Cluster analysis of group integrated cells
Katharina Hembach
8/26/2020
Last updated: 2020-09-02
Checks: 7 0
Knit directory: neural_scRNAseq/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it's best to always run the code in an empty environment.
The command set.seed(20200522) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 043115f. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: ._.DS_Store
Ignored: ._Rplots.pdf
Ignored: .__workflowr.yml
Ignored: ._neural_scRNAseq.Rproj
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: analysis/._.DS_Store
Ignored: analysis/._01-preprocessing.Rmd
Ignored: analysis/._01-preprocessing.html
Ignored: analysis/._02.1-SampleQC.Rmd
Ignored: analysis/._03-filtering.Rmd
Ignored: analysis/._04-clustering.Rmd
Ignored: analysis/._04-clustering.knit.md
Ignored: analysis/._04.1-cell_cycle.Rmd
Ignored: analysis/._05-annotation.Rmd
Ignored: analysis/._Lam-0-NSC_no_integration.Rmd
Ignored: analysis/._Lam-01-NSC_integration.Rmd
Ignored: analysis/._Lam-02-NSC_annotation.Rmd
Ignored: analysis/._NSC-1-clustering.Rmd
Ignored: analysis/._NSC-2-annotation.Rmd
Ignored: analysis/.__site.yml
Ignored: analysis/._additional_filtering.Rmd
Ignored: analysis/._additional_filtering_clustering.Rmd
Ignored: analysis/._index.Rmd
Ignored: analysis/._organoid-01-clustering.Rmd
Ignored: analysis/._organoid-02-integration.Rmd
Ignored: analysis/._organoid-03-cluster_analysis.Rmd
Ignored: analysis/._organoid-04-group_integration.Rmd
Ignored: analysis/._organoid-05-group_integration_cluster_analysis.Rmd
