Bayesian MAR(1) models and structural stability
Matthew A. Barbour
2020-06-23
Last updated: 2020-06-23
Checks: 7 0
Knit directory: genes-to-foodweb-stability/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200205) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: code/.Rhistory
Untracked files:
Untracked: figures/rich-geno-critical-transition.pdf
Unstaged changes:
Modified: figures/rich-geno-critical-transition-v4.pdf
Modified: output/critical-transitions.RData
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 86116c8 | mabarbour | 2020-06-23 | bioRxiv version of code and data. |
| html | 86116c8 | mabarbour | 2020-06-23 | bioRxiv version of code and data. |
Setup
# load data
timeseries_df <- read_csv("output/timeseries_df.csv") %>%
mutate(# this makes the intercept correspond to rich = 1, rather
# than the biologically implausible rich = 0
rich = rich - 1,
# now rich and temp coefficients will correspond to +1 genotype and +1 C
temp = ifelse(temp == "20 C", 0, 3),
uniq = paste(Cage, temp, com, Week_match, sep = "-"),
Week_match.1p = 1 + Week_match) # analysis doesn't like initial Week_match = 0, so I just added 1
# filter dataset for multivariate analysis.
# I only retain data for which all species had positive abundances at the previous time step
full_df <- filter(timeseries_df, BRBR_t > 0, LYER_t > 0, Ptoid_t > 0) ## source in useful functions
# functions for plotting feasibility domains and calculating normalized angles from critical boundary
source("code/plot-feasibility-domain.R")
# functions for non-equilibrium simulation
source("code/simulate-community-dynamics.R")Full model
This model corresponds to equation 1 in the Supplementary Material of the paper. Note that BRBR = aphid Brevicoryne brassicae, LYER = aphid Lipaphis erysimi, and Ptoid = parasitoid Diaeretiella rapae.
# BRBR
full.mv.norm.BRBR.bf <- bf(log1p(BRBR_t1) ~
0 + intercept + offset(log1p(BRBR_t)) +
(log1p(BRBR_t) + log1p(LYER_t) + log1p(Ptoid_t))*(temp + rich) +
log(Biomass_g_t1) +
(1 | Cage) +
ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE))
# LYER
full.mv.norm.LYER.bf <- bf(log1p(LYER_t1) ~
0 + intercept + offset(log1p(LYER_t)) +
(log1p(BRBR_t) + log1p(LYER_t) + log1p(Ptoid_t))*(temp + rich) +
log(Biomass_g_t1) +
(1 | Cage) +
ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE))
# Ptoid
full.mv.norm.Ptoid.bf <- bf(log1p(Ptoid_t1) ~
0 + intercept + offset(log1p(Ptoid_t)) +
(log1p(BRBR_t) + log1p(LYER_t) + log1p(Ptoid_t))*(temp + rich) +
log(Biomass_g_t1) +
(1 | Cage) +
ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE))Set priors
Intrinsic growth rates
# from Jahan et al. 2014, Journal of Insect Science
# Table 4 lambda (finite rate of increase, discrete time analogue of intrinsic growth rate)
# calculated on a per-day basis and not logged. This is why I multiply by 7 and then take the natural logarithm
Jahan.r.BRBR <- log(c(1.42, 1.36, 1.32, 1.35, 1.40, 1.33, 1.38, 1.37) * 7)
mean(Jahan.r.BRBR) # 2.26[1] 2.257713sd(Jahan.r.BRBR) # 0.02[1] 0.02468356# visualize prior
hist(rnorm(1000, mean(Jahan.r.BRBR), sd = 1))
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
prior.r.BRBR <- "normal(2.26, 1)"
# from Taghizadeh 2019, J. Agr. Sci. Tech.
# Table 2 lambda (finite rate of increase, discrete time analogue of intrinsic growth rate)
# calculated on a per-day basis and not logged. This is why I multiply by 7 and then take the natural logarithm
Tag.r.LYER <- log(c(1.35, 1.30, 1.26, 1.23) * 7)
mean(Tag.r.LYER) # 2.20[1] 2.196059sd(Tag.r.LYER) # 0.04[1] 0.04028153# visualize prior
hist(rnorm(1000, mean(Tag.r.LYER), sd = 1))
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
prior.r.LYER <- "normal(2.20, 1)"
# random effects prior based on variance among cultivars
# I'm just going to use this for all of them
mean.r.sd <- mean(c(sd(Jahan.r.BRBR), sd(Tag.r.LYER)))
# visualize prior
hist(rnorm(1000, mean = mean.r.sd, sd = 0.5))
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
prior.random.effects <- "normal(0.03, 0.5)"
# I don't have a great sense for the growth rate of the parasitoid, other than that it should be negative
# this seems like a reasonable starting point
# visualize prior
hist(rnorm(1000, mean = -1.5, sd = 1))
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
prior.r.Ptoid <- "normal(-1.5, 1)"Intra- and interspecific interactions
I assume they are weak, i.e. much less than \(|1|\). I also assume that all species exhibit intraspecific competition, aphids have negative interspecific effects with each other, but positive interspecific effects on the parasitoid. I also assume parasitoids have negative interspecific effects on the aphids.
## intraspecific, 0 = no density dependence. this occurs because of offset in model.
# visualize prior
hist(rnorm(1000, mean = -0.1, sd = 0.5))
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
prior.intra.BRBR <- "normal(-0.1, 0.5)"
prior.intra.LYER <- "normal(-0.1, 0.5)"
prior.intra.Ptoid <- "normal(-0.1, 0.5)"
## negative interspecific, 0 = no interaction
# visualize prior
hist(rnorm(1000, mean = -0.1, sd = 0.5))
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# most of these values are less than 1, which
# is indicative of saturating effects
prior.LYERonBRBR <- "normal(-0.1, 0.5)"
prior.PtoidonBRBR <- "normal(-0.1, 0.5)"
prior.BRBRonLYER <- "normal(-0.1, 0.5)"
prior.PtoidonLYER <- "normal(-0.1, 0.5)"
## positive interspecific
# visualize prior
hist(rnorm(1000, mean = 0.1, sd = 0.5))
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# most of these values are less than 1, which
# is indicative of saturating effects
prior.BRBRonPtoid <- "normal(0.1, 0.5)"
prior.LYERonPtoid <- "normal(0.1, 0.5)"Genetic richness and genotype effects
It was unclear to me a priori exactly how genetic diversity and specific genotypes would affect species’ growth rates or intra- and interspecific interactions. Therefore, I assumed these effects on specific rates could be positive or negative, but would likely be between -1 and 1 (i.e., not ridiculously large).
prior.rich <- "normal(0, 0.5)"I used prior.rich as the prior for genotype-specific effects as well.
Temperature effects
prior.temp <- "normal(0, 0.5)"Biomass effects
For both aphids, I thought that increasing biomass would increase their intrinsic growth rates, but only weakly, because I didn’t expect biomass to be limiting.
# visualize prior
hist(rnorm(1000, mean = 0.1, sd = 0.5))
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
prior.AphidBiomass <- "normal(0.1, 0.5)"For the parasitoid, it was unclear to me whether increasing biomass would have positive or negative effects. I could imagine both, as increasing biomass may increase the search effort of parasitoids, resulting in a negative effect on their growth rate. Alternatively, more biomass may increase the quality of aphids, which could increase the parasitoid’s growth rate. Therefore, I specified a normal prior with mean = 0 and SD = 0.5, so that most of the distribution lied between -1 and 1 (i.e. saturating negative or positive effects).
# visualize prior
hist(rnorm(1000, mean = 0, sd = 0.5))
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
prior.PtoidBiomass <- "normal(0, 0.5)"Analysis
I first fit a complete model with rich and temperature effects
full.mv.norm.brm <- brm(
data = full_df,
formula = mvbf(full.mv.norm.BRBR.bf, full.mv.norm.LYER.bf, full.mv.norm.Ptoid.bf),
iter = 4000,
prior = c(# growth rates
set_prior(prior.r.BRBR, class = "b", coef = "intercept", resp = "log1pBRBRt1"),
set_prior(prior.r.LYER, class = "b", coef = "intercept", resp = "log1pLYERt1"),
set_prior(prior.r.Ptoid, class = "b", coef = "intercept", resp = "log1pPtoidt1"),
# intraspecific effects
set_prior(prior.intra.BRBR, class = "b", coef = "log1pBRBR_t", resp = "log1pBRBRt1"),
set_prior(prior.intra.LYER, class = "b", coef = "log1pLYER_t", resp = "log1pLYERt1"),
set_prior(prior.intra.LYER, class = "b", coef = "log1pPtoid_t", resp = "log1pPtoidt1"),
# negative interspecific effects
set_prior(prior.LYERonBRBR, class = "b", coef = "log1pLYER_t", resp = "log1pBRBRt1"),
set_prior(prior.BRBRonLYER, class = "b", coef = "log1pBRBR_t", resp = "log1pLYERt1"),
set_prior(prior.PtoidonBRBR, class = "b", coef = "log1pPtoid_t", resp = "log1pBRBRt1"),
set_prior(prior.PtoidonLYER, class = "b", coef = "log1pPtoid_t", resp = "log1pLYERt1"),
# positive interspecific effects
set_prior(prior.BRBRonPtoid, class = "b", coef = "log1pBRBR_t", resp = "log1pPtoidt1"),
set_prior(prior.LYERonPtoid, class = "b", coef = "log1pLYER_t", resp = "log1pPtoidt1"),
# rich effects
set_prior(prior.rich, class = "b", coef = "rich", resp = "log1pBRBRt1"),
set_prior(prior.rich, class = "b", coef = "rich", resp = "log1pLYERt1"),
set_prior(prior.rich, class = "b", coef = "rich", resp = "log1pPtoidt1"),
set_prior(prior.rich, class = "b", coef = "log1pBRBR_t:rich", resp = "log1pBRBRt1"),
set_prior(prior.rich, class = "b", coef = "log1pBRBR_t:rich", resp = "log1pLYERt1"),
set_prior(prior.rich, class = "b", coef = "log1pBRBR_t:rich", resp = "log1pPtoidt1"),
set_prior(prior.rich, class = "b", coef = "log1pLYER_t:rich", resp = "log1pBRBRt1"),
set_prior(prior.rich, class = "b", coef = "log1pLYER_t:rich", resp = "log1pLYERt1"),
set_prior(prior.rich, class = "b", coef = "log1pLYER_t:rich", resp = "log1pPtoidt1"),
set_prior(prior.rich, class = "b", coef = "log1pPtoid_t:rich", resp = "log1pBRBRt1"),
set_prior(prior.rich, class = "b", coef = "log1pPtoid_t:rich", resp = "log1pLYERt1"),
set_prior(prior.rich, class = "b", coef = "log1pPtoid_t:rich", resp = "log1pPtoidt1"),
# temp effects
set_prior(prior.temp, class = "b", coef = "temp", resp = "log1pBRBRt1"),
set_prior(prior.temp, class = "b", coef = "temp", resp = "log1pLYERt1"),
set_prior(prior.temp, class = "b", coef = "temp", resp = "log1pPtoidt1"),
set_prior(prior.temp, class = "b", coef = "log1pBRBR_t:temp", resp = "log1pBRBRt1"),
set_prior(prior.temp, class = "b", coef = "log1pBRBR_t:temp", resp = "log1pLYERt1"),
set_prior(prior.temp, class = "b", coef = "log1pBRBR_t:temp", resp = "log1pPtoidt1"),
set_prior(prior.temp, class = "b", coef = "log1pLYER_t:temp", resp = "log1pBRBRt1"),
set_prior(prior.temp, class = "b", coef = "log1pLYER_t:temp", resp = "log1pLYERt1"),
set_prior(prior.temp, class = "b", coef = "log1pLYER_t:temp", resp = "log1pPtoidt1"),
set_prior(prior.temp, class = "b", coef = "log1pPtoid_t:temp", resp = "log1pBRBRt1"),
set_prior(prior.temp, class = "b", coef = "log1pPtoid_t:temp", resp = "log1pLYERt1"),
set_prior(prior.temp, class = "b", coef = "log1pPtoid_t:temp", resp = "log1pPtoidt1"),
# biomass effects
set_prior(prior.AphidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pBRBRt1"),
set_prior(prior.AphidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pLYERt1"),
set_prior(prior.PtoidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pPtoidt1"),
# random effects
set_prior(prior.random.effects, class = "sd", resp = "log1pBRBRt1"),
set_prior(prior.random.effects, class = "sd", resp = "log1pLYERt1"),
set_prior(prior.random.effects, class = "sd", resp = "log1pPtoidt1")),
file = "output/full.mv.norm.brm.rds")
# print summary
summary(full.mv.norm.brm) Family: MV(gaussian, gaussian, gaussian)
Links: mu = identity; sigma = identity
mu = identity; sigma = identity
mu = identity; sigma = identity
Formula: log1p(BRBR_t1) ~ 0 + intercept + offset(log1p(BRBR_t)) + (log1p(BRBR_t) + log1p(LYER_t) + log1p(Ptoid_t)) * (temp + rich) + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE)
log1p(LYER_t1) ~ 0 + intercept + offset(log1p(LYER_t)) + (log1p(BRBR_t) + log1p(LYER_t) + log1p(Ptoid_t)) * (temp + rich) + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE)
log1p(Ptoid_t1) ~ 0 + intercept + offset(log1p(Ptoid_t)) + (log1p(BRBR_t) + log1p(LYER_t) + log1p(Ptoid_t)) * (temp + rich) + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE)
Data: full_df (Number of observations: 264)
Samples: 4 chains, each with iter = 4000; warmup = 2000; thin = 1;
total post-warmup samples = 8000
Correlation Structures:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
ar_log1pBRBRt1[1] -0.45 0.10 -0.63 -0.25 1.00 3525 5191
ar_log1pLYERt1[1] -0.12 0.11 -0.32 0.10 1.00 7178 6071
ar_log1pPtoidt1[1] -0.49 0.08 -0.65 -0.32 1.00 11047 6451
Group-Level Effects:
~Cage (Number of levels: 60)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS
sd(log1pBRBRt1_Intercept) 0.31 0.11 0.06 0.52 1.00 852
sd(log1pLYERt1_Intercept) 0.13 0.08 0.01 0.31 1.00 1928
sd(log1pPtoidt1_Intercept) 0.04 0.03 0.00 0.12 1.00 4679
Tail_ESS
sd(log1pBRBRt1_Intercept) 1308
sd(log1pLYERt1_Intercept) 3987
sd(log1pPtoidt1_Intercept) 3766
