 |
Flip alleles for selected variants
Flip alleles for 10 variants with abs(z-scores) > 2
seed = 1
set.seed(seed)
flip_index <- sample(which(sumstats.locus$zscore > 2), 10)
sumstats.locus.flip <- sumstats.locus
sumstats.locus.flip$zscore[flip_index] <- -sumstats.locus$zscore[flip_index]
sumstats.locus.flip[flip_index, ]# A tibble: 10 × 10
chr pos beta se a0 a1 snp pval zscore locus
<int> <dbl> <dbl> <dbl> <chr> <chr> <chr> <dbl> <dbl> <dbl>
1 2 102985950 0.0746 0.0108 T C rs3771171 11.2 -6.89 193
2 2 102854882 0.0282 0.0105 C T rs3755282 2.13 -2.67 193
3 2 102839199 0.0271 0.0104 C T rs6715919 2.04 -2.61 193
4 2 103194558 0.0537 0.0124 A G rs74263644 4.83 -4.33 193
5 2 103247758 0.0703 0.0213 T C rs76605545 3.00 -3.29 193
6 2 102959080 0.0381 0.0177 G A rs13016771 1.50 -2.15 193
7 2 102945755 0.0283 0.0100 G T rs150341880 2.33 -2.83 193
8 2 103237631 0.0363 0.0102 T C rs2012454 3.41 -3.55 193
9 2 102918018 0.0262 0.0119 G A rs4577297 1.56 -2.21 193
10 2 102965332 0.0765 0.0108 G C rs17027006 11.8 -7.05 193cat(length(flip_index), "Allele switched variants:", sort(sumstats.locus.flip$snp[flip_index]), "\n")10 Allele switched variants: rs13016771 rs150341880 rs17027006 rs2012454 rs3755282 rs3771171 rs4577297 rs6715919 rs74263644 rs76605545 Run SuSiE including variants with flipped alleles
LD_matrices <- list(R.locus)
names(LD_matrices) <- locus
susie.locus.res <- run_finemapping(sumstats.locus.flip, LD_matrices = LD_matrices, priortype = 'uniform', n = n, L = 10)Finemapping locus 193...
