Last updated: 2022-01-14
Checks: 7 0
Knit directory: cacoaAnalysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it's best to always run the code in an empty environment.
The command set.seed(20211123) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 03d3952. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.Rhistory
Ignored: analysis/figure_cluster_based_de_bio.nb.html
Ignored: analysis/figure_cluster_free_expression.nb.html
Ignored: analysis/figure_compositional.nb.html
Ignored: analysis/figure_compositional_cf.nb.html
Ignored: analysis/figure_expression_shifts.nb.html
Ignored: analysis/figure_heterogeneity.nb.html
Ignored: analysis/figure_interpretation.nb.html
Ignored: analysis/prepare_cacoa_results.nb.html
Ignored: analysis/preprocess.nb.html
Ignored: analysis/report_asd.nb.html
Ignored: analysis/report_az.nb.html
Ignored: analysis/report_ep.nb.html
Ignored: analysis/report_ms.nb.html
Ignored: analysis/report_pf.nb.html
Ignored: analysis/report_scc.nb.html
Ignored: analysis/simulation_distances.nb.html
Ignored: analysis/simulation_variance.nb.html
Ignored: cache/
Ignored: data/ASD/
Ignored: data/AZ/
Ignored: data/EP/
Ignored: data/MS/
Ignored: data/PF/
Ignored: data/SCC/
Ignored: man/
Ignored: output/figures/
Unstaged changes:
Modified: analysis/index.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/figure_interpretation.Rmd) and HTML (docs/figure_interpretation.html) files. If you've configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 03d3952 | viktor_petukhov | 2022-01-14 | Updated interpretation figure |
