Last updated: 2021-12-12
Checks: 7 0
Knit directory: cacoaAnalysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20211123) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 17f5b67. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/simulation_ns_nc.nb.html
Ignored: analysis/simulation_types.nb.html
Ignored: cache/
Ignored: data/ASD/
Ignored: man/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/simulation_ns_nc.Rmd) and HTML (docs/simulation_ns_nc.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 17f5b67 | Viktor Petukhov | 2021-12-12 | Renamed simulation_ns_nc.Rmd |
| Rmd | 55525bf | Viktor Petukhov | 2021-12-12 | Simulation notebooks |
In these simulations we used muscat to generate artificial data from the Autism dataset. It allowed us to vary individual covariates, fixing the amount of the actual expression change. For each set of parameters we performed 30 repeats and estimated median distance and p-value (when available) for corresponding metrics.
# Requires running simulation_types.Rmd first
sce <- read_rds(CachePath('asd_sim_sces.rds'))$`IN-PV`$prepDependency on the number of cells and samples
Number of samples per cell type
n_samples <- c(3, 5, 7, 9)
sims_samples <- generateSims(
sce, n.cells=N_CELLS, de.frac=DE_FRAC, n.cores=N_CORES, lfc=LFC,
n.samples=n_samples, n.repeats=N_REPEATS, verbose=FALSE
)
cao_samples <- cacoaFromSim(sims_samples, n.cores=N_CORES)
cao_samples$estimateExpressionShiftMagnitudes(verbose=TRUE, n.permutations=N_PERMUTS, min.samp.per.type=MIN_SAMPS)
cao_samples$estimateExpressionShiftMagnitudes(
verbose=TRUE, n.permutations=N_PERMUTS, top.n.genes=TOP_N_GENES, n.pcs=N_PCS,
min.samp.per.type=MIN_SAMPS, name='es.top.de'
)
cao_samples$estimateDEPerCellType(independent.filtering=TRUE, verbose=TRUE)The raw expression distance does not depend on the number of samples:
p_df <- cao_samples$test.results$expression.shifts %>%
prepareExpressionShiftSimDf(sims=sims_samples) %>% mutate(ns=as.factor(ns))
plotExpressionShiftSimDf(p_df, x.col='ns', norm.dist=FALSE, adj.list=xlab("Num. samples"))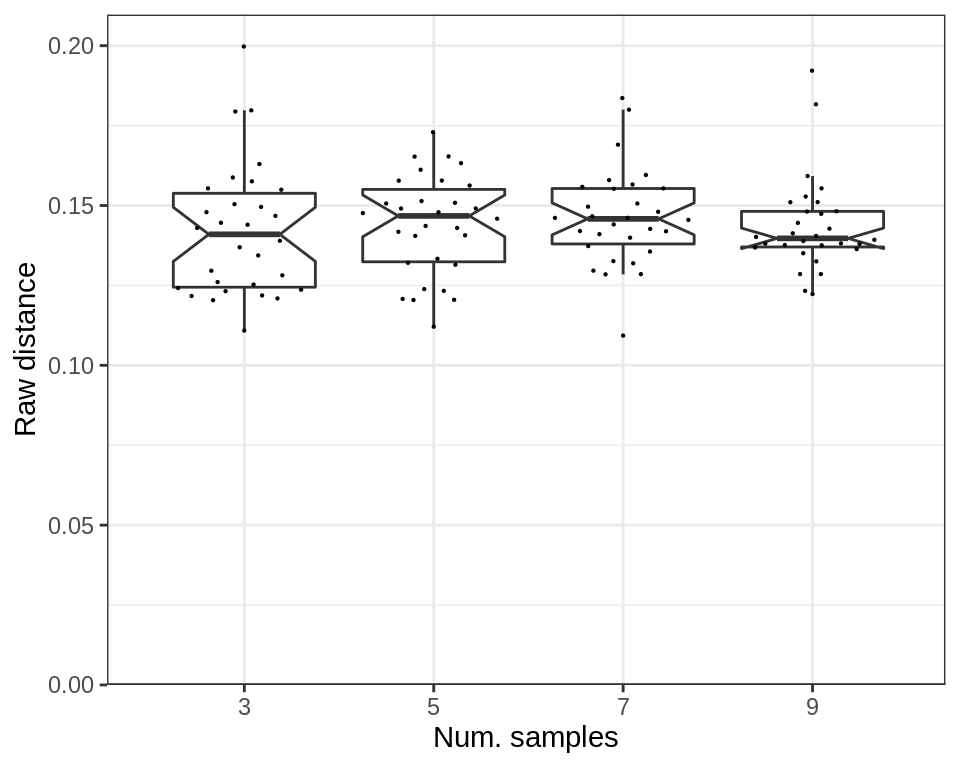
The normalized distance does not depend either, however the statisticsl power of the test depends a lot:
plotExpressionShiftSimDf(p_df, x.col='ns', norm.dist=TRUE, adj.list=xlab("Num. samples"))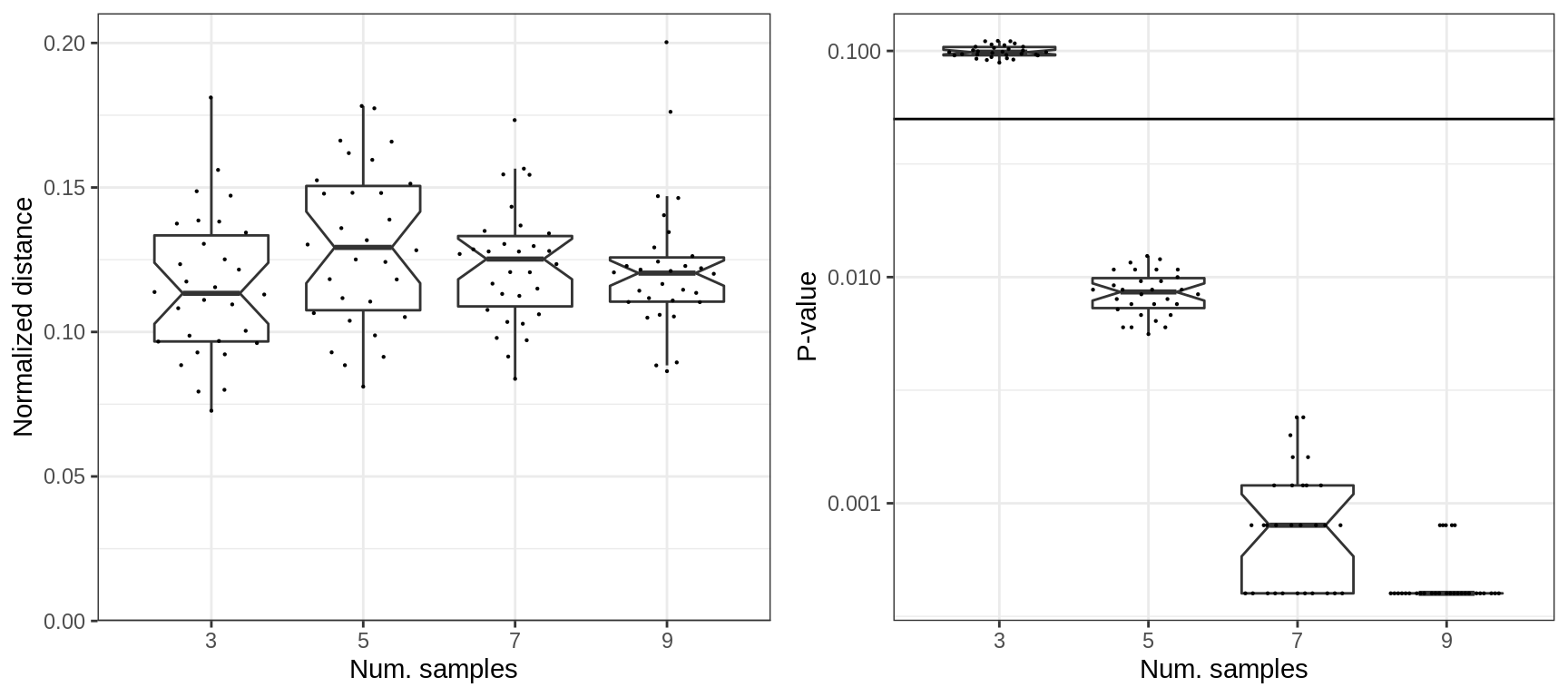
Estimating expression shifts over top DE introduce slight dependency though. Probably, because of the quality of the selected DE genes.
cao_samples$test.results$es.top.de %>%
prepareExpressionShiftSimDf(sims=sims_samples) %>% mutate(ns=as.factor(ns)) %>%
plotExpressionShiftSimDf(x.col='ns', norm.dist=TRUE, adj.list=xlab("Num. samples"))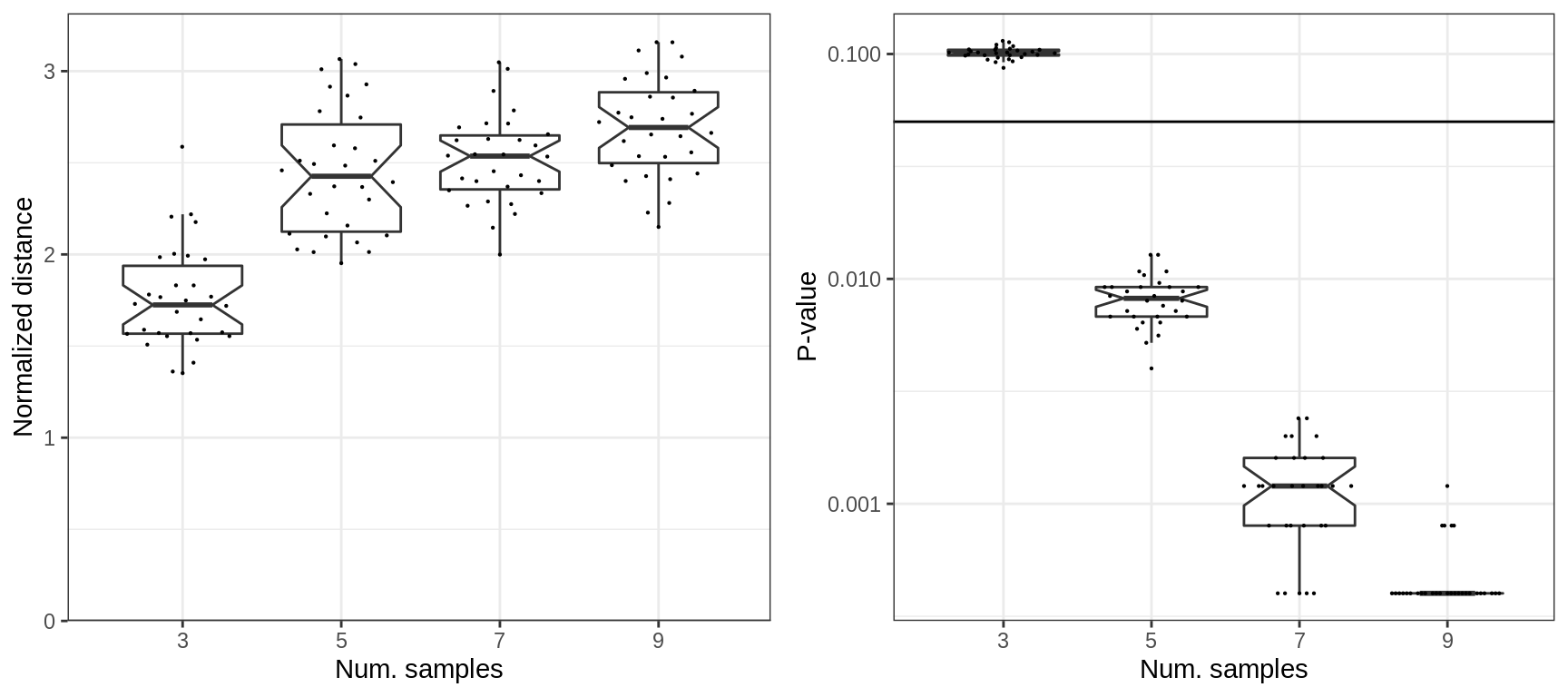
Cluster-free estimates are similar to cluster-based in this regard:
cons_samples <- generateConsForClustFree(sims_samples, n.cores=N_CORES)cao_cf_per_type_samples <- cons_samples %$%
generateCacoaFromConsForClustFree(con.per.type, sim, n.cores=N_CORES)plotClusterFreeShiftSimulations(cao_cf_per_type_samples, params=cons_samples$sim$params,
x.col='ns', adj.list=list(xlab('Num. samples')))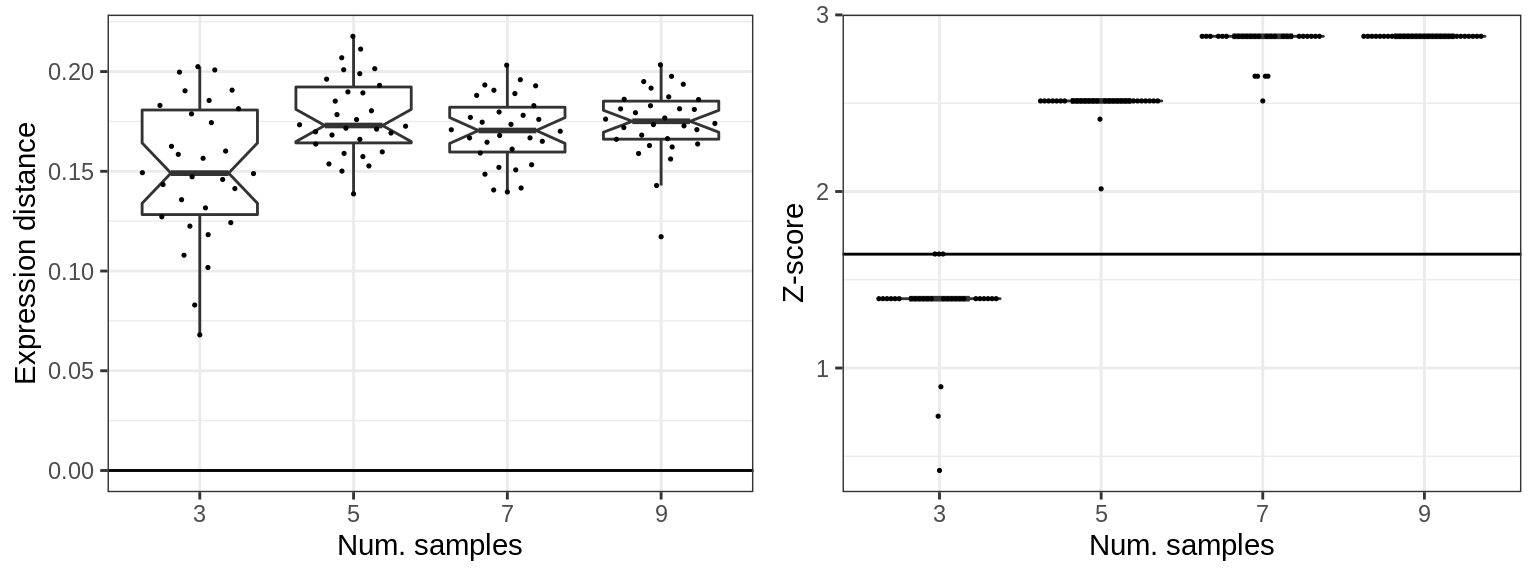
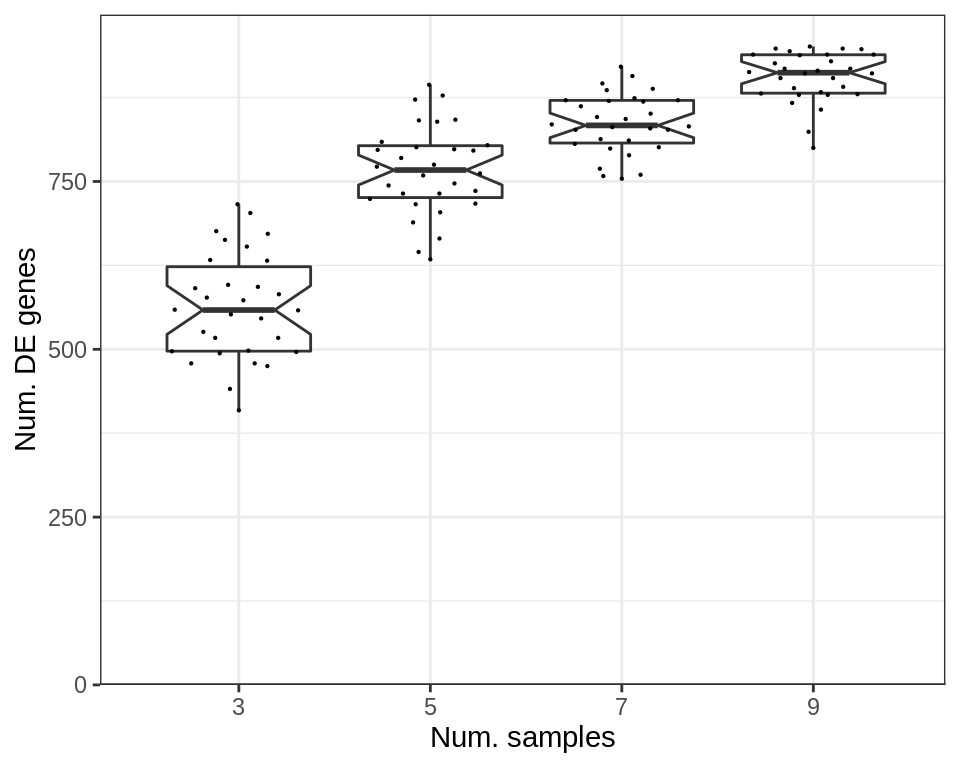
And the number of significant DE genes depends on the number of samples, just like the significance tests for expression shifts above:
p_df$NumDE <- cao_samples$test.results$de %>%