Run susie_rss...Warning in susie_suff_stat(XtX = XtX, Xty = Xty, n = n, yty = (n - 1) * : IBSS algorithm did not converge in 100 iterations!
Please check consistency between summary statistics and LD matrix.
See https://stephenslab.github.io/susieR/articles/susierss_diagnostic.htmlsusie.locus.res[[1]]$sets$cs
$cs$L1
[1] 768
$cs$L2
[1] 766
$cs$L5
[1] 764
$purity
min.abs.corr mean.abs.corr median.abs.corr
L1 1 1 1
L2 1 1 1
L5 1 1 1
$cs_index
[1] 1 2 5
$coverage
[1] 1 1 1
$requested_coverage
[1] 0.95susie_plot(susie.locus.res[[1]], y='PIP')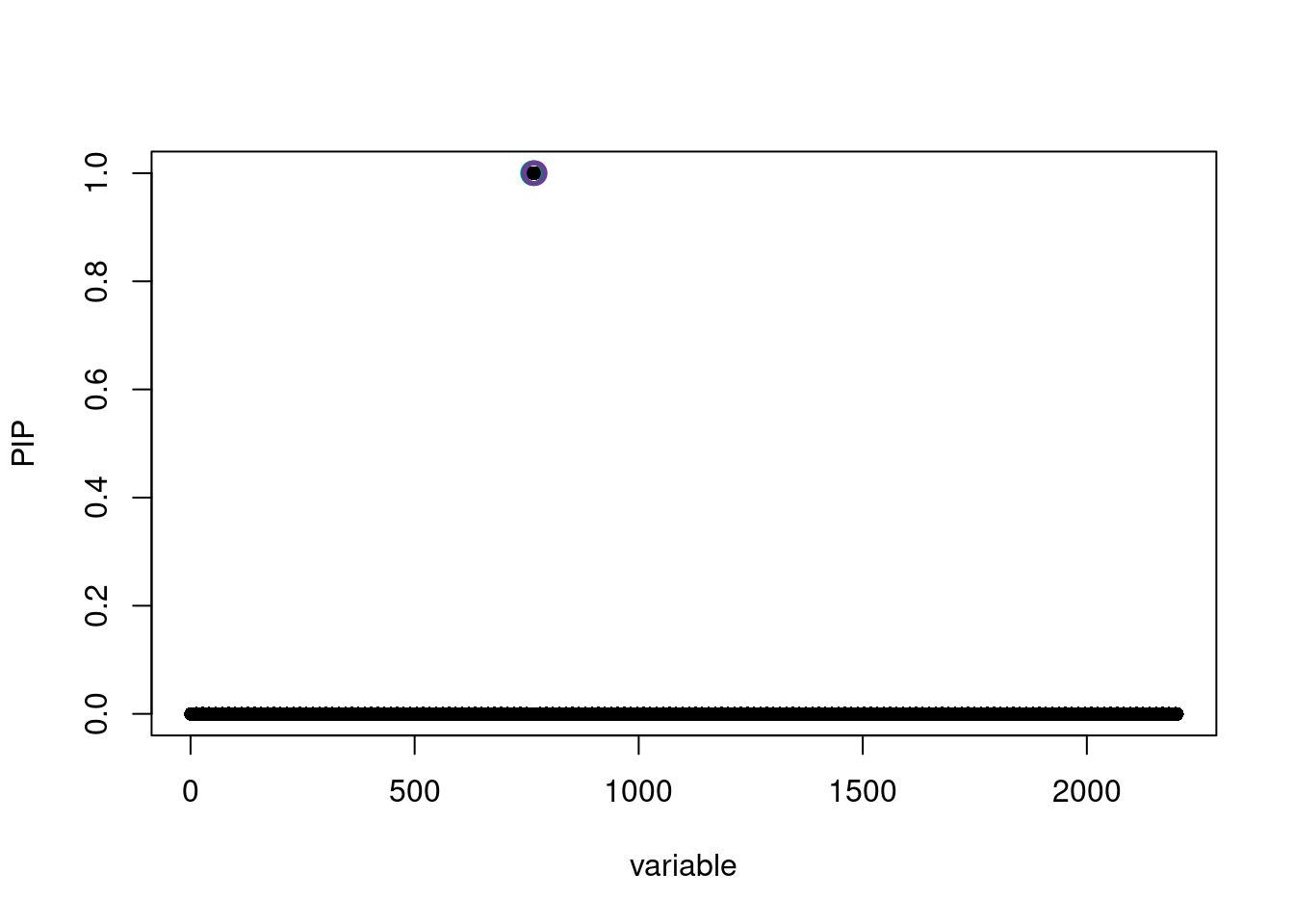
susie.locus.sumstats <- merge_susie_sumstats(susie.locus.res, sumstats.locus)Compares observed z scores vs the expected z scores
condz <- LD_diagnosis_rss(sumstats.locus.flip$zscore, R = R.locus, n = n)Estimate consistency between the z-scores and LD matrix in susie_rss model using regularized LD ...
Estimated lambda = 0.3195599
Compute expected z-scores based on conditional distribution of other z-scores ...# condz$plotDetect possible allele switched variants (logLR > 2 and abs(z) > 2).
detected_index <- which(condz$conditional_dist$logLR > 2 & abs(condz$conditional_dist$z) > 2)
cat(sprintf("Detected %d variants with possible allele switched", length(detected_index)), "\n")Detected 10 variants with possible allele switched cat("Possible allele switched variants:", sort(sumstats.locus.flip$snp[detected_index]), "\n")Possible allele switched variants: rs13016771 rs150341880 rs17027006 rs2012454 rs3755282 rs3771171 rs4577297 rs6715919 rs74263644 rs76605545 condz$conditional_dist$flipped <- 0