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat
log1pBRBRt1_intercept 1.39 0.63 0.17 2.63 1.00
log1pBRBRt1_log1pBRBR_t 0.04 0.13 -0.22 0.30 1.00
log1pBRBRt1_log1pLYER_t 0.03 0.15 -0.26 0.32 1.00
log1pBRBRt1_log1pPtoid_t -0.93 0.07 -1.06 -0.79 1.00
log1pBRBRt1_temp -0.30 0.28 -0.87 0.25 1.00
log1pBRBRt1_rich 0.03 0.35 -0.66 0.73 1.00
log1pBRBRt1_logBiomass_g_t1 -0.12 0.19 -0.49 0.26 1.00
log1pBRBRt1_log1pBRBR_t:temp -0.08 0.05 -0.18 0.03 1.00
log1pBRBRt1_log1pBRBR_t:rich -0.06 0.06 -0.18 0.06 1.00
log1pBRBRt1_log1pLYER_t:temp 0.00 0.06 -0.11 0.11 1.00
log1pBRBRt1_log1pLYER_t:rich 0.03 0.07 -0.10 0.16 1.00
log1pBRBRt1_log1pPtoid_t:temp 0.11 0.04 0.04 0.19 1.00
log1pBRBRt1_log1pPtoid_t:rich 0.04 0.05 -0.06 0.14 1.00
log1pLYERt1_intercept 2.73 0.62 1.52 3.99 1.00
log1pLYERt1_log1pBRBR_t 0.39 0.11 0.17 0.60 1.00
log1pLYERt1_log1pLYER_t -0.67 0.14 -0.95 -0.40 1.00
log1pLYERt1_log1pPtoid_t -0.70 0.07 -0.84 -0.57 1.00
log1pLYERt1_temp 0.12 0.25 -0.37 0.62 1.00
log1pLYERt1_rich 0.26 0.33 -0.39 0.90 1.00
log1pLYERt1_logBiomass_g_t1 0.27 0.17 -0.06 0.60 1.00
log1pLYERt1_log1pBRBR_t:temp -0.04 0.05 -0.13 0.05 1.00
log1pLYERt1_log1pBRBR_t:rich -0.12 0.05 -0.22 -0.01 1.00
log1pLYERt1_log1pLYER_t:temp 0.02 0.05 -0.08 0.12 1.00
log1pLYERt1_log1pLYER_t:rich 0.06 0.06 -0.06 0.17 1.00
log1pLYERt1_log1pPtoid_t:temp -0.03 0.04 -0.10 0.04 1.00
log1pLYERt1_log1pPtoid_t:rich 0.01 0.04 -0.08 0.09 1.00
log1pPtoidt1_intercept -2.81 0.49 -3.77 -1.84 1.00
log1pPtoidt1_log1pBRBR_t 0.47 0.08 0.31 0.63 1.00
log1pPtoidt1_log1pLYER_t 0.24 0.10 0.05 0.43 1.00
log1pPtoidt1_log1pPtoid_t -0.04 0.05 -0.14 0.06 1.00
log1pPtoidt1_temp 0.45 0.23 0.01 0.90 1.00
log1pPtoidt1_rich -0.13 0.31 -0.73 0.47 1.00
log1pPtoidt1_logBiomass_g_t1 -0.48 0.13 -0.73 -0.24 1.00
log1pPtoidt1_log1pBRBR_t:temp -0.18 0.04 -0.25 -0.11 1.00
log1pPtoidt1_log1pBRBR_t:rich -0.06 0.04 -0.13 0.01 1.00
log1pPtoidt1_log1pLYER_t:temp 0.05 0.04 -0.03 0.12 1.00
log1pPtoidt1_log1pLYER_t:rich 0.07 0.05 -0.02 0.17 1.00
log1pPtoidt1_log1pPtoid_t:temp 0.02 0.03 -0.04 0.08 1.00
log1pPtoidt1_log1pPtoid_t:rich 0.01 0.04 -0.06 0.08 1.00
Bulk_ESS Tail_ESS
log1pBRBRt1_intercept 8958 6192
log1pBRBRt1_log1pBRBR_t 4440 4605
log1pBRBRt1_log1pLYER_t 3941 4878
log1pBRBRt1_log1pPtoid_t 8029 5772
log1pBRBRt1_temp 8151 5968
log1pBRBRt1_rich 8200 6098
log1pBRBRt1_logBiomass_g_t1 9358 6174
log1pBRBRt1_log1pBRBR_t:temp 7094 6330
log1pBRBRt1_log1pBRBR_t:rich 7707 6285
log1pBRBRt1_log1pLYER_t:temp 6942 6010
log1pBRBRt1_log1pLYER_t:rich 6892 5878
log1pBRBRt1_log1pPtoid_t:temp 7172 6399
log1pBRBRt1_log1pPtoid_t:rich 8596 6372
log1pLYERt1_intercept 7495 6509
log1pLYERt1_log1pBRBR_t 6752 4912
log1pLYERt1_log1pLYER_t 5984 5991
log1pLYERt1_log1pPtoid_t 8024 6620
log1pLYERt1_temp 8112 6310
log1pLYERt1_rich 9047 6188
log1pLYERt1_logBiomass_g_t1 12846 5477
log1pLYERt1_log1pBRBR_t:temp 7169 5455
log1pLYERt1_log1pBRBR_t:rich 9261 5894
log1pLYERt1_log1pLYER_t:temp 6889 5984
log1pLYERt1_log1pLYER_t:rich 7706 6119
log1pLYERt1_log1pPtoid_t:temp 8851 6469
log1pLYERt1_log1pPtoid_t:rich 8328 6615
log1pPtoidt1_intercept 7110 6276
log1pPtoidt1_log1pBRBR_t 9065 5996
log1pPtoidt1_log1pLYER_t 7049 6085
log1pPtoidt1_log1pPtoid_t 8160 6448
log1pPtoidt1_temp 8378 6562
log1pPtoidt1_rich 7032 5692
log1pPtoidt1_logBiomass_g_t1 13747 6410
log1pPtoidt1_log1pBRBR_t:temp 9783 6497
log1pPtoidt1_log1pBRBR_t:rich 11800 5788
log1pPtoidt1_log1pLYER_t:temp 8403 6467
log1pPtoidt1_log1pLYER_t:rich 7642 6381
log1pPtoidt1_log1pPtoid_t:temp 9237 6362
log1pPtoidt1_log1pPtoid_t:rich 7580 6576
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma_log1pBRBRt1 1.10 0.06 0.99 1.23 1.00 2349 4577
sigma_log1pLYERt1 0.93 0.04 0.85 1.02 1.00 9800 6189
sigma_log1pPtoidt1 0.77 0.04 0.70 0.84 1.00 15372 5800
Residual Correlations:
Estimate Est.Error l-95% CI u-95% CI Rhat
rescor(log1pBRBRt1,log1pLYERt1) 0.52 0.05 0.42 0.62 1.00
rescor(log1pBRBRt1,log1pPtoidt1) -0.04 0.07 -0.18 0.10 1.00
rescor(log1pLYERt1,log1pPtoidt1) 0.06 0.06 -0.06 0.19 1.00
Bulk_ESS Tail_ESS
rescor(log1pBRBRt1,log1pLYERt1) 7359 5636
rescor(log1pBRBRt1,log1pPtoidt1) 10955 6627
rescor(log1pLYERt1,log1pPtoidt1) 13533 5975
Samples were drawn using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).Inspect credible intervals
Below, I inspect which parameters may be safely omitted from the model. It seemed reasonable that if 80% of the posterior probability distribution of the parameter included zero, then I could safely drop it from the model. Therefore, I proceeded with this criteria, starting with the highest-order terms:
# higher-order temperature effects
bayesplot::mcmc_intervals(full.mv.norm.brm, regex_pars = "_t:temp$", prob = 0.80, prob_outer = 0.95) # drop LYER:temp effect on BRBR; all Species:temp effects on LYER; and (LYER and Ptoid):temp effects on Ptoid
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# higher-order rich effects
bayesplot::mcmc_intervals(full.mv.norm.brm, regex_pars = "_t:rich$", prob = 0.80, prob_outer = 0.95) # drop all Species:rich effect on BRBR; drop (LYER and Ptoid):rich effects on LYER; drop Ptoid:rich effect on Ptoid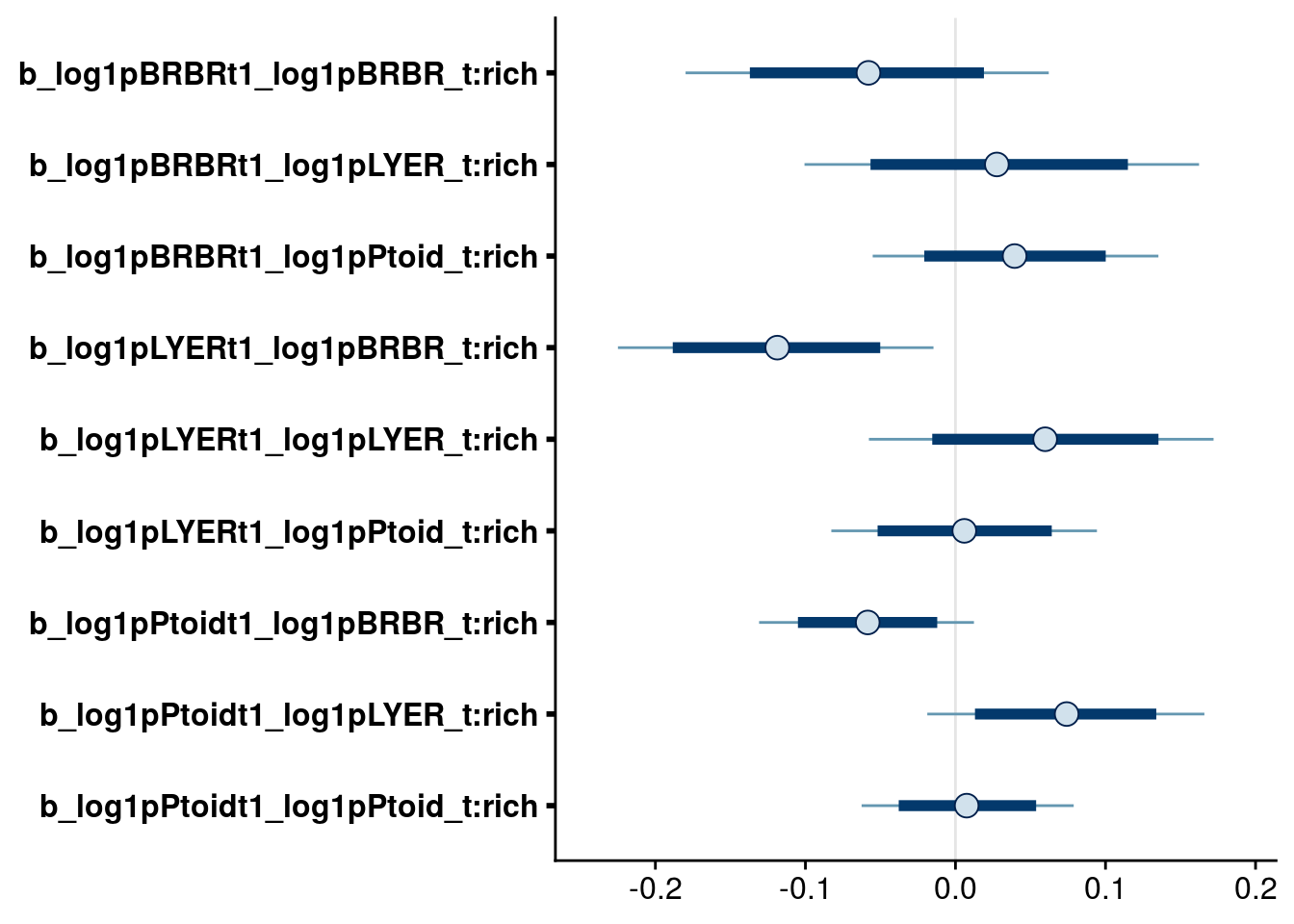
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
Model selection
Reduced model 1
Drop terms
Based on the above plots, I dropped the following higher-order terms:
Effects on BRBR_t1:
(log1p(LYER_t) + log1p(BRBR_t) + log1p(Ptoid_t)):richlog1p(LYER_t):temp
Effects on LYER_t1:
(log1p(LYER_t) + log1p(BRBR_t) + log1p(Ptoid_t)):temp(log1p(LYER_t) + log1p(Ptoid_t)):rich
Effects on Ptoid_t1
(log1p(LYER_t) + log1p(Ptoid_t)):templog1p(Ptoid_t):rich
Refit model
# update formulas
reduced.1.BRBR.bf <- update(full.mv.norm.BRBR.bf, .~. -(log1p(LYER_t) + log1p(BRBR_t) + log1p(Ptoid_t)):rich -log1p(LYER_t):temp)
reduced.1.LYER.bf <- update(full.mv.norm.LYER.bf, .~. -(log1p(LYER_t) + log1p(BRBR_t) + log1p(Ptoid_t)):temp -(log1p(LYER_t) + log1p(Ptoid_t)):rich)
reduced.1.Ptoid.bf <- update(full.mv.norm.Ptoid.bf, .~. -(log1p(LYER_t) + log1p(Ptoid_t)):temp -log1p(Ptoid_t):rich)
# fit update model
reduced.1.brm <- brm(
data = full_df,
formula = mvbf(reduced.1.BRBR.bf, reduced.1.LYER.bf, reduced.1.Ptoid.bf),
iter = 4000,
prior = c(# growth rates
set_prior(prior.r.BRBR, class = "b", coef = "intercept", resp = "log1pBRBRt1"),
set_prior(prior.r.LYER, class = "b", coef = "intercept", resp = "log1pLYERt1"),
set_prior(prior.r.Ptoid, class = "b", coef = "intercept", resp = "log1pPtoidt1"),
# intraspecific effects
set_prior(prior.intra.BRBR, class = "b", coef = "log1pBRBR_t", resp = "log1pBRBRt1"),
set_prior(prior.intra.LYER, class = "b", coef = "log1pLYER_t", resp = "log1pLYERt1"),
set_prior(prior.intra.LYER, class = "b", coef = "log1pPtoid_t", resp = "log1pPtoidt1"),
# negative interspecific effects
set_prior(prior.LYERonBRBR, class = "b", coef = "log1pLYER_t", resp = "log1pBRBRt1"),
set_prior(prior.BRBRonLYER, class = "b", coef = "log1pBRBR_t", resp = "log1pLYERt1"),
set_prior(prior.PtoidonBRBR, class = "b", coef = "log1pPtoid_t", resp = "log1pBRBRt1"),
set_prior(prior.PtoidonLYER, class = "b", coef = "log1pPtoid_t", resp = "log1pLYERt1"),
# positive interspecific effects
set_prior(prior.BRBRonPtoid, class = "b", coef = "log1pBRBR_t", resp = "log1pPtoidt1"),
set_prior(prior.LYERonPtoid, class = "b", coef = "log1pLYER_t", resp = "log1pPtoidt1"),
# rich effects
set_prior(prior.rich, class = "b", coef = "rich", resp = "log1pBRBRt1"),
set_prior(prior.rich, class = "b", coef = "rich", resp = "log1pLYERt1"),
set_prior(prior.rich, class = "b", coef = "rich", resp = "log1pPtoidt1"),
set_prior(prior.rich, class = "b", coef = "log1pBRBR_t:rich", resp = "log1pLYERt1"),
set_prior(prior.rich, class = "b", coef = "log1pBRBR_t:rich", resp = "log1pPtoidt1"),
set_prior(prior.rich, class = "b", coef = "log1pLYER_t:rich", resp = "log1pPtoidt1"),
# temp effects
set_prior(prior.temp, class = "b", coef = "temp", resp = "log1pBRBRt1"),
set_prior(prior.temp, class = "b", coef = "temp", resp = "log1pLYERt1"),
set_prior(prior.temp, class = "b", coef = "temp", resp = "log1pPtoidt1"),
set_prior(prior.temp, class = "b", coef = "log1pBRBR_t:temp", resp = "log1pBRBRt1"),
set_prior(prior.temp, class = "b", coef = "log1pBRBR_t:temp", resp = "log1pPtoidt1"),
set_prior(prior.temp, class = "b", coef = "log1pPtoid_t:temp", resp = "log1pBRBRt1"),
# biomass effects
set_prior(prior.AphidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pBRBRt1"),
set_prior(prior.AphidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pLYERt1"),
set_prior(prior.PtoidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pPtoidt1"),
# random effects
set_prior(prior.random.effects, class = "sd", resp = "log1pBRBRt1"),
set_prior(prior.random.effects, class = "sd", resp = "log1pLYERt1"),
set_prior(prior.random.effects, class = "sd", resp = "log1pPtoidt1")),
file = "output/reduced.1.brm.rds")
# print model summary
summary(reduced.1.brm) Family: MV(gaussian, gaussian, gaussian)
Links: mu = identity; sigma = identity
mu = identity; sigma = identity
mu = identity; sigma = identity
Formula: log1p(BRBR_t1) ~ intercept + log1p(BRBR_t) + log1p(LYER_t) + log1p(Ptoid_t) + temp + rich + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + log1p(BRBR_t):temp + log1p(Ptoid_t):temp + offset(log1p(BRBR_t)) - 1
log1p(LYER_t1) ~ intercept + log1p(BRBR_t) + log1p(LYER_t) + log1p(Ptoid_t) + temp + rich + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + log1p(BRBR_t):rich + offset(log1p(LYER_t)) - 1
log1p(Ptoid_t1) ~ intercept + log1p(BRBR_t) + log1p(LYER_t) + log1p(Ptoid_t) + temp + rich + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + log1p(BRBR_t):temp + log1p(BRBR_t):rich + log1p(LYER_t):rich + offset(log1p(Ptoid_t)) - 1
Data: full_df (Number of observations: 264)
Samples: 4 chains, each with iter = 4000; warmup = 2000; thin = 1;
total post-warmup samples = 8000
Correlation Structures:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
ar_log1pBRBRt1[1] -0.44 0.09 -0.62 -0.25 1.00 3203 5067
ar_log1pLYERt1[1] -0.12 0.10 -0.31 0.09 1.00 5093 5356
ar_log1pPtoidt1[1] -0.48 0.08 -0.64 -0.32 1.00 10699 6081
Group-Level Effects:
~Cage (Number of levels: 60)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS
sd(log1pBRBRt1_Intercept) 0.30 0.11 0.05 0.50 1.00 777
sd(log1pLYERt1_Intercept) 0.14 0.09 0.01 0.33 1.00 1594
sd(log1pPtoidt1_Intercept) 0.04 0.03 0.00 0.12 1.00 5208
Tail_ESS
sd(log1pBRBRt1_Intercept) 1122
sd(log1pLYERt1_Intercept) 2957
sd(log1pPtoidt1_Intercept) 4103
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat
log1pBRBRt1_intercept 1.48 0.57 0.35 2.59 1.00
log1pBRBRt1_log1pBRBR_t -0.03 0.10 -0.23 0.18 1.00
log1pBRBRt1_log1pLYER_t 0.06 0.11 -0.15 0.28 1.00
log1pBRBRt1_log1pPtoid_t -0.90 0.05 -1.00 -0.79 1.00
log1pBRBRt1_temp -0.37 0.22 -0.80 0.07 1.00
log1pBRBRt1_rich 0.01 0.07 -0.14 0.15 1.00
log1pBRBRt1_logBiomass_g_t1 -0.11 0.20 -0.50 0.28 1.00
log1pBRBRt1_log1pBRBR_t:temp -0.07 0.04 -0.14 0.00 1.00
log1pBRBRt1_log1pPtoid_t:temp 0.13 0.04 0.06 0.20 1.00
log1pLYERt1_intercept 2.72 0.55 1.65 3.81 1.00
log1pLYERt1_log1pBRBR_t 0.28 0.09 0.11 0.46 1.00
log1pLYERt1_log1pLYER_t -0.57 0.11 -0.78 -0.37 1.00
log1pLYERt1_log1pPtoid_t -0.71 0.05 -0.82 -0.61 1.00
log1pLYERt1_temp -0.01 0.06 -0.12 0.11 1.00
log1pLYERt1_rich 0.37 0.20 -0.01 0.76 1.00
log1pLYERt1_logBiomass_g_t1 0.26 0.17 -0.06 0.59 1.00
log1pLYERt1_log1pBRBR_t:rich -0.07 0.04 -0.15 0.01 1.00
log1pPtoidt1_intercept -3.08 0.45 -3.94 -2.21 1.00
log1pPtoidt1_log1pBRBR_t 0.43 0.07 0.29 0.58 1.00
log1pPtoidt1_log1pLYER_t 0.31 0.08 0.16 0.47 1.00
log1pPtoidt1_log1pPtoid_t -0.01 0.04 -0.09 0.06 1.00
log1pPtoidt1_temp 0.67 0.12 0.44 0.89 1.00
log1pPtoidt1_rich -0.08 0.23 -0.53 0.36 1.00
log1pPtoidt1_logBiomass_g_t1 -0.48 0.13 -0.73 -0.24 1.00
log1pPtoidt1_log1pBRBR_t:temp -0.17 0.02 -0.21 -0.12 1.00
log1pPtoidt1_log1pBRBR_t:rich -0.06 0.04 -0.13 0.01 1.00
log1pPtoidt1_log1pLYER_t:rich 0.07 0.04 -0.01 0.15 1.00
Bulk_ESS Tail_ESS
log1pBRBRt1_intercept 5598 6048
log1pBRBRt1_log1pBRBR_t 2235 4429
log1pBRBRt1_log1pLYER_t 2053 4966
log1pBRBRt1_log1pPtoid_t 6157 5569
log1pBRBRt1_temp 4344 5359
log1pBRBRt1_rich 7948 5953
log1pBRBRt1_logBiomass_g_t1 7212 6460
log1pBRBRt1_log1pBRBR_t:temp 4940 6025
log1pBRBRt1_log1pPtoid_t:temp 5606 6303
log1pLYERt1_intercept 4807 5502
log1pLYERt1_log1pBRBR_t 3822 5195
log1pLYERt1_log1pLYER_t 3686 4966
log1pLYERt1_log1pPtoid_t 6158 6400
log1pLYERt1_temp 4654 5050
log1pLYERt1_rich 4967 5317
log1pLYERt1_logBiomass_g_t1 9131 5925
log1pLYERt1_log1pBRBR_t:rich 4932 5486
log1pPtoidt1_intercept 5773 5903
log1pPtoidt1_log1pBRBR_t 5298 5790
log1pPtoidt1_log1pLYER_t 6125 6055
log1pPtoidt1_log1pPtoid_t 10201 6054