Ignored: analysis/01-preprocessing_cache/
Ignored: analysis/02-1-SampleQC_cache/
Ignored: analysis/02-quality_control_cache/
Ignored: analysis/02.1-SampleQC_cache/
Ignored: analysis/03-filtering_cache/
Ignored: analysis/04-clustering_cache/
Ignored: analysis/04.1-cell_cycle_cache/
Ignored: analysis/05-annotation_cache/
Ignored: analysis/Lam-01-NSC_integration_cache/
Ignored: analysis/Lam-02-NSC_annotation_cache/
Ignored: analysis/NSC-1-clustering_cache/
Ignored: analysis/NSC-2-annotation_cache/
Ignored: analysis/additional_filtering_cache/
Ignored: analysis/additional_filtering_clustering_cache/
Ignored: analysis/organoid-01-clustering_cache/
Ignored: analysis/organoid-02-integration_cache/
Ignored: analysis/organoid-03-cluster_analysis_cache/
Ignored: analysis/organoid-04-group_integration_cache/
Ignored: analysis/sample5_QC_cache/
Ignored: data/.DS_Store
Ignored: data/._.DS_Store
Ignored: data/._.smbdeleteAAA17ed8b4b
Ignored: data/._Lam_figure2_markers.R
Ignored: data/._known_NSC_markers.R
Ignored: data/._known_cell_type_markers.R
Ignored: data/._metadata.csv
Ignored: data/data_sushi/
Ignored: data/filtered_feature_matrices/
Ignored: output/.DS_Store
Ignored: output/._.DS_Store
Ignored: output/._NSC_cluster1_marker_genes.txt
Ignored: output/Lam-01-clustering.rds
Ignored: output/NSC_1_clustering.rds
Ignored: output/NSC_cluster1_marker_genes.txt
Ignored: output/NSC_cluster2_marker_genes.txt
Ignored: output/NSC_cluster3_marker_genes.txt
Ignored: output/NSC_cluster4_marker_genes.txt