condz$conditional_dist$flipped[flip_index] <- 1
condz$conditional_dist$detected <- 0
condz$conditional_dist$detected[detected_index] <- 1
cat(sprintf("%d out of %d flipped variants detected with logLR > 2 and abs(z) > 2. \n",
length(intersect(detected_index, flip_index)), length(flip_index)))10 out of 10 flipped variants detected with logLR > 2 and abs(z) > 2. condz$conditional_dist[union(flip_index, detected_index),] z condmean condvar z_std_diff logLR flipped detected
820 -6.885113 6.714560 0.3269829 -23.782961 9.627679 1 1
458 -2.674648 2.647015 0.3673281 -8.780521 8.338206 1 1
393 -2.609874 2.609901 0.3229037 -9.185765 8.360083 1 1
1515 -4.332197 3.929706 0.3614657 -13.741893 8.300118 1 1
1605 -3.291488 2.690636 0.4585908 -8.833702 7.726878 1 1
743 -2.150657 2.265469 0.3324752 -7.658820 8.257533 1 1
697 -2.828630 2.753038 0.3239093 -9.807364 8.379124 1 1
1592 -3.545367 3.522010 0.3320191 -12.265259 8.542509 1 1
579 -2.205385 2.007778 0.3299552 -7.334673 8.182472 1 1
766 -7.054485 6.753350 0.3287578 -24.081724 9.521240 1 1ggplot(condz$conditional_dist, aes(x = condmean, y = z, col = factor(flipped))) +
geom_point() +
scale_colour_manual(values = c("0" = "black", "1" = "red")) +
labs(x = "Expected", y = "Observed z scores", color = "Allele flipped") +
theme_bw()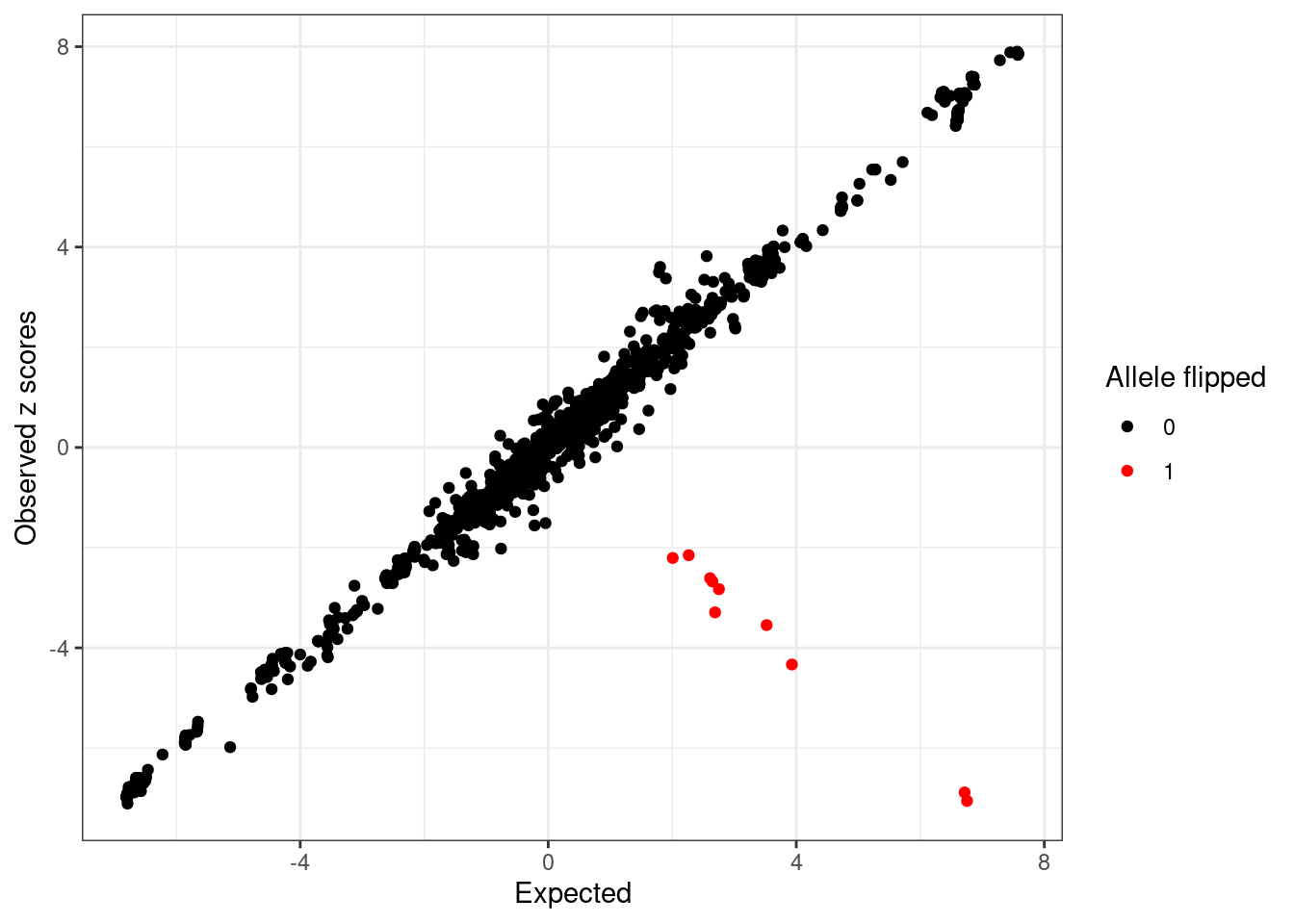