| Rmd | c4d4786 | viktor_petukhov | 2021-12-27 | Interpretation figure |
Epithelial cells
Extract
cao_ept <- readOrCreate(CachePath("cao_pf_ept.rds"), function() {
cao.pf <- read_rds(DataPath("PF/cao.rds")) %>% Cacoa$new()
epithelial.types <- c(
"AT1", "Transitional AT2", "AT2", "Basal", "KRT5-/KRT17+", "MUC5AC+ High", "MUC5B+",
"Proliferating Epithelial Cells", "SCGB3A2+", "SCGB3A2+ SCGB1A1+"
)
ept.cms <- lapply(cao.pf$data.object$samples, function(p2) {
p2$misc$rawCounts %>% .[cao.pf$cell.groups[rownames(.)] %in% epithelial.types,] %>% t()
}) %>% .[sapply(., ncol) > 80]
ept.p2s <- plapply(ept.cms, createPagoda, n.pcs=50, n.cores=N_CORES, progress=TRUE,
mc.preschedule=TRUE)
if ("value" %in% names(ept.p2s)) ept.p2s <- ept.p2s$value
ept.con <- conos::Conos$new(ept.p2s, n.cores=N_CORES)
ept.con$buildGraph(k=30, k.self.weight=0.5)
ept.con$embedGraph(min.prob.lower=1e-4, method="UMAP", verbose=FALSE)
cao.ept <- Cacoa$new(
ept.con, cell.groups=cao.pf$cell.groups[names(ept.con$getDatasetPerCell())],
sample.groups=cao.pf$sample.groups[names(ept.con$samples)],
ref.level=cao.pf$ref.level, target.level=cao.pf$target.level, n.cores=N_CORES
)
cao.ept$plot.theme %<>% `+`(theme(legend.background=element_blank()))
cao.ept$estimateDEPerCellType(independent.filtering=TRUE, test="DESeq2.Wald")
cao.ept$estimateOntology(org.db=org.db, type='GSEA')
return(cao.ept)
}) %>% Cacoa$new()Cluster-based