sapply(function(de) sum(de$res$padj < 0.05)) %>% .[p_df$Type]
plotExpressionShiftSimDf(
p_df, x.col='ns', dist.col='NumDE', adj.list=labs(x="Num. samples", y="Num. DE genes")
)
Number of cells per cell type
n_cells <- c(25, 50, 75, 100, 125, 150)
sims_cells <- generateSims(
sce, n.cells=n_cells, de.frac=DE_FRAC, n.cores=N_CORES, lfc=LFC,
n.samples=N_SAMPLES, n.repeats=N_REPEATS, verbose=FALSE
)
cao_cells <- cacoaFromSim(sims_cells, n.cores=N_CORES)
cao_cells$estimateExpressionShiftMagnitudes(verbose=FALSE, n.permutations=N_PERMUTS, min.samp.per.type=MIN_SAMPS)
cao_cells$estimateExpressionShiftMagnitudes(
verbose=TRUE, n.permutations=N_PERMUTS, top.n.genes=TOP_N_GENES, n.pcs=N_PCS,
min.samp.per.type=MIN_SAMPS, name='es.top.de'
)
cao_cells$estimateDEPerCellType(independent.filtering=TRUE, verbose=TRUE)The raw expression distances depend on the number of samples:
p_df <- cao_cells$test.results$expression.shifts %>%
prepareExpressionShiftSimDf(sims=sims_cells) %>% mutate(nc=as.factor(nc))
plotExpressionShiftSimDf(p_df, x.col='nc', norm.dist=FALSE, adj.list=xlab("Num. cells"))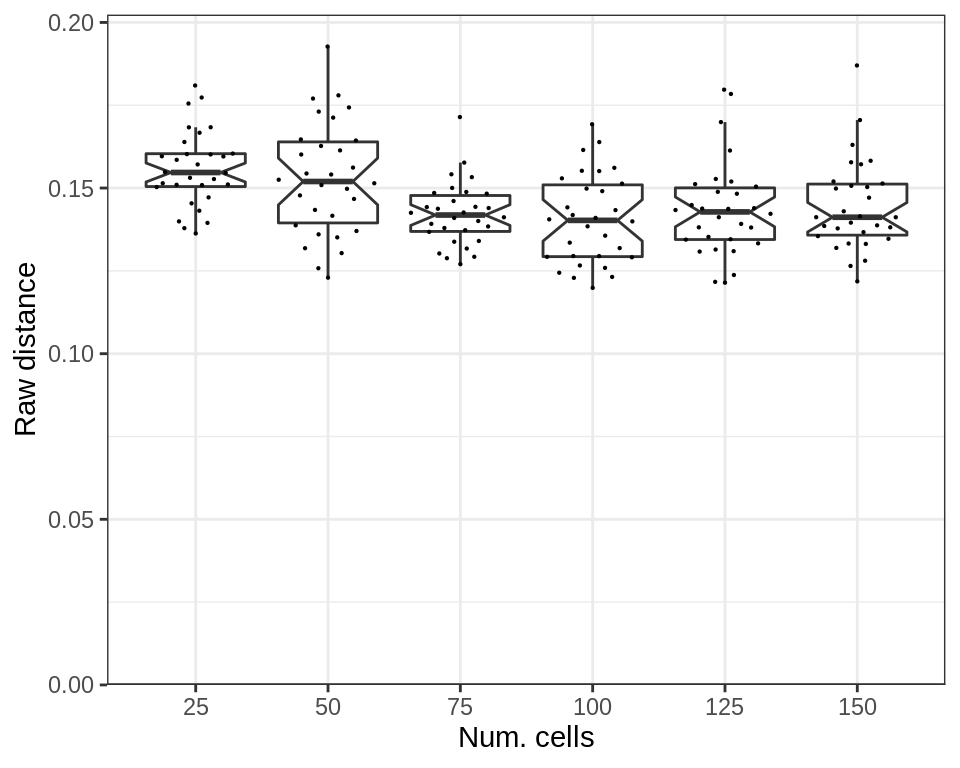