| Version | Author | Date |
|---|---|---|
| b117f93 | kevinlkx | 2023-11-06 |
ggplot(condz$conditional_dist, aes(x = condmean, y = z, col = factor(detected))) +
geom_point() +
scale_colour_manual(values = c("0" = "black", "1" = "red")) +
labs(x = "Expected", y = "Observed z scores", color = "Possible allele switched") +
theme_bw()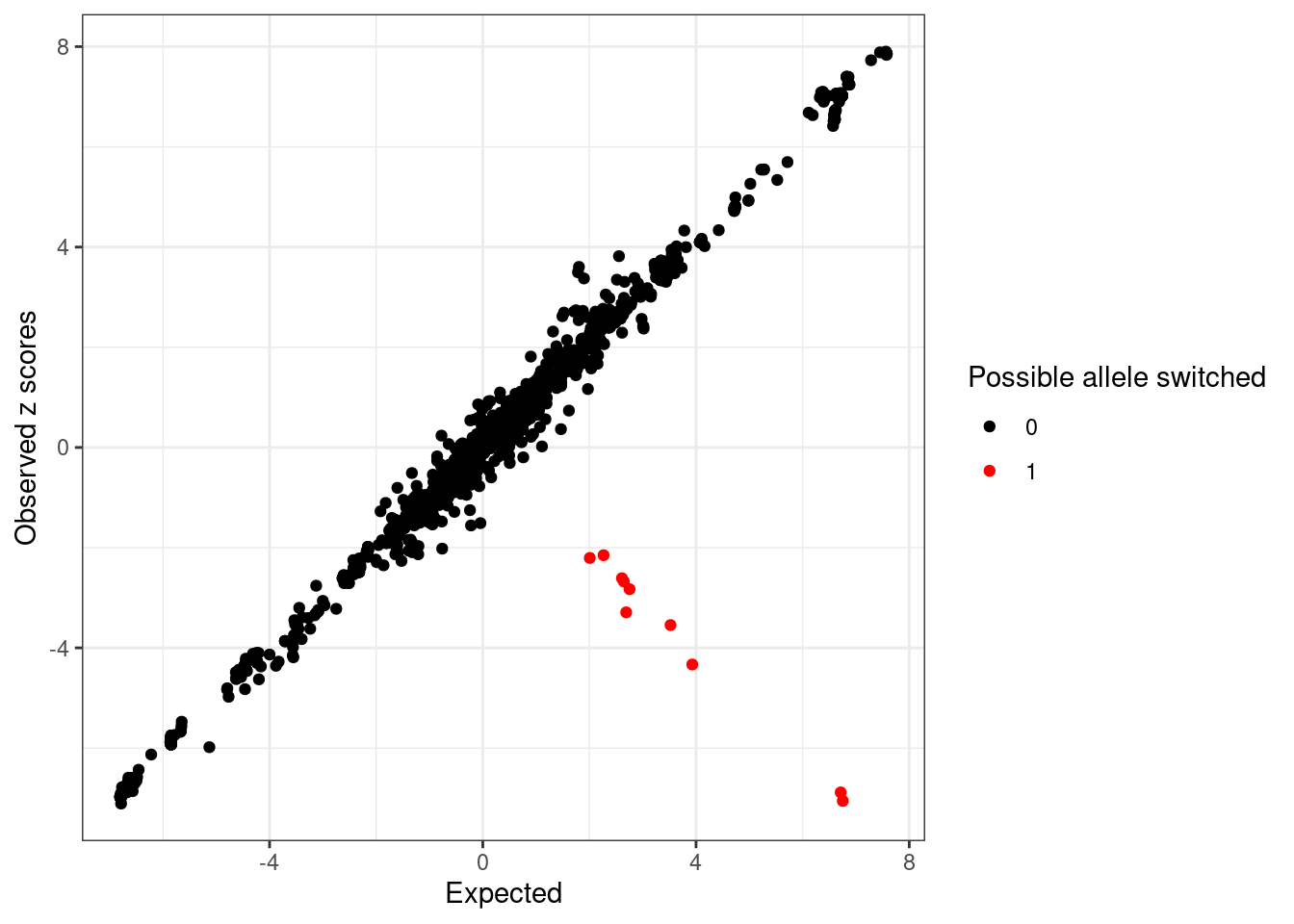
| Version | Author | Date |
|---|---|---|
| b117f93 | kevinlkx | 2023-11-06 |
sessionInfo()R version 4.2.0 (2022-04-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.3.13-el7-x86_64/lib/libopenblas_haswellp-r0.3.13.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=C
[4] LC_COLLATE=C LC_MONETARY=C LC_MESSAGES=C
[7] LC_PAPER=C LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=C LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] susieR_0.12.27 forcats_1.0.0 stringr_1.5.0 dplyr_1.1.0
[5] purrr_1.0.1 readr_2.1.4 tidyr_1.3.0 tibble_3.1.8
[9] ggplot2_3.4.1 tidyverse_1.3.2 mapgen_0.5.6 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] googledrive_2.0.0 colorspace_2.1-0
[3] rjson_0.2.21 ellipsis_0.3.2
[5] rprojroot_2.0.3 XVector_0.38.0
[7] GenomicRanges_1.48.0 fs_1.6.1
[9] rstudioapi_0.14 farver_2.1.1
[11] bit64_4.0.5 fansi_1.0.4
[13] lubridate_1.9.2 xml2_1.3.3
[15] codetools_0.2-18 cachem_1.0.6
[17] knitr_1.42 jsonlite_1.8.4
[19] Rsamtools_2.12.0 broom_1.0.3
[21] dbplyr_2.3.0 compiler_4.2.0
[23] httr_1.4.4 backports_1.4.1
[25] RcppZiggurat_0.1.6 assertthat_0.2.1
