Last updated: 2021-11-30
Checks: 7 0
Knit directory: cacoaAnalysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it's best to always run the code in an empty environment.
The command set.seed(20211123) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 70d2759. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.Rhistory
Ignored: analysis/MS.preProcess.nb.html
Ignored: analysis/figure/
Ignored: analysis/preprocess.nb.html
Ignored: analysis/preprocess__ms.nb.html
Ignored: analysis/site_libs/
Ignored: analysis/test.nb.html
Ignored: data/ASD/
Ignored: data/AZ/
Ignored: data/EP/
Ignored: data/MS/
Ignored: data/PF/
Ignored: data/SCC/
Untracked files:
Untracked: analysis/MS.preProcess.Rmd
Untracked: analysis/SCC.preProcess.Rmd
Untracked: analysis/autism.preProcess.Rmd
Untracked: analysis/autism_peter.Rmd
Untracked: analysis/ms_peter.Rmd
Untracked: analysis/test.Rmd
Untracked: man/
Unstaged changes:
Modified: analysis/about.Rmd
Modified: analysis/index.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/preprocess.Rmd) and HTML (docs/preprocess.html) files. If you've configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 70d2759 | viktor_petukhov | 2021-11-30 | Preprocessing notebook |
Libraries
library(tidyverse)
library(magrittr)
library(sccore)
library(pagoda2)
library(conos)
library(dataorganizer)
library(Matrix)
library(reticulate)
devtools::load_all()
N_CORES <- 45virtualenv_create("r-scrublet")virtualenv: r-scrubletvirtualenv_install("r-scrublet", c("scrublet", "matplotlib"))Using virtual environment 'r-scrublet' ...use_virtualenv("r-scrublet")
mpl <- import("matplotlib")
mpl$use('Agg') # Otherwise it shows Qt error in RStudio
scrublet <- import("scrublet")
estimateDoubletInfo <- function(mats, progress=FALSE) {
dub.info <- sccore::plapply(mats, function(m) {