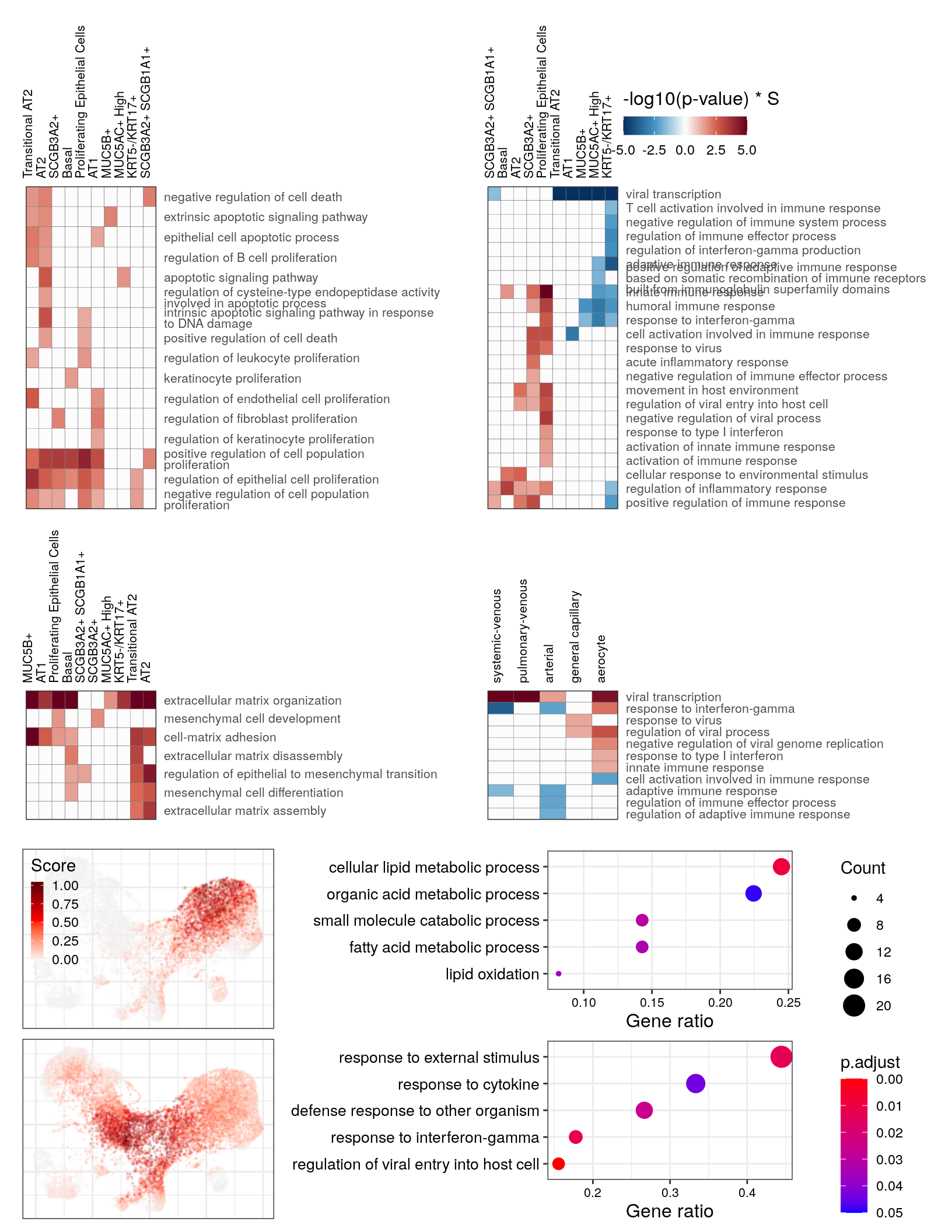
cao_ept$estimateOntology(org.db=org.db, type='GSEA') # TODO: remove thisgg_at_apopt <- cao_ept$plotOntologyHeatmap(
name='GSEA', genes="up", description.regex='death|apopt|proliferation', min.genes=10,
description.exclude.regex='neur', max.log.p=5
)
gg_at_apopt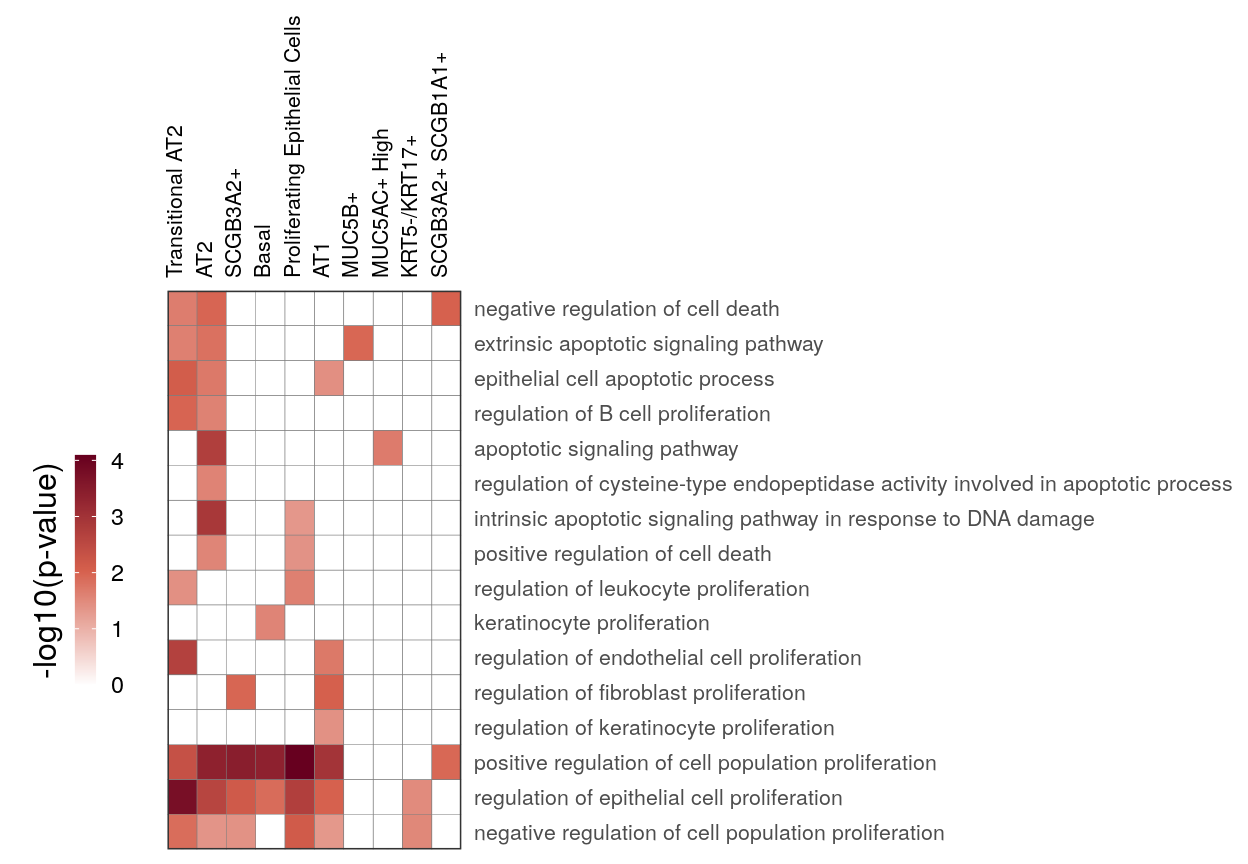
immune_regex <- 'vir|immune|interferon|inflam'
gg_at_immune <- cao_ept$plotOntologyHeatmap(
name='GSEA', genes="all", legend.title='-log10(p-value) * S',
description.regex=immune_regex, min.genes=10, max.log.p=5
)
gg_at_immune
gg_at_matrix <- cao_ept$plotOntologyHeatmap(
name='GSEA', genes="up", description.regex='matrix|mesen', min.genes=10, max.log.p=5
)
gg_at_matrix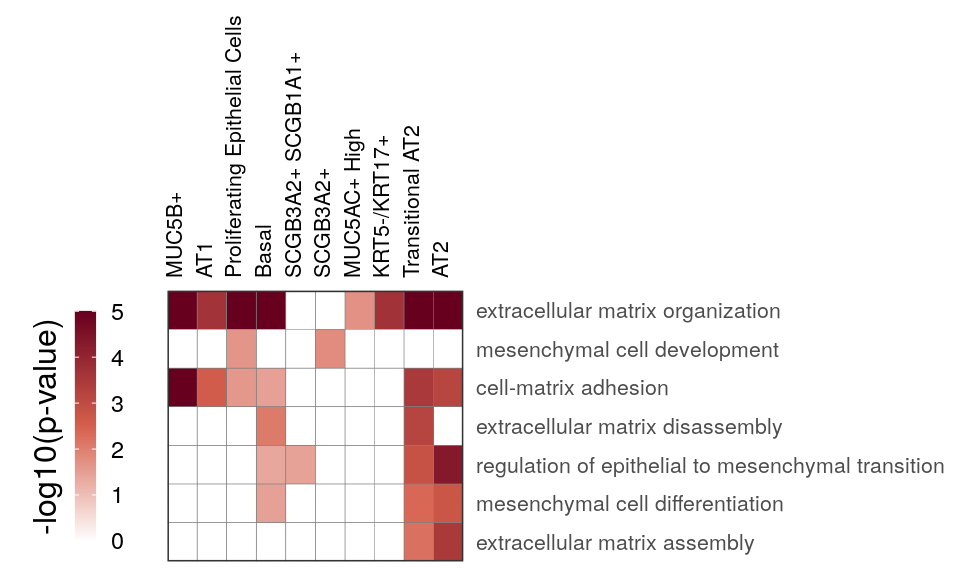
Cluster-free
cao_ept$estimateClusterFreeDE(n.top.genes=1000, min.expr.frac=0.01, adjust.pvalues=TRUE,
smooth=TRUE)Estimating cluster-free Z-scores for 1000 most expressed genes0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
***************************************************Gene programs
cao_ept$estimateGenePrograms(n.programs=9, z.adj=TRUE, abs.scores=TRUE, smooth=FALSE, verbose=FALSE)cao_ept$plotGeneProgramScores(legend.position=c(0, 1), size=0.1, alpha=0.5, plot.na=FALSE,
adj.list=theme(plot.margin=margin()))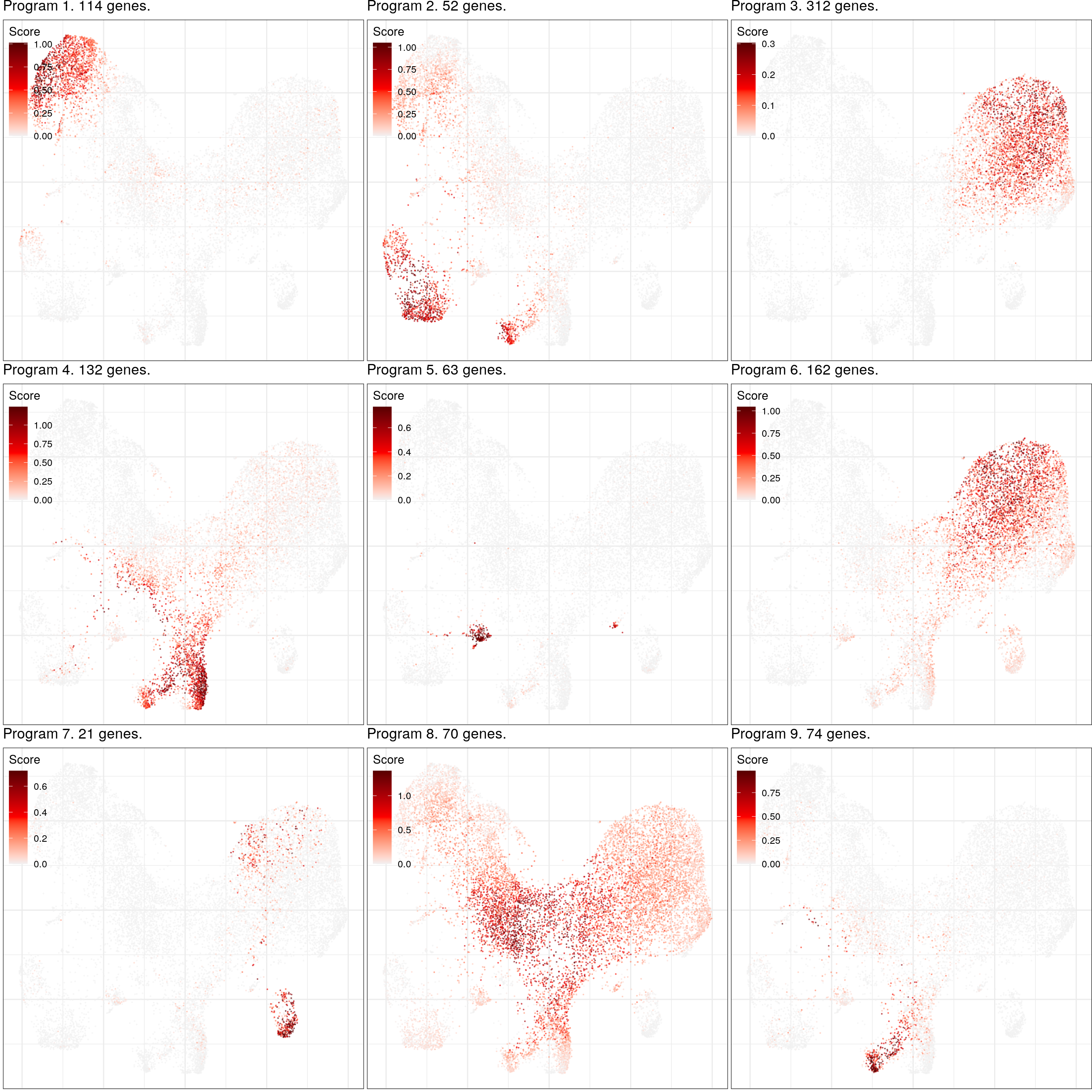
ggs_cf_scores <- cao_ept$plotGeneProgramScores(
prog.ids=c(6, 8), legend.position=c(0, 1), size=0.1, alpha=0.3, build.panel=FALSE,