Ignored: output/NSC_cluster5_marker_genes.txt
Ignored: output/NSC_cluster6_marker_genes.txt
Ignored: output/NSC_cluster7_marker_genes.txt
Ignored: output/additional_filtering.rds
Ignored: output/figures/
Ignored: output/sce_01_preprocessing.rds
Ignored: output/sce_02_quality_control.rds
Ignored: output/sce_03_filtering.rds
Ignored: output/sce_organoid-01-clustering.rds
Ignored: output/sce_preprocessing.rds
Ignored: output/so_04-group_integration.rds
Ignored: output/so_04_1_cell_cycle.rds
Ignored: output/so_04_clustering.rds
Ignored: output/so_additional_filtering_clustering.rds
Ignored: output/so_integrated_organoid-02-integration.rds
Ignored: output/so_merged_organoid-02-integration.rds
Ignored: output/so_organoid-01-clustering.rds
Ignored: output/so_sample_organoid-01-clustering.rds
Untracked files:
Untracked: Rplots.pdf
Untracked: analysis/Lam-0-NSC_no_integration.Rmd
Untracked: analysis/additional_filtering.Rmd
Untracked: analysis/additional_filtering_clustering.Rmd
Untracked: analysis/sample5_QC.Rmd
Untracked: data/Homo_sapiens.GRCh38.98.sorted.gtf
Untracked: data/Kanton_et_al/
Untracked: data/Lam_et_al/
Untracked: scripts/
Unstaged changes:
Modified: analysis/Lam-02-NSC_annotation.Rmd
Modified: analysis/_site.yml
Modified: analysis/organoid-02-integration.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/organoid-05-group_integration_cluster_analysis.Rmd) and HTML (docs/organoid-05-group_integration_cluster_analysis.html) files. If you've configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 043115f | khembach | 2020-09-02 | group organoid integration cluster abundances |