suppressMessages(scrublet$Scrublet(t(m), random_state=as.integer(42))$scrub_doublets()) %>%
lapply(setNames, colnames(m))
}, n.cores=1, progress=progress)
lapply(c(scores=1, mask=2), function(i) {
lapply(dub.info, `[[`, i) %>% unname() %>% unlist()
})
}Alzheimer
Cells are already filtered by mit. fraction, and doublets are removed. We filter only by minimum 500 UMIs per cell and scrublet scores.
con <- readOrCreate(DataPath('ASD/con.rds'), function() {
mat <- DataPath("AZ/cell_counts.csv") %>% data.table::fread(sep=",") %>%
{set_rownames(mltools::sparsify(.[, 2:ncol(.)]), .$V1)} %>%
.[rowSums(. > 0) >= 10,]
cell_metadata <- DataPath("AZ/cell_metadata.csv") %>% read_delim(delim='\t') %>%
rename(cell=sampleID, sample=patient) %>%
select(cell, batch, sample, sex, cellType, subclustID) %>%
filter(!grepl("un", sample), !(cellType %in% c('doublet', 'unID')))
sample_metadata <- group_by(cell_metadata, batch, sample, sex) %>%
summarise(n=n()) %>% select(-n) %>% lapply(setNames, .$sample) %>% ungroup()
cell_metadata %<>% lapply(setNames, .$cell)
mat_per_samp <- splitMatrixByFactor(mat, cell_metadata$sample)
dub_info <- estimateDoubletInfo(mat_per_samp)
p2s <- plapply(mat_per_samp, createPagoda, min.transcripts.per.cell=500, dub.scores=dub_info$scores,
dub.threshold=0.3, mc.preschedule=TRUE, n.cores=N_CORES, progress=FALSE)
createConos(p2s, sample.meta=sample_metadata, cell.meta=cell_metadata, n.cores=N_CORES)
}) %>% Conos$new()
con$plotGraph(groups=con$misc$cell_metadata$cellType, size=0.2, alpha=0.2)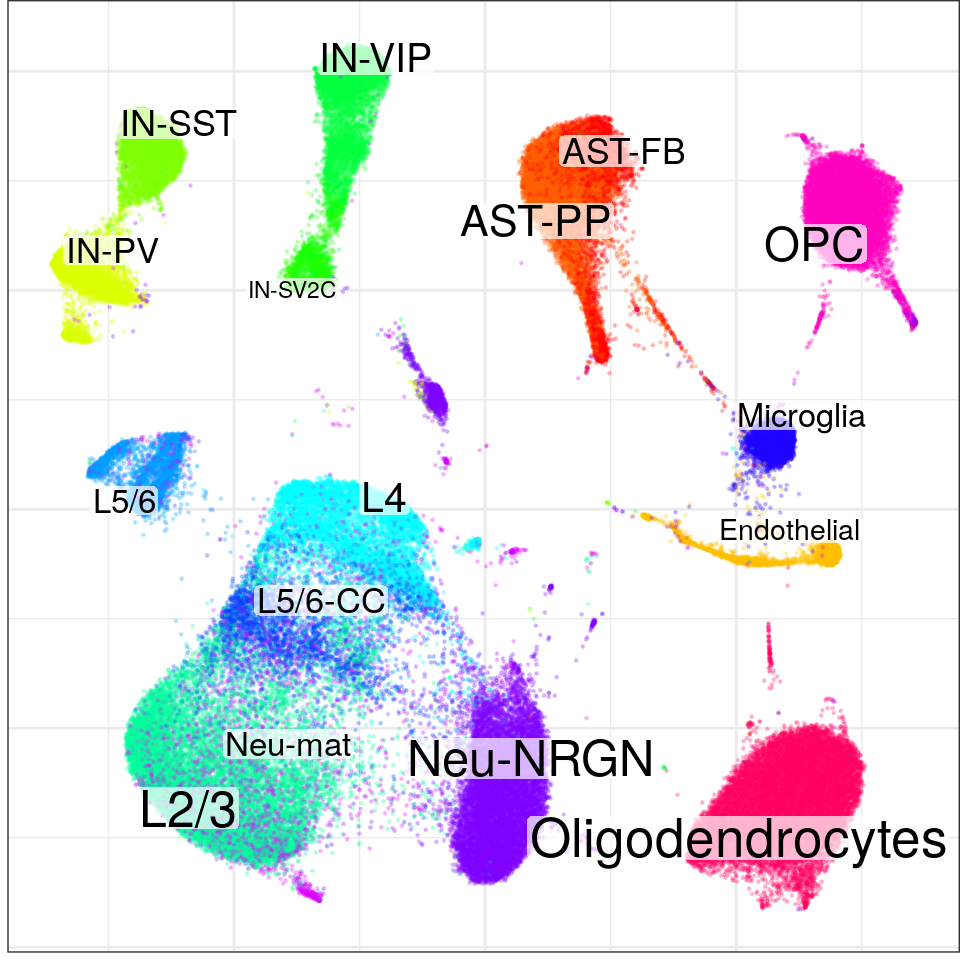
rm(con); gc(); used (Mb) gc trigger (Mb) max used (Mb)
Ncells 4383678 234.2 8419145 449.7 8419145 449.7
Vcells 8764166 66.9 744343347 5678.9 906778991 6918.2Autism
Cells are already filtered by mitochondrial fraction of 0.05 and UMI threshold ~500, no additional filtration is needed.