log1pPtoidt1_temp 4968 5418
log1pPtoidt1_rich 6823 6122
log1pPtoidt1_logBiomass_g_t1 12595 6135
log1pPtoidt1_log1pBRBR_t:temp 5374 5712
log1pPtoidt1_log1pBRBR_t:rich 7666 6098
log1pPtoidt1_log1pLYER_t:rich 6024 6090
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma_log1pBRBRt1 1.10 0.06 0.99 1.23 1.00 2177 4140
sigma_log1pLYERt1 0.92 0.04 0.84 1.02 1.00 7751 5609
sigma_log1pPtoidt1 0.77 0.04 0.70 0.84 1.00 13386 5869
Residual Correlations:
Estimate Est.Error l-95% CI u-95% CI Rhat
rescor(log1pBRBRt1,log1pLYERt1) 0.52 0.05 0.42 0.62 1.00
rescor(log1pBRBRt1,log1pPtoidt1) -0.04 0.07 -0.18 0.11 1.00
rescor(log1pLYERt1,log1pPtoidt1) 0.07 0.07 -0.06 0.20 1.00
Bulk_ESS Tail_ESS
rescor(log1pBRBRt1,log1pLYERt1) 7033 6115
rescor(log1pBRBRt1,log1pPtoidt1) 10078 5917
rescor(log1pLYERt1,log1pPtoidt1) 10885 5789
Samples were drawn using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).Inspect credible intervals
# check highest-order temp effects
bayesplot::mcmc_intervals(reduced.1.brm, regex_pars = ":temp$", prob = 0.80, prob_outer = 0.95) # retain all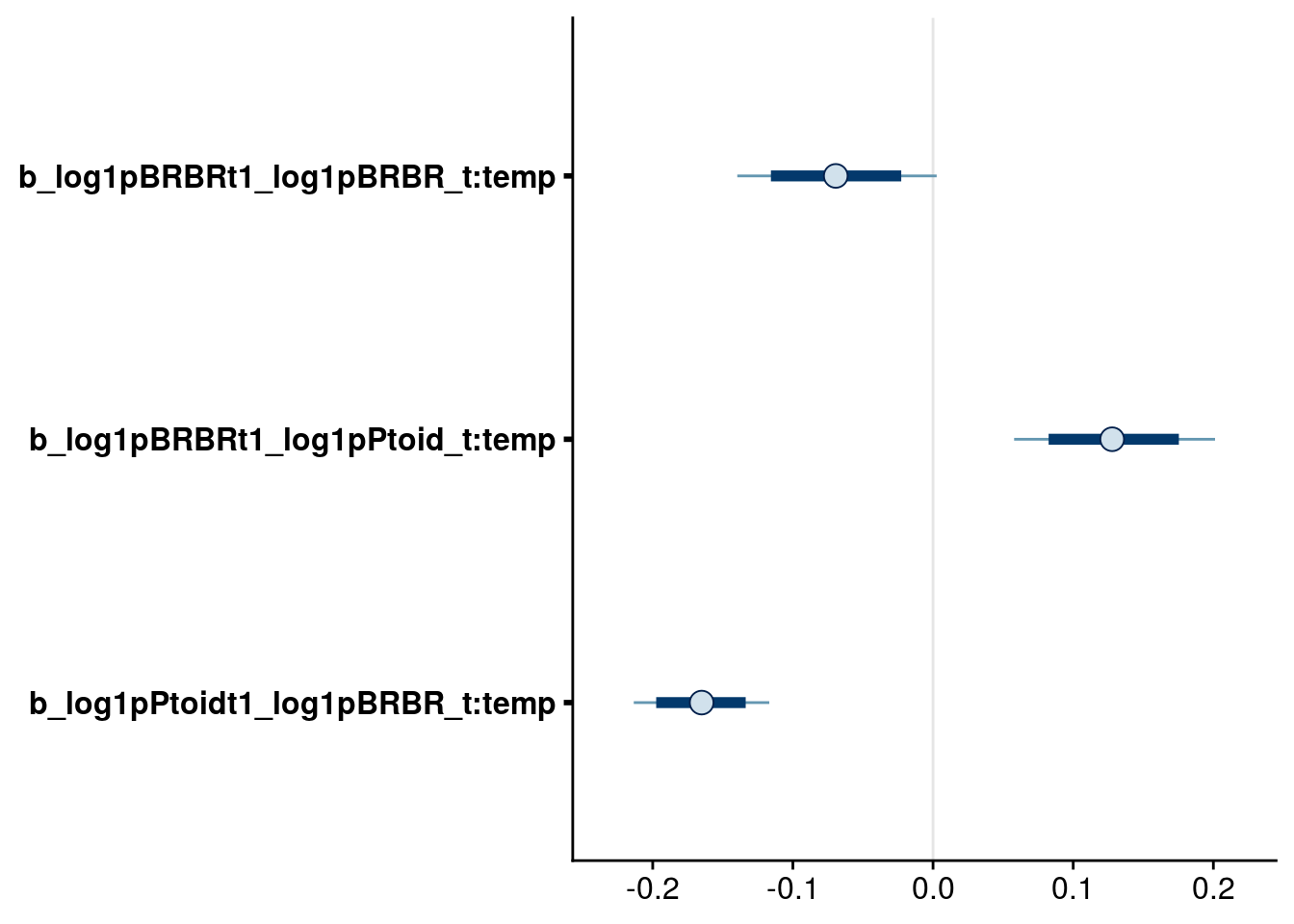
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# check main temp effect
bayesplot::mcmc_intervals(reduced.1.brm, regex_pars = "_temp$", prob = 0.80, prob_outer = 0.95) # drop temp effect on LYER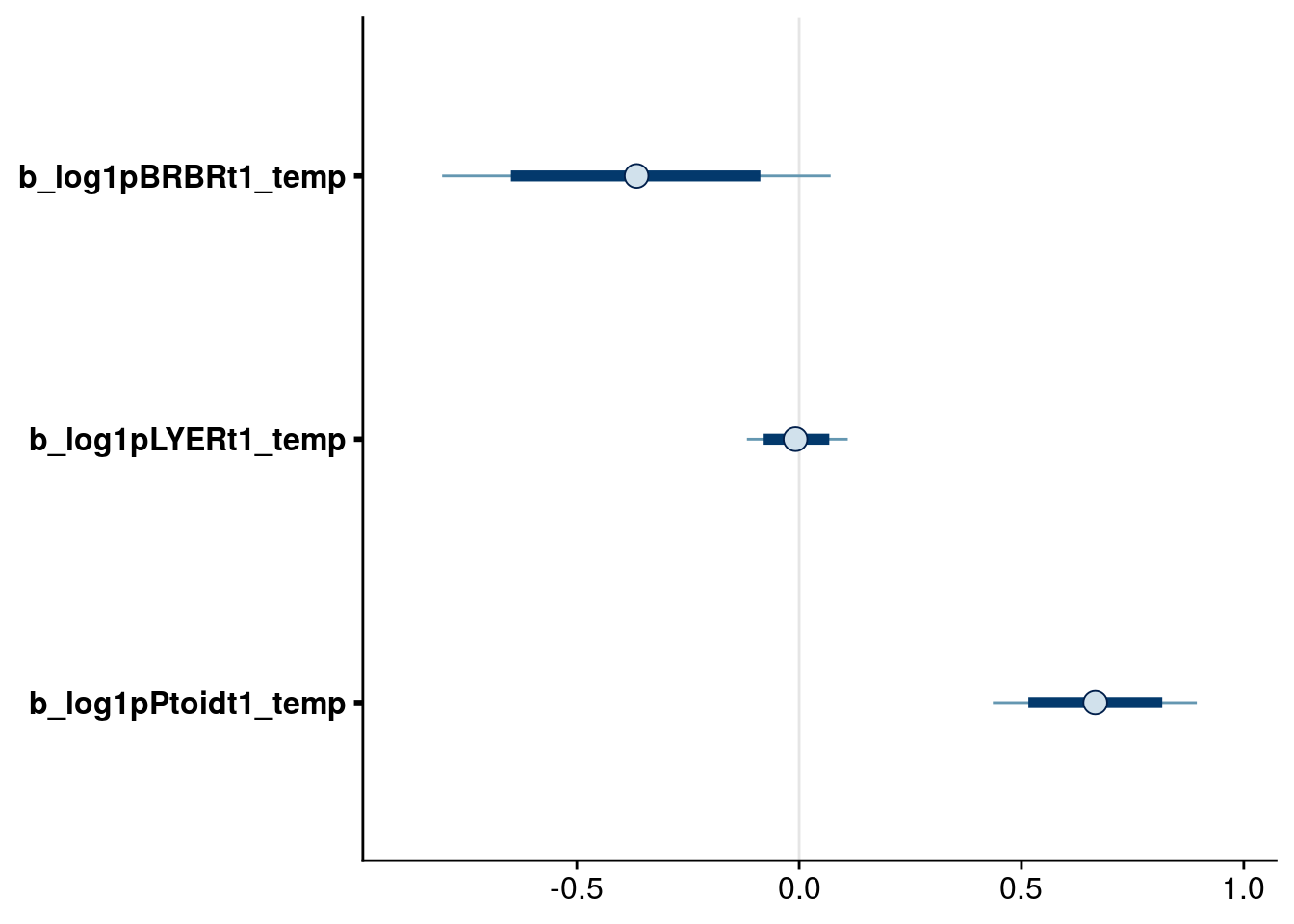
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# check highest-order rich effects
bayesplot::mcmc_intervals(reduced.1.brm, regex_pars = ":rich$", prob = 0.80, prob_outer = 0.95) # retain all
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# check main rich effect
bayesplot::mcmc_intervals(reduced.1.brm, regex_pars = "_rich$", prob = 0.80, prob_outer = 0.95) # drop rich effect on BRBR, keep others to preserve marginality in higher-order terms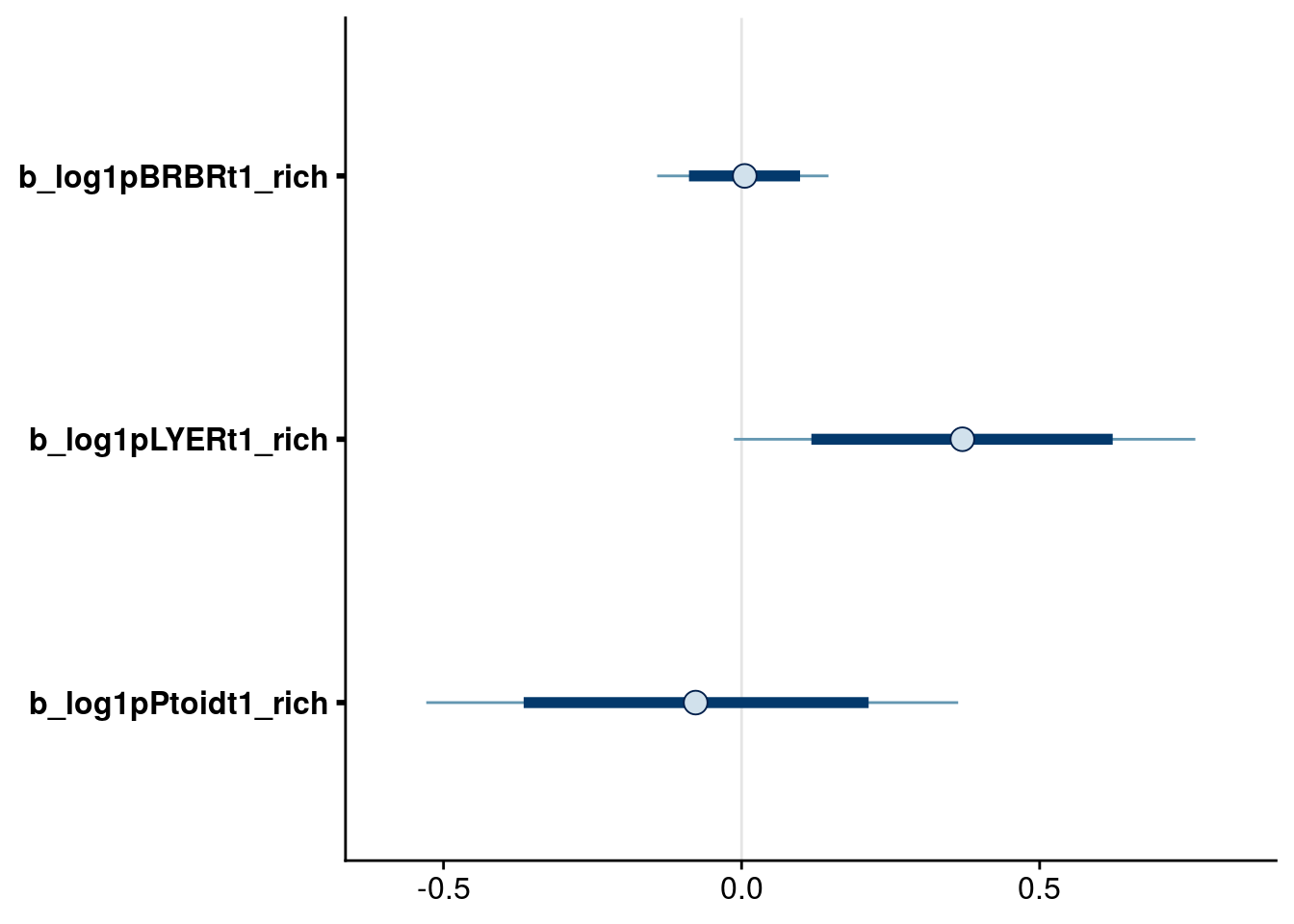
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# check biomass effects
bayesplot::mcmc_intervals(reduced.1.brm, regex_pars = "logBiomass_g_t1$", prob = 0.80, prob_outer = 0.95) # drop biomass effect on BRBR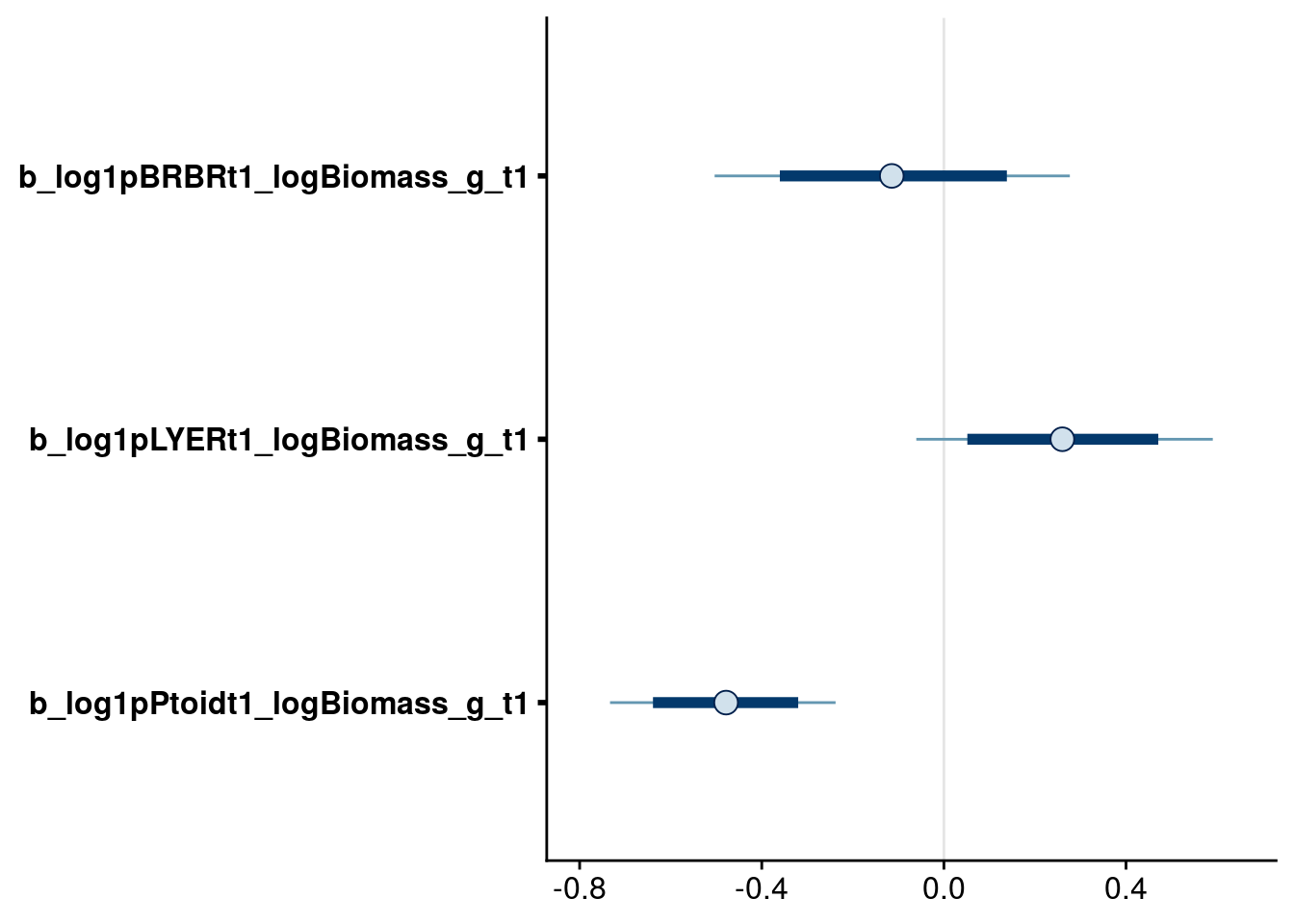
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
## check interactions (at 20 C in average monoculture)
# BRBR effect
bayesplot::mcmc_intervals(reduced.1.brm, regex_pars = "_log1pBRBR_t$", prob = 0.80, prob_outer = 0.95) # note that I keep intraspecific BRBR effects to preserve marginality with higher-order BRBR:temp effect on BRBR.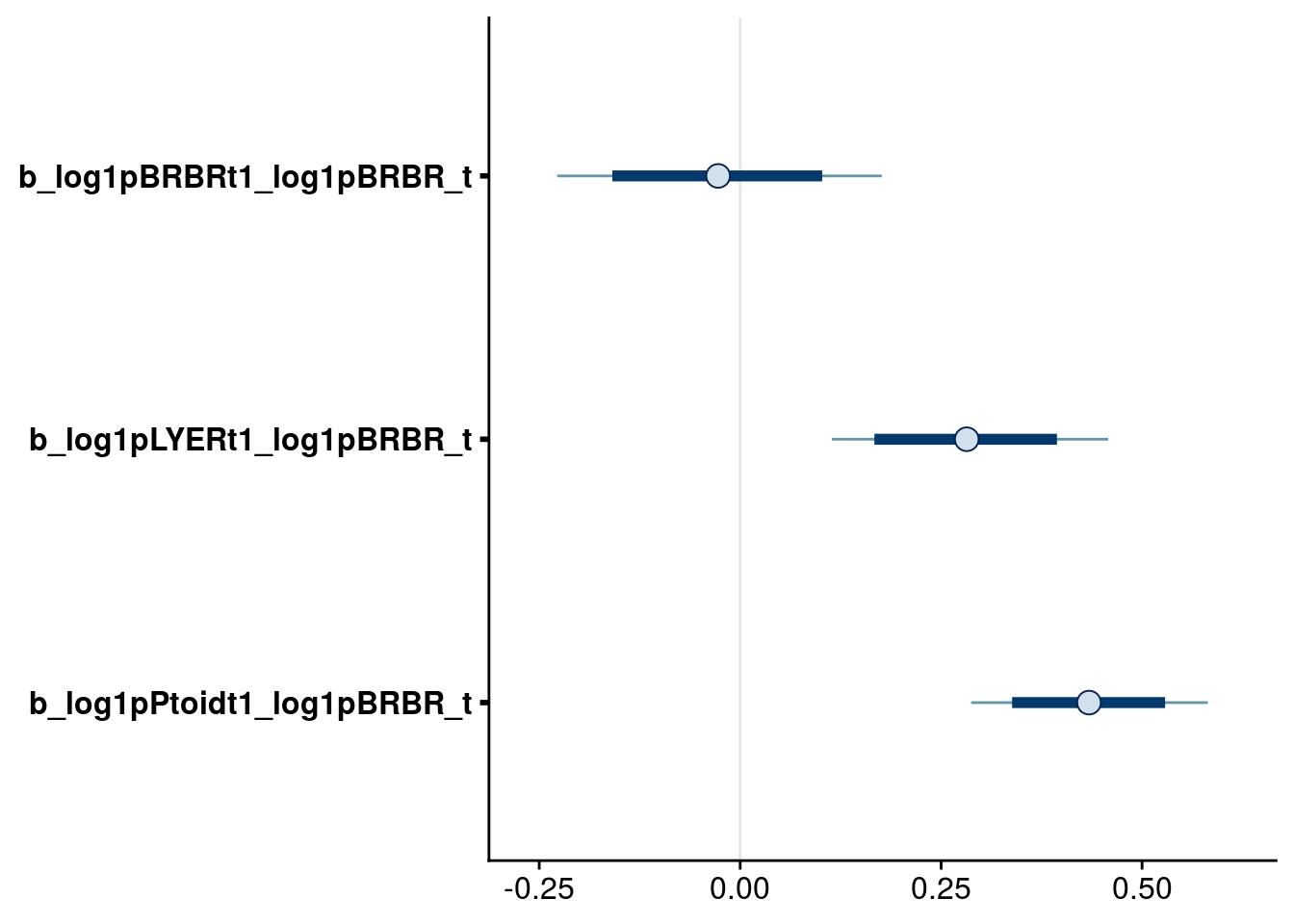
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# LYER effect
bayesplot::mcmc_intervals(reduced.1.brm, regex_pars = "_log1pLYER_t$", prob = 0.80, prob_outer = 0.95) # drop LYER effect on BRBR
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# Ptoid effect
bayesplot::mcmc_intervals(reduced.1.brm, regex_pars = "_log1pPtoid_t$", prob = 0.80, prob_outer = 0.95) # drop Ptoid effect on itself
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
Reduced model 2
Drop terms
Based on the above plots, I dropped the following terms:
Effects on BRBR_t1:
richlog1p(LYER_t)log(Biomass_g_t1)
Effects on LYER_t1:
temp
Effects on Ptoid_t1:
log1p(Ptoid_t)
Refit model
# update formulas
reduced.2.BRBR.bf <- update(reduced.1.BRBR.bf, .~. -rich -log1p(LYER_t) -log(Biomass_g_t1))
reduced.2.LYER.bf <- update(reduced.1.LYER.bf, .~. -temp)
reduced.2.Ptoid.bf <- update(reduced.1.Ptoid.bf, .~. -log1p(Ptoid_t))
# fit new model
reduced.2.brm <- brm(
data = full_df,
formula = mvbf(reduced.2.BRBR.bf, reduced.2.LYER.bf, reduced.2.Ptoid.bf),
iter = 4000,
prior = c(# growth rates
set_prior(prior.r.BRBR, class = "b", coef = "intercept", resp = "log1pBRBRt1"),
set_prior(prior.r.LYER, class = "b", coef = "intercept", resp = "log1pLYERt1"),
set_prior(prior.r.Ptoid, class = "b", coef = "intercept", resp = "log1pPtoidt1"),
# intraspecific effects
set_prior(prior.intra.BRBR, class = "b", coef = "log1pBRBR_t", resp = "log1pBRBRt1"),
set_prior(prior.intra.LYER, class = "b", coef = "log1pLYER_t", resp = "log1pLYERt1"),
# negative interspecific effects
set_prior(prior.BRBRonLYER, class = "b", coef = "log1pBRBR_t", resp = "log1pLYERt1"),
set_prior(prior.PtoidonBRBR, class = "b", coef = "log1pPtoid_t", resp = "log1pBRBRt1"),
set_prior(prior.PtoidonLYER, class = "b", coef = "log1pPtoid_t", resp = "log1pLYERt1"),
# positive interspecific effects
set_prior(prior.BRBRonPtoid, class = "b", coef = "log1pBRBR_t", resp = "log1pPtoidt1"),
set_prior(prior.LYERonPtoid, class = "b", coef = "log1pLYER_t", resp = "log1pPtoidt1"),
# rich effects
set_prior(prior.rich, class = "b", coef = "rich", resp = "log1pLYERt1"),
set_prior(prior.rich, class = "b", coef = "rich", resp = "log1pPtoidt1"),
set_prior(prior.rich, class = "b", coef = "log1pBRBR_t:rich", resp = "log1pLYERt1"),
set_prior(prior.rich, class = "b", coef = "log1pBRBR_t:rich", resp = "log1pPtoidt1"),
set_prior(prior.rich, class = "b", coef = "log1pLYER_t:rich", resp = "log1pPtoidt1"),
# temp effects
set_prior(prior.temp, class = "b", coef = "temp", resp = "log1pBRBRt1"),
set_prior(prior.temp, class = "b", coef = "temp", resp = "log1pPtoidt1"),
set_prior(prior.temp, class = "b", coef = "log1pBRBR_t:temp", resp = "log1pBRBRt1"),
set_prior(prior.temp, class = "b", coef = "log1pBRBR_t:temp", resp = "log1pPtoidt1"),
set_prior(prior.temp, class = "b", coef = "log1pPtoid_t:temp", resp = "log1pBRBRt1"),
# biomass effects
set_prior(prior.AphidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pLYERt1"),
set_prior(prior.PtoidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pPtoidt1"),
# random effects
set_prior(prior.random.effects, class = "sd", resp = "log1pBRBRt1"),
set_prior(prior.random.effects, class = "sd", resp = "log1pLYERt1"),
set_prior(prior.random.effects, class = "sd", resp = "log1pPtoidt1")),
file = "output/reduced.2.brm.rds")
# print model summary
summary(reduced.2.brm) Family: MV(gaussian, gaussian, gaussian)
Links: mu = identity; sigma = identity
mu = identity; sigma = identity
mu = identity; sigma = identity
Formula: log1p(BRBR_t1) ~ intercept + log1p(BRBR_t) + log1p(Ptoid_t) + temp + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + log1p(BRBR_t):temp + log1p(Ptoid_t):temp + offset(log1p(BRBR_t)) - 1
log1p(LYER_t1) ~ intercept + log1p(BRBR_t) + log1p(LYER_t) + log1p(Ptoid_t) + rich + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + log1p(BRBR_t):rich + offset(log1p(LYER_t)) - 1
log1p(Ptoid_t1) ~ intercept + log1p(BRBR_t) + log1p(LYER_t) + temp + rich + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + log1p(BRBR_t):temp + log1p(BRBR_t):rich + log1p(LYER_t):rich + offset(log1p(Ptoid_t)) - 1
Data: full_df (Number of observations: 264)
Samples: 4 chains, each with iter = 4000; warmup = 2000; thin = 1;
total post-warmup samples = 8000
Correlation Structures:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
ar_log1pBRBRt1[1] -0.44 0.09 -0.61 -0.26 1.00 4459 5718
ar_log1pLYERt1[1] -0.12 0.10 -0.31 0.07 1.00 6657 6198
ar_log1pPtoidt1[1] -0.50 0.08 -0.65 -0.34 1.00 12922 7042
Group-Level Effects:
~Cage (Number of levels: 60)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS
sd(log1pBRBRt1_Intercept) 0.24 0.10 0.02 0.42 1.00 1059
sd(log1pLYERt1_Intercept) 0.14 0.09 0.01 0.31 1.00 1762
sd(log1pPtoidt1_Intercept) 0.04 0.03 0.00 0.11 1.00 6103
Tail_ESS
sd(log1pBRBRt1_Intercept) 1514
sd(log1pLYERt1_Intercept) 4050
sd(log1pPtoidt1_Intercept) 4428
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat
log1pBRBRt1_intercept 1.47 0.46 0.56 2.38 1.00
log1pBRBRt1_log1pBRBR_t 0.03 0.08 -0.12 0.18 1.00
log1pBRBRt1_log1pPtoid_t -0.90 0.05 -1.00 -0.79 1.00
log1pBRBRt1_temp -0.32 0.22 -0.76 0.10 1.00
log1pBRBRt1_log1pBRBR_t:temp -0.07 0.04 -0.14 0.00 1.00
log1pBRBRt1_log1pPtoid_t:temp 0.13 0.04 0.06 0.20 1.00
log1pLYERt1_intercept 2.73 0.54 1.67 3.81 1.00
log1pLYERt1_log1pBRBR_t 0.30 0.07 0.16 0.44 1.00
log1pLYERt1_log1pLYER_t -0.58 0.09 -0.75 -0.42 1.00
log1pLYERt1_log1pPtoid_t -0.71 0.05 -0.82 -0.62 1.00
log1pLYERt1_rich 0.38 0.19 -0.01 0.76 1.00
log1pLYERt1_logBiomass_g_t1 0.30 0.15 0.01 0.59 1.00
log1pLYERt1_log1pBRBR_t:rich -0.07 0.04 -0.15 0.01 1.00
log1pPtoidt1_intercept -3.16 0.38 -3.90 -2.41 1.00
log1pPtoidt1_log1pBRBR_t 0.44 0.07 0.29 0.58 1.00
log1pPtoidt1_log1pLYER_t 0.32 0.08 0.17 0.47 1.00
log1pPtoidt1_temp 0.67 0.12 0.44 0.90 1.00
log1pPtoidt1_rich -0.09 0.23 -0.53 0.36 1.00
log1pPtoidt1_logBiomass_g_t1 -0.48 0.13 -0.72 -0.24 1.00
log1pPtoidt1_log1pBRBR_t:temp -0.16 0.02 -0.21 -0.12 1.00
log1pPtoidt1_log1pBRBR_t:rich -0.06 0.04 -0.13 0.01 1.00
log1pPtoidt1_log1pLYER_t:rich 0.07 0.04 -0.01 0.15 1.00
Bulk_ESS Tail_ESS
log1pBRBRt1_intercept 5086 5637
log1pBRBRt1_log1pBRBR_t 5879 6205
log1pBRBRt1_log1pPtoid_t 6050 5878
log1pBRBRt1_temp 5766 5973
log1pBRBRt1_log1pBRBR_t:temp 6211 6633
log1pBRBRt1_log1pPtoid_t:temp 7802 6128
log1pLYERt1_intercept 5579 5882
log1pLYERt1_log1pBRBR_t 8269 6543
log1pLYERt1_log1pLYER_t 5428 6233
log1pLYERt1_log1pPtoid_t 7915 6689
log1pLYERt1_rich 7974 6381
log1pLYERt1_logBiomass_g_t1 13788 6820
log1pLYERt1_log1pBRBR_t:rich 7925 5781
log1pPtoidt1_intercept 8135 6326
log1pPtoidt1_log1pBRBR_t 7555 6151
log1pPtoidt1_log1pLYER_t 8498 6495
log1pPtoidt1_temp 6863 6310
log1pPtoidt1_rich 9087 6182
log1pPtoidt1_logBiomass_g_t1 15373 6540
log1pPtoidt1_log1pBRBR_t:temp 7205 6642
log1pPtoidt1_log1pBRBR_t:rich 10300 6535
log1pPtoidt1_log1pLYER_t:rich 8488 6476
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma_log1pBRBRt1 1.12 0.06 1.01 1.23 1.00 3659 5831
sigma_log1pLYERt1 0.92 0.04 0.84 1.01 1.00 10094 6942
sigma_log1pPtoidt1 0.76 0.03 0.70 0.84 1.00 16970 5296
Residual Correlations:
Estimate Est.Error l-95% CI u-95% CI Rhat
rescor(log1pBRBRt1,log1pLYERt1) 0.51 0.05 0.41 0.60 1.00
rescor(log1pBRBRt1,log1pPtoidt1) -0.03 0.07 -0.17 0.11 1.00
rescor(log1pLYERt1,log1pPtoidt1) 0.07 0.07 -0.06 0.20 1.00
Bulk_ESS Tail_ESS
rescor(log1pBRBRt1,log1pLYERt1) 8918 6339
rescor(log1pBRBRt1,log1pPtoidt1) 12543 6766
rescor(log1pLYERt1,log1pPtoidt1) 15340 6311
Samples were drawn using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).Inspect credible intervals
# check highest-order temp effects
bayesplot::mcmc_intervals(reduced.2.brm, regex_pars = "_t:temp$", prob = 0.80, prob_outer = 0.95)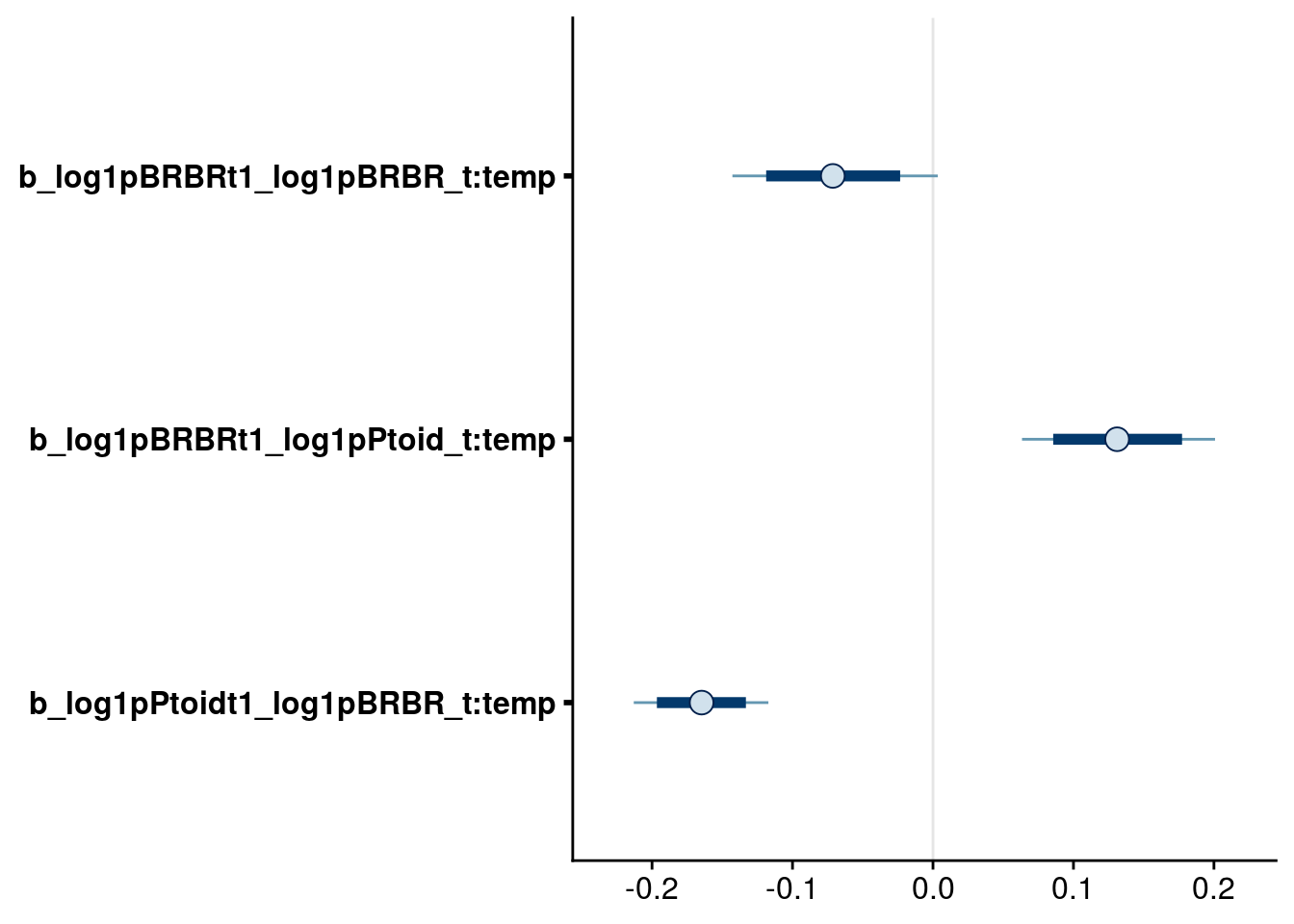
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# check temp effects on growth rates
bayesplot::mcmc_intervals(reduced.2.brm, regex_pars = "_temp$", prob = 0.80, prob_outer = 0.95)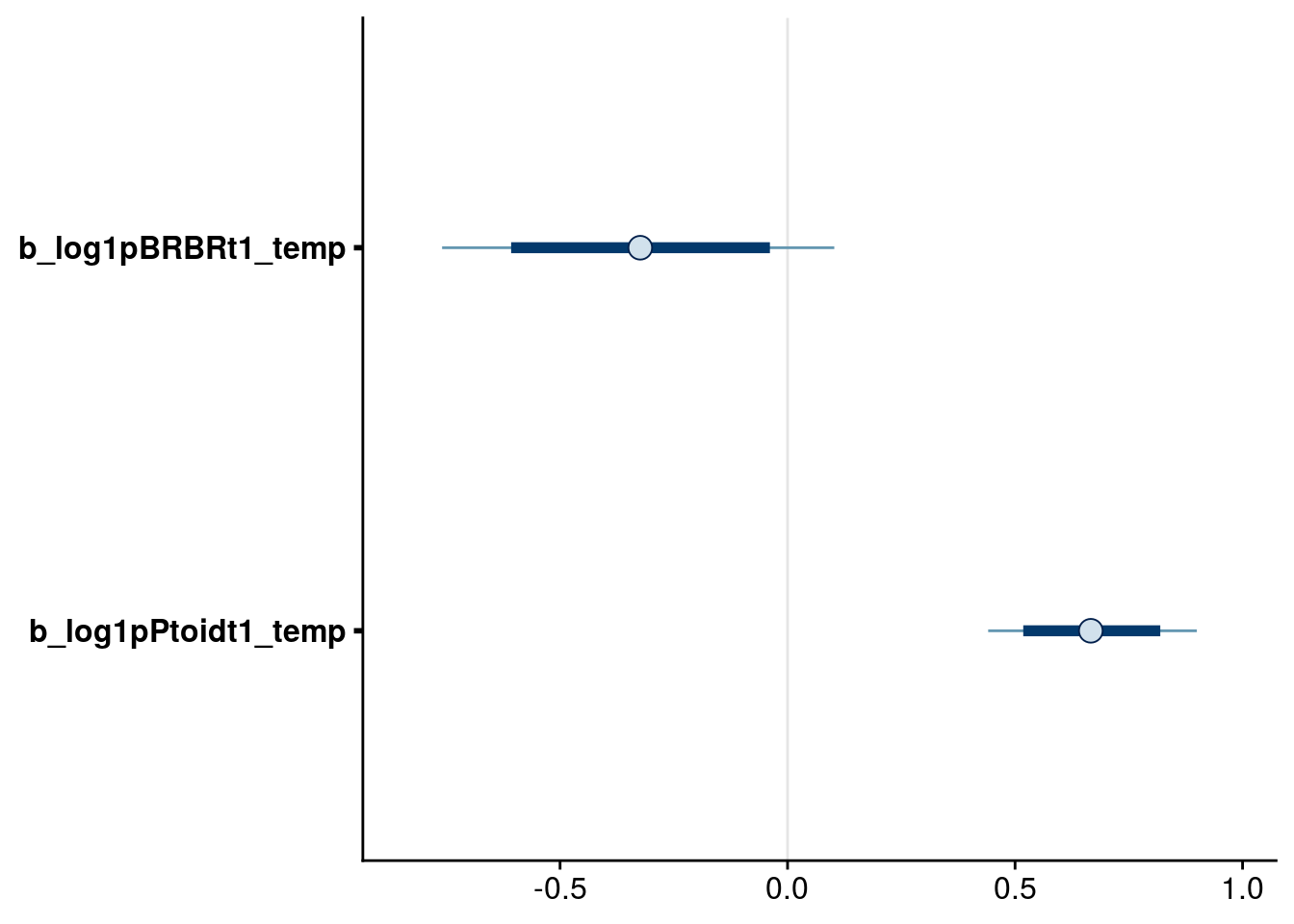
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# check highest-order rich effects
bayesplot::mcmc_intervals(reduced.2.brm, regex_pars = "_t:rich$", prob = 0.80, prob_outer = 0.95) # all remaining higher-order rich effects have 95% credible intervals that overlap zero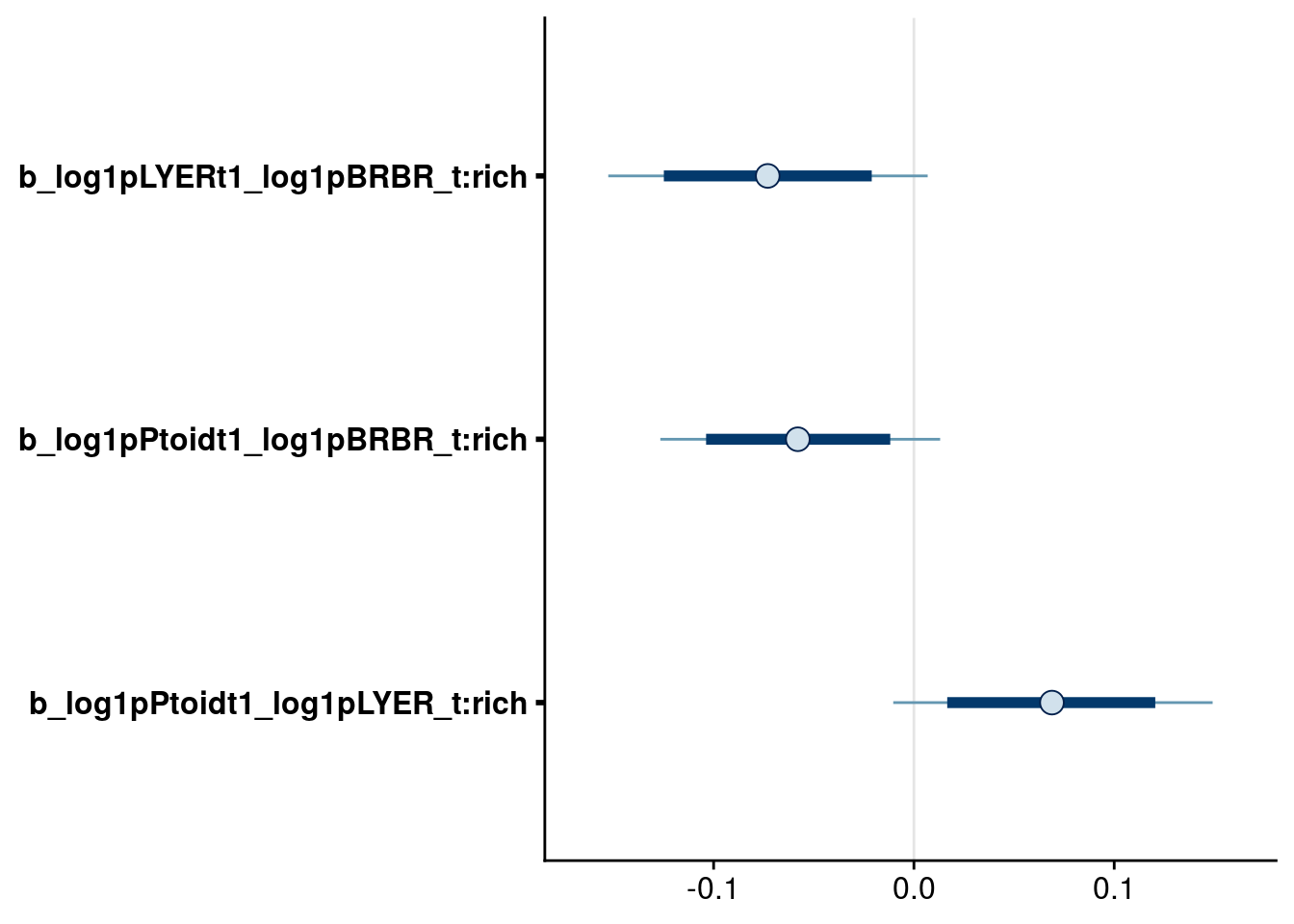
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# check rich effects on growth rates
bayesplot::mcmc_intervals(reduced.2.brm, regex_pars = "_rich$", prob = 0.80, prob_outer = 0.95) # note that I keep rich effect on Ptoid to preserve marginality with higher-order rich effect
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# check biomass effects
bayesplot::mcmc_intervals(reduced.2.brm, regex_pars = "logBiomass_g_t1$", prob = 0.80, prob_outer = 0.95)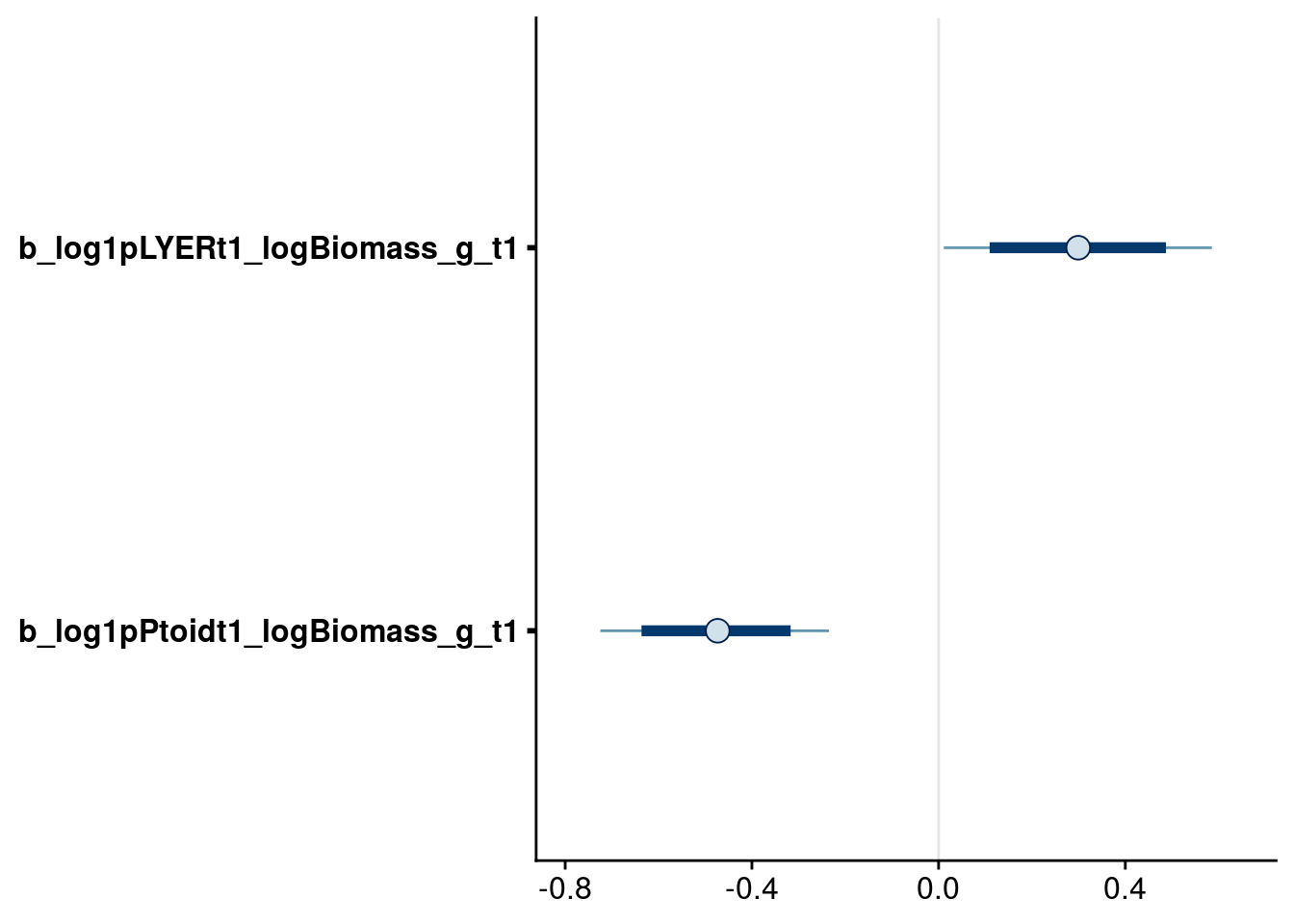
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# check intrinsic growth rates (at 20 C in average monoculture)
bayesplot::mcmc_intervals(reduced.2.brm, regex_pars = "intercept$", prob = 0.80, prob_outer = 0.95)
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# check interactions (at 20 C in average monoculture)
bayesplot::mcmc_intervals(reduced.2.brm, regex_pars = "_log1pBRBR_t$", prob = 0.80, prob_outer = 0.95) # note that I keep intraspecific BRBR effects to preserve marginality with higher-order BRBR:temp effect on itself.
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
bayesplot::mcmc_intervals(reduced.2.brm, regex_pars = "_log1pLYER_t$", prob = 0.80, prob_outer = 0.95)
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
bayesplot::mcmc_intervals(reduced.2.brm, regex_pars = "_log1pPtoid_t$", prob = 0.80, prob_outer = 0.95)
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
Now, all higher-order terms have 80% credible intervals that do not overlap zero.
Reduced model 3
Drop terms
It is unclear how to progress now, although there are a few terms with less clear effects (still above 80% criteria used previously), including:
Effects on LYER_t1:
log1p(BRBR_t):rich
Effects on Ptoid_t1:
log1p(LYER_t):richlog1p(BRBR_t):rich
I’m going to try dropping log1p(LYER_t):rich from the previous model.