Load packages
library(ComplexHeatmap)
library(cowplot)
library(ggplot2)
library(dplyr)
library(muscat)
library(RColorBrewer)
library(Seurat)
library(SingleCellExperiment)Load data & convert to SCE
so <- readRDS(file.path("output", "so_04-group_integration.rds"))
sce <- as.SingleCellExperiment(so, assay = "RNA")
colData(sce) <- as.data.frame(colData(sce)) %>%
mutate_if(is.character, as.factor) %>%
DataFrame(row.names = colnames(sce))Cluster-sample counts
# set cluster IDs to resolution 0.4 clustering
so <- SetIdent(so, value = "integrated_snn_res.0.4")
so@meta.data$cluster_id <- Idents(so)
sce$cluster_id <- Idents(so)
(n_cells <- table(sce$cluster_id, sce$sample_id))
1NSC 2NSC 3NC52 4NC52 5NC96 6NC96 H9 409b2
0 17 16 5165 4290 1352 2047 2722 2391
1 4357 4307 281 193 421 49 64 78
2 11 12 1307 1316 802 1321 1787 2672
3 3111 3232 30 17 7 2 414 395
4 0 0 393 337 90 96 2483 3335
5 1 0 7 31 6 7 3656 1244
6 35 22 780 522 360 565 475 1016
7 0 0 0 0 0 0 2017 1708
8 0 0 1 0 0 0 2409 817
9 4 1 9 9 12 4 1866 1174
10 3 3 28 12 7 8 1619 1351
11 5 7 39 40 3 6 1472 1382
12 539 553 270 232 271 247 215 358
13 0 2 0 0 0 1 811 1149
14 1 3 191 162 51 101 666 754
15 244 245 186 277 156 141 127 170
16 3 5 0 0 0 0 423 278Relative cluster-abundances
fqs <- prop.table(n_cells, margin = 2)
mat <- as.matrix(unclass(fqs))
Heatmap(mat,
col = rev(brewer.pal(11, "RdGy")[-6]),
name = "Frequency",
cluster_rows = FALSE,
cluster_columns = FALSE,