con <- readOrCreate(DataPath('ASD/con.rds'), function() {
mat <- Seurat::Read10X(DataPath("ASD")) %>% .[rowSums(. > 0) >= 10,]
meta <- read_delim(DataPath("ASD/meta.txt"), delim='\t') %>%
rename(cellType=cluster, PMI=`post-mortem interval (hours)`) %>%
mutate(cellType=gsub("-I(I)?", "", cellType))
sample_metadata <- meta %>%
group_by(sample, individual, region, age, sex, diagnosis, Capbatch, Seqbatch) %>%
summarise(PMI=median(PMI)) %>% ungroup() %>% lapply(setNames, .$sample)
sample_metadata$region_hr <- sample_metadata$sample %>% strsplit('_') %>% sapply(`[[`, 2)
cell_metadata <- meta %>% lapply(setNames, .$cell)
mat_per_samp <- splitMatrixByFactor(mat, cell_metadata$sample)
mat_per_cap <- splitMatrixByFactor(mat, cell_metadata$Capbatch)
dub_info <- estimateDoubletInfo(mat_per_cap)
p2s <- plapply(mat_per_samp, createPagoda, dub.scores=dub_info$scores, dub.threshold=0.17,
mc.preschedule=TRUE, n.cores=N_CORES, progress=FALSE)
createConos(p2s, sample.meta=sample_metadata, cell.meta=cell_metadata, n.cores=N_CORES)
}) %>% Conos$new()
con$plotGraph(groups=con$misc$cell_metadata$cellType, size=0.1, alpha=0.1)
rm(con); gc(); used (Mb) gc trigger (Mb) max used (Mb)
Ncells 4383811 234.2 8419145 449.7 8419145 449.7
Vcells 8764587 66.9 716263197 5464.7 906778991 6918.2Epilepsy