Refit model
# update formulas
reduced.3.BRBR.bf <- reduced.2.BRBR.bf
reduced.3.LYER.bf <- reduced.2.LYER.bf
reduced.3.Ptoid.bf <- update(reduced.2.Ptoid.bf, .~. -log1p(LYER_t):rich)
# fit new model
reduced.3.brm <- brm(
data = full_df,
formula = mvbf(reduced.3.BRBR.bf, reduced.3.LYER.bf, reduced.3.Ptoid.bf),
iter = 4000,
prior = c(# growth rates
set_prior(prior.r.BRBR, class = "b", coef = "intercept", resp = "log1pBRBRt1"),
set_prior(prior.r.LYER, class = "b", coef = "intercept", resp = "log1pLYERt1"),
set_prior(prior.r.Ptoid, class = "b", coef = "intercept", resp = "log1pPtoidt1"),
# intraspecific effects
set_prior(prior.intra.BRBR, class = "b", coef = "log1pBRBR_t", resp = "log1pBRBRt1"),
set_prior(prior.intra.LYER, class = "b", coef = "log1pLYER_t", resp = "log1pLYERt1"),
# negative interspecific effects
set_prior(prior.BRBRonLYER, class = "b", coef = "log1pBRBR_t", resp = "log1pLYERt1"),
set_prior(prior.PtoidonBRBR, class = "b", coef = "log1pPtoid_t", resp = "log1pBRBRt1"),
set_prior(prior.PtoidonLYER, class = "b", coef = "log1pPtoid_t", resp = "log1pLYERt1"),
# positive interspecific effects
set_prior(prior.BRBRonPtoid, class = "b", coef = "log1pBRBR_t", resp = "log1pPtoidt1"),
set_prior(prior.LYERonPtoid, class = "b", coef = "log1pLYER_t", resp = "log1pPtoidt1"),
# rich effects
set_prior(prior.rich, class = "b", coef = "rich", resp = "log1pLYERt1"),
set_prior(prior.rich, class = "b", coef = "rich", resp = "log1pPtoidt1"),
set_prior(prior.rich, class = "b", coef = "log1pBRBR_t:rich", resp = "log1pLYERt1"),
set_prior(prior.rich, class = "b", coef = "log1pBRBR_t:rich", resp = "log1pPtoidt1"),
# temp effects
set_prior(prior.temp, class = "b", coef = "temp", resp = "log1pBRBRt1"),
set_prior(prior.temp, class = "b", coef = "temp", resp = "log1pPtoidt1"),
set_prior(prior.temp, class = "b", coef = "log1pBRBR_t:temp", resp = "log1pBRBRt1"),
set_prior(prior.temp, class = "b", coef = "log1pBRBR_t:temp", resp = "log1pPtoidt1"),
set_prior(prior.temp, class = "b", coef = "log1pPtoid_t:temp", resp = "log1pBRBRt1"),
# biomass effects
set_prior(prior.AphidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pLYERt1"),
set_prior(prior.PtoidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pPtoidt1"),
# random effects
set_prior(prior.random.effects, class = "sd", resp = "log1pBRBRt1"),
set_prior(prior.random.effects, class = "sd", resp = "log1pLYERt1"),
set_prior(prior.random.effects, class = "sd", resp = "log1pPtoidt1")),
file = "output/reduced.3.brm.rds")
# print model summary
summary(reduced.3.brm) Family: MV(gaussian, gaussian, gaussian)
Links: mu = identity; sigma = identity
mu = identity; sigma = identity
mu = identity; sigma = identity
Formula: log1p(BRBR_t1) ~ intercept + log1p(BRBR_t) + log1p(Ptoid_t) + temp + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + log1p(BRBR_t):temp + log1p(Ptoid_t):temp + offset(log1p(BRBR_t)) - 1
log1p(LYER_t1) ~ intercept + log1p(BRBR_t) + log1p(LYER_t) + log1p(Ptoid_t) + rich + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + log1p(BRBR_t):rich + offset(log1p(LYER_t)) - 1
log1p(Ptoid_t1) ~ intercept + log1p(BRBR_t) + log1p(LYER_t) + temp + rich + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + log1p(BRBR_t):temp + log1p(BRBR_t):rich + offset(log1p(Ptoid_t)) - 1
Data: full_df (Number of observations: 264)
Samples: 4 chains, each with iter = 4000; warmup = 2000; thin = 1;
total post-warmup samples = 8000
Correlation Structures:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
ar_log1pBRBRt1[1] -0.44 0.09 -0.62 -0.26 1.00 3843 6249
ar_log1pLYERt1[1] -0.12 0.10 -0.30 0.07 1.00 6062 6048
ar_log1pPtoidt1[1] -0.50 0.08 -0.66 -0.34 1.00 9303 5957
Group-Level Effects:
~Cage (Number of levels: 60)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS
sd(log1pBRBRt1_Intercept) 0.24 0.10 0.03 0.43 1.00 1093
sd(log1pLYERt1_Intercept) 0.14 0.08 0.01 0.31 1.00 1737
sd(log1pPtoidt1_Intercept) 0.04 0.03 0.00 0.11 1.00 5475
Tail_ESS
sd(log1pBRBRt1_Intercept) 1963
sd(log1pLYERt1_Intercept) 2994
sd(log1pPtoidt1_Intercept) 4459
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat
log1pBRBRt1_intercept 1.48 0.46 0.59 2.40 1.00
log1pBRBRt1_log1pBRBR_t 0.03 0.08 -0.12 0.18 1.00
log1pBRBRt1_log1pPtoid_t -0.90 0.05 -1.01 -0.79 1.00
log1pBRBRt1_temp -0.32 0.21 -0.75 0.09 1.00
log1pBRBRt1_log1pBRBR_t:temp -0.07 0.04 -0.14 0.00 1.00
log1pBRBRt1_log1pPtoid_t:temp 0.13 0.04 0.06 0.20 1.00
log1pLYERt1_intercept 2.74 0.54 1.70 3.79 1.00
log1pLYERt1_log1pBRBR_t 0.30 0.07 0.16 0.44 1.00
log1pLYERt1_log1pLYER_t -0.58 0.08 -0.76 -0.42 1.00
log1pLYERt1_log1pPtoid_t -0.72 0.05 -0.82 -0.61 1.00
log1pLYERt1_rich 0.38 0.20 -0.01 0.76 1.00
log1pLYERt1_logBiomass_g_t1 0.30 0.15 0.01 0.58 1.00
log1pLYERt1_log1pBRBR_t:rich -0.07 0.04 -0.15 0.01 1.00
log1pPtoidt1_intercept -3.40 0.36 -4.10 -2.70 1.00
log1pPtoidt1_log1pBRBR_t 0.40 0.07 0.26 0.54 1.00
log1pPtoidt1_log1pLYER_t 0.40 0.06 0.28 0.52 1.00
log1pPtoidt1_temp 0.65 0.12 0.42 0.88 1.00
log1pPtoidt1_rich 0.18 0.17 -0.14 0.51 1.00
log1pPtoidt1_logBiomass_g_t1 -0.49 0.12 -0.73 -0.25 1.00
log1pPtoidt1_log1pBRBR_t:temp -0.16 0.02 -0.21 -0.12 1.00
log1pPtoidt1_log1pBRBR_t:rich -0.04 0.03 -0.11 0.03 1.00
Bulk_ESS Tail_ESS
log1pBRBRt1_intercept 4031 5396
log1pBRBRt1_log1pBRBR_t 5076 5933
log1pBRBRt1_log1pPtoid_t 4404 5489
log1pBRBRt1_temp 4269 5087
log1pBRBRt1_log1pBRBR_t:temp 4835 5526
log1pBRBRt1_log1pPtoid_t:temp 5559 5716
log1pLYERt1_intercept 4928 5071
log1pLYERt1_log1pBRBR_t 6863 6090
log1pLYERt1_log1pLYER_t 4690 5519
log1pLYERt1_log1pPtoid_t 5838 6263
log1pLYERt1_rich 5960 5619
log1pLYERt1_logBiomass_g_t1 10203 6204
log1pLYERt1_log1pBRBR_t:rich 5916 5539
log1pPtoidt1_intercept 6774 5597
log1pPtoidt1_log1pBRBR_t 5859 5853
log1pPtoidt1_log1pLYER_t 9124 4922
log1pPtoidt1_temp 5450 5166
log1pPtoidt1_rich 6268 5242
log1pPtoidt1_logBiomass_g_t1 10418 6393
log1pPtoidt1_log1pBRBR_t:temp 5668 5454
log1pPtoidt1_log1pBRBR_t:rich 6250 5271
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma_log1pBRBRt1 1.11 0.06 1.00 1.23 1.00 3337 4780
sigma_log1pLYERt1 0.92 0.04 0.84 1.01 1.00 7022 5783
sigma_log1pPtoidt1 0.77 0.03 0.70 0.84 1.00 10358 5625
Residual Correlations:
Estimate Est.Error l-95% CI u-95% CI Rhat
rescor(log1pBRBRt1,log1pLYERt1) 0.51 0.05 0.41 0.61 1.00
rescor(log1pBRBRt1,log1pPtoidt1) -0.03 0.07 -0.17 0.11 1.00
rescor(log1pLYERt1,log1pPtoidt1) 0.07 0.06 -0.05 0.20 1.00
Bulk_ESS Tail_ESS
rescor(log1pBRBRt1,log1pLYERt1) 6159 5704
rescor(log1pBRBRt1,log1pPtoidt1) 9267 6352
rescor(log1pLYERt1,log1pPtoidt1) 11538 6463
Samples were drawn using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).Inspect credible intervals
Note that this is the final model used to assess the structural stability of the initial food web and the dominant food chain, so I illustrate the credible intervals of its modelled parameters.
# check highest-order temp effects
bayesplot::mcmc_intervals(reduced.3.brm, regex_pars = "_t:temp$", prob = 0.80, prob_outer = 0.95)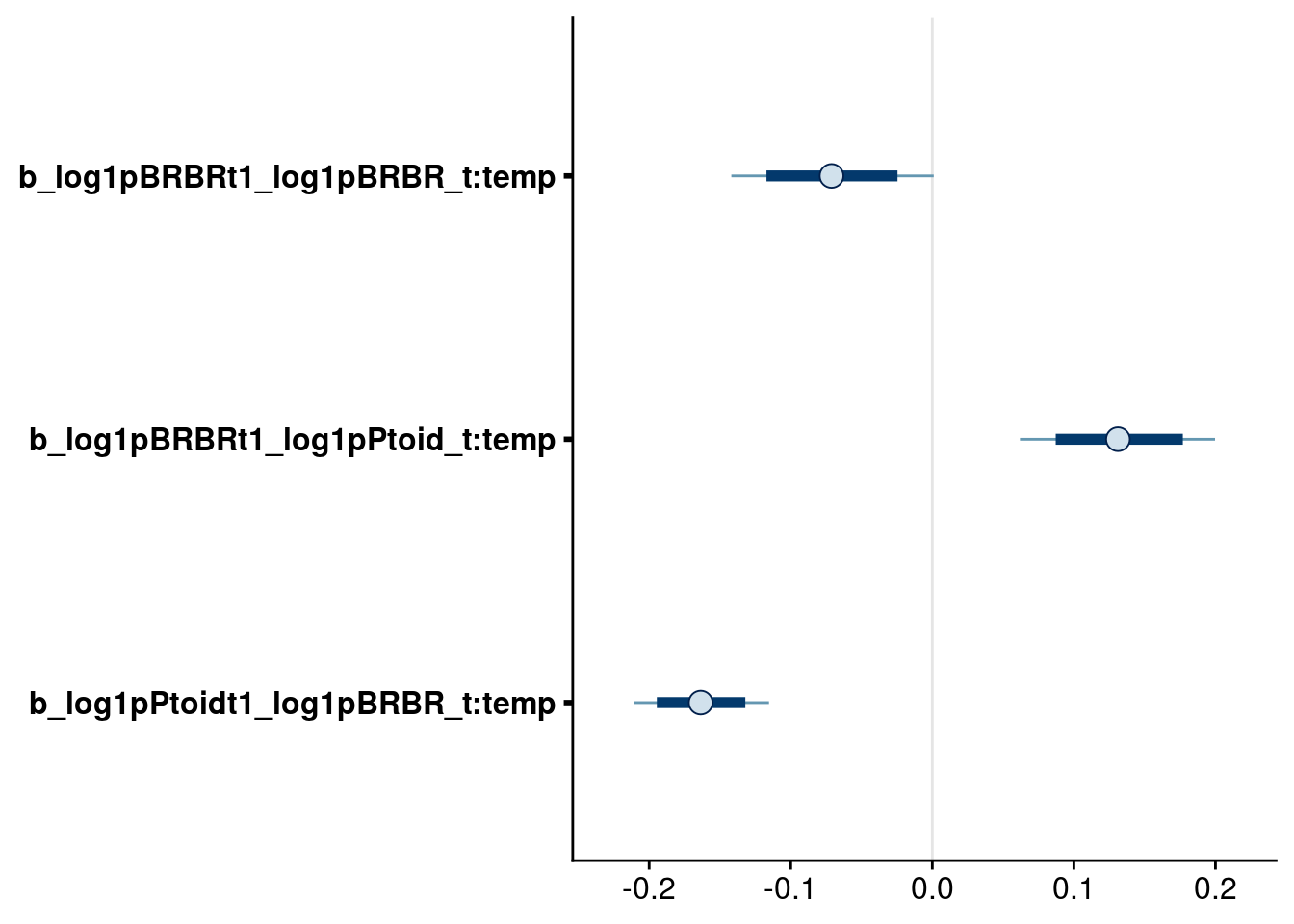
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# check highest-order rich effects
bayesplot::mcmc_intervals(reduced.3.brm, regex_pars = "_t:rich$", prob = 0.80, prob_outer = 0.95) # higher-order BRBR:rich effect on Ptoid no longer meets 80% cutoff.
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# check temp effects on growth rates
bayesplot::mcmc_intervals(reduced.3.brm, regex_pars = "_temp$", prob = 0.80, prob_outer = 0.95)
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# check rich effects on growth rates
bayesplot::mcmc_intervals(reduced.3.brm, regex_pars = "_rich$", prob = 0.80, prob_outer = 0.95)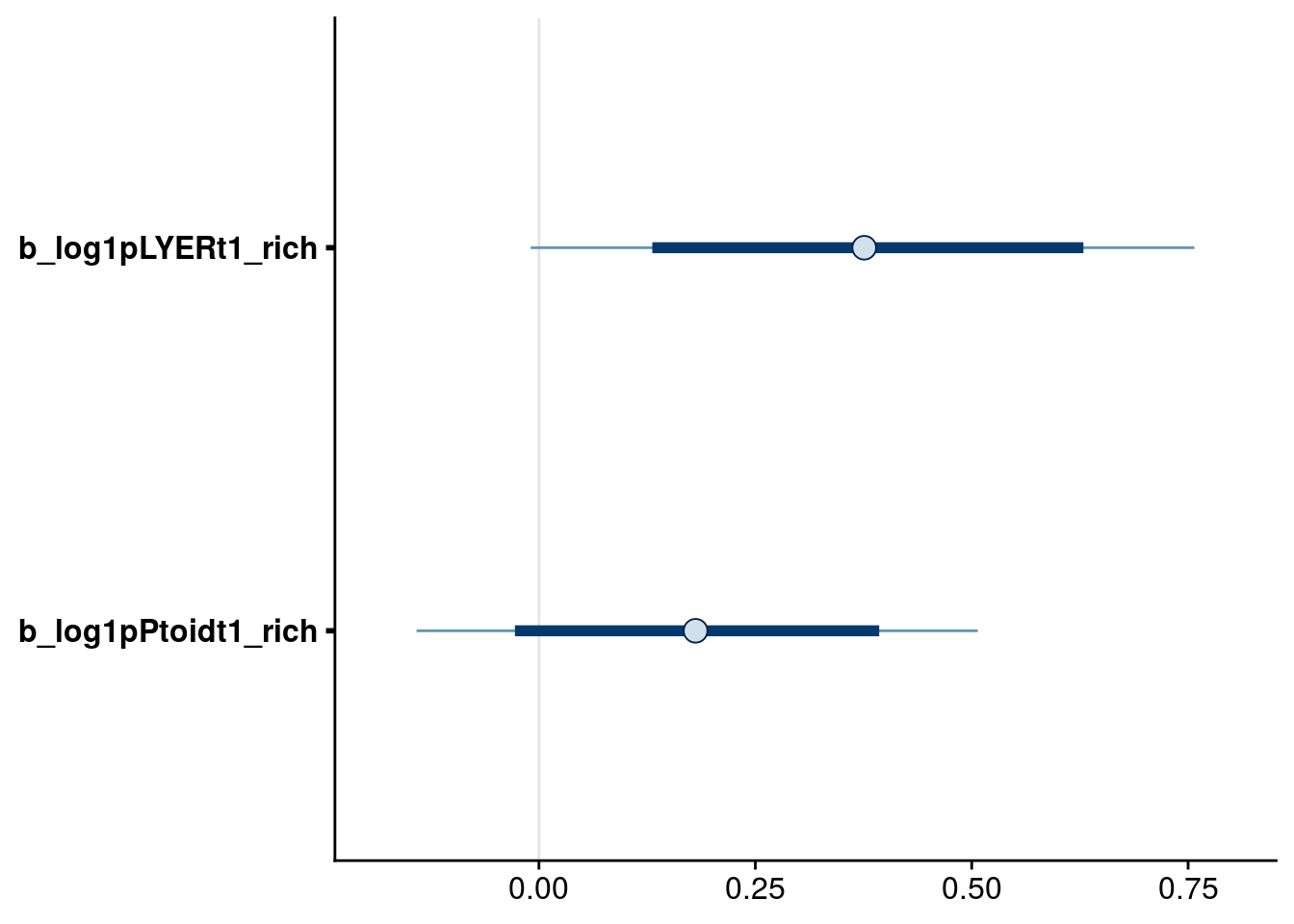
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# check biomass effects
bayesplot::mcmc_intervals(reduced.3.brm, regex_pars = "logBiomass_g_t1$", prob = 0.80, prob_outer = 0.95)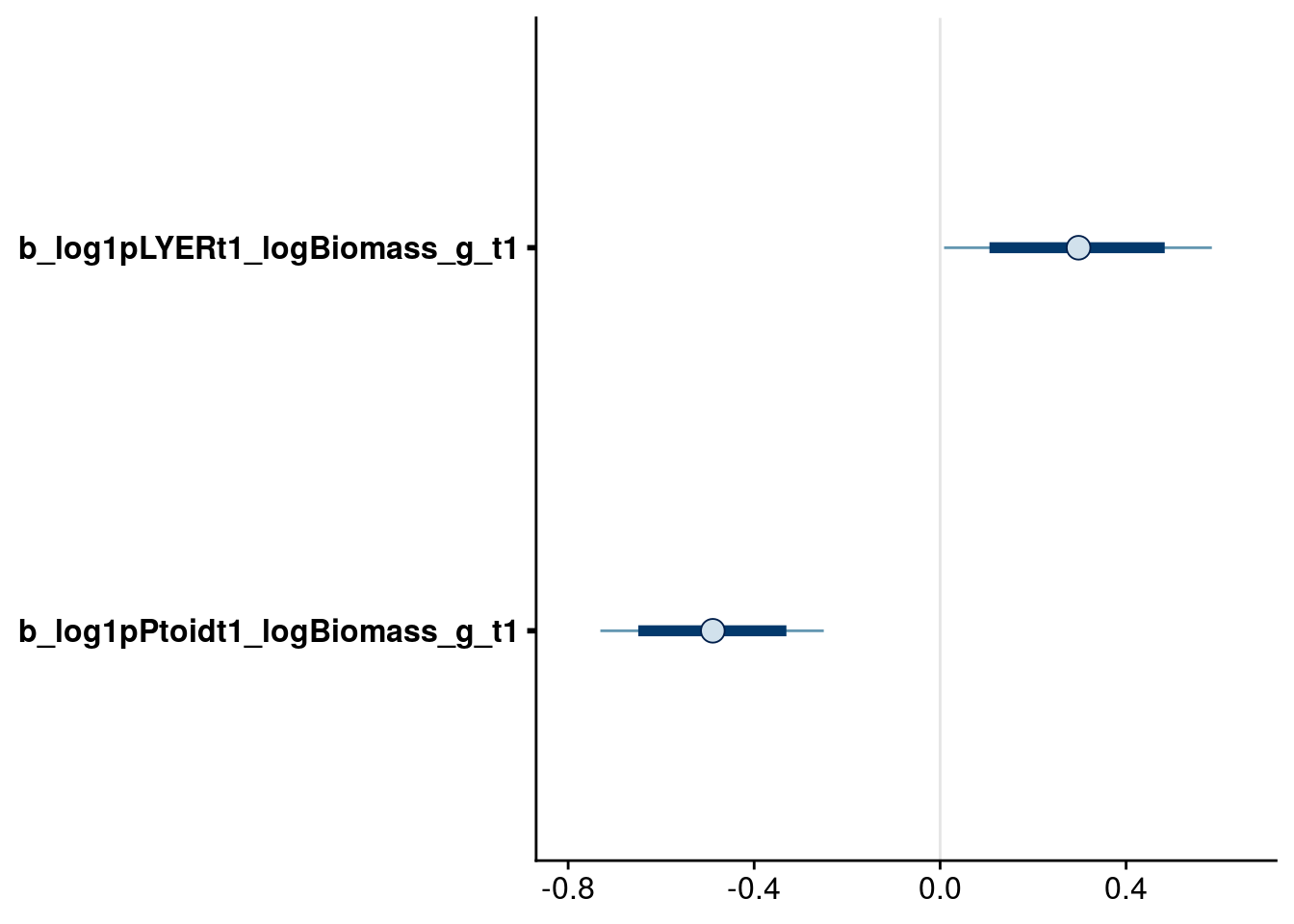
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# check intrinsic growth rates (at 20 C in average monoculture)
bayesplot::mcmc_intervals(reduced.3.brm, regex_pars = "intercept$", prob = 0.80, prob_outer = 0.95)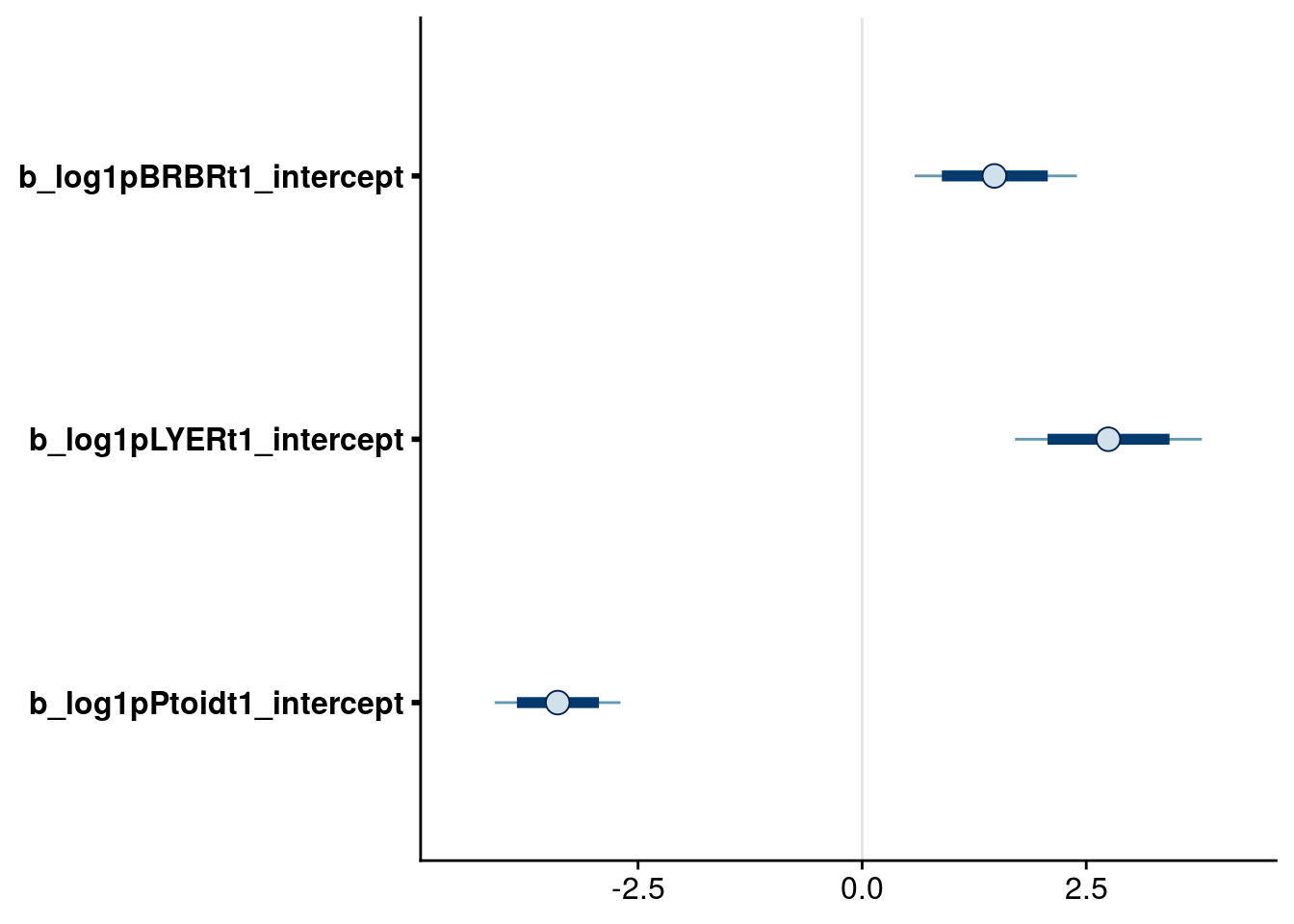
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# check interactions (at 20 C in average monoculture)
bayesplot::mcmc_intervals(reduced.3.brm, regex_pars = "_log1pBRBR_t$", prob = 0.80, prob_outer = 0.95)
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
bayesplot::mcmc_intervals(reduced.3.brm, regex_pars = "_log1pLYER_t$", prob = 0.80, prob_outer = 0.95)
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
bayesplot::mcmc_intervals(reduced.3.brm, regex_pars = "_log1pPtoid_t$", prob = 0.80, prob_outer = 0.95)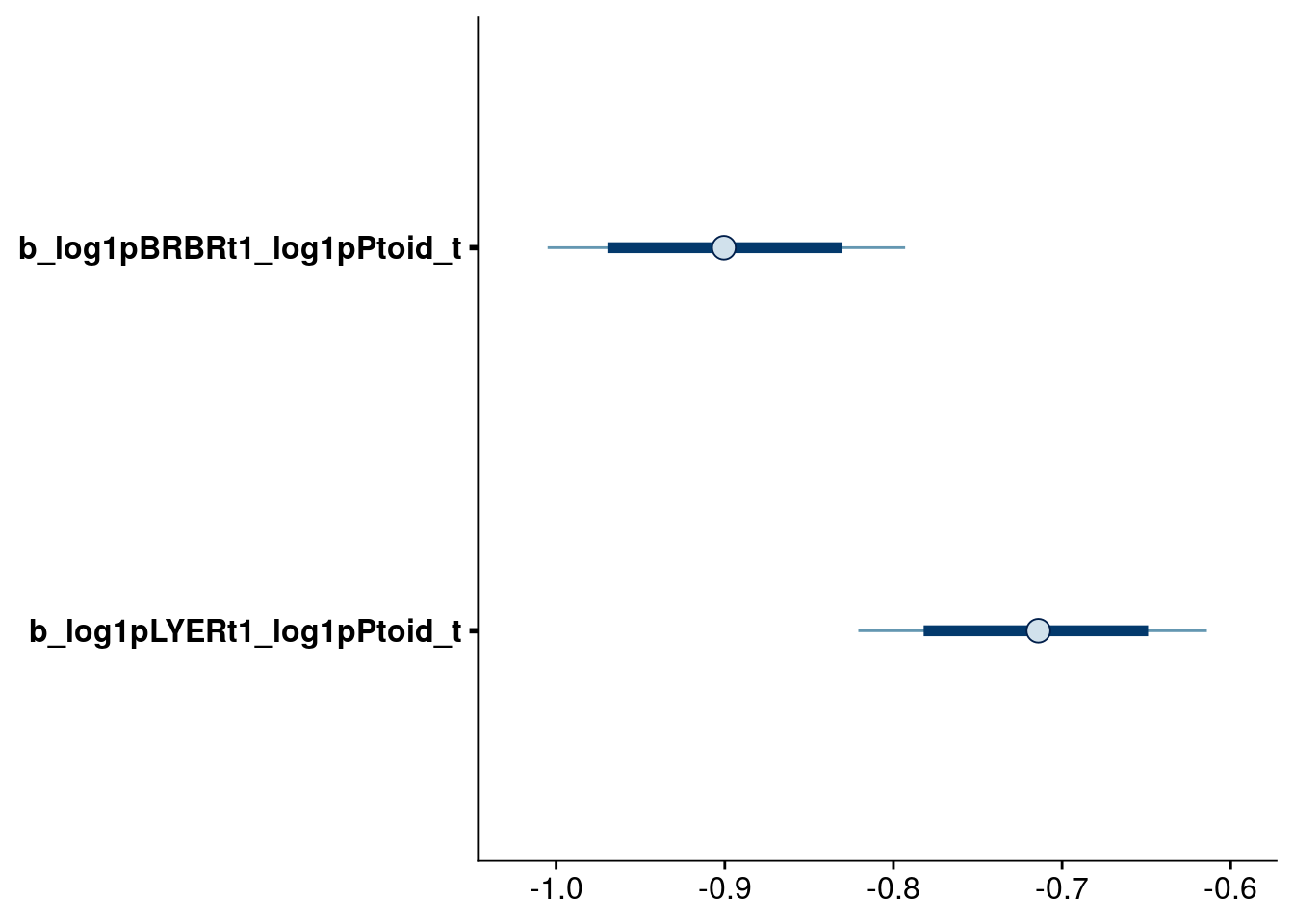
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
Reduced model 4
Drop terms
Now try dropping log1p(BRBR_t):rich effect on Ptoid_t1 instead.