plot.na=FALSE, adj.list=theme(plot.margin=margin(), plot.title=element_blank())
)
plot_grid(plotlist=ggs_cf_scores)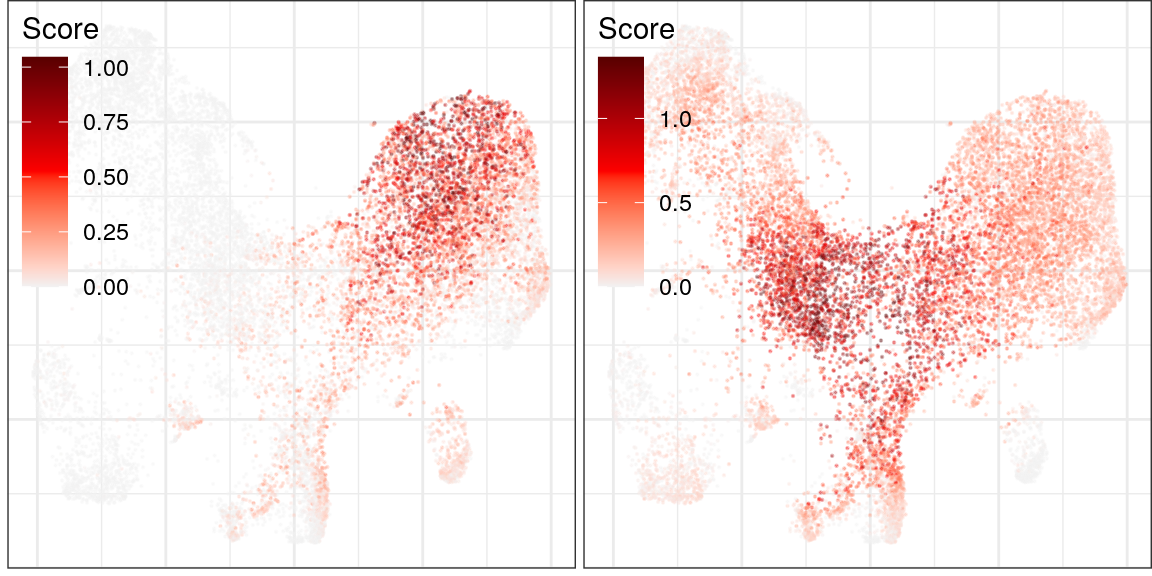
go_env <- cao_ept$getGOEnvironment(org.db=org.db)Using stored GO environment. Use `ignore.cache=TRUE` if you want to re-estimate it. Set `ignore.cache=FALSE` to suppress this message.gene_universe_global <- colnames(cao_ept$test.results$cluster.free.de$z.adj) %>%
cacoa:::mapGeneIds(org.db)
length(gene_universe_global)[1] 932t_scores <- c(6, 8) %>%
{setNames(cao_ept$test.results$gene.programs$sim.scores[.], .)} %>%
lapply(function(x) x[x > 0.5])
sapply(t_scores, length) 6 8
104 47 go_global <- lapply(t_scores, function(x) head(names(x[x > 0.5]), 50)) %>%
lapply(cacoa:::mapGeneIds, org.db) %>%
cacoa:::estimateEnrichedGO(org.db=org.db, go.environment=go_env, universe=gene_universe_global)
go_dfs <- lapply(go_global$BP, function(r) filter(r@result, p.adjust < 0.05)) %>%
.[sapply(., nrow) > 0]
sapply(go_dfs, nrow) 6 8
11 25 AT2 -> AT1
go_dfs$`6` %$% setNames(strsplit(geneID, "/"), Description) %>%
cacoa:::estimateClusterPerGO(cut.h=0.4) %>% {split(names(.), .)} %>%
sapply(paste, collapse='"; "') %>% {paste0('"', ., '"\n')} %>% cat()"cellular lipid metabolic process"; "lipid metabolic process"
"small molecule metabolic process"; "organic acid metabolic process"; "carboxylic acid metabolic process"; "oxoacid metabolic process"
"small molecule catabolic process"
"fatty acid metabolic process"