row_names_side = "left",
row_title = "cluster_id",
column_title = "sample_id",
column_title_side = "bottom",
rect_gp = gpar(col = "white"),
cell_fun = function(i, j, x, y, width, height, fill)
grid.text(round(mat[j, i] * 100, 2), x = x, y = y,
gp = gpar(col = "white", fontsize = 10)))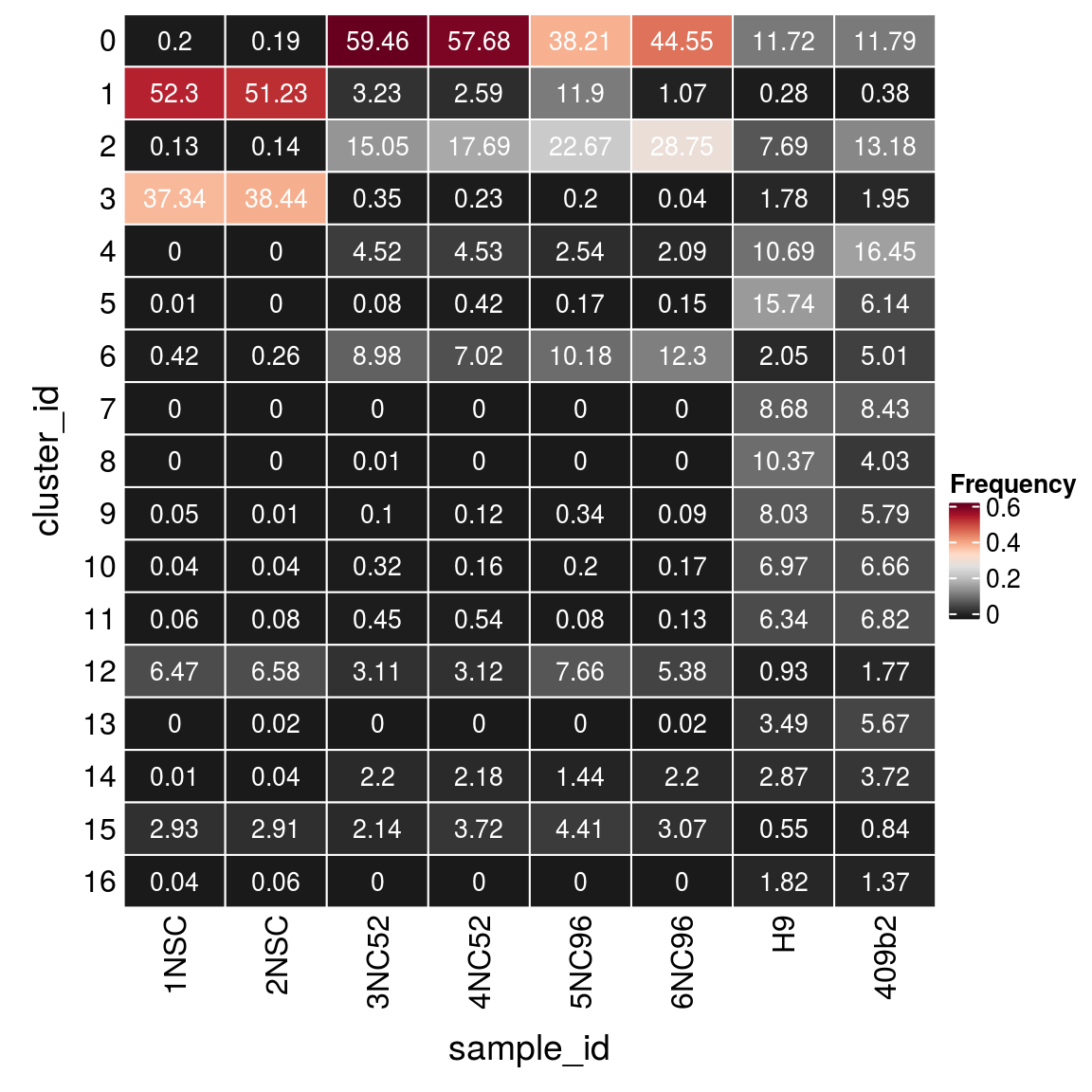
(n_cells_group <- table(sce$cluster_id, sce$group_id))
P22 D52 D96 iPSCs EB Neuroectoderm Neuroepithelium Organoid-1M
0 33 9455 3399 0 0 0 0 266
1 8664 474 470 0 0 0 1 18
2 23 2623 2123 0 0 0 0 5
3 6343 47 9 8 16 4 29 297
4 0 730 186 0 0 0 0 0
5 1 38 13 0 0 0 2 67
6 57 1302 925 0 0 1 0 753
7 0 0 0 3656 16 53 0 0
8 0 1 0 16 3203 7 0 0
9 5 18 16 19 6 2635 375 5
10 6 40 15 0 0 1 7 2350
11 12 79 9 0 0 0 0 49
12 1092 502 518 0 0 2 7 161
13 2 0 1 11 15 9 913 1012
14 4 353 152 0 0 0 0 9
15 489 463 297 0 0 4 4 78
16 8 0 0 590 44 41 25 0
Organoid-2M Organoid-4M
0 3878 969
1 102 21
2 3139 1315
3 398 57
4 3553 2265
5 1476 3355
6 735 2
7 0 0
8 0 0
9 0 0
10 607 5
11 1955 850
12 241 162
13 0 0
14 983 428
15 152 59
16 1 0fqs <- prop.table(n_cells_group, margin = 2)
mat <- as.matrix(unclass(fqs))
Heatmap(mat,
col = rev(brewer.pal(11, "RdGy")[-6]),
name = "Frequency",
cluster_rows = FALSE,
cluster_columns = FALSE,
row_names_side = "left",
row_title = "cluster_id",
column_title = "group_id",
column_title_side = "bottom",
rect_gp = gpar(col = "white"),
cell_fun = function(i, j, x, y, width, height, fill)
grid.text(round(mat[j, i] * 100, 2), x = x, y = y,