Refit model
# update formulas
# note that I'm updating model 2, not model 3
reduced.4.BRBR.bf <- reduced.2.BRBR.bf
reduced.4.LYER.bf <- reduced.2.LYER.bf
reduced.4.Ptoid.bf <- update(reduced.2.Ptoid.bf, .~. -log1p(BRBR_t):rich)
# fit new model
reduced.4.brm <- brm(
data = full_df,
formula = mvbf(reduced.4.BRBR.bf, reduced.4.LYER.bf, reduced.4.Ptoid.bf),
iter = 4000,
prior = c(# growth rates
set_prior(prior.r.BRBR, class = "b", coef = "intercept", resp = "log1pBRBRt1"),
set_prior(prior.r.LYER, class = "b", coef = "intercept", resp = "log1pLYERt1"),
set_prior(prior.r.Ptoid, class = "b", coef = "intercept", resp = "log1pPtoidt1"),
# intraspecific effects
set_prior(prior.intra.BRBR, class = "b", coef = "log1pBRBR_t", resp = "log1pBRBRt1"),
set_prior(prior.intra.LYER, class = "b", coef = "log1pLYER_t", resp = "log1pLYERt1"),
# negative interspecific effects
set_prior(prior.BRBRonLYER, class = "b", coef = "log1pBRBR_t", resp = "log1pLYERt1"),
set_prior(prior.PtoidonBRBR, class = "b", coef = "log1pPtoid_t", resp = "log1pBRBRt1"),
set_prior(prior.PtoidonLYER, class = "b", coef = "log1pPtoid_t", resp = "log1pLYERt1"),
# positive interspecific effects
set_prior(prior.BRBRonPtoid, class = "b", coef = "log1pBRBR_t", resp = "log1pPtoidt1"),
set_prior(prior.LYERonPtoid, class = "b", coef = "log1pLYER_t", resp = "log1pPtoidt1"),
# rich effects
set_prior(prior.rich, class = "b", coef = "rich", resp = "log1pLYERt1"),
set_prior(prior.rich, class = "b", coef = "rich", resp = "log1pPtoidt1"),
set_prior(prior.rich, class = "b", coef = "log1pBRBR_t:rich", resp = "log1pLYERt1"),
set_prior(prior.rich, class = "b", coef = "log1pLYER_t:rich", resp = "log1pPtoidt1"),
# temp effects
set_prior(prior.temp, class = "b", coef = "temp", resp = "log1pBRBRt1"),
set_prior(prior.temp, class = "b", coef = "temp", resp = "log1pPtoidt1"),
set_prior(prior.temp, class = "b", coef = "log1pBRBR_t:temp", resp = "log1pBRBRt1"),
set_prior(prior.temp, class = "b", coef = "log1pBRBR_t:temp", resp = "log1pPtoidt1"),
set_prior(prior.temp, class = "b", coef = "log1pPtoid_t:temp", resp = "log1pBRBRt1"),
# biomass effects
set_prior(prior.AphidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pLYERt1"),
set_prior(prior.PtoidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pPtoidt1"),
# random effects
set_prior(prior.random.effects, class = "sd", resp = "log1pBRBRt1"),
set_prior(prior.random.effects, class = "sd", resp = "log1pLYERt1"),
set_prior(prior.random.effects, class = "sd", resp = "log1pPtoidt1")),
file = "output/reduced.4.brm.rds")
# print model summary
summary(reduced.4.brm) Family: MV(gaussian, gaussian, gaussian)
Links: mu = identity; sigma = identity
mu = identity; sigma = identity
mu = identity; sigma = identity
Formula: log1p(BRBR_t1) ~ intercept + log1p(BRBR_t) + log1p(Ptoid_t) + temp + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + log1p(BRBR_t):temp + log1p(Ptoid_t):temp + offset(log1p(BRBR_t)) - 1
log1p(LYER_t1) ~ intercept + log1p(BRBR_t) + log1p(LYER_t) + log1p(Ptoid_t) + rich + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + log1p(BRBR_t):rich + offset(log1p(LYER_t)) - 1
log1p(Ptoid_t1) ~ intercept + log1p(BRBR_t) + log1p(LYER_t) + temp + rich + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + log1p(BRBR_t):temp + log1p(LYER_t):rich + offset(log1p(Ptoid_t)) - 1
Data: full_df (Number of observations: 264)
Samples: 4 chains, each with iter = 4000; warmup = 2000; thin = 1;
total post-warmup samples = 8000
Correlation Structures:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
ar_log1pBRBRt1[1] -0.45 0.09 -0.62 -0.26 1.00 4747 6121
ar_log1pLYERt1[1] -0.12 0.10 -0.30 0.07 1.00 6624 6554
ar_log1pPtoidt1[1] -0.50 0.08 -0.66 -0.34 1.00 13790 6929
Group-Level Effects:
~Cage (Number of levels: 60)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS
sd(log1pBRBRt1_Intercept) 0.25 0.10 0.03 0.43 1.00 1349
sd(log1pLYERt1_Intercept) 0.13 0.09 0.01 0.31 1.00 1988
sd(log1pPtoidt1_Intercept) 0.04 0.03 0.00 0.11 1.00 5637
Tail_ESS
sd(log1pBRBRt1_Intercept) 2018
sd(log1pLYERt1_Intercept) 3924
sd(log1pPtoidt1_Intercept) 4762
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat
log1pBRBRt1_intercept 1.48 0.47 0.55 2.38 1.00
log1pBRBRt1_log1pBRBR_t 0.03 0.08 -0.12 0.18 1.00
log1pBRBRt1_log1pPtoid_t -0.90 0.05 -1.01 -0.79 1.00
log1pBRBRt1_temp -0.32 0.22 -0.74 0.11 1.00
log1pBRBRt1_log1pBRBR_t:temp -0.07 0.04 -0.14 0.00 1.00
log1pBRBRt1_log1pPtoid_t:temp 0.13 0.04 0.06 0.20 1.00
log1pLYERt1_intercept 2.76 0.55 1.70 3.84 1.00
log1pLYERt1_log1pBRBR_t 0.29 0.07 0.16 0.43 1.00
log1pLYERt1_log1pLYER_t -0.58 0.08 -0.75 -0.42 1.00
log1pLYERt1_log1pPtoid_t -0.71 0.05 -0.82 -0.62 1.00
log1pLYERt1_rich 0.35 0.20 -0.03 0.73 1.00
log1pLYERt1_logBiomass_g_t1 0.30 0.15 0.00 0.59 1.00
log1pLYERt1_log1pBRBR_t:rich -0.07 0.04 -0.15 0.01 1.00
log1pPtoidt1_intercept -3.04 0.38 -3.78 -2.31 1.00
log1pPtoidt1_log1pBRBR_t 0.37 0.06 0.25 0.49 1.00
log1pPtoidt1_log1pLYER_t 0.35 0.07 0.21 0.50 1.00
log1pPtoidt1_temp 0.69 0.12 0.46 0.92 1.00
log1pPtoidt1_rich -0.24 0.20 -0.64 0.16 1.00
log1pPtoidt1_logBiomass_g_t1 -0.47 0.13 -0.72 -0.22 1.00
log1pPtoidt1_log1pBRBR_t:temp -0.17 0.02 -0.22 -0.12 1.00
log1pPtoidt1_log1pLYER_t:rich 0.05 0.04 -0.03 0.12 1.00
Bulk_ESS Tail_ESS
log1pBRBRt1_intercept 4795 5644
log1pBRBRt1_log1pBRBR_t 5547 6061
log1pBRBRt1_log1pPtoid_t 6059 5661
log1pBRBRt1_temp 5384 5468
log1pBRBRt1_log1pBRBR_t:temp 5527 6037
log1pBRBRt1_log1pPtoid_t:temp 7658 5942
log1pLYERt1_intercept 6093 5968
log1pLYERt1_log1pBRBR_t 7264 5423
log1pLYERt1_log1pLYER_t 6798 6071
log1pLYERt1_log1pPtoid_t 8210 7051
log1pLYERt1_rich 6865 5295
log1pLYERt1_logBiomass_g_t1 12129 5633
log1pLYERt1_log1pBRBR_t:rich 6788 5810
log1pPtoidt1_intercept 7589 6544
log1pPtoidt1_log1pBRBR_t 7144 6251
log1pPtoidt1_log1pLYER_t 7816 6150
log1pPtoidt1_temp 6741 5841
log1pPtoidt1_rich 7441 6258
log1pPtoidt1_logBiomass_g_t1 13801 6286
log1pPtoidt1_log1pBRBR_t:temp 7264 5826
log1pPtoidt1_log1pLYER_t:rich 7523 6249
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma_log1pBRBRt1 1.11 0.06 1.00 1.23 1.00 3881 6502
sigma_log1pLYERt1 0.92 0.04 0.84 1.01 1.00 8520 6614
sigma_log1pPtoidt1 0.77 0.03 0.70 0.84 1.00 15660 5320
Residual Correlations:
Estimate Est.Error l-95% CI u-95% CI Rhat
rescor(log1pBRBRt1,log1pLYERt1) 0.51 0.05 0.41 0.61 1.00
rescor(log1pBRBRt1,log1pPtoidt1) -0.02 0.07 -0.16 0.12 1.00
rescor(log1pLYERt1,log1pPtoidt1) 0.08 0.07 -0.05 0.20 1.00
Bulk_ESS Tail_ESS
rescor(log1pBRBRt1,log1pLYERt1) 9039 6468
rescor(log1pBRBRt1,log1pPtoidt1) 11723 6652
rescor(log1pLYERt1,log1pPtoidt1) 13786 5964
Samples were drawn using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).Inspect credible intervals
# check highest-order rich effects
bayesplot::mcmc_intervals(reduced.4.brm, regex_pars = "_t:rich$", prob = 0.80, prob_outer = 0.95) # higher-order LYER:rich effect on Ptoid no longer meets 80% cutoff.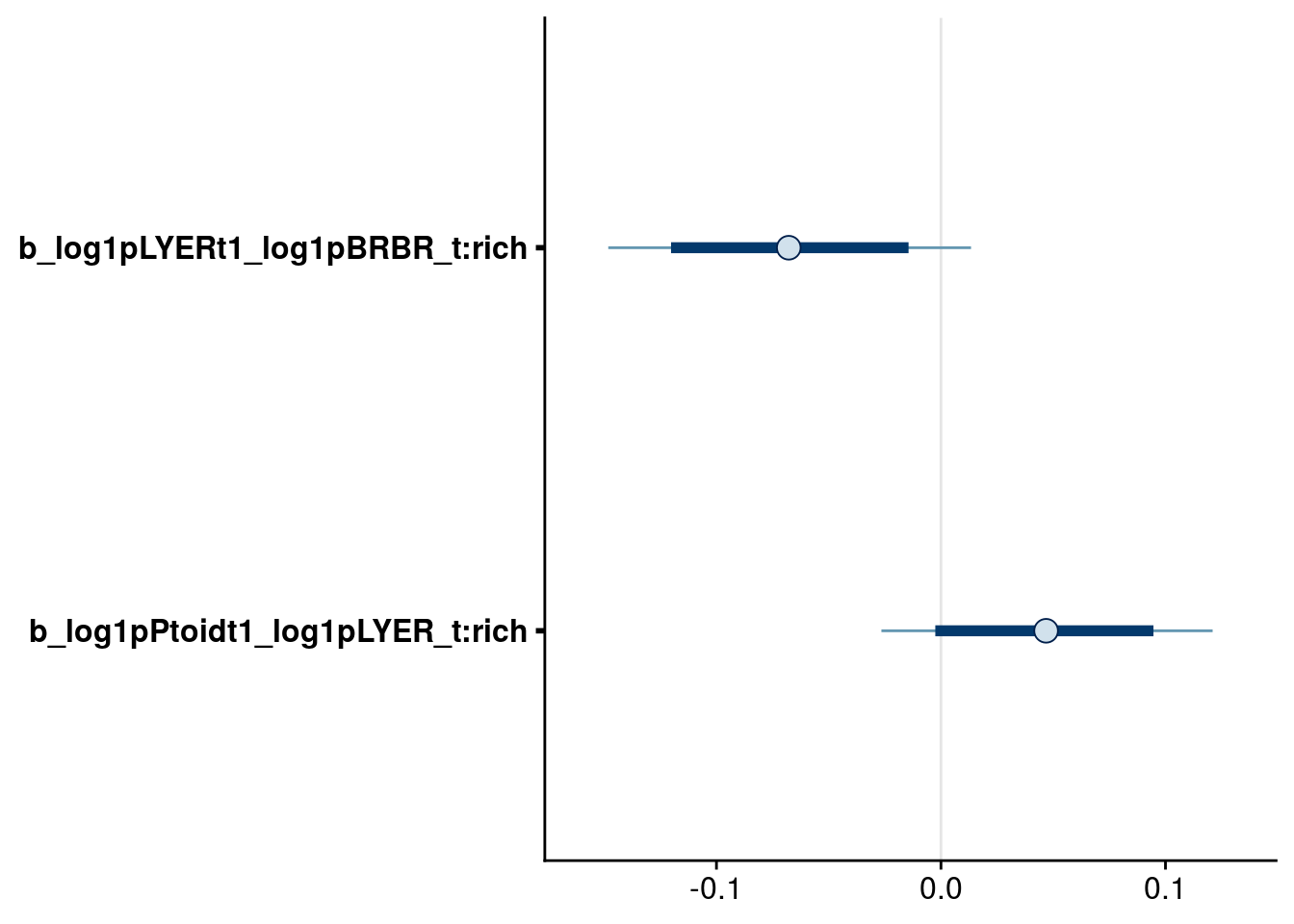
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
Reduced model 5
Drop terms
For both reduced model 3 and 4, once I drop the higher-order interaction with rich, the other higher-order term doesn’t meet the 80% interval cutoff, so I will try dropping both from the model.
After dropping this term, it also appears that rich effect on Ptoid intrinsic growth rate is no longer needed (80% interval criterion, model not shown), so I will drop that now too.
This model effectively removes all of the rich effects on Ptoid_t1.
Refit model
# update formulas
# note that I'm updating model 2
reduced.5.BRBR.bf <- reduced.2.BRBR.bf
reduced.5.LYER.bf <- reduced.2.LYER.bf
reduced.5.Ptoid.bf <- update(reduced.2.Ptoid.bf, .~. -log1p(BRBR_t):rich -log1p(LYER_t):rich -rich)
# fit new model
reduced.5.brm <- brm(
data = full_df,
formula = mvbf(reduced.5.BRBR.bf, reduced.5.LYER.bf, reduced.5.Ptoid.bf),
iter = 4000,
prior = c(# growth rates
set_prior(prior.r.BRBR, class = "b", coef = "intercept", resp = "log1pBRBRt1"),
set_prior(prior.r.LYER, class = "b", coef = "intercept", resp = "log1pLYERt1"),
set_prior(prior.r.Ptoid, class = "b", coef = "intercept", resp = "log1pPtoidt1"),
# intraspecific effects
set_prior(prior.intra.BRBR, class = "b", coef = "log1pBRBR_t", resp = "log1pBRBRt1"),
set_prior(prior.intra.LYER, class = "b", coef = "log1pLYER_t", resp = "log1pLYERt1"),
# negative interspecific effects
set_prior(prior.BRBRonLYER, class = "b", coef = "log1pBRBR_t", resp = "log1pLYERt1"),
set_prior(prior.PtoidonBRBR, class = "b", coef = "log1pPtoid_t", resp = "log1pBRBRt1"),
set_prior(prior.PtoidonLYER, class = "b", coef = "log1pPtoid_t", resp = "log1pLYERt1"),
# positive interspecific effects
set_prior(prior.BRBRonPtoid, class = "b", coef = "log1pBRBR_t", resp = "log1pPtoidt1"),
set_prior(prior.LYERonPtoid, class = "b", coef = "log1pLYER_t", resp = "log1pPtoidt1"),
# rich effects
set_prior(prior.rich, class = "b", coef = "rich", resp = "log1pLYERt1"),
set_prior(prior.rich, class = "b", coef = "log1pBRBR_t:rich", resp = "log1pLYERt1"),
# temp effects
set_prior(prior.temp, class = "b", coef = "temp", resp = "log1pBRBRt1"),
set_prior(prior.temp, class = "b", coef = "temp", resp = "log1pPtoidt1"),
set_prior(prior.temp, class = "b", coef = "log1pBRBR_t:temp", resp = "log1pBRBRt1"),
set_prior(prior.temp, class = "b", coef = "log1pBRBR_t:temp", resp = "log1pPtoidt1"),
set_prior(prior.temp, class = "b", coef = "log1pPtoid_t:temp", resp = "log1pBRBRt1"),
# biomass effects
set_prior(prior.AphidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pLYERt1"),
set_prior(prior.PtoidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pPtoidt1"),
# random effects
set_prior(prior.random.effects, class = "sd", resp = "log1pBRBRt1"),
set_prior(prior.random.effects, class = "sd", resp = "log1pLYERt1"),
set_prior(prior.random.effects, class = "sd", resp = "log1pPtoidt1")),
file = "output/reduced.5.brm.rds")
# print model summary
summary(reduced.5.brm) Family: MV(gaussian, gaussian, gaussian)
Links: mu = identity; sigma = identity
mu = identity; sigma = identity
mu = identity; sigma = identity
Formula: log1p(BRBR_t1) ~ intercept + log1p(BRBR_t) + log1p(Ptoid_t) + temp + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + log1p(BRBR_t):temp + log1p(Ptoid_t):temp + offset(log1p(BRBR_t)) - 1
log1p(LYER_t1) ~ intercept + log1p(BRBR_t) + log1p(LYER_t) + log1p(Ptoid_t) + rich + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + log1p(BRBR_t):rich + offset(log1p(LYER_t)) - 1
log1p(Ptoid_t1) ~ intercept + log1p(BRBR_t) + log1p(LYER_t) + temp + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + log1p(BRBR_t):temp + offset(log1p(Ptoid_t)) - 1
Data: full_df (Number of observations: 264)
Samples: 4 chains, each with iter = 4000; warmup = 2000; thin = 1;
total post-warmup samples = 8000
Correlation Structures:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
ar_log1pBRBRt1[1] -0.45 0.09 -0.62 -0.26 1.00 4900 5318
ar_log1pLYERt1[1] -0.12 0.10 -0.31 0.07 1.00 7305 7164
ar_log1pPtoidt1[1] -0.50 0.08 -0.66 -0.35 1.00 11431 5980
Group-Level Effects:
~Cage (Number of levels: 60)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS
sd(log1pBRBRt1_Intercept) 0.24 0.10 0.03 0.42 1.00 1291
sd(log1pLYERt1_Intercept) 0.14 0.09 0.01 0.31 1.00 2075
sd(log1pPtoidt1_Intercept) 0.04 0.03 0.00 0.11 1.00 4911
Tail_ESS
sd(log1pBRBRt1_Intercept) 2487
sd(log1pLYERt1_Intercept) 4278
sd(log1pPtoidt1_Intercept) 3714
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat
log1pBRBRt1_intercept 1.47 0.46 0.57 2.34 1.00
log1pBRBRt1_log1pBRBR_t 0.03 0.08 -0.11 0.18 1.00
log1pBRBRt1_log1pPtoid_t -0.90 0.05 -1.00 -0.79 1.00
log1pBRBRt1_temp -0.32 0.22 -0.74 0.10 1.00
log1pBRBRt1_log1pBRBR_t:temp -0.07 0.04 -0.14 -0.00 1.00
log1pBRBRt1_log1pPtoid_t:temp 0.13 0.04 0.06 0.20 1.00
log1pLYERt1_intercept 2.75 0.54 1.72 3.83 1.00
log1pLYERt1_log1pBRBR_t 0.29 0.07 0.16 0.43 1.00
log1pLYERt1_log1pLYER_t -0.58 0.08 -0.75 -0.42 1.00
log1pLYERt1_log1pPtoid_t -0.71 0.05 -0.82 -0.62 1.00
log1pLYERt1_rich 0.36 0.20 -0.02 0.74 1.00
log1pLYERt1_logBiomass_g_t1 0.30 0.15 0.01 0.59 1.00
log1pLYERt1_log1pBRBR_t:rich -0.07 0.04 -0.15 0.01 1.00
log1pPtoidt1_intercept -3.25 0.34 -3.91 -2.59 1.00
log1pPtoidt1_log1pBRBR_t 0.36 0.06 0.23 0.48 1.00
log1pPtoidt1_log1pLYER_t 0.41 0.06 0.29 0.52 1.00
log1pPtoidt1_temp 0.67 0.12 0.44 0.90 1.00
log1pPtoidt1_logBiomass_g_t1 -0.48 0.12 -0.72 -0.24 1.00
log1pPtoidt1_log1pBRBR_t:temp -0.17 0.02 -0.22 -0.12 1.00
Bulk_ESS Tail_ESS
log1pBRBRt1_intercept 5167 5003
log1pBRBRt1_log1pBRBR_t 5937 5666
log1pBRBRt1_log1pPtoid_t 5961 5947
log1pBRBRt1_temp 6444 5891
log1pBRBRt1_log1pBRBR_t:temp 6640 6597
log1pBRBRt1_log1pPtoid_t:temp 8100 6339
log1pLYERt1_intercept 6329 5893
log1pLYERt1_log1pBRBR_t 8726 6808
log1pLYERt1_log1pLYER_t 6805 6194
log1pLYERt1_log1pPtoid_t 8296 6771
log1pLYERt1_rich 8477 6143
log1pLYERt1_logBiomass_g_t1 14740 6609
log1pLYERt1_log1pBRBR_t:rich 8112 5788
log1pPtoidt1_intercept 9095 6886
log1pPtoidt1_log1pBRBR_t 8140 6727
log1pPtoidt1_log1pLYER_t 15809 6295
log1pPtoidt1_temp 7216 5719
log1pPtoidt1_logBiomass_g_t1 13607 6868
log1pPtoidt1_log1pBRBR_t:temp 7651 5889
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma_log1pBRBRt1 1.11 0.06 1.00 1.23 1.00 3728 5261
sigma_log1pLYERt1 0.92 0.04 0.84 1.01 1.00 9609 5933
sigma_log1pPtoidt1 0.77 0.03 0.70 0.84 1.00 17124 4977
Residual Correlations:
Estimate Est.Error l-95% CI u-95% CI Rhat
rescor(log1pBRBRt1,log1pLYERt1) 0.51 0.05 0.41 0.61 1.00
rescor(log1pBRBRt1,log1pPtoidt1) -0.02 0.07 -0.16 0.11 1.00
rescor(log1pLYERt1,log1pPtoidt1) 0.08 0.06 -0.05 0.20 1.00
Bulk_ESS Tail_ESS
rescor(log1pBRBRt1,log1pLYERt1) 8854 6597
rescor(log1pBRBRt1,log1pPtoidt1) 12544 6219
rescor(log1pLYERt1,log1pPtoidt1) 15707 6683
Samples were drawn using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).Reduced model 6
Drop terms
Try dropping log1p(BRBR_t):rich effect on LYER_t1 instead of rich effects on Ptoid_t1. I also drop main effect of rich on LYER_t1, because its 80% interval overlapped with zero after removing log1p(BRBR_t):rich (not shown).
Refit model
# update formulas
# note that I'm updating model 2
reduced.6.BRBR.bf <- reduced.2.BRBR.bf
reduced.6.LYER.bf <- update(reduced.2.LYER.bf, .~. -log1p(BRBR_t):rich -rich)
reduced.6.Ptoid.bf <- reduced.2.Ptoid.bf
# fit new model
reduced.6.brm <- brm(
data = full_df,
formula = mvbf(reduced.6.BRBR.bf, reduced.6.LYER.bf, reduced.6.Ptoid.bf),
iter = 4000,
prior = c(# growth rates
set_prior(prior.r.BRBR, class = "b", coef = "intercept", resp = "log1pBRBRt1"),
set_prior(prior.r.LYER, class = "b", coef = "intercept", resp = "log1pLYERt1"),
set_prior(prior.r.Ptoid, class = "b", coef = "intercept", resp = "log1pPtoidt1"),
# intraspecific effects
set_prior(prior.intra.BRBR, class = "b", coef = "log1pBRBR_t", resp = "log1pBRBRt1"),
set_prior(prior.intra.LYER, class = "b", coef = "log1pLYER_t", resp = "log1pLYERt1"),
# negative interspecific effects
set_prior(prior.BRBRonLYER, class = "b", coef = "log1pBRBR_t", resp = "log1pLYERt1"),
set_prior(prior.PtoidonBRBR, class = "b", coef = "log1pPtoid_t", resp = "log1pBRBRt1"),
set_prior(prior.PtoidonLYER, class = "b", coef = "log1pPtoid_t", resp = "log1pLYERt1"),
# positive interspecific effects
set_prior(prior.BRBRonPtoid, class = "b", coef = "log1pBRBR_t", resp = "log1pPtoidt1"),
set_prior(prior.LYERonPtoid, class = "b", coef = "log1pLYER_t", resp = "log1pPtoidt1"),
# rich effects
set_prior(prior.rich, class = "b", coef = "rich", resp = "log1pPtoidt1"),
set_prior(prior.rich, class = "b", coef = "log1pBRBR_t:rich", resp = "log1pPtoidt1"),
set_prior(prior.rich, class = "b", coef = "log1pLYER_t:rich", resp = "log1pPtoidt1"),
# temp effects
set_prior(prior.temp, class = "b", coef = "temp", resp = "log1pBRBRt1"),
set_prior(prior.temp, class = "b", coef = "temp", resp = "log1pPtoidt1"),
set_prior(prior.temp, class = "b", coef = "log1pBRBR_t:temp", resp = "log1pBRBRt1"),
set_prior(prior.temp, class = "b", coef = "log1pBRBR_t:temp", resp = "log1pPtoidt1"),
set_prior(prior.temp, class = "b", coef = "log1pPtoid_t:temp", resp = "log1pBRBRt1"),
# biomass effects
set_prior(prior.AphidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pLYERt1"),
set_prior(prior.PtoidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pPtoidt1"),
# random effects
set_prior(prior.random.effects, class = "sd", resp = "log1pBRBRt1"),
set_prior(prior.random.effects, class = "sd", resp = "log1pLYERt1"),
set_prior(prior.random.effects, class = "sd", resp = "log1pPtoidt1")),
file = "output/reduced.6.brm.rds")
# print model summary
summary(reduced.6.brm) Family: MV(gaussian, gaussian, gaussian)
Links: mu = identity; sigma = identity
mu = identity; sigma = identity
mu = identity; sigma = identity
Formula: log1p(BRBR_t1) ~ intercept + log1p(BRBR_t) + log1p(Ptoid_t) + temp + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + log1p(BRBR_t):temp + log1p(Ptoid_t):temp + offset(log1p(BRBR_t)) - 1
log1p(LYER_t1) ~ intercept + log1p(BRBR_t) + log1p(LYER_t) + log1p(Ptoid_t) + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + offset(log1p(LYER_t)) - 1
log1p(Ptoid_t1) ~ intercept + log1p(BRBR_t) + log1p(LYER_t) + temp + rich + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + log1p(BRBR_t):temp + log1p(BRBR_t):rich + log1p(LYER_t):rich + offset(log1p(Ptoid_t)) - 1
Data: full_df (Number of observations: 264)
Samples: 4 chains, each with iter = 4000; warmup = 2000; thin = 1;
total post-warmup samples = 8000
Correlation Structures:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
ar_log1pBRBRt1[1] -0.45 0.09 -0.62 -0.26 1.00 4136 6337
ar_log1pLYERt1[1] -0.11 0.10 -0.29 0.09 1.00 6446 6468
ar_log1pPtoidt1[1] -0.50 0.08 -0.66 -0.34 1.00 14378 6408
Group-Level Effects:
~Cage (Number of levels: 60)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS
sd(log1pBRBRt1_Intercept) 0.25 0.10 0.03 0.43 1.00 931
sd(log1pLYERt1_Intercept) 0.15 0.09 0.01 0.32 1.00 1684
sd(log1pPtoidt1_Intercept) 0.04 0.03 0.00 0.12 1.00 5768
Tail_ESS
sd(log1pBRBRt1_Intercept) 2133
sd(log1pLYERt1_Intercept) 3186
sd(log1pPtoidt1_Intercept) 4315
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat
log1pBRBRt1_intercept 1.49 0.46 0.61 2.39 1.00
log1pBRBRt1_log1pBRBR_t 0.03 0.08 -0.12 0.18 1.00
log1pBRBRt1_log1pPtoid_t -0.90 0.05 -1.01 -0.79 1.00
log1pBRBRt1_temp -0.35 0.21 -0.77 0.07 1.00
log1pBRBRt1_log1pBRBR_t:temp -0.07 0.04 -0.14 0.00 1.00
log1pBRBRt1_log1pPtoid_t:temp 0.13 0.03 0.07 0.20 1.00
log1pLYERt1_intercept 3.10 0.50 2.13 4.06 1.00
log1pLYERt1_log1pBRBR_t 0.22 0.06 0.11 0.33 1.00
log1pLYERt1_log1pLYER_t -0.58 0.08 -0.75 -0.41 1.00
log1pLYERt1_log1pPtoid_t -0.71 0.05 -0.82 -0.62 1.00
log1pLYERt1_logBiomass_g_t1 0.32 0.15 0.03 0.61 1.00
log1pPtoidt1_intercept -3.13 0.38 -3.88 -2.39 1.00
log1pPtoidt1_log1pBRBR_t 0.43 0.07 0.29 0.58 1.00
log1pPtoidt1_log1pLYER_t 0.32 0.08 0.17 0.47 1.00
log1pPtoidt1_temp 0.67 0.12 0.44 0.89 1.00
log1pPtoidt1_rich -0.11 0.22 -0.55 0.32 1.00
log1pPtoidt1_logBiomass_g_t1 -0.47 0.13 -0.72 -0.23 1.00
log1pPtoidt1_log1pBRBR_t:temp -0.16 0.02 -0.21 -0.12 1.00
log1pPtoidt1_log1pBRBR_t:rich -0.05 0.04 -0.12 0.02 1.00
log1pPtoidt1_log1pLYER_t:rich 0.07 0.04 -0.01 0.15 1.00
Bulk_ESS Tail_ESS
log1pBRBRt1_intercept 4738 6258
log1pBRBRt1_log1pBRBR_t 5596 6180
log1pBRBRt1_log1pPtoid_t 5388 5932
log1pBRBRt1_temp 6293 5811
log1pBRBRt1_log1pBRBR_t:temp 6893 6500
log1pBRBRt1_log1pPtoid_t:temp 8096 6321
log1pLYERt1_intercept 6652 6524
log1pLYERt1_log1pBRBR_t 9583 5333
log1pLYERt1_log1pLYER_t 6256 6106
log1pLYERt1_log1pPtoid_t 8616 7207
log1pLYERt1_logBiomass_g_t1 14140 6550
log1pPtoidt1_intercept 9280 6472
log1pPtoidt1_log1pBRBR_t 8580 6126
log1pPtoidt1_log1pLYER_t 9729 6874
log1pPtoidt1_temp 9010 6390
log1pPtoidt1_rich 11090 6410
log1pPtoidt1_logBiomass_g_t1 14999 5745
log1pPtoidt1_log1pBRBR_t:temp 9452 6571
log1pPtoidt1_log1pBRBR_t:rich 11171 5992
log1pPtoidt1_log1pLYER_t:rich 9422 6786
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma_log1pBRBRt1 1.11 0.06 1.00 1.23 1.00 3871 5616
sigma_log1pLYERt1 0.93 0.04 0.85 1.02 1.00 7979 6487
sigma_log1pPtoidt1 0.76 0.03 0.70 0.84 1.00 17519 5509
Residual Correlations:
Estimate Est.Error l-95% CI u-95% CI Rhat
rescor(log1pBRBRt1,log1pLYERt1) 0.52 0.05 0.42 0.61 1.00
rescor(log1pBRBRt1,log1pPtoidt1) -0.03 0.07 -0.17 0.11 1.00
rescor(log1pLYERt1,log1pPtoidt1) 0.07 0.06 -0.06 0.20 1.00
Bulk_ESS Tail_ESS
rescor(log1pBRBRt1,log1pLYERt1) 9838 7084
rescor(log1pBRBRt1,log1pPtoidt1) 13861 6242
rescor(log1pLYERt1,log1pPtoidt1) 16591 6570
Samples were drawn using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).Reduced model 7
Drop terms
Drop all rich effects, but keep temp effects (for all of which their 95% posterior estimates are different from zero.)