"fatty acid oxidation"; "lipid oxidation"; "lipid modification"gg_go_at <- clusteredOntologyDotplot(go_global$BP$`6`, orderBy='x', cut.h=0.4)
gg_go_at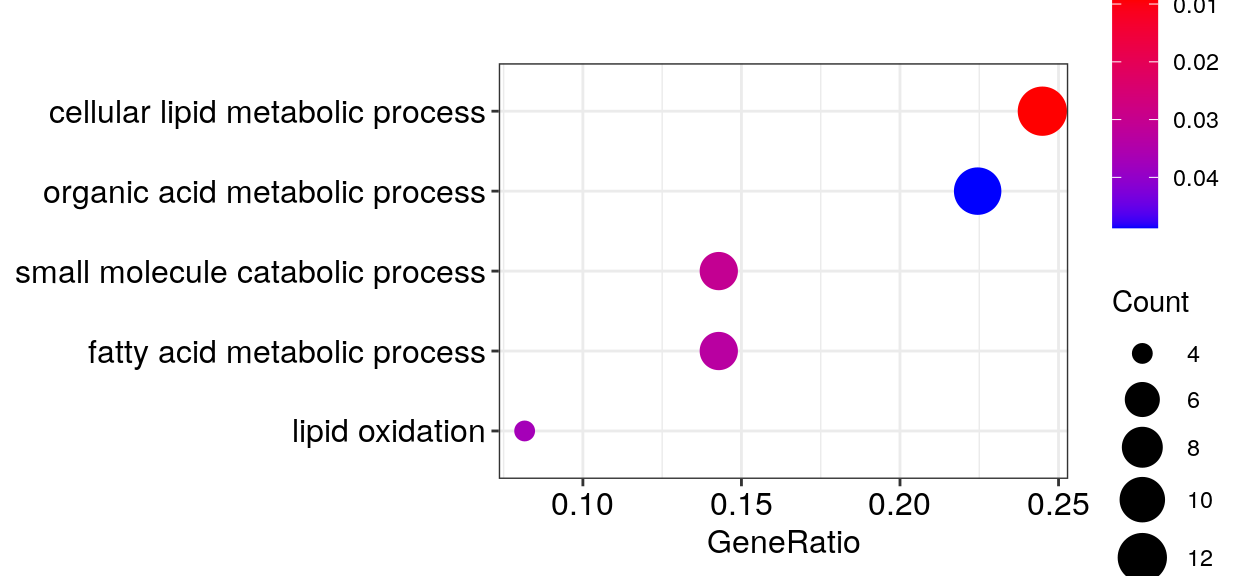
Transitional state program
c_go_clusts <- go_dfs$`8` %$% setNames(strsplit(geneID, "/"), Description) %>%
cacoa:::estimateClusterPerGO(cut.h=0.4) %>% {split(names(.), .)}
c_go_clusts %>% sapply(paste, collapse='"; "') %>% {paste0('"', ., '"\n')} %>% cat"regulation of viral entry into host cell"; "modulation by symbiont of entry into host"; "negative regulation of viral entry into host cell"; "negative regulation of viral life cycle"; "regulation of viral life cycle"; "negative regulation of viral process"; "viral entry into host cell"; "entry into host"; "regulation of viral process"; "regulation of biological process involved in symbiotic interaction"; "movement in host environment"; "biological process involved in interaction with host"
"response to interferon-gamma"
"innate immune response"; "defense response"; "defense response to other organism"; "response to external biotic stimulus"; "response to other organism"
"response to external stimulus"; "immune system process"; "response to stress"; "cellular response to chemical stimulus"
"cytokine-mediated signaling pathway"; "cellular response to cytokine stimulus"; "response to cytokine"gg_go_trans <- clusteredOntologyDotplot(go_global$BP[["8"]], orderBy='x', cut.h=0.4)
gg_go_trans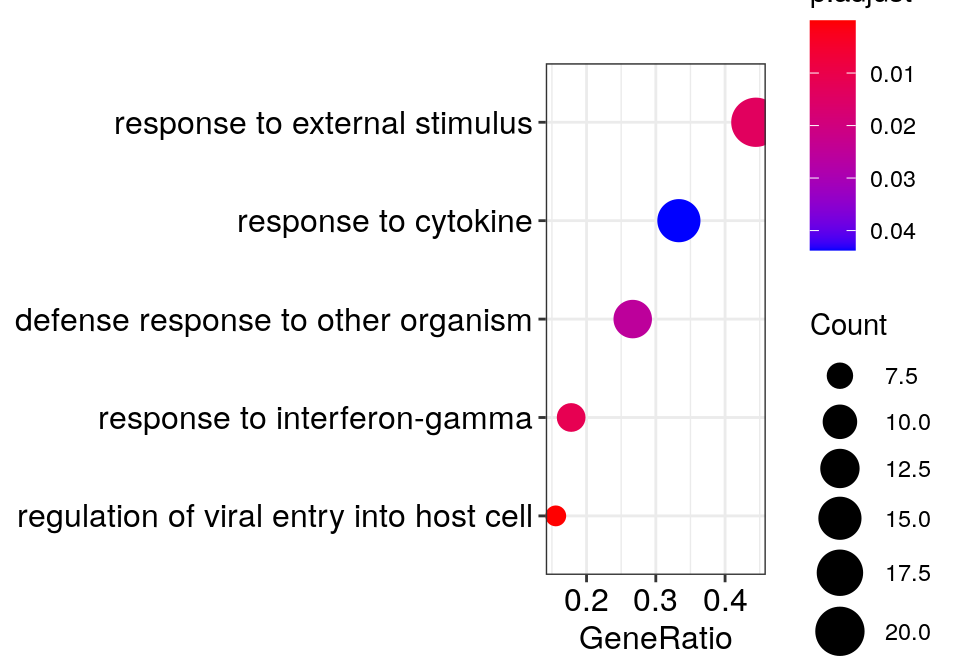