However the normalization fixes it:
plotExpressionShiftSimDf(p_df, x.col='nc', norm.dist=TRUE, adj.list=xlab("Num. cells"))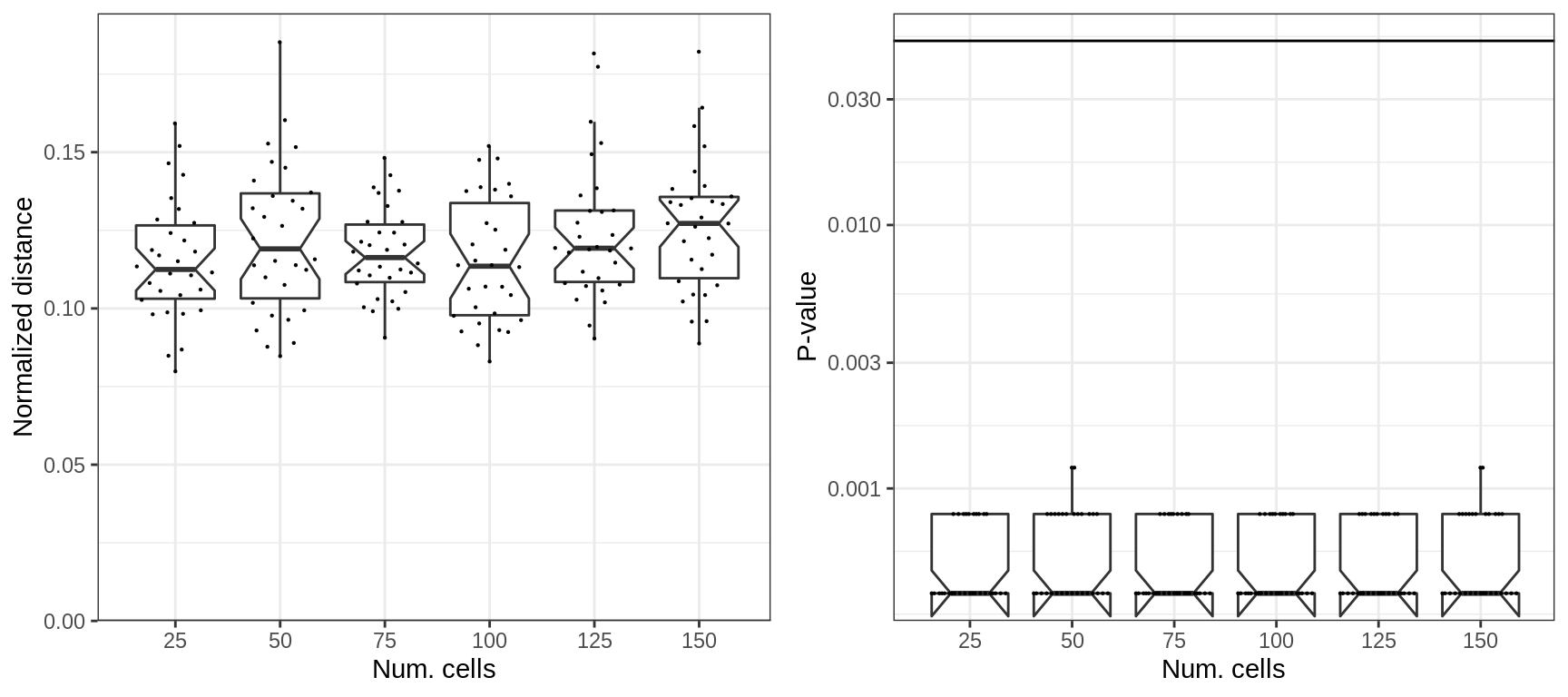
And here, using top-DE genes does not introduce new dependencies:
cao_cells$test.results$es.top.de %>%
prepareExpressionShiftSimDf(sims=sims_cells) %>% mutate(nc=as.factor(nc)) %>%
plotExpressionShiftSimDf(x.col='nc', norm.dist=TRUE, adj.list=xlab("Num. cells"))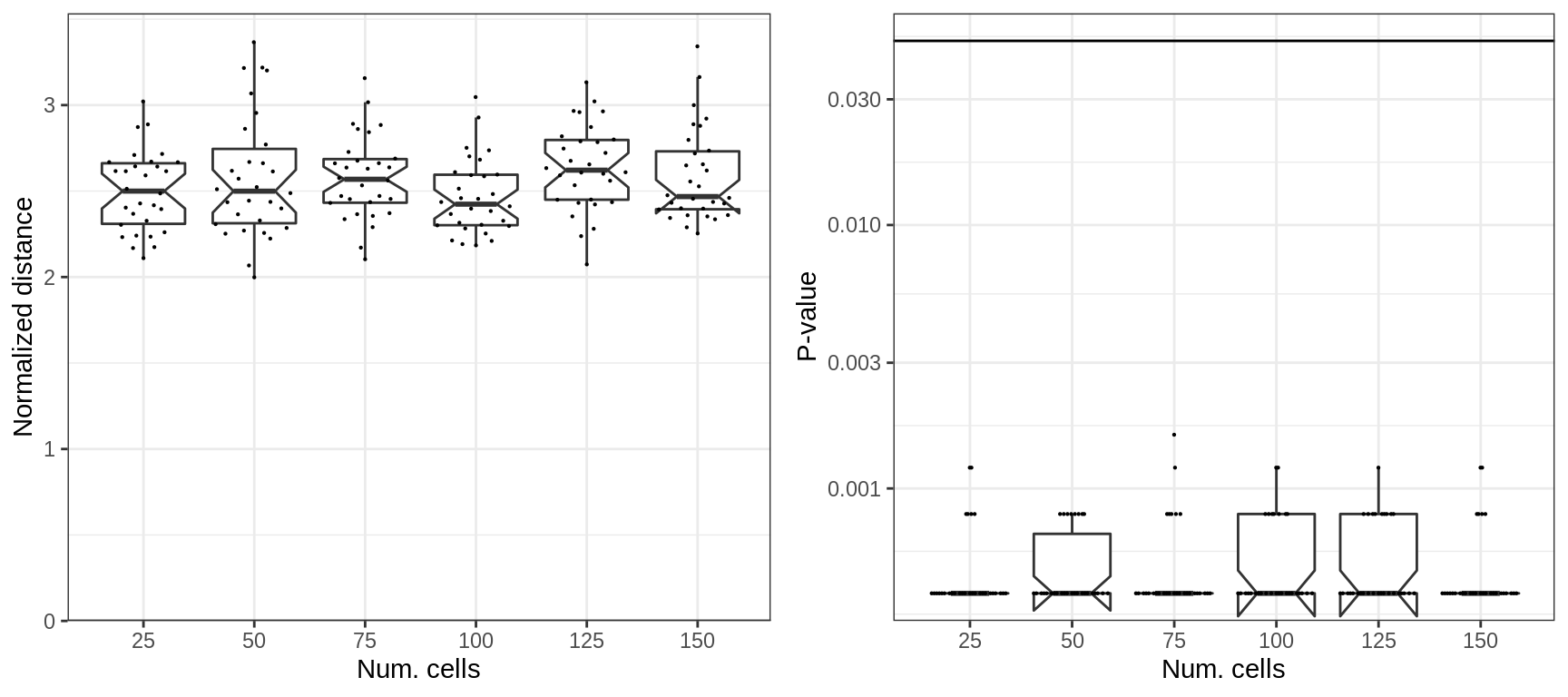
The same with cluster-free:
cons_cells <- generateConsForClustFree(sims_cells, n.cores=N_CORES, ncomps=15, metric="L2")
cao_cf_per_type_cells <- cons_cells %$%
generateCacoaFromConsForClustFree(con.per.type, sim, n.cores=N_CORES)plotClusterFreeShiftSimulations(cao_cf_per_type_cells, params=cons_cells$sim$params,
x.col='nc', adj.list=list(xlab('Num. cells')))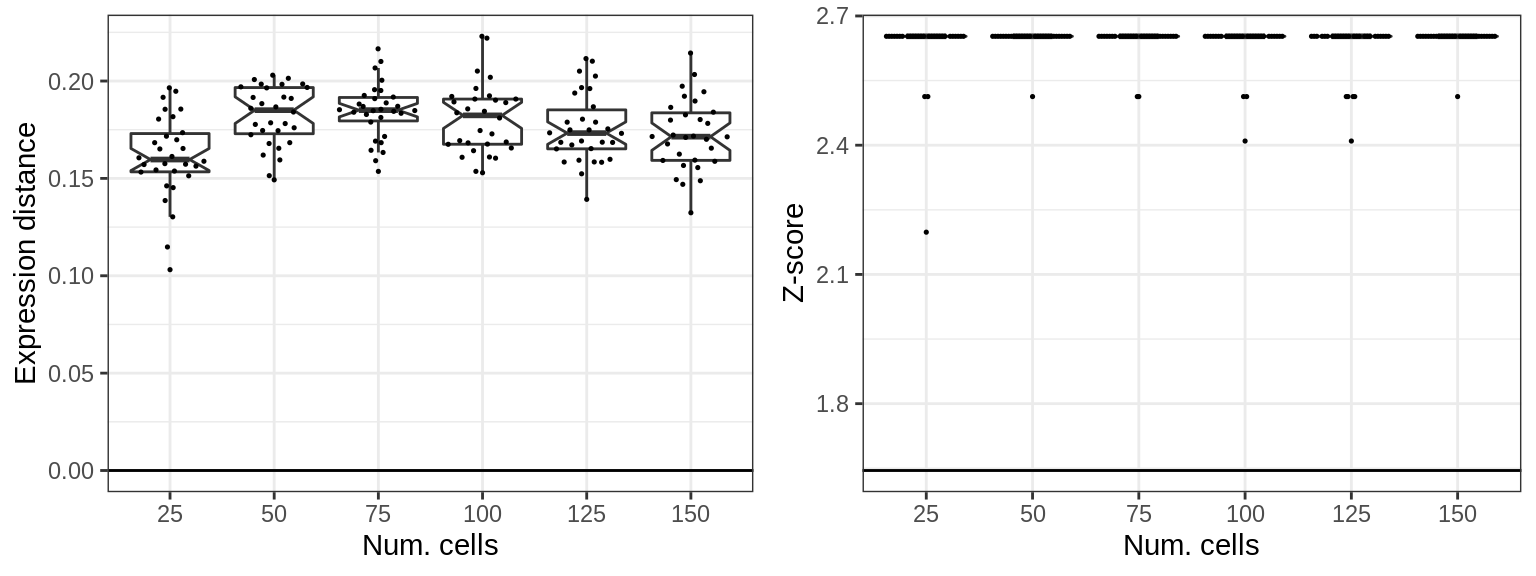
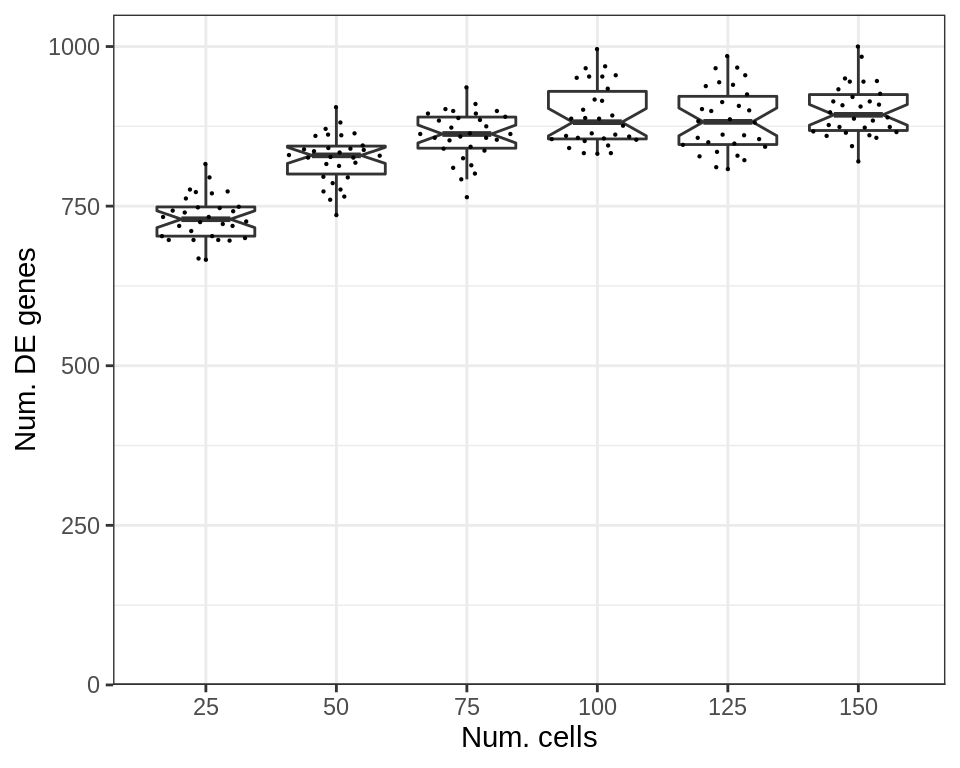
While the number of significant DE genes does depend on the number of cells:
p_df$NumDE <- cao_cells$test.results$de %>%
sapply(function(de) sum(de$res$padj < 0.05)) %>% .[p_df$Type]
plotExpressionShiftSimDf(