con <- readOrCreate(DataPath('EP/con.rds'), function() {
con.pap <- read_rds(DataPath("EP/con_filt_samples.rds")) %>% conos::Conos$new()
cell.metadata <- DataPath("EP/annotation.csv") %>% read_csv() %>% lapply(setNames, .$cell)
sample.metadata <- DataPath("EP/sample_info.csv") %>% read_csv() %>% lapply(setNames, .$Alias)
con.pap$samples %>% lapply(pagoda2::Pagoda2$new) %>%
createConos(sample.meta=sample.metadata, cell.meta=cell.metadata, k=40, n.cores=N_CORES)
}) %>% Conos$new()
con$plotGraph(groups=con$misc$cell_metadata$l4, size=0.1, alpha=0.1, font.size=c(2, 3))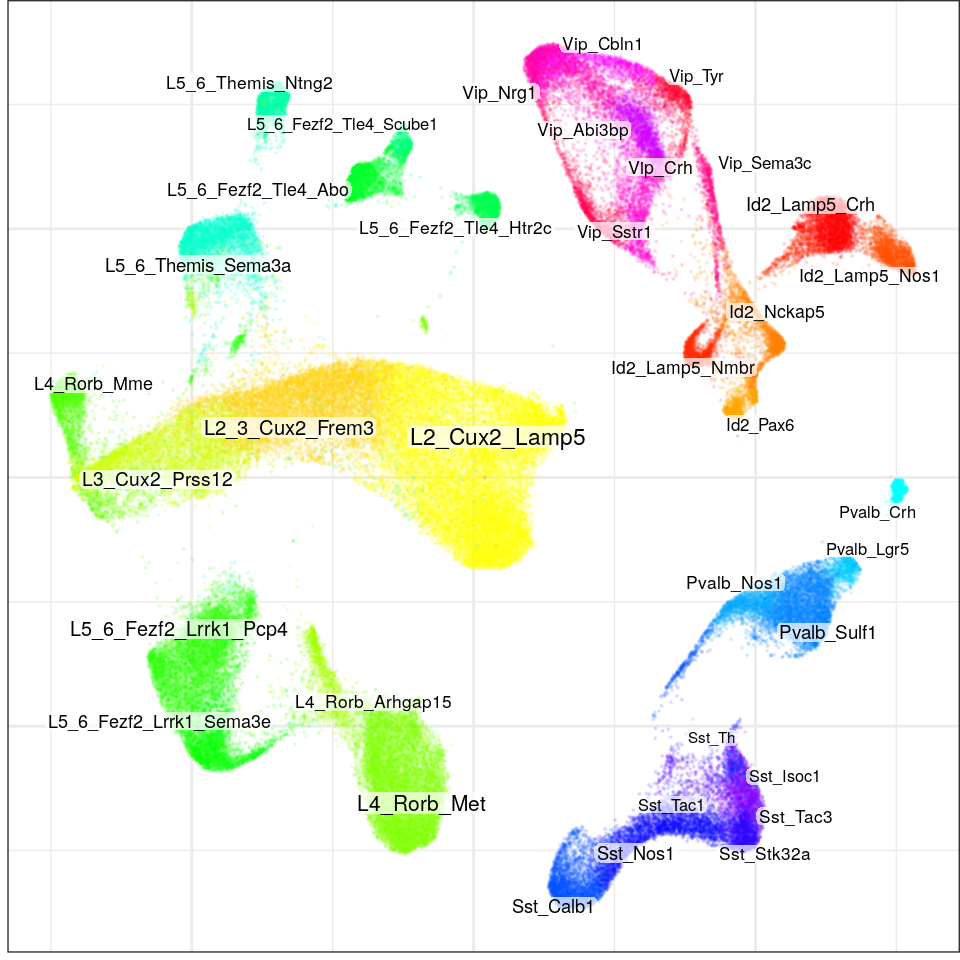
rm(con); gc(); used (Mb) gc trigger (Mb) max used (Mb)
Ncells 4387983 234.4 8419145 449.7 8419145 449.7
Vcells 8894187 67.9 697289774 5319.9 906778991 6918.2MS
The threshold on transcripts here is set only because there was 1 almost empty cell reported, all other cells already had enough transcripts.
con <- readOrCreate(DataPath('MS/con.rds'), function() {
mat <- Seurat::Read10X(DataPath("MS")) %>% .[rowSums(. > 0) >= 10,]
meta <- read_delim(DataPath("MS/meta.txt"), delim='\t') %>%
mutate(cell_type=gsub("-(Cntl|MS(-1|-2)?)", "", x=cell_type))
sample.metadata <- meta[,5:14] %>% split(.$sample) %>% lapply(`[`, 1,) %>%
do.call(rbind, .) %>% lapply(setNames, .$sample)