cao_ept$plot.params <- list(size=0.5, alpha=0.5)
ggs_at_genes <- c("AGER", "HOPX", "SFTPC") %>% sccore::sn() %>% lapply(function(gn) {
cao_ept$plotEmbedding(colors=cao_ept$cache$joint.count.matrix.norm[,gn], legend.title="Expr.",
legend.position=c(0, 1))
})
ggs_at_genes$AGER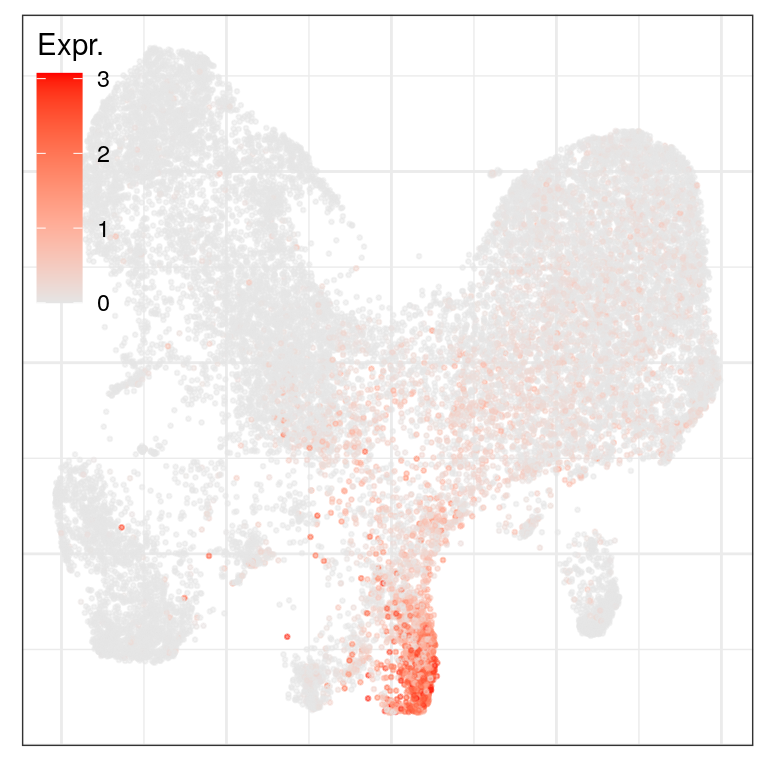
$HOPX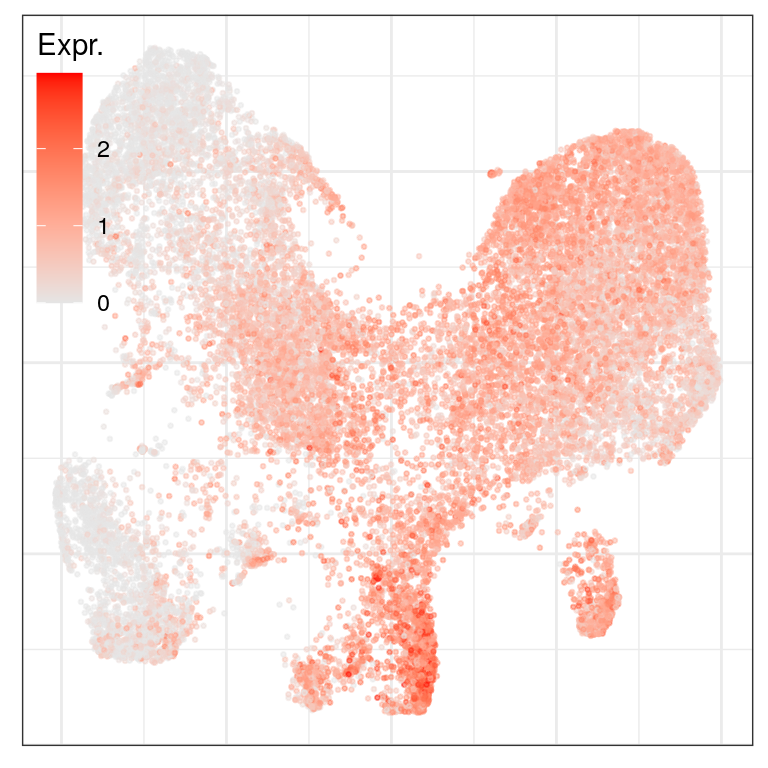
$SFTPC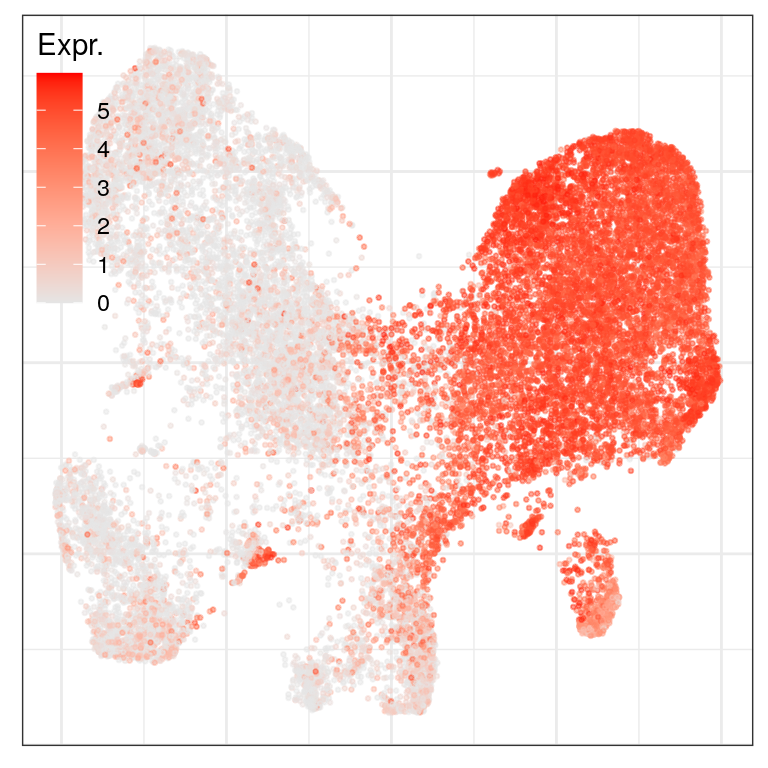
Endothelial cells
# Requires running cluster-free expression figure first
cao_endo <- read_rds(CachePath("cao_pf_endo.rds")) %>% Cacoa$new()
cao_endo$estimateOntology(type="GSEA", org.db=org.Hs.eg.db::org.Hs.eg.db)gg_end_viral <- cao_endo$plotOntologyHeatmap(
name='GSEA', genes="all", legend.title='-log10(p-value) * S',
description.regex='vir|immune|interferon|inflam', min.genes=10, max.log.p=5,
description.exclude.regex='built from' # Remove one super-long GO for cluster name
)
gg_end_viral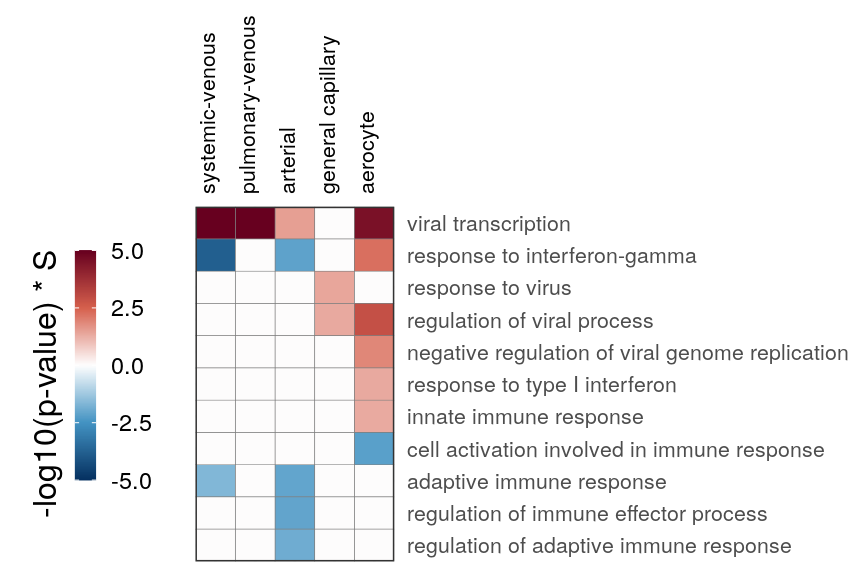
cao_endo$plotOntologyHeatmap(name='GSEA', genes="up", description.regex='matrix|mesen')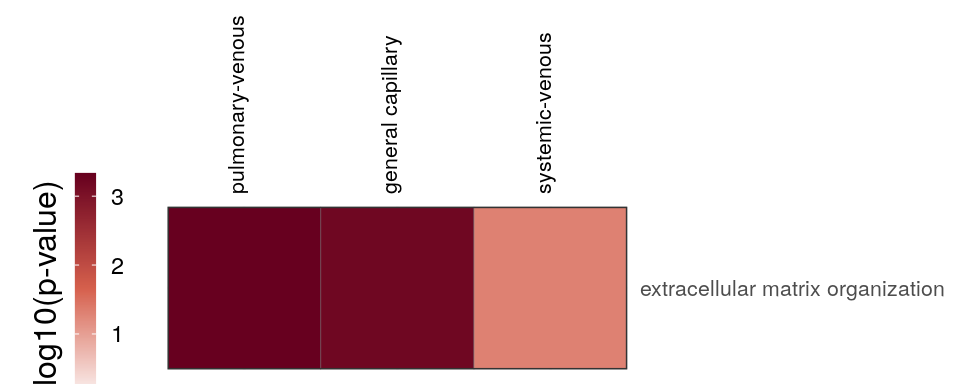
Compile the figure
theme_ax <- theme(
axis.text.x=element_text(size=8),
axis.text.y=element_text(size=8, lineheight=0.75),
plot.title=element_blank(),
plot.margin=margin()
)
fill_guide <- guides(fill=guide_colorbar(
title='-log10(p-value) * S', title.theme=element_text(size=12),
title.position="top"
))
fill_scale <- gg_at_immune$scales$scales[[3]]
plt_list <- list(gg_at_apopt, gg_at_immune, gg_at_matrix, gg_end_viral) %>% lapply(function(gg) {