Refit model
# update formulas
reduced.7.BRBR.bf <- reduced.2.BRBR.bf
reduced.7.LYER.bf <- reduced.6.LYER.bf
reduced.7.Ptoid.bf <- reduced.5.Ptoid.bf
# fit new model
reduced.7.brm <- brm(
data = full_df,
formula = mvbf(reduced.7.BRBR.bf, reduced.7.LYER.bf, reduced.7.Ptoid.bf),
iter = 4000,
prior = c(# growth rates
set_prior(prior.r.BRBR, class = "b", coef = "intercept", resp = "log1pBRBRt1"),
set_prior(prior.r.LYER, class = "b", coef = "intercept", resp = "log1pLYERt1"),
set_prior(prior.r.Ptoid, class = "b", coef = "intercept", resp = "log1pPtoidt1"),
# intraspecific effects
set_prior(prior.intra.BRBR, class = "b", coef = "log1pBRBR_t", resp = "log1pBRBRt1"),
set_prior(prior.intra.LYER, class = "b", coef = "log1pLYER_t", resp = "log1pLYERt1"),
# negative interspecific effects
set_prior(prior.BRBRonLYER, class = "b", coef = "log1pBRBR_t", resp = "log1pLYERt1"),
set_prior(prior.PtoidonBRBR, class = "b", coef = "log1pPtoid_t", resp = "log1pBRBRt1"),
set_prior(prior.PtoidonLYER, class = "b", coef = "log1pPtoid_t", resp = "log1pLYERt1"),
# positive interspecific effects
set_prior(prior.BRBRonPtoid, class = "b", coef = "log1pBRBR_t", resp = "log1pPtoidt1"),
set_prior(prior.LYERonPtoid, class = "b", coef = "log1pLYER_t", resp = "log1pPtoidt1"),
# temp effects
set_prior(prior.temp, class = "b", coef = "temp", resp = "log1pBRBRt1"),
set_prior(prior.temp, class = "b", coef = "temp", resp = "log1pPtoidt1"),
set_prior(prior.temp, class = "b", coef = "log1pBRBR_t:temp", resp = "log1pBRBRt1"),
set_prior(prior.temp, class = "b", coef = "log1pBRBR_t:temp", resp = "log1pPtoidt1"),
set_prior(prior.temp, class = "b", coef = "log1pPtoid_t:temp", resp = "log1pBRBRt1"),
# biomass effects
set_prior(prior.AphidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pLYERt1"),
set_prior(prior.PtoidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pPtoidt1"),
# random effects
set_prior(prior.random.effects, class = "sd", resp = "log1pBRBRt1"),
set_prior(prior.random.effects, class = "sd", resp = "log1pLYERt1"),
set_prior(prior.random.effects, class = "sd", resp = "log1pPtoidt1")),
file = "output/reduced.7.brm.rds")
# print model summary
summary(reduced.7.brm) Family: MV(gaussian, gaussian, gaussian)
Links: mu = identity; sigma = identity
mu = identity; sigma = identity
mu = identity; sigma = identity
Formula: log1p(BRBR_t1) ~ intercept + log1p(BRBR_t) + log1p(Ptoid_t) + temp + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + log1p(BRBR_t):temp + log1p(Ptoid_t):temp + offset(log1p(BRBR_t)) - 1
log1p(LYER_t1) ~ intercept + log1p(BRBR_t) + log1p(LYER_t) + log1p(Ptoid_t) + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + offset(log1p(LYER_t)) - 1
log1p(Ptoid_t1) ~ intercept + log1p(BRBR_t) + log1p(LYER_t) + temp + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + log1p(BRBR_t):temp + offset(log1p(Ptoid_t)) - 1
Data: full_df (Number of observations: 264)
Samples: 4 chains, each with iter = 4000; warmup = 2000; thin = 1;
total post-warmup samples = 8000
Correlation Structures:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
ar_log1pBRBRt1[1] -0.45 0.09 -0.63 -0.26 1.00 3076 4946
ar_log1pLYERt1[1] -0.11 0.10 -0.30 0.08 1.00 4221 5662
ar_log1pPtoidt1[1] -0.50 0.08 -0.66 -0.34 1.00 7546 6064
Group-Level Effects:
~Cage (Number of levels: 60)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS
sd(log1pBRBRt1_Intercept) 0.25 0.10 0.04 0.43 1.00 1078
sd(log1pLYERt1_Intercept) 0.14 0.09 0.01 0.32 1.00 1450
sd(log1pPtoidt1_Intercept) 0.04 0.03 0.00 0.11 1.00 3907
Tail_ESS
sd(log1pBRBRt1_Intercept) 1636
sd(log1pLYERt1_Intercept) 3092
sd(log1pPtoidt1_Intercept) 3511
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat
log1pBRBRt1_intercept 1.51 0.46 0.61 2.41 1.00
log1pBRBRt1_log1pBRBR_t 0.03 0.08 -0.12 0.17 1.00
log1pBRBRt1_log1pPtoid_t -0.90 0.05 -1.00 -0.79 1.00
log1pBRBRt1_temp -0.36 0.22 -0.79 0.06 1.00
log1pBRBRt1_log1pBRBR_t:temp -0.06 0.04 -0.14 0.01 1.00
log1pBRBRt1_log1pPtoid_t:temp 0.13 0.04 0.07 0.20 1.00
log1pLYERt1_intercept 3.09 0.51 2.12 4.12 1.00
log1pLYERt1_log1pBRBR_t 0.22 0.05 0.11 0.33 1.00
log1pLYERt1_log1pLYER_t -0.57 0.09 -0.75 -0.41 1.00
log1pLYERt1_log1pPtoid_t -0.71 0.05 -0.82 -0.62 1.00
log1pLYERt1_logBiomass_g_t1 0.32 0.15 0.04 0.61 1.00
log1pPtoidt1_intercept -3.25 0.33 -3.90 -2.60 1.00
log1pPtoidt1_log1pBRBR_t 0.36 0.06 0.24 0.48 1.00
log1pPtoidt1_log1pLYER_t 0.41 0.06 0.29 0.52 1.00
log1pPtoidt1_temp 0.67 0.12 0.44 0.90 1.00
log1pPtoidt1_logBiomass_g_t1 -0.48 0.12 -0.73 -0.23 1.00
log1pPtoidt1_log1pBRBR_t:temp -0.17 0.02 -0.22 -0.12 1.00
Bulk_ESS Tail_ESS
log1pBRBRt1_intercept 3049 4011
log1pBRBRt1_log1pBRBR_t 3539 4490
log1pBRBRt1_log1pPtoid_t 3432 5041
log1pBRBRt1_temp 3449 4772
log1pBRBRt1_log1pBRBR_t:temp 3807 5110
log1pBRBRt1_log1pPtoid_t:temp 4705 5693
log1pLYERt1_intercept 3482 3866
log1pLYERt1_log1pBRBR_t 5525 5412
log1pLYERt1_log1pLYER_t 3282 4234
log1pLYERt1_log1pPtoid_t 4290 5073
log1pLYERt1_logBiomass_g_t1 6956 6018
log1pPtoidt1_intercept 4888 5499
log1pPtoidt1_log1pBRBR_t 4013 5374
log1pPtoidt1_log1pLYER_t 6339 5703
log1pPtoidt1_temp 3673 4590
log1pPtoidt1_logBiomass_g_t1 7887 5677
log1pPtoidt1_log1pBRBR_t:temp 4025 4950
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma_log1pBRBRt1 1.11 0.06 1.00 1.23 1.00 2927 5205
sigma_log1pLYERt1 0.93 0.04 0.85 1.02 1.00 5090 6179
sigma_log1pPtoidt1 0.77 0.03 0.70 0.84 1.00 8414 6032
Residual Correlations:
Estimate Est.Error l-95% CI u-95% CI Rhat
rescor(log1pBRBRt1,log1pLYERt1) 0.52 0.05 0.42 0.61 1.00
rescor(log1pBRBRt1,log1pPtoidt1) -0.02 0.07 -0.16 0.12 1.00
rescor(log1pLYERt1,log1pPtoidt1) 0.09 0.07 -0.04 0.22 1.00
Bulk_ESS Tail_ESS
rescor(log1pBRBRt1,log1pLYERt1) 5193 5887
rescor(log1pBRBRt1,log1pPtoidt1) 6982 6329
rescor(log1pLYERt1,log1pPtoidt1) 8469 6142
Samples were drawn using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).Reduced model 8
Drop terms
Drop all temp effects now even though most of their 95% posterior estimates are different from zero in the previous models. This will serve as a baseline model without any treatment effects. I expect this to be the worst model.
I also dropped log1p(BRBR_t) effect on BRBR_t1 because it no longer had a widely overlapping confidence interval with zero (not shown).
Refit model
# update formulas
# note that I'm updating model 2 for BRBR and model 5 for the Ptoid
reduced.8.BRBR.bf <- update(reduced.2.BRBR.bf, .~. -log1p(BRBR_t):temp -log1p(Ptoid_t):temp -temp -log1p(BRBR_t))
reduced.8.LYER.bf <- reduced.6.LYER.bf # no temp effects anyway
reduced.8.Ptoid.bf <- update(reduced.5.Ptoid.bf, .~. -log1p(BRBR_t):temp -temp)
# fit new model
reduced.8.brm <- brm(
data = full_df,
formula = mvbf(reduced.8.BRBR.bf, reduced.8.LYER.bf, reduced.8.Ptoid.bf),
iter = 4000,
prior = c(# growth rates
set_prior(prior.r.BRBR, class = "b", coef = "intercept", resp = "log1pBRBRt1"),
set_prior(prior.r.LYER, class = "b", coef = "intercept", resp = "log1pLYERt1"),
set_prior(prior.r.Ptoid, class = "b", coef = "intercept", resp = "log1pPtoidt1"),
# intraspecific effects
set_prior(prior.intra.LYER, class = "b", coef = "log1pLYER_t", resp = "log1pLYERt1"),
# negative interspecific effects
set_prior(prior.BRBRonLYER, class = "b", coef = "log1pBRBR_t", resp = "log1pLYERt1"),
set_prior(prior.PtoidonBRBR, class = "b", coef = "log1pPtoid_t", resp = "log1pBRBRt1"),
set_prior(prior.PtoidonLYER, class = "b", coef = "log1pPtoid_t", resp = "log1pLYERt1"),
# positive interspecific effects
set_prior(prior.BRBRonPtoid, class = "b", coef = "log1pBRBR_t", resp = "log1pPtoidt1"),
set_prior(prior.LYERonPtoid, class = "b", coef = "log1pLYER_t", resp = "log1pPtoidt1"),
# biomass effects
set_prior(prior.AphidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pLYERt1"),
set_prior(prior.PtoidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pPtoidt1"),
# random effects
set_prior(prior.random.effects, class = "sd", resp = "log1pBRBRt1"),
set_prior(prior.random.effects, class = "sd", resp = "log1pLYERt1"),
set_prior(prior.random.effects, class = "sd", resp = "log1pPtoidt1")),
file = "output/reduced.8.brm.rds")
# print model summary
summary(reduced.8.brm) Family: MV(gaussian, gaussian, gaussian)
Links: mu = identity; sigma = identity
mu = identity; sigma = identity
mu = identity; sigma = identity
Formula: log1p(BRBR_t1) ~ intercept + log1p(Ptoid_t) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + offset(log1p(BRBR_t)) - 1
log1p(LYER_t1) ~ intercept + log1p(BRBR_t) + log1p(LYER_t) + log1p(Ptoid_t) + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + offset(log1p(LYER_t)) - 1
log1p(Ptoid_t1) ~ intercept + log1p(BRBR_t) + log1p(LYER_t) + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + offset(log1p(Ptoid_t)) - 1
Data: full_df (Number of observations: 264)
Samples: 4 chains, each with iter = 4000; warmup = 2000; thin = 1;
total post-warmup samples = 8000
Correlation Structures:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
ar_log1pBRBRt1[1] -0.33 0.10 -0.53 -0.12 1.00 1605 1606
ar_log1pLYERt1[1] -0.09 0.09 -0.27 0.10 1.00 4411 5493
ar_log1pPtoidt1[1] -0.52 0.09 -0.69 -0.35 1.00 7548 6085
Group-Level Effects:
~Cage (Number of levels: 60)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS
sd(log1pBRBRt1_Intercept) 0.51 0.13 0.22 0.74 1.00 809
sd(log1pLYERt1_Intercept) 0.11 0.08 0.00 0.29 1.00 1971
sd(log1pPtoidt1_Intercept) 0.04 0.03 0.00 0.11 1.00 4392
Tail_ESS
sd(log1pBRBRt1_Intercept) 646
sd(log1pLYERt1_Intercept) 3347
sd(log1pPtoidt1_Intercept) 3109
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS
log1pBRBRt1_intercept 0.78 0.16 0.45 1.09 1.00 2043
log1pBRBRt1_log1pPtoid_t -0.75 0.06 -0.86 -0.63 1.00 1424
log1pLYERt1_intercept 2.99 0.51 2.01 3.99 1.00 3657
log1pLYERt1_log1pBRBR_t 0.20 0.06 0.09 0.31 1.00 3292
log1pLYERt1_log1pLYER_t -0.53 0.09 -0.71 -0.37 1.00 2871
log1pLYERt1_log1pPtoid_t -0.71 0.05 -0.81 -0.60 1.00 4021
log1pLYERt1_logBiomass_g_t1 0.22 0.15 -0.08 0.52 1.00 7871
log1pPtoidt1_intercept -2.16 0.30 -2.76 -1.56 1.00 5070
log1pPtoidt1_log1pBRBR_t 0.19 0.04 0.12 0.27 1.00 9374
log1pPtoidt1_log1pLYER_t 0.34 0.05 0.23 0.45 1.00 5174
log1pPtoidt1_logBiomass_g_t1 -0.33 0.13 -0.58 -0.07 1.00 6871
Tail_ESS
log1pBRBRt1_intercept 2983
log1pBRBRt1_log1pPtoid_t 1694
log1pLYERt1_intercept 4916
log1pLYERt1_log1pBRBR_t 4337
log1pLYERt1_log1pLYER_t 4312
log1pLYERt1_log1pPtoid_t 5487
log1pLYERt1_logBiomass_g_t1 6358
log1pPtoidt1_intercept 5714
log1pPtoidt1_log1pBRBR_t 5813
log1pPtoidt1_log1pLYER_t 5525
log1pPtoidt1_logBiomass_g_t1 6086
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma_log1pBRBRt1 1.20 0.07 1.08 1.35 1.00 1524 1982
sigma_log1pLYERt1 0.94 0.04 0.85 1.02 1.00 6553 5854
sigma_log1pPtoidt1 0.86 0.04 0.79 0.93 1.00 12643 5691
Residual Correlations:
Estimate Est.Error l-95% CI u-95% CI Rhat
rescor(log1pBRBRt1,log1pLYERt1) 0.50 0.05 0.40 0.59 1.00
rescor(log1pBRBRt1,log1pPtoidt1) 0.11 0.07 -0.04 0.25 1.00
rescor(log1pLYERt1,log1pPtoidt1) 0.13 0.07 -0.00 0.26 1.00
Bulk_ESS Tail_ESS
rescor(log1pBRBRt1,log1pLYERt1) 5449 5784
rescor(log1pBRBRt1,log1pPtoidt1) 6930 6273
rescor(log1pLYERt1,log1pPtoidt1) 9259 5881
Samples were drawn using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).Inspect credible intervals
Only log(Biomass_g_t1) effect on LYER has a 95% posterior interval that includes zero (80% interval doesn’t though). See also summary of model above.
# check biomass effects
bayesplot::mcmc_intervals(reduced.8.brm, regex_pars = "logBiomass_g_t1$", prob = 0.80, prob_outer = 0.95)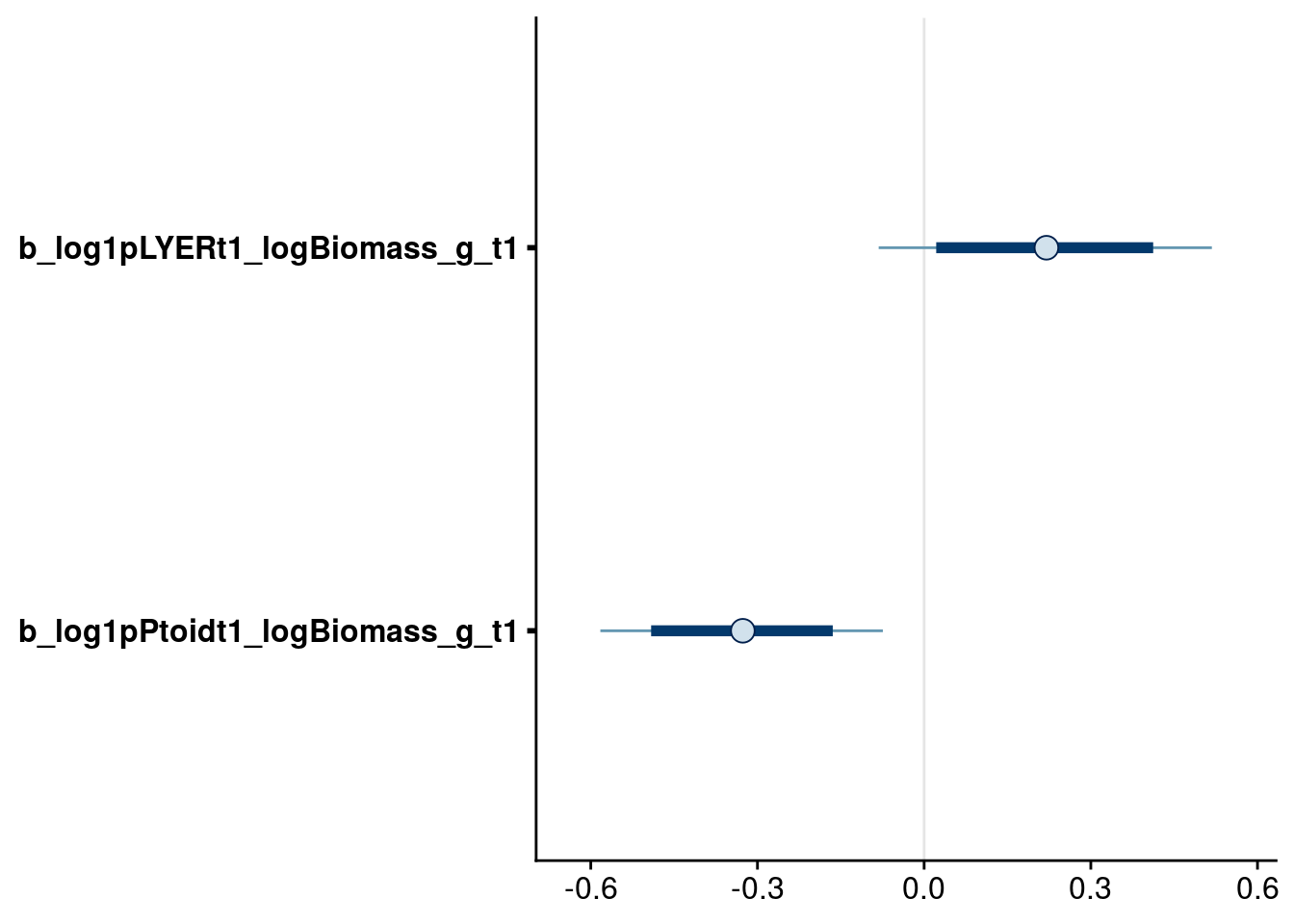
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
Model comparison
# add leave-one-out (loo) cross-validation criterion to each model
full.mv.norm.brm <- add_criterion(full.mv.norm.brm, criterion = "loo", force_save = T, reloo = T)
# reduced.1.brm # intermediate model on way to determine 80% cutoff, I so don't use it for comparison
reduced.2.brm <- add_criterion(reduced.2.brm, criterion = "loo", force_save = T, reloo = T)
reduced.3.brm <- add_criterion(reduced.3.brm, criterion = "loo", force_save = T, reloo = T)
reduced.4.brm <- add_criterion(reduced.4.brm, criterion = "loo", force_save = T, reloo = T)
reduced.5.brm <- add_criterion(reduced.5.brm, criterion = "loo", force_save = T, reloo = T)
reduced.6.brm <- add_criterion(reduced.6.brm, criterion = "loo", force_save = T, reloo = T)
reduced.7.brm <- add_criterion(reduced.7.brm, criterion = "loo", force_save = T) # reloo not needed
reduced.8.brm <- add_criterion(reduced.8.brm, criterion = "loo", force_save = T, reloo = T)
# compare models
loo_compare(full.mv.norm.brm, # full model
#reduced.1.brm, # intermediate model on way to determine 80% cutoff
reduced.2.brm, # all parameters have >80% of their posterior estimates different from zero
reduced.3.brm, # drop log1p(LYER_t):rich on Ptoid from 80% PP cutoff model
reduced.4.brm, # drop log1p(BRBR_t):rich on Ptoid from 80% PP cutoff model
reduced.5.brm, # drop all rich effects on Ptoid from 80% PP cutoff model
reduced.6.brm, # drop all rich effects on LYER from 80% PP cutoff model
reduced.7.brm, # drop all rich effects from 80% PP cutoff model
reduced.8.brm, # drop all rich and temp effects from 80% PP cutoff model
criterion = "loo",
model_names = c("Full","80% PP cutoff","Drop LYER:rich for Ptoid","Drop BRBR:rich for Ptoid","Drop all rich for Ptoid","Drop all rich for LYER","Drop all rich effects","Drop all rich and temp effects")) elpd_diff se_diff
Drop all rich effects 0.0 0.0
Drop all rich for LYER -1.8 2.5
Drop LYER:rich for Ptoid -2.1 3.0
80% PP cutoff -2.3 3.5
Drop BRBR:rich for Ptoid -2.8 2.4
Drop all rich for Ptoid -3.3 3.6
Full -19.2 6.0
Drop all rich and temp effects -64.0 9.9 As you can see, there are several models that provide similar fits to data for the initial food web (i.e., the standard error of the model fit overlaps with the top model). Of these models, however, only reduced.3.brm (i.e., “Drop LYER:rich for Ptoid”) was able to reproduce our observed genotype-specific effects on critical transitions in the dominant food chain (see Genotypic effects). Therefore, I used reduced.3.brm to assess the Structural stability of dominant food chain and Structural stability of initial food web.
Structural stability of dominant food chain
# specify the model for subsequent analysis
rich_model <- reduced.3.brmReproduce Fig. 4
Since our observational data indicated clear effects of genetic diversity on the persistence of the LYER-Ptoid subcommunity, I restrict our subsequent analysis to this effect (i.e. no temperature).
Remember that I set rich = 1 as the zero point for this model. Therefore, the intercept in the model corresponds to when rich = 1, and the effect of rich corresponds to a unit increase in rich (i.e., rich = 2).
Note that I did not specify an effect of richness on the intra- or interspecific interactions in LYER or Ptoid. Therefore, the interaction matrix is the same for all levels of richness.
Also, I did this comparison at an imaginary midpoint of temperature, i.e. temp = 1.5. In this food chain, temperature only had a clear effect on the parasitoid’s intrinsic growth rate, so I only add the temperature effect here.
coef.rich <- fixef(rich_model)[,"Estimate"]
# value of rich to condition
temp.cond <- 1.5 # 1.5 = 21.5 C
# I chose 1.5 to condition on
# an imaginary average between temperature treatments
# note that setting it to zero, represents temp = 20 C
# interaction matrix for all levels of rich
rich1.mat <- matrix(c(coef.rich["log1pLYERt1_log1pLYER_t"],
coef.rich["log1pLYERt1_log1pPtoid_t"],
coef.rich["log1pPtoidt1_log1pLYER_t"],
0), # set to zero because there was no clear evidence for intraspecific Ptoid effect
ncol = 2, byrow = TRUE)
rich1.mat [,1] [,2]
[1,] -0.5837297 -0.7150802
[2,] 0.3990346 0.0000000rich2.mat <- rich1.mat
rich4.mat <- rich1.mat
# growth rates when rich = 1
rich1.IGR <- matrix(c(coef.rich["log1pLYERt1_intercept"],
coef.rich["log1pPtoidt1_intercept"] + coef.rich["log1pPtoidt1_temp"]*temp.cond),
ncol = 1)
rich1.IGR [,1]
[1,] 2.744906
[2,] -2.419228# growth rates when rich = 2
rich2.IGR <- matrix(c(coef.rich["log1pLYERt1_intercept"] + coef.rich["log1pLYERt1_rich"],
coef.rich["log1pPtoidt1_intercept"] + coef.rich["log1pPtoidt1_rich"] + coef.rich["log1pPtoidt1_temp"]*temp.cond),
ncol = 1)
rich2.IGR [,1]
[1,] 3.122174
[2,] -2.237049# growth rates when rich = 4
rich4.IGR <- matrix(c(coef.rich["log1pLYERt1_intercept"] + coef.rich["log1pLYERt1_rich"]*3,
coef.rich["log1pPtoidt1_intercept"] + coef.rich["log1pPtoidt1_rich"]*3 + coef.rich["log1pPtoidt1_temp"]*temp.cond),
ncol = 1)
rich4.IGR [,1]
[1,] 3.876709
[2,] -1.872689Plot the effect of genetic diversity on structural stability.