cell.metadata <- meta %>% lapply(setNames, .$cell)
mat.per.samp <- splitMatrixByFactor(mat, cell.metadata$sample)
mat.per.cap <- splitMatrixByFactor(mat, cell.metadata$Capbatch)
dub.info <- estimateDoubletInfo(mat.per.cap)
p2s <- plapply(mat.per.samp, createPagoda, min.transcripts.per.cell=800, dub.scores=dub.info$scores,
dub.threshold=0.2, mc.preschedule=TRUE, n.cores=N_CORES, progress=FALSE)
createConos(p2s, sample.meta=sample.metadata, cell.meta=cell.metadata, n.cores=N_CORES)
}) %>% Conos$new()
con$plotGraph(groups=con$misc$cell_metadata$cell_type, size=0.1, alpha=0.1)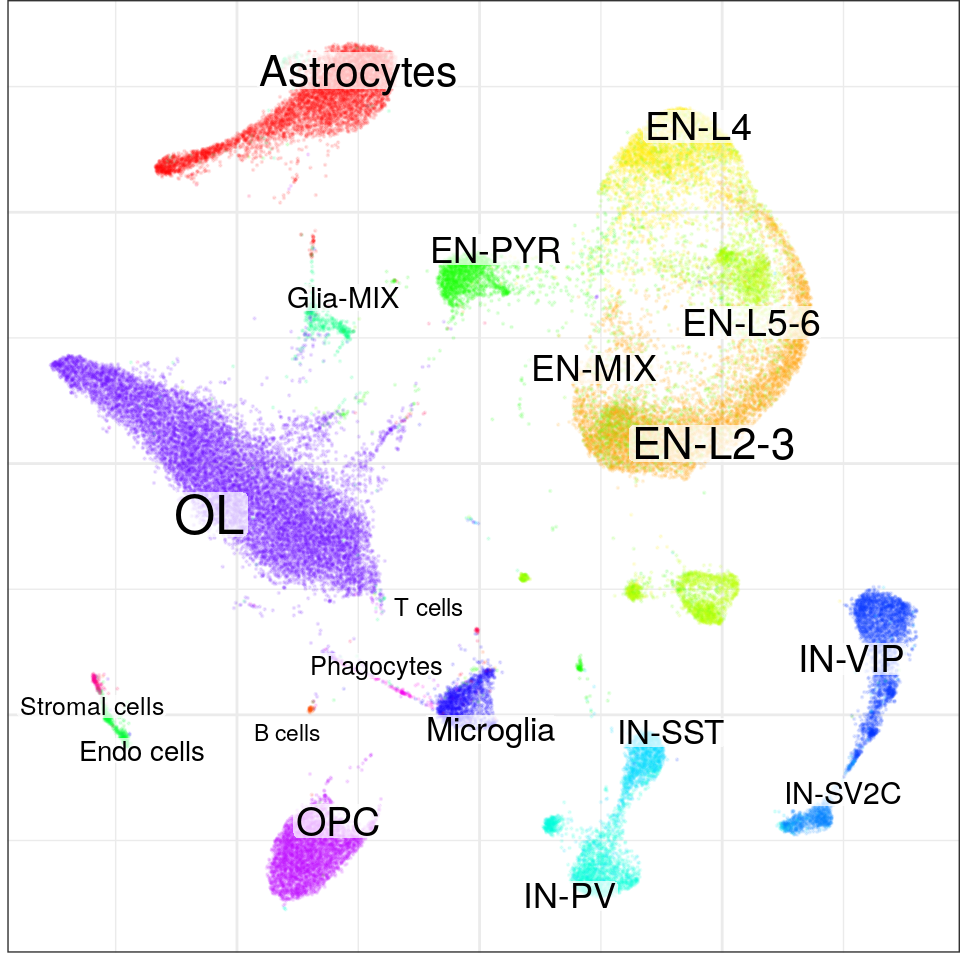
rm(con); gc(); used (Mb) gc trigger (Mb) max used (Mb)
Ncells 4332336 231.4 8419145 449.7 8419145 449.7
Vcells 8062399 61.6 557831820 4256.0 906778991 6918.2PF
The paper already performed the filtration
con <- readOrCreate(DataPath('PF/con.rds'), function() {
cell.metadata <- read_csv(DataPath("PF/cell_metadata.csv")) %>% rename(cell=X1) %>%
filter(Diagnosis %in% c("Control", "IPF")) %>%
rename(sample=Sample_Name, cellType=celltype)
sample.metadata <- cell.metadata %>%
group_by(sample, Sample_Source, Diagnosis, Status, orig.ident) %>%
summarise(n=n()) %>% select(-n) %>%
lapply(setNames, .$sample)
mat <- DataPath("PF") %>% Seurat::Read10X(gene.column=1) %>%