levels(gg$data$G1) %<>% str_wrap(50)
gg <- gg + theme_ax + fill_guide + fill_scale + theme_legend_position("none")
gg
})Scale for 'fill' is already present. Adding another scale for 'fill', which
will replace the existing scale.
Scale for 'fill' is already present. Adding another scale for 'fill', which
will replace the existing scale.
Scale for 'fill' is already present. Adding another scale for 'fill', which
will replace the existing scale.
Scale for 'fill' is already present. Adding another scale for 'fill', which
will replace the existing scale.plt_list[[2]] %<>% {. + theme(
legend.position=c(2.3, 1.3), legend.justification=c(1, 1),
legend.direction="horizontal", legend.margin=margin(),
legend.box.margin=margin(),
legend.key.height=unit(12, "pt"), legend.key.width=unit(16, "pt")
)}
go_fill_scale <- scale_color_continuous(low="red", high="blue", limits=c(0, 0.05),
guide=guide_colorbar(reverse=TRUE))
go_size_scale <- scale_size_continuous(limits=c(4, 20))
gg_go_at %<>% {. + go_fill_scale + go_size_scale + xlab("Gene ratio")}Scale for 'colour' is already present. Adding another scale for 'colour',
which will replace the existing scale.Scale for 'size' is already present. Adding another scale for 'size', which
will replace the existing scale.gg_go_trans %<>% {. + go_fill_scale + go_size_scale + xlab("Gene ratio")}Scale for 'colour' is already present. Adding another scale for 'colour',
which will replace the existing scale.