gp = gpar(col = "white", fontsize = 10)))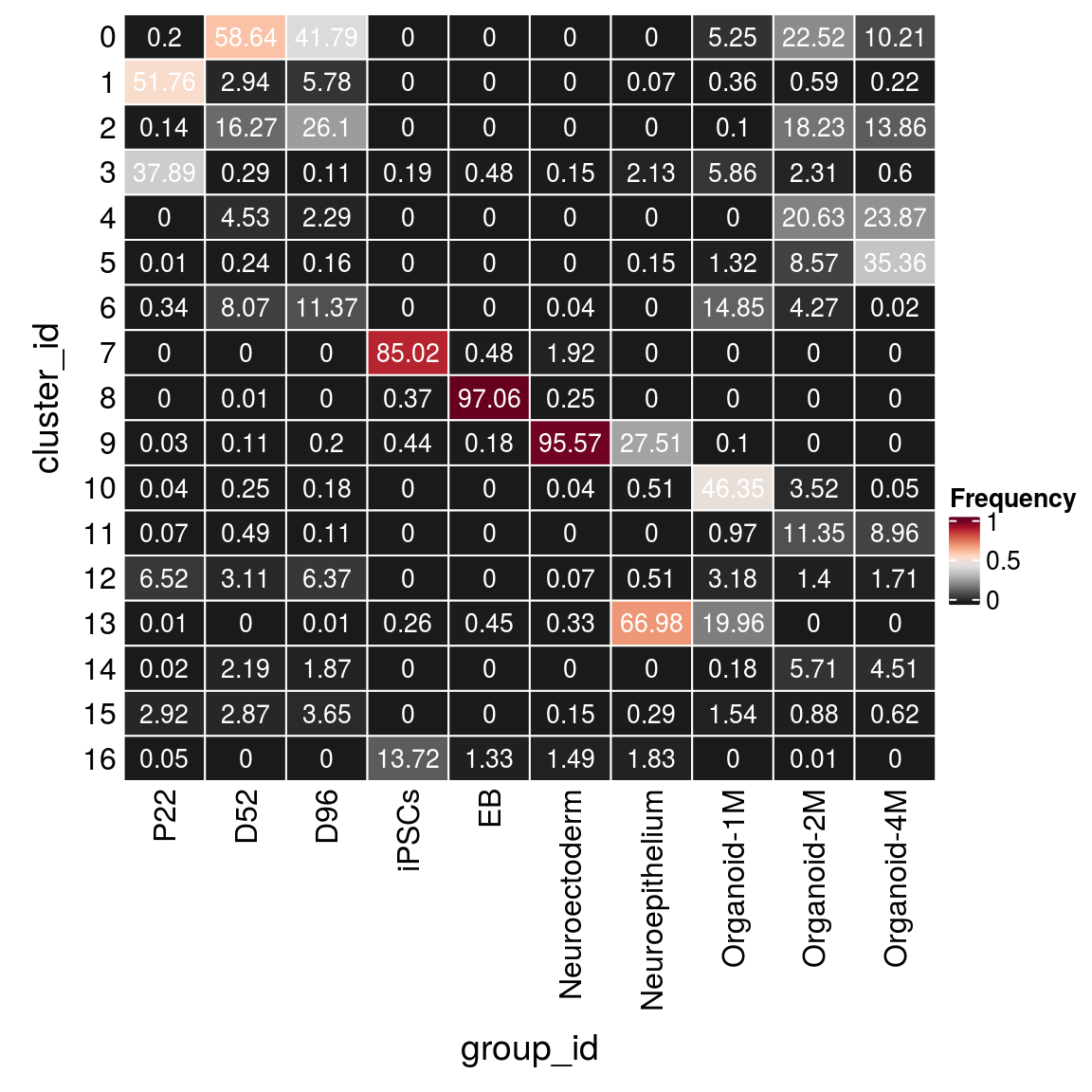
n_cells_lineage <- table(sce$cluster_id, sce$cl_FullLineage)
fqs <- prop.table(n_cells_lineage, margin = 2)
mat <- as.matrix(unclass(fqs))
cn <- colnames(mat)
Heatmap(mat,
col = rev(brewer.pal(11, "RdGy")[-6]),
name = "Frequency",
cluster_rows = FALSE,
cluster_columns = FALSE,
show_column_names = FALSE,
row_names_side = "left",
row_title = "cluster_id",
column_title = "cl_FullLineage",
column_title_side = "bottom",
rect_gp = gpar(col = "white"),
cell_fun = function(i, j, x, y, width, height, fill)
grid.text(round(mat[j, i] * 100, 1), x = x, y = y,
gp = gpar(col = "white", fontsize = 10)),
bottom_annotation = HeatmapAnnotation(
text = anno_text(cn, rot = 80, just = "right")))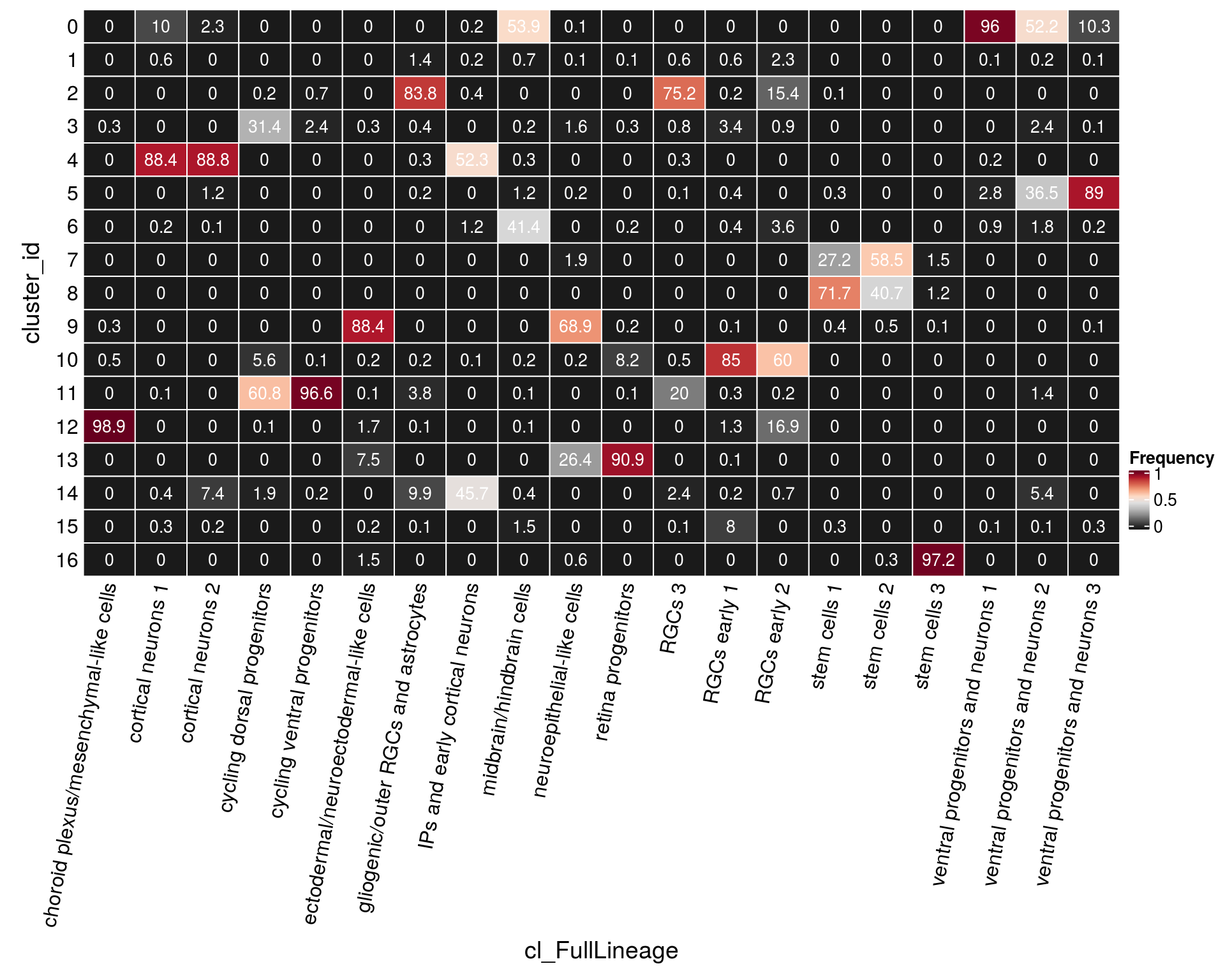
n_cells_lineage <- table(sce$cl_FullLineage, sce$cluster_id)
fqs <- prop.table(n_cells_lineage, margin = 2)
mat <- as.matrix(unclass(fqs))
Heatmap(mat,
col = rev(brewer.pal(11, "RdGy")[-6]),
name = "Frequency",
cluster_rows = FALSE,
cluster_columns = FALSE,
row_names_side = "left",
row_title = "cl_FullLineage",
row_names_rot = 10,
column_title = "cluster_id",
column_title_side = "bottom",
rect_gp = gpar(col = "white"),
cell_fun = function(i, j, x, y, width, height, fill)
grid.text(round(mat[j, i] * 100, 1), x = x, y = y,
gp = gpar(col = "white", fontsize = 10)))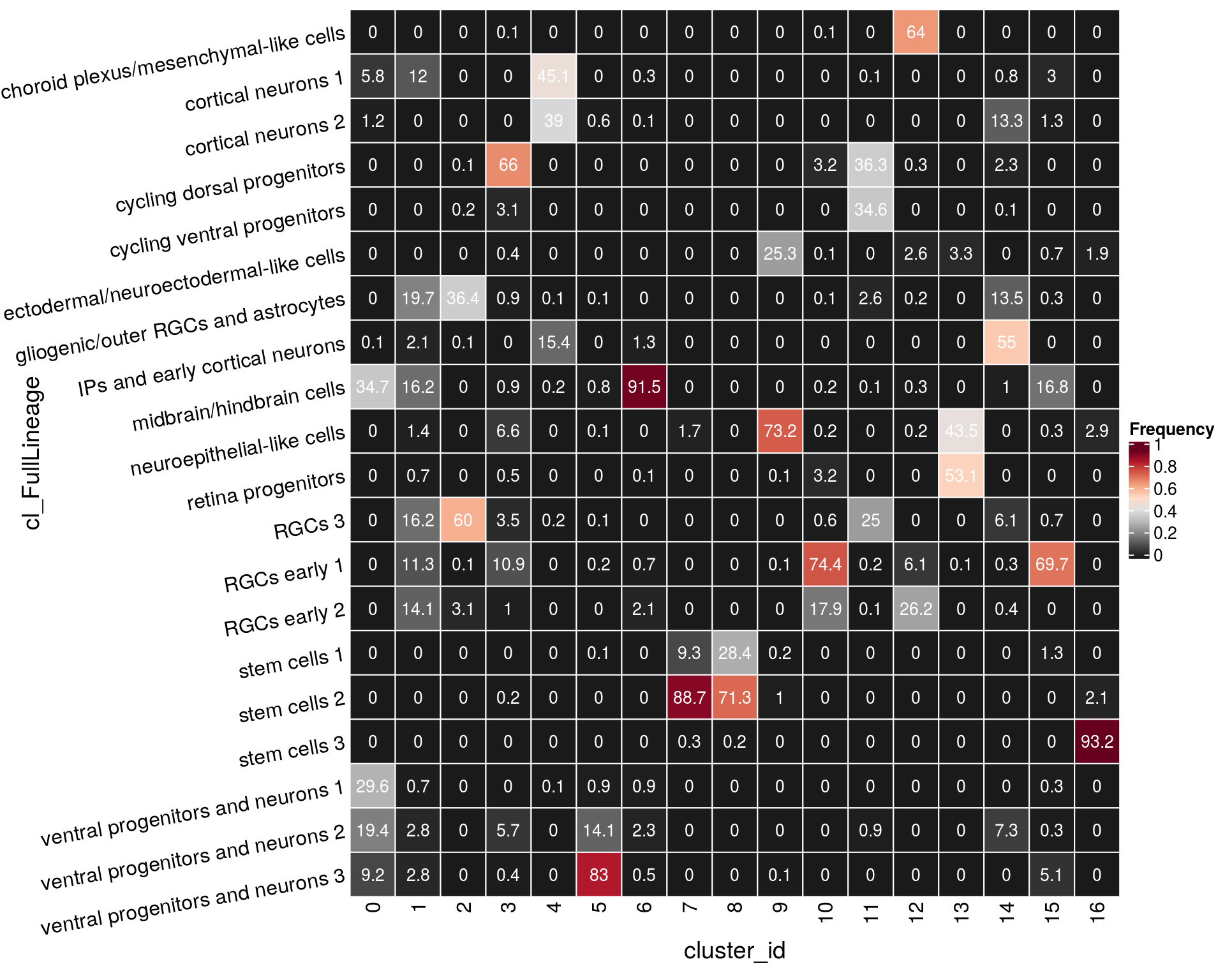
sessionInfo()R version 4.0.0 (2020-04-24)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 16.04.6 LTS
Matrix products: default