p_df, x.col='nc', dist.col='NumDE', adj.list=labs(x="Num. cells", y="Num. DE genes")
)
sessionInfo()R version 4.1.1 (2021-08-10)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 18.04.6 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.7.1
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.7.1
locale:
[1] C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] cacoaAnalysis_0.1.0 sccore_1.0.0 dataorganizer_0.1.0
[4] conos_1.4.4 igraph_1.2.9 cacoa_0.2.0
[7] Matrix_1.3-4 cowplot_1.1.1 forcats_0.5.1
[10] stringr_1.4.0 dplyr_1.0.7 purrr_0.3.4
[13] readr_2.0.1 tidyr_1.1.4 tibble_3.1.6
[16] ggplot2_3.3.5 tidyverse_1.3.1 magrittr_2.0.1
[19] workflowr_1.6.2
loaded via a namespace (and not attached):
[1] utf8_1.2.2 R.utils_2.10.1
[3] tidyselect_1.1.1 RSQLite_2.2.8
[5] AnnotationDbi_1.54.1 grid_4.1.1
[7] BiocParallel_1.26.2 Rtsne_0.15
[9] devtools_2.4.2 munsell_0.5.0
[11] codetools_0.2-18 withr_2.4.3
[13] colorspace_2.0-2 Biobase_2.52.0
[15] highr_0.9 knitr_1.36
[17] rstudioapi_0.13 stats4_4.1.1
[19] pbmcapply_1.5.0 labeling_0.4.2
[21] MatrixGenerics_1.4.3 git2r_0.29.0
[23] urltools_1.7.3 GenomeInfoDbData_1.2.6