[27] Matrix_1.5-3 fastmap_1.1.0
[29] gargle_1.3.0 cli_3.6.0
[31] later_1.3.0 htmltools_0.5.4
[33] tools_4.2.0 gtable_0.3.1
[35] glue_1.6.2 GenomeInfoDbData_1.2.9
[37] Rcpp_1.0.10 Biobase_2.58.0
[39] cellranger_1.1.0 jquerylib_0.1.4
[41] vctrs_0.5.2 Biostrings_2.66.0
[43] rtracklayer_1.58.0 xfun_0.37
[45] plyranges_1.18.0 ps_1.7.2
[47] rvest_1.0.3 timechange_0.2.0
[49] lifecycle_1.0.3 irlba_2.3.5
[51] restfulr_0.0.15 XML_3.99-0.13
[53] googlesheets4_1.0.1 getPass_0.2-2
[55] zlibbioc_1.44.0 scales_1.2.1
[57] vroom_1.6.1 hms_1.1.2
[59] promises_1.2.0.1 MatrixGenerics_1.10.0
[61] parallel_4.2.0 SummarizedExperiment_1.28.0
[63] yaml_2.3.7 sass_0.4.5
[65] reshape_0.8.9 stringi_1.7.12
[67] highr_0.10 BiocIO_1.8.0
[69] S4Vectors_0.36.1 BiocGenerics_0.44.0
[71] BiocParallel_1.32.5 GenomeInfoDb_1.34.9
[73] rlang_1.0.6 pkgconfig_2.0.3
[75] bitops_1.0-7 matrixStats_0.63.0
[77] evaluate_0.20 lattice_0.20-45
[79] labeling_0.4.2 GenomicAlignments_1.34.0
[81] Rfast_2.0.6 bit_4.0.5
[83] processx_3.8.0 tidyselect_1.2.0
[85] plyr_1.8.7 magrittr_2.0.3
[87] R6_2.5.1 IRanges_2.32.0
[89] generics_0.1.3 DelayedArray_0.24.0
[91] DBI_1.1.3 pillar_1.8.1
[93] haven_2.5.1 whisker_0.4
[95] withr_2.5.0 RCurl_1.98-1.10
[97] mixsqp_0.3-43 modelr_0.1.10
[99] crayon_1.5.2 utf8_1.2.3
[101] tzdb_0.3.0 rmarkdown_2.20
[103] grid_4.2.0 readxl_1.4.2
[105] data.table_1.14.6 callr_3.7.3
[107] git2r_0.30.1 reprex_2.0.2
[109] digest_0.6.31 httpuv_1.6.5
[111] stats4_4.2.0 munsell_0.5.0
[113] bslib_0.4.2