BLAS: /usr/local/R/R-4.0.0/lib/libRblas.so
LAPACK: /usr/local/R/R-4.0.0/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 grid stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] SingleCellExperiment_1.10.1 SummarizedExperiment_1.18.1
[3] DelayedArray_0.14.0 matrixStats_0.56.0
[5] Biobase_2.48.0 GenomicRanges_1.40.0
[7] GenomeInfoDb_1.24.2 IRanges_2.22.2
[9] S4Vectors_0.26.1 BiocGenerics_0.34.0
[11] Seurat_3.1.5 RColorBrewer_1.1-2
[13] muscat_1.2.1 dplyr_1.0.0
[15] ggplot2_3.3.2 cowplot_1.0.0
[17] ComplexHeatmap_2.4.2 workflowr_1.6.2
loaded via a namespace (and not attached):
[1] backports_1.1.8 circlize_0.4.10
[3] blme_1.0-4 igraph_1.2.5
[5] plyr_1.8.6 lazyeval_0.2.2
[7] TMB_1.7.16 splines_4.0.0
[9] BiocParallel_1.22.0 listenv_0.8.0
[11] scater_1.16.2 digest_0.6.25
[13] foreach_1.5.0 htmltools_0.5.0
[15] viridis_0.5.1 gdata_2.18.0
[17] lmerTest_3.1-2 magrittr_1.5
[19] memoise_1.1.0 cluster_2.1.0
[21] doParallel_1.0.15 ROCR_1.0-11
[23] limma_3.44.3 globals_0.12.5
[25] annotate_1.66.0 prettyunits_1.1.1
[27] colorspace_1.4-1 rappdirs_0.3.1
[29] ggrepel_0.8.2 blob_1.2.1
[31] xfun_0.15 jsonlite_1.7.0
[33] crayon_1.3.4 RCurl_1.98-1.2
[35] genefilter_1.70.0 lme4_1.1-23
[37] zoo_1.8-8 ape_5.4
[39] survival_3.2-3 iterators_1.0.12
[41] glue_1.4.1 gtable_0.3.0
[43] zlibbioc_1.34.0 XVector_0.28.0
[45] leiden_0.3.3 GetoptLong_1.0.1
[47] BiocSingular_1.4.0 future.apply_1.6.0
[49] shape_1.4.4 scales_1.1.1
[51] DBI_1.1.0 edgeR_3.30.3
[53] Rcpp_1.0.4.6 viridisLite_0.3.0
[55] xtable_1.8-4 progress_1.2.2