[25] farver_2.1.0 bit64_4.0.5
[27] rprojroot_2.0.2 Matrix.utils_0.9.8
[29] vctrs_0.3.8 generics_0.1.1
[31] xfun_0.28 R6_2.5.1
[33] doParallel_1.0.16 GenomeInfoDb_1.28.4
[35] ggbeeswarm_0.6.0 clue_0.3-59
[37] locfit_1.5-9.4 bitops_1.0-7
[39] cachem_1.0.6 DelayedArray_0.18.0
[41] assertthat_0.2.1 promises_1.2.0.1
[43] scales_1.1.1 beeswarm_0.4.0
[45] gtable_0.3.0 Cairo_1.5-12.2
[47] processx_3.5.2 drat_0.2.1
[49] rlang_0.4.12 genefilter_1.74.0
[51] GlobalOptions_0.1.2 splines_4.1.1
[53] broom_0.7.10 brew_1.0-6
[55] yaml_2.2.1 reshape2_1.4.4
[57] modelr_0.1.8 backports_1.4.0
[59] httpuv_1.6.3 tools_4.1.1
[61] usethis_2.0.1 ellipsis_0.3.2
[63] jquerylib_0.1.4 RColorBrewer_1.1-2
[65] BiocGenerics_0.38.0 sessioninfo_1.1.1
[67] Rcpp_1.0.7 plyr_1.8.6
[69] zlibbioc_1.38.0 RCurl_1.98-1.4
[71] ps_1.6.0 prettyunits_1.1.1
[73] dendsort_0.3.4 GetoptLong_1.0.5
[75] S4Vectors_0.30.0 SummarizedExperiment_1.22.0
[77] grr_0.9.5 haven_2.4.3
[79] ggrepel_0.9.1 cluster_2.1.2
[81] fs_1.5.0 circlize_0.4.13
[83] triebeard_0.3.0 reprex_2.0.1
[85] whisker_0.4 matrixStats_0.61.0
[87] pkgload_1.2.4 hms_1.1.0
[89] evaluate_0.14 xtable_1.8-4