# get raw data for manually making plot
get_FD.2sp <- FeasibilityDomain2sp(A = list(rich1.mat, rich2.mat, rich4.mat),
r = list(rich1.IGR, rich2.IGR, rich4.IGR),
labels = c("1", "2", "4"),
normalize = TRUE) %>%
rename(rich = A_ID)
# Draw polygon for feasibility domain
# from: https://stackoverflow.com/questions/12794596/how-fill-part-of-a-circle-using-ggplot2
# define the circle; add a point at the center if the 'pie slice' if the shape is to be filled
circleFun <- function(center=c(0,0), diameter=1, npoints=100, start=0, end=2, filled=TRUE){
tt <- seq(start*pi, end*pi, length.out=npoints)
df <- data.frame(
x = center[1] + diameter / 2 * cos(tt),
y = center[2] + diameter / 2 * sin(tt)
)
if(filled==TRUE) { #add a point at the center so the whole 'pie slice' is filled
df <- rbind(df, center)
}
return(df)
}
# new version
ggplot(filter(get_FD.2sp, Type == "r"), aes(x = Sp_1, y = Sp_2, size = rich)) +
# draw intrinsic growth rate vectors
geom_segment(aes(x = 0, y = 0, xend = Sp_1, yend = Sp_2),
arrow = arrow(length = unit(0.3,"cm"))) +
# draw critical boundary
#geom_segment(data = filter(get_FD.2sp, Type == "A")[1,], # just need one lower bound
# aes(x = 0, y = 0, xend = Sp_1, yend = Sp_2, linetype = "Critical\nboundary"),
# color = "black",
# size = 0.5) +
#scale_linetype_manual(values = "dotted", name = "") +
xlab("Aphid growth rate (normalized)") +
ylab("Parasitoid growth rate (normalized)") +
# illustrate circular nature of feasibility domain
coord_cartesian(xlim = c(0,1), ylim = c(0.0,-0.75), expand = F) +
scale_size_manual(values = c(0.25,0.5,1), labels = c("1 genotype","2 genotypes","4 genotypes"), name = "") +
# adjusted until critical boundary was correct
geom_polygon(data=circleFun(c(0,0), diameter = 2, start=0, end=-0.191, npoints=100, filled=TRUE),
aes(x,y), alpha = 0.1, inherit.aes = F) +
theme_cowplot(font_size = 18, line_size = 1)
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
ggsave("figures/rich-structural-stability-R-v2.pdf", width = 8, height = 8, units = "in") I then used Keynote to manually add images and text to create the final version presented in Figure 4 of the main text.
And just as a quick check, the above plot suggests that the community is only feasible (all three species have positive abundances at equilibrium) when there are 4 genotypes, which is true:
-inv(rich1.mat)%*%rich1.IGR # Ptoid goes extinct [,1]
[1,] 6.062704
[2,] -1.110468-inv(rich2.mat)%*%rich2.IGR # Ptoid goes extinct [,1]
[1,] 5.6061526
[2,] -0.2101917-inv(rich4.mat)%*%rich4.IGR # Coexistence [,1]
[1,] 4.693051
[2,] 1.590362Non-equilibrium simulation (Fig. S6)
The above plot illustrates the effect of genetic diversity on the structural stability of the equilibrium abundances of species. I can explore whether our results hold in a non-equilibrium scenario that better characterizes our observational data.
To do this, I look at the the effect of genetic diversity across a range of initial conditions for the abundances of LYER and Ptoid. I get this data by simulating community dynamics with the observed effects of genetic diversity across a range of intial conditions. I restricted our simulation to 10 time steps, as BRBR went extinct commonly at week 7 (experiment was 17 weeks long). I also set an extinction threshold of log(abundance) = 1.
LP_duration <- 10
LP_threshold <- 1
res <- 0.1
LP_test_df <- expand.grid(LYER = seq(1, 6, by = res), Ptoid = seq(1, 6, by = res))
## simulate population dynamics and determine which species goes extinct
# rich = 1
FE_LP_rich1 <- list()
for(i in 1:length(LP_test_df$LYER)){
FE_LP_rich1[[i]] <- first_extinction_2sp(Initial.States = c(LYER = LP_test_df[i,"LYER"], Ptoid = LP_test_df[i,"Ptoid"]),
Interaction.Matrix = rich1.mat + diag(2),
IGR.Vector = rich1.IGR,
Duration = LP_duration,
threshold = LP_threshold)
}
FE_LP_rich1_df <- bind_cols(LP_test_df, plyr::ldply(FE_LP_rich1)) %>%
mutate(rich = 1)
# rich = 2
FE_LP_rich2 <- list()
for(i in 1:length(LP_test_df$LYER)){
FE_LP_rich2[[i]] <- first_extinction_2sp(Initial.States = c(LYER = LP_test_df[i,"LYER"], Ptoid = LP_test_df[i,"Ptoid"]),
Interaction.Matrix = rich2.mat + diag(2),
IGR.Vector = rich2.IGR,
Duration = LP_duration,
threshold = LP_threshold)
}
FE_LP_rich2_df <- bind_cols(LP_test_df, plyr::ldply(FE_LP_rich2)) %>%
mutate(rich = 2)
# rich = 4
FE_LP_rich4 <- list()
for(i in 1:length(LP_test_df$LYER)){
FE_LP_rich4[[i]] <- first_extinction_2sp(Initial.States = c(LYER = LP_test_df[i,"LYER"], Ptoid = LP_test_df[i,"Ptoid"]),
Interaction.Matrix = rich4.mat + diag(2),
IGR.Vector = rich4.IGR,
Duration = LP_duration,
threshold = LP_threshold)
}
FE_LP_rich4_df <- bind_cols(LP_test_df, plyr::ldply(FE_LP_rich4)) %>%
mutate(rich = 4)
# get observed data on initial abundances of LYER and Ptoid after BRBR went extinct
LP_actual_df <- timeseries_df %>%
filter(BRBR_t == 0, LYER_t > 0, Ptoid_t > 0) %>%
group_by(rich, Cage) %>%
summarise_at(vars(LYER_t, Ptoid_t), list(first = first)) %>%
ungroup() %>%
mutate(rich = rich + 1,
log1p_LYER_t_first = log1p(LYER_t_first),
log1p_Ptoid_t_first = log1p(Ptoid_t_first)) %>%
as.data.frame()The graph below shows a couple of useful things. First, our predictions hold for outside of equilibrium. That is, there is a greater likelihood of LYER-Ptoid persistence with increasing genetic diversity. Note the threshold between rich = 2 and rich = 4. This likely reflects the fact that the equilibrium community was only feasible when rich = 4.
It’s also important to note that there is a region of parameter space where LYER goes extinct before Ptoid, which would eventually lead to the collapse of the Ptoid since it has lost its resource. This is not possible if I were to assume the community is at equilibrium. It also suggests that the likelihood of LYER going extinct before the Ptoid is more likely at lower levels of genetic diversity. I also observe this in our empirical data, as LYER going extinct before the Ptoid never occurred when rich = 4.
cbPalette <- viridis::viridis(4)
bind_rows(FE_LP_rich1_df, FE_LP_rich2_df, FE_LP_rich4_df) %>%
mutate(rich = factor(rich, labels = c("1 genotype","2 genotypes","4 genotypes"))) %>%
mutate(species = ifelse(is.na(species) == T, "Food chain persists", species),
fspecies = factor(species, levels = c("LYER","Ptoid","Food chain persists"), labels = c("Complete collapse","Aphid only","Food chain persists"))) %>%
ggplot(., aes(x = LYER, y = Ptoid, fill = fspecies)) +
geom_tile() +
facet_grid(~rich) +
scale_fill_manual(name = "Critical transition", values = cbPalette[1:3]) +
coord_cartesian(xlim = c(1, max(LP_actual_df$log1p_LYER_t_first)),
ylim = c(1, max(LP_actual_df$log1p_Ptoid_t_first))) +
xlab("Aphid initial abundance (log scale)") +
ylab("Parasitoid initial abundance (log scale)") +
theme(strip.background = element_blank())
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
Remember though that our real communities are also stochastic, which is something I did not include in this non-equilibrium simulation. Below, I explicitly address stochasticity by estimating the uncertainty in our observed “mean” effects of genetic diversity on structural stability.
Posterior estimates (Fig. S5)
The above is a nice visualization of the central tendency, but it doesn’t illustrate the uncertainty in the effect of genetic richness on structural stability. To get at this, I need to use the posterior samples from our model.
# get posterior predictions
pp.rich_model <- posterior_samples(rich_model, pars = "^b")
# calculate structural stability when rich = 1
rich1_stability <- apply(
pp.rich_model,
MARGIN = 1,
FUN = function(x) {
tmp.mat = matrix(c(x["b_log1pLYERt1_log1pLYER_t"], x["b_log1pLYERt1_log1pPtoid_t"],
x["b_log1pPtoidt1_log1pLYER_t"], 0),
ncol = 2, byrow = TRUE)
tmp.r = matrix(c(x["b_log1pLYERt1_intercept"],
x["b_log1pPtoidt1_intercept"] + x["b_log1pPtoidt1_temp"]*temp.cond),
ncol = 1)
FeasibilityBoundaryLYER.Ptoid = as.numeric(BoundaryLYER.Ptoid(tmp.mat, tmp.r)["boundary"])
c(FeasibilityBoundaryLYER.Ptoid = FeasibilityBoundaryLYER.Ptoid)
})
rich1_stability.df <- data.frame(
rich = 1,
posterior_sample = 1:nrow(pp.rich_model),
FeasibilityBoundaryLYER.Ptoid = rich1_stability
)
# calculate structural stability when rich = 2
rich2_stability <- apply(
pp.rich_model,
MARGIN = 1,
FUN = function(x) {
tmp.mat = matrix(c(x["b_log1pLYERt1_log1pLYER_t"], x["b_log1pLYERt1_log1pPtoid_t"],
x["b_log1pPtoidt1_log1pLYER_t"], 0),
ncol = 2, byrow = TRUE)
tmp.r = matrix(c(x["b_log1pLYERt1_intercept"] + x["b_log1pLYERt1_rich"],
x["b_log1pPtoidt1_intercept"] + x["b_log1pPtoidt1_rich"] + x["b_log1pPtoidt1_temp"]*temp.cond),
ncol = 1)
FeasibilityBoundaryLYER.Ptoid = as.numeric(BoundaryLYER.Ptoid(tmp.mat, tmp.r)["boundary"])
c(FeasibilityBoundaryLYER.Ptoid = FeasibilityBoundaryLYER.Ptoid)
})
rich2_stability.df <- data.frame(
rich = 2,
posterior_sample = 1:nrow(pp.rich_model),
FeasibilityBoundaryLYER.Ptoid = rich2_stability
)
# calculate structural stability when rich = 4
rich4_stability <- apply(
pp.rich_model,
MARGIN = 1,
FUN = function(x) {
tmp.mat = matrix(c(x["b_log1pLYERt1_log1pLYER_t"], x["b_log1pLYERt1_log1pPtoid_t"],
x["b_log1pPtoidt1_log1pLYER_t"], 0),
ncol = 2, byrow = TRUE)
tmp.r = matrix(c(x["b_log1pLYERt1_intercept"] + x["b_log1pLYERt1_rich"]*3,
x["b_log1pPtoidt1_intercept"] + x["b_log1pPtoidt1_rich"]*3 + x["b_log1pPtoidt1_temp"]*temp.cond),
ncol = 1)
FeasibilityBoundaryLYER.Ptoid = as.numeric(BoundaryLYER.Ptoid(tmp.mat, tmp.r)["boundary"])
c(FeasibilityBoundaryLYER.Ptoid = FeasibilityBoundaryLYER.Ptoid)
})
rich4_stability.df <- data.frame(
rich = 4,
posterior_sample = 1:nrow(pp.rich_model),
FeasibilityBoundaryLYER.Ptoid = rich4_stability
)
# combine data
all.rich_stability.df <- bind_rows(rich1_stability.df, rich2_stability.df, rich4_stability.df)Reproduce Fig. S5
# subsample 1/8 of the posterior to make it easier to visualize
set.seed(38)
rsamp <- sample(1:8000, size = 1000)
# plot
all.rich_stability.df %>%
filter(posterior_sample %in% rsamp) %>%
ggplot(., aes(x = rich, y = FeasibilityBoundaryLYER.Ptoid)) +
geom_line(aes(group = posterior_sample), alpha = 0.1) +
stat_summary(fun.y = mean, geom = "line", color = "red") +
theme_minimal_hgrid() +
ylab("Normalized angle from critical boundary") +
xlab("Genetic diversity (no. of genotypes)")
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
Calculate the percentage of posterior estimates where genetic diversity increased the structural stability of the community.
# organize data
rich_LPbound <- all.rich_stability.df %>%
select(rich, posterior_sample, FeasibilityBoundaryLYER.Ptoid) %>%
spread(rich, FeasibilityBoundaryLYER.Ptoid) %>%
mutate(rich_effect = `2` - `1`)
# calculate percentage
mean(rich_LPbound$rich_effect > 0)[1] 0.97775Genotypic effects
I first modify rich_model by replacing rich with (Col + gsm1 + AOP2 + AOP2.gsoh), which are the components of the rich effect.
## formulas
geno.rich_model.BRBR.bf <- reduced.3.BRBR.bf
geno.rich_model.LYER.bf <- update(reduced.3.LYER.bf, .~.
-rich + (Col + gsm1 + AOP2 + AOP2.gsoh)
-log1p(BRBR_t):rich + log1p(BRBR_t):(Col + gsm1 + AOP2 + AOP2.gsoh))
geno.rich_model.Ptoid.bf <- update(reduced.3.Ptoid.bf, .~.
-rich + (Col + gsm1 + AOP2 + AOP2.gsoh)
-log1p(BRBR_t):rich + log1p(BRBR_t):(Col + gsm1 + AOP2 + AOP2.gsoh))
## fit new model
geno.rich_model.brm <- brm(
data = full_df,
mvbf(geno.rich_model.BRBR.bf, geno.rich_model.LYER.bf, geno.rich_model.Ptoid.bf),
iter = 4000,
prior = c(# growth rates
set_prior(prior.r.BRBR, class = "b", coef = "intercept", resp = "log1pBRBRt1"),
set_prior(prior.r.LYER, class = "b", coef = "intercept", resp = "log1pLYERt1"),
set_prior(prior.r.Ptoid, class = "b", coef = "intercept", resp = "log1pPtoidt1"),
# intraspecific effects
set_prior(prior.intra.BRBR, class = "b", coef = "log1pBRBR_t", resp = "log1pBRBRt1"),
set_prior(prior.intra.LYER, class = "b", coef = "log1pLYER_t", resp = "log1pLYERt1"),
# negative interspecific effects
set_prior(prior.BRBRonLYER, class = "b", coef = "log1pBRBR_t", resp = "log1pLYERt1"),
set_prior(prior.PtoidonBRBR, class = "b", coef = "log1pPtoid_t", resp = "log1pBRBRt1"),
set_prior(prior.PtoidonLYER, class = "b", coef = "log1pPtoid_t", resp = "log1pLYERt1"),
# positive interspecific effects
set_prior(prior.BRBRonPtoid, class = "b", coef = "log1pBRBR_t", resp = "log1pPtoidt1"),
set_prior(prior.LYERonPtoid, class = "b", coef = "log1pLYER_t", resp = "log1pPtoidt1"),
# rich effects
set_prior(prior.rich, class = "b", coef = "Col", resp = "log1pLYERt1"),
set_prior(prior.rich, class = "b", coef = "log1pBRBR_t:Col", resp = "log1pLYERt1"),
set_prior(prior.rich, class = "b", coef = "gsm1", resp = "log1pLYERt1"),
set_prior(prior.rich, class = "b", coef = "log1pBRBR_t:gsm1", resp = "log1pLYERt1"),
set_prior(prior.rich, class = "b", coef = "AOP2", resp = "log1pLYERt1"),
set_prior(prior.rich, class = "b", coef = "log1pBRBR_t:AOP2", resp = "log1pLYERt1"),
set_prior(prior.rich, class = "b", coef = "AOP2.gsoh", resp = "log1pLYERt1"),
set_prior(prior.rich, class = "b", coef = "log1pBRBR_t:AOP2.gsoh", resp = "log1pLYERt1"),
set_prior(prior.rich, class = "b", coef = "Col", resp = "log1pPtoidt1"),
set_prior(prior.rich, class = "b", coef = "log1pBRBR_t:Col", resp = "log1pPtoidt1"),
set_prior(prior.rich, class = "b", coef = "gsm1", resp = "log1pPtoidt1"),
set_prior(prior.rich, class = "b", coef = "log1pBRBR_t:gsm1", resp = "log1pPtoidt1"),
set_prior(prior.rich, class = "b", coef = "AOP2", resp = "log1pPtoidt1"),
set_prior(prior.rich, class = "b", coef = "log1pBRBR_t:AOP2", resp = "log1pPtoidt1"),
set_prior(prior.rich, class = "b", coef = "AOP2.gsoh", resp = "log1pPtoidt1"),
set_prior(prior.rich, class = "b", coef = "log1pBRBR_t:AOP2.gsoh", resp = "log1pPtoidt1"),
# temp effects
set_prior(prior.temp, class = "b", coef = "temp", resp = "log1pBRBRt1"),
set_prior(prior.temp, class = "b", coef = "temp", resp = "log1pPtoidt1"),
set_prior(prior.temp, class = "b", coef = "log1pBRBR_t:temp", resp = "log1pBRBRt1"),
set_prior(prior.temp, class = "b", coef = "log1pBRBR_t:temp", resp = "log1pPtoidt1"),
set_prior(prior.temp, class = "b", coef = "log1pPtoid_t:temp", resp = "log1pBRBRt1"),
# biomass effects
set_prior(prior.AphidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pLYERt1"),
set_prior(prior.PtoidBiomass, class = "b", coef = "logBiomass_g_t1", resp = "log1pPtoidt1"),
# random effects
set_prior(prior.random.effects, class = "sd", resp = "log1pBRBRt1"),
set_prior(prior.random.effects, class = "sd", resp = "log1pLYERt1"),
set_prior(prior.random.effects, class = "sd", resp = "log1pPtoidt1")),
file = "output/geno.rich_model.brm.rds")
# print summary
summary(geno.rich_model.brm) Family: MV(gaussian, gaussian, gaussian)
Links: mu = identity; sigma = identity
mu = identity; sigma = identity
mu = identity; sigma = identity
Formula: log1p(BRBR_t1) ~ intercept + log1p(BRBR_t) + log1p(Ptoid_t) + temp + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + log1p(BRBR_t):temp + log1p(Ptoid_t):temp + offset(log1p(BRBR_t)) - 1
log1p(LYER_t1) ~ intercept + log1p(BRBR_t) + log1p(LYER_t) + log1p(Ptoid_t) + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + Col + gsm1 + AOP2 + AOP2.gsoh + log1p(BRBR_t):Col + log1p(BRBR_t):gsm1 + log1p(BRBR_t):AOP2 + log1p(BRBR_t):AOP2.gsoh + offset(log1p(LYER_t)) - 1
log1p(Ptoid_t1) ~ intercept + log1p(BRBR_t) + log1p(LYER_t) + temp + log(Biomass_g_t1) + (1 | Cage) + ar(time = Week_match.1p, gr = Cage, p = 1, cov = FALSE) + Col + gsm1 + AOP2 + AOP2.gsoh + log1p(BRBR_t):temp + log1p(BRBR_t):Col + log1p(BRBR_t):gsm1 + log1p(BRBR_t):AOP2 + log1p(BRBR_t):AOP2.gsoh + offset(log1p(Ptoid_t)) - 1
Data: full_df (Number of observations: 264)
Samples: 4 chains, each with iter = 4000; warmup = 2000; thin = 1;
total post-warmup samples = 8000
Correlation Structures:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
ar_log1pBRBRt1[1] -0.45 0.09 -0.63 -0.26 1.00 3923 5401
ar_log1pLYERt1[1] -0.11 0.10 -0.29 0.09 1.00 4849 5799
ar_log1pPtoidt1[1] -0.49 0.08 -0.65 -0.33 1.00 10963 6664
Group-Level Effects:
~Cage (Number of levels: 60)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS
sd(log1pBRBRt1_Intercept) 0.26 0.10 0.04 0.44 1.01 1281
sd(log1pLYERt1_Intercept) 0.14 0.09 0.01 0.32 1.00 1850
sd(log1pPtoidt1_Intercept) 0.04 0.03 0.00 0.11 1.00 5278
Tail_ESS
sd(log1pBRBRt1_Intercept) 1390
sd(log1pLYERt1_Intercept) 3358
sd(log1pPtoidt1_Intercept) 4207
Population-Level Effects:
Estimate Est.Error l-95% CI u-95% CI Rhat
log1pBRBRt1_intercept 1.46 0.45 0.59 2.35 1.00
log1pBRBRt1_log1pBRBR_t 0.04 0.07 -0.11 0.18 1.00
log1pBRBRt1_log1pPtoid_t -0.90 0.05 -1.00 -0.80 1.00
log1pBRBRt1_temp -0.30 0.22 -0.72 0.12 1.00
log1pBRBRt1_log1pBRBR_t:temp -0.07 0.04 -0.14 -0.00 1.00
log1pBRBRt1_log1pPtoid_t:temp 0.13 0.04 0.06 0.20 1.00
log1pLYERt1_intercept 3.07 0.59 1.88 4.27 1.00
log1pLYERt1_log1pBRBR_t 0.31 0.09 0.14 0.48 1.00
log1pLYERt1_log1pLYER_t -0.65 0.09 -0.83 -0.48 1.00
log1pLYERt1_log1pPtoid_t -0.73 0.05 -0.83 -0.63 1.00
log1pLYERt1_logBiomass_g_t1 0.08 0.18 -0.27 0.44 1.00
log1pLYERt1_Col 0.37 0.31 -0.24 0.98 1.00
log1pLYERt1_gsm1 0.26 0.31 -0.35 0.85 1.00
log1pLYERt1_AOP2 0.03 0.31 -0.58 0.64 1.00
log1pLYERt1_AOP2.gsoh 0.40 0.31 -0.20 0.99 1.00
log1pLYERt1_log1pBRBR_t:Col -0.03 0.07 -0.16 0.09 1.00
log1pLYERt1_log1pBRBR_t:gsm1 -0.03 0.07 -0.16 0.10 1.00
log1pLYERt1_log1pBRBR_t:AOP2 -0.02 0.07 -0.15 0.11 1.00
log1pLYERt1_log1pBRBR_t:AOP2.gsoh -0.11 0.07 -0.24 0.02 1.00
log1pPtoidt1_intercept -3.11 0.45 -3.98 -2.23 1.00
log1pPtoidt1_log1pBRBR_t 0.40 0.09 0.23 0.57 1.00
log1pPtoidt1_log1pLYER_t 0.35 0.07 0.22 0.47 1.00
log1pPtoidt1_temp 0.65 0.12 0.42 0.89 1.00
log1pPtoidt1_logBiomass_g_t1 -0.69 0.17 -1.03 -0.35 1.00
log1pPtoidt1_Col 0.25 0.26 -0.27 0.76 1.00
log1pPtoidt1_gsm1 0.37 0.27 -0.17 0.90 1.00
log1pPtoidt1_AOP2 -0.08 0.27 -0.60 0.45 1.00
log1pPtoidt1_AOP2.gsoh -0.09 0.26 -0.62 0.42 1.00
log1pPtoidt1_log1pBRBR_t:temp -0.16 0.02 -0.21 -0.12 1.00
log1pPtoidt1_log1pBRBR_t:Col -0.04 0.06 -0.15 0.07 1.00
log1pPtoidt1_log1pBRBR_t:gsm1 -0.05 0.06 -0.16 0.06 1.00
log1pPtoidt1_log1pBRBR_t:AOP2 -0.00 0.06 -0.11 0.11 1.00
log1pPtoidt1_log1pBRBR_t:AOP2.gsoh 0.00 0.06 -0.11 0.11 1.00
Bulk_ESS Tail_ESS
log1pBRBRt1_intercept 3724 4984
log1pBRBRt1_log1pBRBR_t 4418 5634
log1pBRBRt1_log1pPtoid_t 4714 5542
log1pBRBRt1_temp 4335 5259
log1pBRBRt1_log1pBRBR_t:temp 4672 5686
log1pBRBRt1_log1pPtoid_t:temp 6553 6331
log1pLYERt1_intercept 4620 5364
log1pLYERt1_log1pBRBR_t 5168 5142
log1pLYERt1_log1pLYER_t 4768 6014
log1pLYERt1_log1pPtoid_t 5903 6396
log1pLYERt1_logBiomass_g_t1 9638 6338
log1pLYERt1_Col 6748 6225
log1pLYERt1_gsm1 6627 5751
log1pLYERt1_AOP2 6444 5962
log1pLYERt1_AOP2.gsoh 6522 5999
log1pLYERt1_log1pBRBR_t:Col 6878 5683
log1pLYERt1_log1pBRBR_t:gsm1 6418 6253
log1pLYERt1_log1pBRBR_t:AOP2 6490 5787
log1pLYERt1_log1pBRBR_t:AOP2.gsoh 6460 5579
log1pPtoidt1_intercept 5157 5723
log1pPtoidt1_log1pBRBR_t 4762 5676
log1pPtoidt1_log1pLYER_t 9179 5794
log1pPtoidt1_temp 6193 5885
log1pPtoidt1_logBiomass_g_t1 9417 6608
log1pPtoidt1_Col 6463 6296
log1pPtoidt1_gsm1 6713 6047
log1pPtoidt1_AOP2 7189 5968
log1pPtoidt1_AOP2.gsoh 6689 5908
log1pPtoidt1_log1pBRBR_t:temp 6394 5833
log1pPtoidt1_log1pBRBR_t:Col 6303 5942
log1pPtoidt1_log1pBRBR_t:gsm1 6666 5749
log1pPtoidt1_log1pBRBR_t:AOP2 7457 6175
log1pPtoidt1_log1pBRBR_t:AOP2.gsoh 6615 5715
Family Specific Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma_log1pBRBRt1 1.11 0.06 1.00 1.23 1.00 3767 5221
sigma_log1pLYERt1 0.92 0.04 0.84 1.01 1.00 8728 5993
sigma_log1pPtoidt1 0.76 0.03 0.70 0.83 1.00 14675 5618
Residual Correlations:
Estimate Est.Error l-95% CI u-95% CI Rhat
rescor(log1pBRBRt1,log1pLYERt1) 0.52 0.05 0.42 0.61 1.00
rescor(log1pBRBRt1,log1pPtoidt1) -0.04 0.07 -0.18 0.10 1.00
rescor(log1pLYERt1,log1pPtoidt1) 0.06 0.07 -0.08 0.19 1.00
Bulk_ESS Tail_ESS
rescor(log1pBRBRt1,log1pLYERt1) 8262 6288
rescor(log1pBRBRt1,log1pPtoidt1) 9931 6720
rescor(log1pLYERt1,log1pPtoidt1) 12325 6163
Samples were drawn using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).Posterior estimates
# get posterior predictions
pp.geno.rich_model.norm <- posterior_samples(geno.rich_model.brm, pars = "^b")
# intercept term corresponds to a hypothetical plant population where each genotype is absent
int.mv_stability <- apply(
pp.geno.rich_model.norm,
MARGIN = 1,
FUN = function(x) {
tmp.mat = matrix(c(x["b_log1pLYERt1_log1pLYER_t"], x["b_log1pLYERt1_log1pPtoid_t"],
x["b_log1pPtoidt1_log1pLYER_t"], 0),
ncol = 2, byrow = TRUE)
tmp.r = matrix(c(x["b_log1pLYERt1_intercept"],
x["b_log1pPtoidt1_intercept"] + x["b_log1pPtoidt1_temp"]*temp.cond),
ncol = 1)
FeasibilityBoundaryLYER.Ptoid = as.numeric(BoundaryLYER.Ptoid(tmp.mat, tmp.r)["boundary"])
c(FeasibilityBoundaryLYER.Ptoid = FeasibilityBoundaryLYER.Ptoid)
})
int.mv_stability.df <- data.frame(
geno = "int",
posterior_sample = 1:nrow(pp.geno.rich_model.norm),
FeasibilityBoundaryLYER.Ptoid = int.mv_stability
)
# Col effect on structural stability
Col.mv_stability <- apply(
pp.geno.rich_model.norm,
MARGIN = 1,
FUN = function(x) {
tmp.mat = matrix(c(x["b_log1pLYERt1_log1pLYER_t"], x["b_log1pLYERt1_log1pPtoid_t"],
x["b_log1pPtoidt1_log1pLYER_t"], 0),
ncol = 2, byrow = TRUE)
tmp.r = matrix(c(x["b_log1pLYERt1_intercept"] + x["b_log1pLYERt1_Col"],
x["b_log1pPtoidt1_intercept"] + x["b_log1pPtoidt1_Col"] + x["b_log1pPtoidt1_temp"]*temp.cond),
ncol = 1)
FeasibilityBoundaryLYER.Ptoid = as.numeric(BoundaryLYER.Ptoid(tmp.mat, tmp.r)["boundary"])
c(FeasibilityBoundaryLYER.Ptoid = FeasibilityBoundaryLYER.Ptoid)
})
Col.mv_stability.df <- data.frame(
geno = "Col",
posterior_sample = 1:nrow(pp.geno.rich_model.norm),
FeasibilityBoundaryLYER.Ptoid = Col.mv_stability
)
# gsm1 effect on structural stability
gsm1.mv_stability <- apply(
pp.geno.rich_model.norm,
MARGIN = 1,
FUN = function(x) {
tmp.mat = matrix(c(x["b_log1pLYERt1_log1pLYER_t"], x["b_log1pLYERt1_log1pPtoid_t"],
x["b_log1pPtoidt1_log1pLYER_t"], 0),
ncol = 2, byrow = TRUE)
tmp.r = matrix(c(x["b_log1pLYERt1_intercept"] + x["b_log1pLYERt1_gsm1"],
x["b_log1pPtoidt1_intercept"] + x["b_log1pPtoidt1_gsm1"] + x["b_log1pPtoidt1_temp"]*temp.cond),
ncol = 1)
FeasibilityBoundaryLYER.Ptoid = as.numeric(BoundaryLYER.Ptoid(tmp.mat, tmp.r)["boundary"])
c(FeasibilityBoundaryLYER.Ptoid = FeasibilityBoundaryLYER.Ptoid)
})
gsm1.mv_stability.df <- data.frame(
geno = "gsm1",
posterior_sample = 1:nrow(pp.geno.rich_model.norm),
FeasibilityBoundaryLYER.Ptoid = gsm1.mv_stability
)
# AOP2 effect on structural stability
AOP2.mv_stability <- apply(
pp.geno.rich_model.norm,
MARGIN = 1,
FUN = function(x) {
tmp.mat = matrix(c(x["b_log1pLYERt1_log1pLYER_t"], x["b_log1pLYERt1_log1pPtoid_t"],
x["b_log1pPtoidt1_log1pLYER_t"], 0),
ncol = 2, byrow = TRUE)
tmp.r = matrix(c(x["b_log1pLYERt1_intercept"] + x["b_log1pLYERt1_AOP2"],
x["b_log1pPtoidt1_intercept"] + x["b_log1pPtoidt1_AOP2"] + x["b_log1pPtoidt1_temp"]*temp.cond),
ncol = 1)
FeasibilityBoundaryLYER.Ptoid = as.numeric(BoundaryLYER.Ptoid(tmp.mat, tmp.r)["boundary"])
c(FeasibilityBoundaryLYER.Ptoid = FeasibilityBoundaryLYER.Ptoid)
})
AOP2.mv_stability.df <- data.frame(
geno = "AOP2",
posterior_sample = 1:nrow(pp.geno.rich_model.norm),
FeasibilityBoundaryLYER.Ptoid = AOP2.mv_stability
)
# AOP2/gsoh effect on structural stability
AOP2.gsoh.mv_stability <- apply(
pp.geno.rich_model.norm,
MARGIN = 1,
FUN = function(x) {
tmp.mat = matrix(c(x["b_log1pLYERt1_log1pLYER_t"], x["b_log1pLYERt1_log1pPtoid_t"],
x["b_log1pPtoidt1_log1pLYER_t"], 0),
ncol = 2, byrow = TRUE)
tmp.r = matrix(c(x["b_log1pLYERt1_intercept"] + x["b_log1pLYERt1_AOP2.gsoh"],
x["b_log1pPtoidt1_intercept"] + x["b_log1pPtoidt1_AOP2.gsoh"] + x["b_log1pPtoidt1_temp"]*temp.cond),
ncol = 1)
FeasibilityBoundaryLYER.Ptoid = as.numeric(BoundaryLYER.Ptoid((tmp.mat), tmp.r)["boundary"])
c(FeasibilityBoundaryLYER.Ptoid = FeasibilityBoundaryLYER.Ptoid)
})
AOP2.gsoh.mv_stability.df <- data.frame(
geno = "AOP2.gsoh",
posterior_sample = 1:nrow(pp.geno.rich_model.norm),
FeasibilityBoundaryLYER.Ptoid = AOP2.gsoh.mv_stability
)
# combine data
all.geno.mv_stability.df <- bind_rows(
int.mv_stability.df,
Col.mv_stability.df,
gsm1.mv_stability.df,
AOP2.mv_stability.df,
AOP2.gsoh.mv_stability.df)Reproduce Fig. S7
Col_df <- bind_rows(int.mv_stability.df, Col.mv_stability.df) %>% mutate(compare = "Col", geno = ifelse(geno == "int", 0, 1))
gsm1_df <- bind_rows(int.mv_stability.df, gsm1.mv_stability.df) %>% mutate(compare = "gsm1", geno = ifelse(geno == "int", 0, 1))
AOP2_df <- bind_rows(int.mv_stability.df, AOP2.mv_stability.df) %>% mutate(compare = "AOP2", geno = ifelse(geno == "int", 0, 1))
AOP2.gsoh_df <- bind_rows(int.mv_stability.df, AOP2.gsoh.mv_stability.df) %>% mutate(compare = "AOP2.gsoh", geno = ifelse(geno == "int", 0, 1))
bind_rows(Col_df, gsm1_df, AOP2_df, AOP2.gsoh_df) %>%
filter(posterior_sample %in% rsamp) %>%
mutate(compare = factor(compare, levels = c("Col","gsm1","AOP2","AOP2.gsoh"), labels = c("Col-0","gsm1","AOP2","AOP2/gsoh"))) %>%
ggplot(., aes(x = geno, y = FeasibilityBoundaryLYER.Ptoid, color = compare)) +
geom_line(aes(group = posterior_sample), alpha = 0.05) +
stat_summary(fun.y = mean, geom = "line", size = 1.5) +
facet_wrap(~compare, nrow = 1) +
geom_hline(yintercept = 0, linetype = 'dotted') +
ylab("Normalized angle from feasibility boundary") +
scale_x_continuous(expand = c(0.1, 0.1), name = "", breaks = c(0,1), labels = c("Absent", "Present")) +
scale_color_manual(values = c("darkgreen","steelblue","darkorange","firebrick1")) +
theme_minimal_hgrid(font_size = 11) +
theme(legend.position = "none")
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
I then calculate the percentage of posterior samples where each genotype increases the structural stability of the community.