.[,cell.metadata$cell] %>% .[rowSums(. > 0) >= 10,]
cell.metadata %<>% lapply(setNames, .$cell)
mat.per.samp <- splitMatrixByFactor(mat, cell.metadata$sample)
p2s <- plapply(mat.per.samp, createPagoda, mc.preschedule=TRUE, n.cores=N_CORES, progress=FALSE)
createConos(p2s, sample.meta=sample.metadata, cell.meta=cell.metadata, n.cores=N_CORES)
}) %>% Conos$new()
con$plotGraph(groups=con$misc$cell_metadata$cellType, size=0.1, alpha=0.1)Warning: ggrepel: 1 unlabeled data points (too many overlaps). Consider
increasing max.overlaps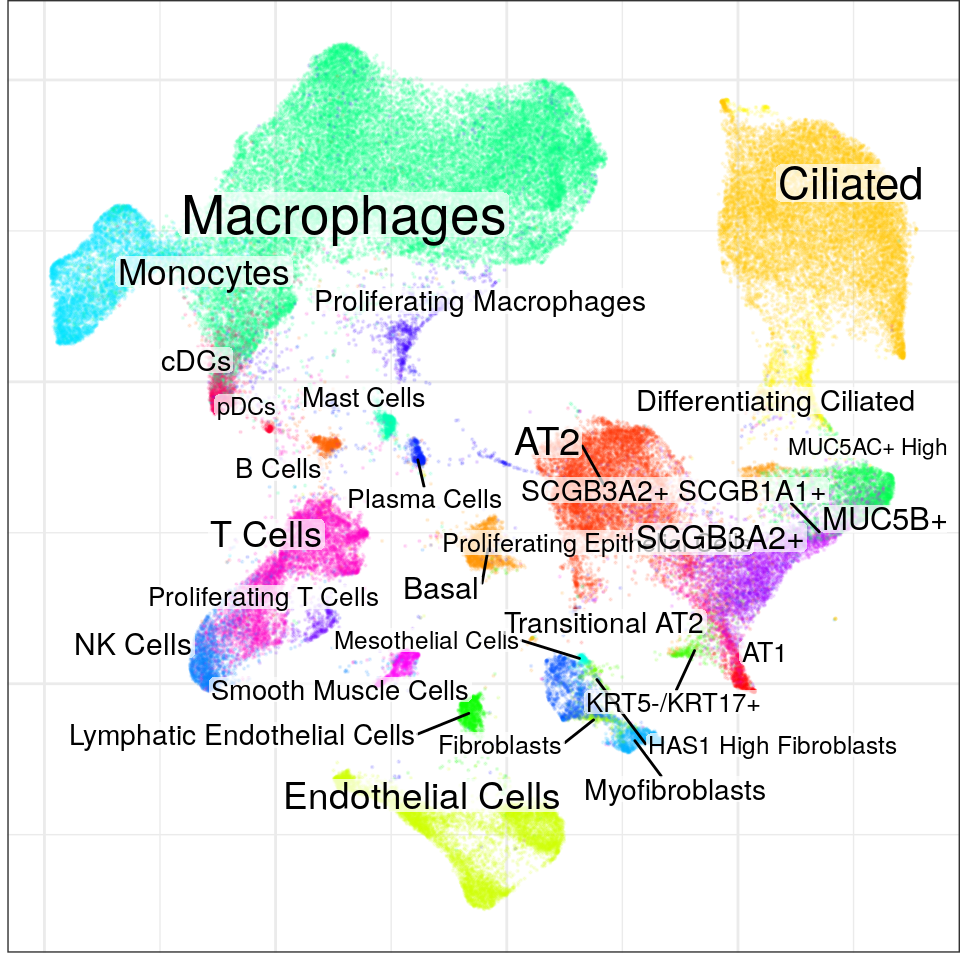
rm(con); gc(); used (Mb) gc trigger (Mb) max used (Mb)
Ncells 4380209 234.0 8419145 449.7 8419145 449.7
Vcells 8739996 66.7 659446623 5031.2 906778991 6918.2SCC
con <- readOrCreate(DataPath('SCC/con.rds'), function() {
mat <- data.table::fread(DataPath("SCC/counts.txt"), sep="\t") %>%
{set_rownames(mltools::sparsify(.[3:nrow(.), 2:ncol(.)]), .$V1[3:nrow(.)])}
cell.metadata <- read_delim(DataPath('SCC/cell_metadata.txt'), delim='\t') %>%
filter(!(level3_celltype %in% c('Multiplet', 'Keratinocyte'))) %>% rename(cell=nCount_RNA)
cell.metadata$sample <- cell.metadata$cell %>% strsplit("_") %>%
sapply(function(x) paste(x[1:2], collapse='_'))
cell.metadata$level3_celltype %<>% gsub("(Normal|Tumor)_", "", .)