Scale for 'size' is already present. Adding another scale for 'size', which
will replace the existing scale.go_leg_grob <- ggpubr::get_legend(gg_go_at)
ggs_cf_scores_rast <- lapply(ggs_cf_scores, ggrastr::rasterise, dev="ragg_png", dpi=100)
plot_grid(
plot_grid(
plotlist=plt_list,
ncol=2, rel_heights=c(1, 0.6), align="hv", scale=0.95
),
plot_grid(
plot_grid(
ggs_cf_scores_rast[[1]] +
theme(legend.key.width=unit(8, "pt"), legend.key.height=unit(10, "pt")),
ggs_cf_scores_rast[[2]] + theme(legend.position="none"),
ncol=1, scale=0.97
),
plot_grid(
gg_go_at + theme_ax + theme(legend.position="none", axis.text.y=element_text(size=10)),
gg_go_trans + theme_ax + theme(legend.position="none", axis.text.y=element_text(size=10)),
ncol=1, align="v", scale=0.95
),
go_leg_grob,
nrow=1, rel_widths=c(1, 2, 0.5)
),
ncol=1, rel_heights=c(7.5, 3.5), scale=0.97
)
ggsave(figurePath("7_functional_interpretation.pdf"))Saving 8.5 x 11 in image
sessionInfo()R version 4.0.3 (2020-10-10)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 18.04.6 LTS
Matrix products: default
BLAS: /usr/local/R/R-4.0.3/lib/R/lib/libRblas.so
LAPACK: /usr/local/R/R-4.0.3/lib/R/lib/libRlapack.so
locale:
[1] C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] cacoaAnalysis_0.1.0 dataorganizer_0.1.0 sccore_1.0.1
[4] cacoa_0.2.0 cowplot_1.1.1 conos_1.4.4
[7] igraph_1.2.6 Matrix_1.2-18 magrittr_2.0.1
[10] forcats_0.5.1 stringr_1.4.0 dplyr_1.0.7
[13] purrr_0.3.4 readr_1.4.0 tidyr_1.1.4
[16] tibble_3.1.5 ggplot2_3.3.5 tidyverse_1.3.0
[19] workflowr_1.6.2
loaded via a namespace (and not attached):
[1] utf8_1.2.2 reticulate_1.22 R.utils_2.10.1
[4] tidyselect_1.1.1 RSQLite_2.2.8 AnnotationDbi_1.52.0
[7] grid_4.0.3 BiocParallel_1.24.1 Rtsne_0.15
[10] scatterpie_0.1.5 devtools_2.3.2 munsell_0.5.0
[13] ragg_0.4.1 codetools_0.2-16 withr_2.4.2
[16] GOSemSim_2.16.1 colorspace_2.0-2 Biobase_2.50.0
[19] highr_0.9 knitr_1.36 rstudioapi_0.13
[22] stats4_4.0.3 ggsignif_0.6.1 pbmcapply_1.5.0
[25] DOSE_3.16.0 labeling_0.4.2 git2r_0.27.1
[28] urltools_1.7.3 polyclip_1.10-0 farver_2.1.0
[31] bit64_4.0.5 downloader_0.4 rprojroot_2.0.2
[34] Matrix.utils_0.9.8 vctrs_0.3.8 generics_0.1.0
[37] xfun_0.26 R6_2.5.1 doParallel_1.0.16
[40] graphlayouts_0.7.1 ggbeeswarm_0.6.0 clue_0.3-59
[43] fgsea_1.16.0 cachem_1.0.6 assertthat_0.2.1
[46] promises_1.1.1 scales_1.1.1 ggraph_2.0.4
[49] enrichplot_1.10.1 beeswarm_0.4.0 gtable_0.3.0
[52] processx_3.4.5 tidygraph_1.2.0 drat_0.1.8
[55] rlang_0.4.11 systemfonts_1.0.0 GlobalOptions_0.1.2
[58] splines_4.0.3 rstatix_0.7.0 broom_0.7.9
[61] brew_1.0-6 BiocManager_1.30.10 yaml_2.2.1
[64] reshape2_1.4.4 abind_1.4-5 modelr_0.1.8
[67] backports_1.2.1 httpuv_1.5.4 qvalue_2.22.0
[70] clusterProfiler_3.18.0 tools_4.0.3 usethis_1.6.3
[73] ellipsis_0.3.2 jquerylib_0.1.4 RColorBrewer_1.1-2
[76] BiocGenerics_0.36.1 sessioninfo_1.1.1 Rcpp_1.0.7
[79] plyr_1.8.6 ps_1.4.0 prettyunits_1.1.1
[82] ggpubr_0.4.0 dendsort_0.3.3 viridis_0.6.1
[85] GetoptLong_1.0.5 S4Vectors_0.28.1 grr_0.9.5
[88] haven_2.4.1 ggrepel_0.9.1 cluster_2.1.0
[91] fs_1.5.0 data.table_1.14.2 DO.db_2.9
[94] openxlsx_4.2.3 circlize_0.4.13 triebeard_0.3.0
[97] reprex_0.3.0 whisker_0.4 matrixStats_0.61.0
[100] pkgload_1.2.1 hms_1.1.1 evaluate_0.14
[103] rio_0.5.26 RMTstat_0.3 readxl_1.3.1
[106] N2R_0.1.1 IRanges_2.24.1 gridExtra_2.3
[109] shape_1.4.6 testthat_3.0.0 compiler_4.0.3
[112] shadowtext_0.0.7 crayon_1.4.1 R.oo_1.24.0
[115] htmltools_0.5.2 mgcv_1.8-33 later_1.1.0.1
[118] lubridate_1.7.9.2 DBI_1.1.1 tweenr_1.0.1
[121] dbplyr_2.0.0 pagoda2_1.0.7 ComplexHeatmap_2.9.4
[124] MASS_7.3-53 car_3.0-10 cli_3.0.1
[127] R.methodsS3_1.8.1 parallel_4.0.3 pkgconfig_2.0.3
[130] rvcheck_0.1.8 foreign_0.8-80 xml2_1.3.2
[133] foreach_1.5.1 vipor_0.4.5 leidenAlg_0.1.0
[136] rvest_0.3.6 callr_3.5.1 digest_0.6.28
[139] fastmatch_1.1-0 rmarkdown_2.11 cellranger_1.1.0
[142] Rook_1.1-1 curl_4.3.2 rjson_0.2.20
[145] lifecycle_1.0.1 nlme_3.1-149 jsonlite_1.7.2
[148] carData_3.0-4 viridisLite_0.4.0 desc_1.3.0
[151] fansi_0.5.0 pillar_1.6.3 lattice_0.20-41
[154] GO.db_3.12.1 ggrastr_1.0.1 fastmap_1.1.0
[157] httr_1.4.2 pkgbuild_1.1.0 glue_1.4.2
[160] remotes_2.2.0 zip_2.2.0 png_0.1-7
[163] iterators_1.0.13 bit_4.0.4 ggforce_0.3.2
[166] stringi_1.7.5 blob_1.2.2 textshaping_0.2.1
[169] org.Hs.eg.db_3.12.0 memoise_2.0.0 irlba_2.3.3
[172] ape_5.5