[57] clue_0.3-57 reticulate_1.16
[59] bit_1.1-15.2 rsvd_1.0.3
[61] tsne_0.1-3 htmlwidgets_1.5.1
[63] httr_1.4.1 gplots_3.0.4
[65] ellipsis_0.3.1 ica_1.0-2
[67] pkgconfig_2.0.3 XML_3.99-0.4
[69] uwot_0.1.8 locfit_1.5-9.4
[71] tidyselect_1.1.0 rlang_0.4.6
[73] reshape2_1.4.4 later_1.1.0.1
[75] AnnotationDbi_1.50.1 munsell_0.5.0
[77] tools_4.0.0 generics_0.0.2
[79] RSQLite_2.2.0 ggridges_0.5.2
[81] evaluate_0.14 stringr_1.4.0
[83] yaml_2.2.1 knitr_1.29
[85] bit64_0.9-7 fs_1.4.2
[87] fitdistrplus_1.1-1 caTools_1.18.0
[89] RANN_2.6.1 purrr_0.3.4
[91] pbapply_1.4-2 future_1.17.0
[93] nlme_3.1-148 whisker_0.4
[95] pbkrtest_0.4-8.6 compiler_4.0.0
[97] plotly_4.9.2.1 beeswarm_0.2.3
[99] png_0.1-7 variancePartition_1.18.2
[101] tibble_3.0.1 statmod_1.4.34
[103] geneplotter_1.66.0 stringi_1.4.6
[105] lattice_0.20-41 Matrix_1.2-18
[107] nloptr_1.2.2.2 vctrs_0.3.1
[109] pillar_1.4.4 lifecycle_0.2.0
[111] lmtest_0.9-37 GlobalOptions_0.1.2
[113] RcppAnnoy_0.0.16 BiocNeighbors_1.6.0
[115] data.table_1.12.8 bitops_1.0-6
[117] irlba_2.3.3 patchwork_1.0.1
[119] httpuv_1.5.4 colorRamps_2.3
[121] R6_2.4.1 promises_1.1.1
[123] KernSmooth_2.23-17 gridExtra_2.3
[125] vipor_0.4.5 codetools_0.2-16
[127] boot_1.3-25 MASS_7.3-51.6
[129] gtools_3.8.2 DESeq2_1.28.1
[131] rprojroot_1.3-2 rjson_0.2.20
[133] withr_2.2.0 sctransform_0.2.1
[135] GenomeInfoDbData_1.2.3 hms_0.5.3
[137] tidyr_1.1.0 glmmTMB_1.0.2.1
[139] minqa_1.2.4 rmarkdown_2.3
[141] DelayedMatrixStats_1.10.1 Rtsne_0.15
[143] git2r_0.27.1 numDeriv_2016.8-1.1
[145] ggbeeswarm_0.6.0