cell.metadata %<>% lapply(setNames, .$cell)
mat <- mat[rowSums(mat > 0) >= 10, cell.metadata$cell]
mat.per.samp <- splitMatrixByFactor(mat, cell.metadata$sample)
p2s <- plapply(mat.per.samp, createPagoda, min.transcripts.per.cell=800,
mc.preschedule=TRUE, n.cores=N_CORES, progress=FALSE)
createConos(p2s, sample.meta=sample.metadata, cell.meta=cell.metadata, n.cores=N_CORES)
}) %>% Conos$new()
con$plotGraph(groups=con$misc$cell_metadata$level3_celltype, size=0.1, alpha=0.1)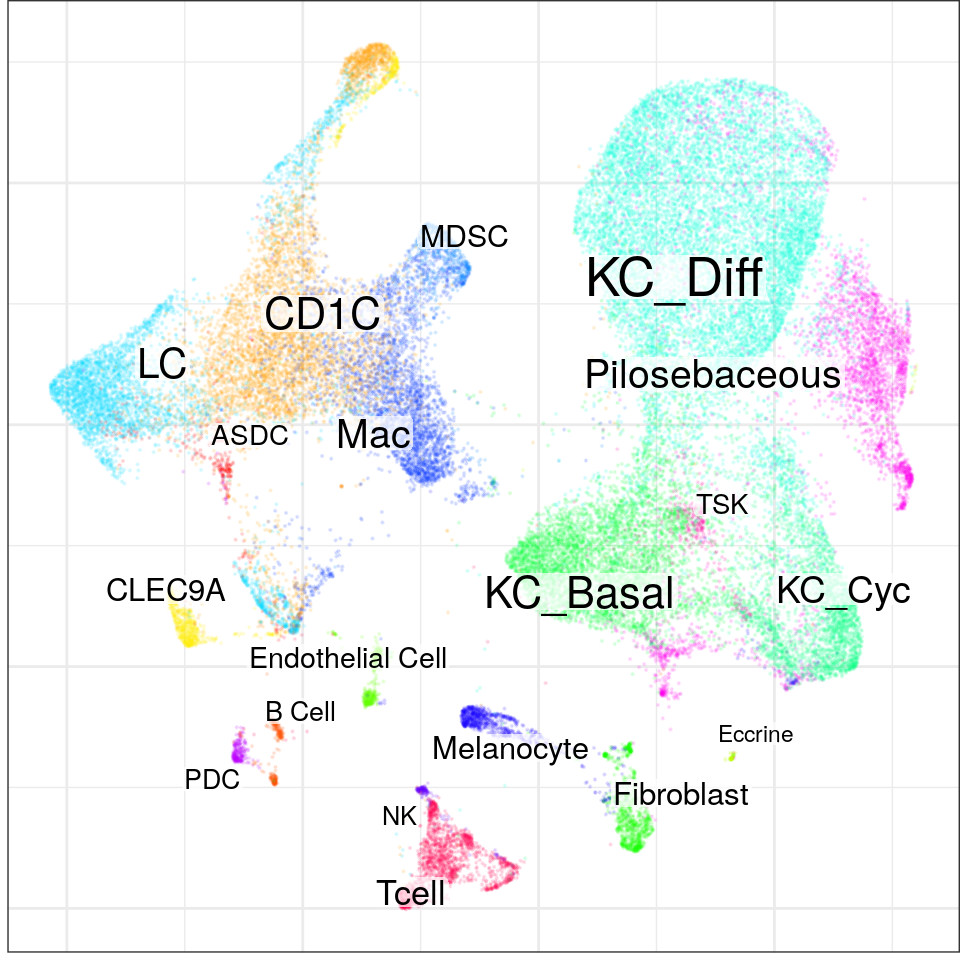
rm(con); gc(); used (Mb) gc trigger (Mb) max used (Mb)
Ncells 4330713 231.3 8419145 449.7 8419145 449.7
Vcells 7972766 60.9 527557299 4025.0 906778991 6918.2
sessionInfo()R version 4.0.3 (2020-10-10)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 18.04.6 LTS
Matrix products: default
BLAS: /usr/local/R/R-4.0.3/lib/R/lib/libRblas.so
LAPACK: /usr/local/R/R-4.0.3/lib/R/lib/libRlapack.so
locale:
[1] C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] cacoaAnalysis_0.1.0 reticulate_1.22 dataorganizer_0.1.0
[4] conos_1.4.4 pagoda2_1.0.7 igraph_1.2.6
[7] Matrix_1.2-18 sccore_1.0.0 magrittr_2.0.1
[10] forcats_0.5.1 stringr_1.4.0 dplyr_1.0.7
[13] purrr_0.3.4 readr_1.4.0 tidyr_1.1.4
[16] tibble_3.1.5 ggplot2_3.3.5 tidyverse_1.3.0
[19] workflowr_1.6.2
loaded via a namespace (and not attached):
[1] readxl_1.3.1 N2R_0.1.1 backports_1.2.1
[4] circlize_0.4.13 splines_4.0.3 usethis_1.6.3
[7] urltools_1.7.3 digest_0.6.28 foreach_1.5.1
[10] htmltools_0.5.2 fansi_0.5.0 RMTstat_0.3
[13] memoise_2.0.0 cluster_2.1.0 doParallel_1.0.16
[16] remotes_2.2.0 ComplexHeatmap_2.9.4 modelr_0.1.8