[91] XML_3.99-0.7 RMTstat_0.3
[93] readxl_1.3.1 N2R_0.1.1
[95] IRanges_2.26.0 gridExtra_2.3
[97] shape_1.4.6 testthat_3.0.4
[99] compiler_4.1.1 crayon_1.4.2
[101] R.oo_1.24.0 htmltools_0.5.2
[103] mgcv_1.8-37 later_1.3.0
[105] tzdb_0.1.2 geneplotter_1.70.0
[107] lubridate_1.8.0 DBI_1.1.1
[109] dbplyr_2.1.1 pagoda2_1.0.7
[111] ComplexHeatmap_2.8.0 MASS_7.3-54
[113] cli_3.1.0 R.methodsS3_1.8.1
[115] parallel_4.1.1 GenomicRanges_1.44.0
[117] pkgconfig_2.0.3 xml2_1.3.3
[119] foreach_1.5.1 annotate_1.70.0
[121] vipor_0.4.5 bslib_0.3.0
[123] XVector_0.32.0 leidenAlg_0.1.1
[125] rvest_1.0.2 callr_3.7.0
[127] digest_0.6.29 Biostrings_2.60.2
[129] rmarkdown_2.11 cellranger_1.1.0
[131] Rook_1.1-1 rjson_0.2.20
[133] lifecycle_1.0.1 nlme_3.1-152
[135] jsonlite_1.7.2 desc_1.4.0
[137] fansi_0.5.0 pillar_1.6.4
[139] lattice_0.20-44 survival_3.2-13
[141] KEGGREST_1.32.0 ggrastr_0.2.3
[143] fastmap_1.1.0 httr_1.4.2
[145] pkgbuild_1.2.0 glue_1.5.1
[147] remotes_2.4.0 png_0.1-7
[149] iterators_1.0.13 bit_4.0.4
[151] stringi_1.7.6 sass_0.4.0
[153] blob_1.2.2 DESeq2_1.32.0
[155] memoise_2.0.0 irlba_2.3.3
[157] ape_5.5