[19] matrixStats_0.61.0 R.utils_2.10.1 prettyunits_1.1.1
[22] colorspace_2.0-2 rappdirs_0.3.3 rvest_0.3.6
[25] ggrepel_0.9.1 haven_2.4.1 xfun_0.26
[28] callr_3.5.1 crayon_1.4.1 jsonlite_1.7.2
[31] brew_1.0-6 iterators_1.0.13 glue_1.4.2
[34] gtable_0.3.0 GetoptLong_1.0.5 leidenAlg_0.1.0
[37] pkgbuild_1.1.0 Rook_1.1-1 shape_1.4.6
[40] BiocGenerics_0.36.1 scales_1.1.1 DBI_1.1.1
[43] Rcpp_1.0.7 clue_0.3-59 stats4_4.0.3
[46] httr_1.4.2 RColorBrewer_1.1-2 ellipsis_0.3.2
[49] farver_2.1.0 pkgconfig_2.0.3 R.methodsS3_1.8.1
[52] dbplyr_2.0.0 here_1.0.1 utf8_1.2.2
[55] labeling_0.4.2 tidyselect_1.1.1 rlang_0.4.11
[58] later_1.1.0.1 munsell_0.5.0 cellranger_1.1.0
[61] tools_4.0.3 cachem_1.0.6 cli_3.0.1
[64] generics_0.1.0 devtools_2.3.2 broom_0.7.9
[67] evaluate_0.14 fastmap_1.1.0 yaml_2.2.1
[70] processx_3.4.5 knitr_1.36 fs_1.5.0
[73] nlme_3.1-149 whisker_0.4 ggrastr_0.2.1
[76] R.oo_1.24.0 grr_0.9.5 xml2_1.3.2
[79] compiler_4.0.3 rstudioapi_0.13 beeswarm_0.4.0
[82] png_0.1-7 testthat_3.0.0 reprex_0.3.0
[85] stringi_1.7.5 highr_0.9 ps_1.4.0
[88] drat_0.1.8 desc_1.3.0 lattice_0.20-41
[91] vctrs_0.3.8 pillar_1.6.3 lifecycle_1.0.1
[94] triebeard_0.3.0 jquerylib_0.1.4 GlobalOptions_0.1.2
[97] irlba_2.3.3 Matrix.utils_0.9.8 httpuv_1.5.4
[100] R6_2.5.1 promises_1.1.1 gridExtra_2.3
[103] vipor_0.4.5 IRanges_2.24.1 sessioninfo_1.1.1
[106] codetools_0.2-16 MASS_7.3-53 assertthat_0.2.1
[109] pkgload_1.2.1 rprojroot_2.0.2 rjson_0.2.20
[112] withr_2.4.2 S4Vectors_0.28.1 mgcv_1.8-33
[115] parallel_4.0.3 hms_1.1.1 grid_4.0.3
[118] rmarkdown_2.11 dendsort_0.3.3 Rtsne_0.15
[121] git2r_0.27.1 lubridate_1.7.9.2 ggbeeswarm_0.6.0