# gather data to calculate genotype-specific effects
geno_LPbound <- all.geno.mv_stability.df %>%
select(geno, posterior_sample, FeasibilityBoundaryLYER.Ptoid) %>%
spread(geno, FeasibilityBoundaryLYER.Ptoid) %>%
mutate(Col_effect = (Col - int),
gsm1_effect = (gsm1 - int),
AOP2_effect = (AOP2 - int),
AOP2.gsoh_effect = (AOP2.gsoh - int))
# effect of Col
mean(geno_LPbound$Col_effect > 0)[1] 0.924125# effect of gsm1
mean(geno_LPbound$gsm1_effect > 0)[1] 0.937125# effect of AOP2
mean(geno_LPbound$AOP2_effect > 0)[1] 0.432375# effect of AOP2/gsoh
mean(geno_LPbound$AOP2.gsoh_effect > 0)[1] 0.695125Structural stability of initial food web
I use the same model as I did for the effect of genetic diversity rich_model. Note that there were other models that did as good of, if not better, a job at predicting the dynamics of the initial food web. However, it was only reduced.3.brm (now rich_model) that could also reproduce the genotype-specific effects (see @ref{genotype-effects}).
temp_model <- rich_modelReproduce Fig. S1
coef.temp <- fixef(temp_model)[,"Estimate"]
# value of rich to condition
rich.cond <- 1.5 # 1.5 = 2.5 genotypes
# I chose 1.5, as with temp, to condition on
# an imaginary average between treatments
# note that setting it to zero, represents temp
# effect in plant monoculture
# interaction matrix when temp = 20 C
temp20.mat <- matrix(c(0, # constrained to biologically reasonable value of zero, rather than the positive estimate # coef.temp["log1pBRBRt1_log1pBRBR_t"],
0,
coef.temp["log1pBRBRt1_log1pPtoid_t"],
coef.temp["log1pLYERt1_log1pBRBR_t"] + coef.temp["log1pLYERt1_log1pBRBR_t:rich"]*rich.cond,
coef.temp["log1pLYERt1_log1pLYER_t"],
coef.temp["log1pLYERt1_log1pPtoid_t"],
coef.temp["log1pPtoidt1_log1pBRBR_t"] + coef.temp["log1pPtoidt1_log1pBRBR_t:rich"]*rich.cond,
coef.temp["log1pPtoidt1_log1pLYER_t"],
0),
ncol = 3, byrow = TRUE)
# growth rates when temp = 20 C
temp20.IGR <- matrix(c(coef.temp["log1pBRBRt1_intercept"],
coef.temp["log1pLYERt1_intercept"] + coef.temp["log1pLYERt1_rich"]*rich.cond,
coef.temp["log1pPtoidt1_intercept"] + coef.temp["log1pPtoidt1_rich"]*rich.cond),
ncol = 1)
# interaction matrix when temp = 23 C
temp23.mat <- matrix(c(coef.temp["log1pBRBRt1_log1pBRBR_t"] + coef.temp["log1pBRBRt1_log1pBRBR_t:temp"]*3,
0,
coef.temp["log1pBRBRt1_log1pPtoid_t"] + coef.temp["log1pBRBRt1_log1pPtoid_t:temp"]*3,
coef.temp["log1pLYERt1_log1pBRBR_t"] + coef.temp["log1pLYERt1_log1pBRBR_t:rich"]*rich.cond,
coef.temp["log1pLYERt1_log1pLYER_t"],
coef.temp["log1pLYERt1_log1pPtoid_t"],
0, # constrained to biologically reasonable value of zero, rather than negative estimate # coef.temp["log1pPtoidt1_log1pBRBR_t"] + coef.temp["log1pPtoidt1_log1pBRBR_t:rich"]*rich.cond + coef.temp["log1pPtoidt1_log1pBRBR_t:temp"]*3,
coef.temp["log1pPtoidt1_log1pLYER_t"],
0),
ncol = 3, byrow = TRUE)
# growth rates when temp = 23 C
temp23.IGR <- matrix(c(coef.temp["log1pBRBRt1_intercept"] + coef.temp["log1pBRBRt1_temp"]*3,
coef.temp["log1pLYERt1_intercept"] + coef.temp["log1pLYERt1_rich"]*rich.cond,
coef.temp["log1pPtoidt1_intercept"] + coef.temp["log1pPtoidt1_rich"]*rich.cond + coef.temp["log1pPtoidt1_temp"]*3),
ncol = 1)
# assess feasibility
-1*inv(temp20.mat) %*% temp20.IGR # all species persist [,1]
[1,] 3.533528
[2,] 4.794988
[3,] 1.642139-1*inv(temp23.mat) %*% temp23.IGR # BRBR goes extinct [,1]
[1,] -2.008388
[2,] 2.928847
[3,] 1.712592# plot structural stability
constraints.matrix <- matrix(c(-1,0,0,
0,-1,0,
0,0,1),
ncol = 3, byrow = T)
PlotFeasibilityDomain3sp(A = list(temp20.mat, temp23.mat, constraints.matrix),
r = list(temp20.IGR, temp23.IGR, temp23.IGR), # IGR doesn't matter for constraints
A.color = c("steelblue","firebrick1","grey"),
r.color = c("steelblue","firebrick1","grey"),
normalize = TRUE,
sphere.alpha = 0,
arc.width = c(2,2,2),
barb.n = 2,
species.labels = c("B","L","P"))
# add axes
rgl.lines(x = c(0,1.1), y = c(0,0), z = c(0,0), color = "black", lwd = 3)
rgl.lines(x = c(0,0), y = c(1.1,0), z = c(0,0), color = "black", lwd = 3)
rgl.lines(x = c(0,0), y = c(0,0), z = c(-1.1,0), color = "black", lwd = 3)
# visualize in .html file
# note that I reproduce this visualization in `code/temperature-structural-stability-fig.R`
scene3d()RGL scene containing:
material: default material properties
objects: list of 40 object(s):
surface text text text linestrip linestrip linestrip linestrip linestrip linestrip linestrip points text text text linestrip linestrip linestrip linestrip linestrip linestrip linestrip points text text text linestrip linestrip linestrip linestrip linestrip linestrip linestrip points lines lines lines light background background
root: a root subscenerglwidget()From the above figure, it appears that the L-P boundary determines the feasibility of the community (alpha.A23 below). Therefore, I focus on estimating the normalized angle of the intrinsic growth rate from this critical boundary.
# calculate angle from critical boundary L-P for 23 C community
FeasibilityBoundary(temp23.mat, temp23.IGR)["alpha.A23"] alpha.A23
-6.335992 Posterior estimates
# get posterior predictions
pp.temp_model <- posterior_samples(temp_model, pars = "^b")Inspect parameter estimates to ensure they are biologically reasonable.
# some BRBR intrinsic growth rates became negative with warming
pp.temp_model %>%
mutate(pp_ID = 1:nrow(.),
r_BRBR_20 = b_log1pBRBRt1_intercept,
r_BRBR_23 = b_log1pBRBRt1_intercept + b_log1pBRBRt1_temp*3) %>%
select(pp_ID, r_BRBR_20, r_BRBR_23) %>%
gather(key = temp, value = r, -pp_ID) %>%
filter(pp_ID %in% rsamp) %>% # subsample to avoid crowding plot
ggplot(., aes(x = temp, y = r)) +
geom_line(aes(group = pp_ID), alpha = 0.1) +
stat_summary(fun.y = mean, geom = "point", col = "red", size = 2) +
geom_hline(yintercept = 0, linetype = "dotted", color = "red") 
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# positive intraspecific interactions were sometimes estimated,
# even though this is biologically improbable
pp.temp_model %>%
mutate(pp_ID = 1:nrow(.),
intra_BRBR_20 = b_log1pBRBRt1_log1pBRBR_t,
intra_BRBR_23 = b_log1pBRBRt1_log1pBRBR_t + `b_log1pBRBRt1_log1pBRBR_t:temp`*3) %>%
select(pp_ID, intra_BRBR_20, intra_BRBR_23) %>%
gather(key = temp, value = intra, -pp_ID) %>%
filter(pp_ID %in% rsamp) %>% # subsample to avoid crowding plot
ggplot(., aes(x = temp, y = intra)) +
geom_line(aes(group = pp_ID), alpha = 0.1) +
stat_summary(fun.y = mean, geom = "point", col = "red", size = 2) +
geom_hline(yintercept = 0, linetype = "dotted", color = "red")
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# the effect of BRBR on the parasitoid at warmer temperatures apparently
# was reduced to zero. Rather than setting it to zero, I used a more conservative
# approach, by constraining the lower bound to zero, allowing positive effects
# in the posterior samples.
pp.temp_model %>%
mutate(pp_ID = 1:nrow(.),
BRBR_Ptoid_20 = b_log1pPtoidt1_log1pBRBR_t,
BRBR_Ptoid_23 = b_log1pPtoidt1_log1pBRBR_t + `b_log1pPtoidt1_log1pBRBR_t:temp`*3 + `b_log1pPtoidt1_log1pBRBR_t:rich`*rich.cond) %>%
select(pp_ID, BRBR_Ptoid_20, BRBR_Ptoid_23) %>%
gather(key = temp, value = inter, -pp_ID) %>%
filter(pp_ID %in% rsamp) %>% # subsample to avoid crowding plot
ggplot(., aes(x = temp, y = inter)) +
geom_line(aes(group = pp_ID), alpha = 0.1) +
stat_summary(fun.y = mean, geom = "point", col = "red", size = 2) +
geom_hline(yintercept = 0, linetype = "dotted", color = "red")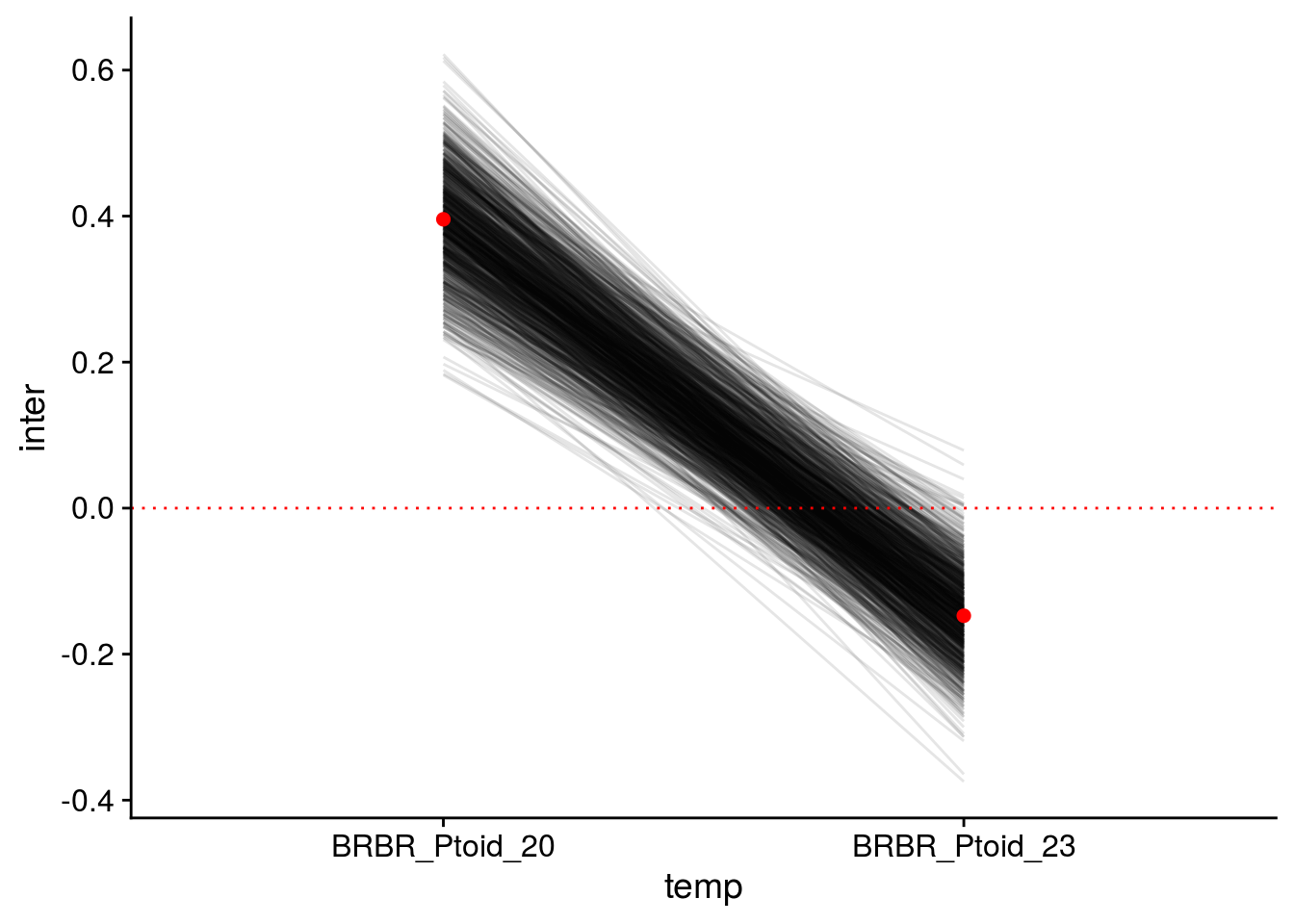
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
# rarely is the parasitoid's intrinsic growth rate estimated as positive,
# althought it should be negative
pp.temp_model %>%
mutate(pp_ID = 1:nrow(.),
r_BRBR_20 = b_log1pPtoidt1_intercept + b_log1pPtoidt1_rich*rich.cond,
r_BRBR_23 = b_log1pPtoidt1_intercept + b_log1pPtoidt1_rich*rich.cond + b_log1pPtoidt1_temp*3) %>%
select(pp_ID, r_BRBR_20, r_BRBR_23) %>%
gather(key = temp, value = r, -pp_ID) %>%
# use all samples to see occasional unrealistic estimate # filter(pp_ID %in% rsamp) %>% # subsample to avoid crowding plot
ggplot(., aes(x = temp, y = r)) +
geom_line(aes(group = pp_ID), alpha = 0.1) +
stat_summary(fun.y = mean, geom = "point", col = "red", size = 2) +
geom_hline(yintercept = 0, linetype = "dotted", color = "red") 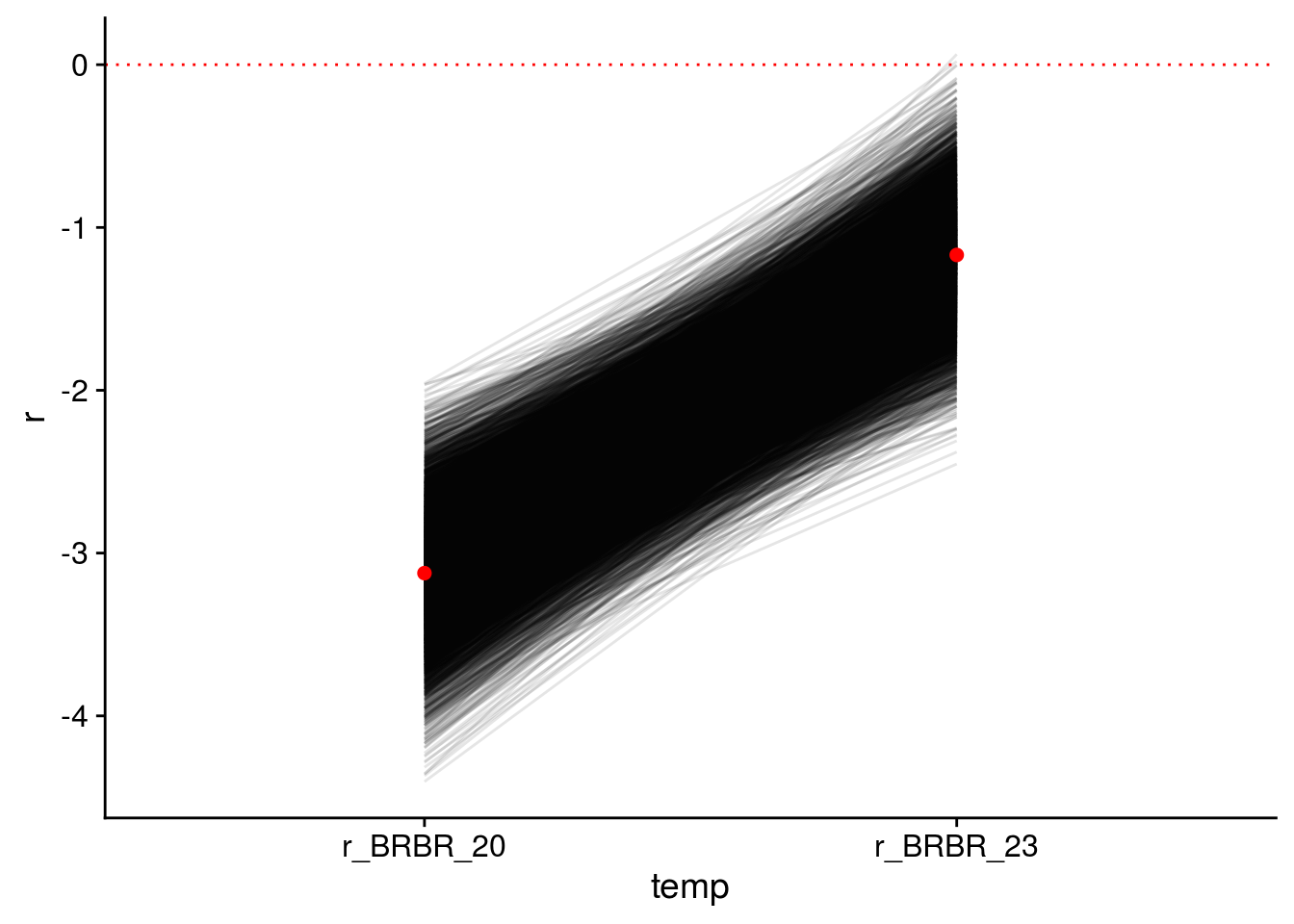
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
Note that some of the posterior samples can be biologically unfeasible, such as aphids with negative intrinsic growth rates. I restrict our inference to those parameters that are feasible. For example, if an aphid’s intrinsic growth rate was estimated as negative, I manually set it to a small positive value of 0.1. In terms of the interactions, I ensured that any apparent negative effects of aphids on the parasitoids was unrealistic, so I constrained this parameter to be zero or positive.
Note that if I went ahead without this biological restriction, our results remain qualitatively the same.
# get posterior samples of structural stability
temp20C_stability <- apply(
pp.temp_model,
MARGIN = 1,
FUN = function(x) {
tmp.mat = matrix(c(x["b_log1pBRBRt1_log1pBRBR_t"],
0,
x["b_log1pBRBRt1_log1pPtoid_t"],
x["b_log1pLYERt1_log1pBRBR_t"] + x["b_log1pLYERt1_log1pBRBR_t:rich"]*rich.cond,
x["b_log1pLYERt1_log1pLYER_t"],
x["b_log1pLYERt1_log1pPtoid_t"],
x["b_log1pPtoidt1_log1pBRBR_t"] + x["b_log1pPtoidt1_log1pBRBR_t:rich"]*rich.cond,
x["b_log1pPtoidt1_log1pLYER_t"],
0),
ncol = 3, byrow = TRUE)
# constrain intraspecific BRBR effect to a max of zero
if(tmp.mat[1,1] > 0){
tmp.mat[1,1] <- 0
}
# constrain BRBR-Ptoid interaction to a min of zero
if(tmp.mat[3,1] < 0){
tmp.mat[3,1] <- 0
}
tmp.r = matrix(c(x["b_log1pBRBRt1_intercept"],
x["b_log1pLYERt1_intercept"] + x["b_log1pLYERt1_rich"]*rich.cond,
x["b_log1pPtoidt1_intercept"] + x["b_log1pPtoidt1_rich"]*rich.cond),
ncol = 1)
# constrain BRBR intrinsic growth rate to positive values
if(tmp.r[1] < 0){
tmp.r[1] <- 0.1 # small positive value
}
# constrain Ptoid intrinsic growth rate to negative values
if(tmp.r[3] > 0){
tmp.r[3] <- -0.1 # small negative value
}
Feasibility = as.numeric(FeasibilityBoundary(tmp.mat, tmp.r)["feasibility"])
FeasibilityBoundary23 = as.numeric(FeasibilityBoundary(tmp.mat, tmp.r)["alpha.A23"])
Extinctions = paste0(which(-1*inv(tmp.mat) %*% tmp.r < 0), collapse = ",")
Resilience = max(Re(eigen((tmp.mat + diag(3)))$values))
list(tmp.mat = tmp.mat,
tmp.r = tmp.r,
Feasibility = Feasibility,
FeasibilityBoundary23 = FeasibilityBoundary23,
Extinctions = Extinctions,
Resilience = Resilience)
})
temp20C_stability.df <- data.frame(
temp = "20 C",
posterior_sample = 1:nrow(pp.temp_model),
Feasibility = unlist(lapply(temp20C_stability, FUN = function(x) x$Feasibility)),
FeasibilityBoundary23 = unlist(lapply(temp20C_stability, FUN = function(x) x$FeasibilityBoundary23)),
Extinctions = unlist(lapply(temp20C_stability, FUN = function(x) x$Extinctions)),
Resilience = unlist(lapply(temp20C_stability, FUN = function(x) x$Resilience))
)
temp23C_stability <- apply(
pp.temp_model,
MARGIN = 1,
FUN = function(x) {
tmp.mat = matrix(c(x["b_log1pBRBRt1_log1pBRBR_t"] + x["b_log1pBRBRt1_log1pBRBR_t:temp"]*3,
0,
x["b_log1pBRBRt1_log1pPtoid_t"] + x["b_log1pBRBRt1_log1pPtoid_t:temp"]*3,
x["b_log1pLYERt1_log1pBRBR_t"] + x["b_log1pLYERt1_log1pBRBR_t:rich"]*rich.cond,
x["b_log1pLYERt1_log1pLYER_t"],
x["b_log1pLYERt1_log1pPtoid_t"],
x["b_log1pPtoidt1_log1pBRBR_t"] + x["b_log1pPtoidt1_log1pBRBR_t:rich"]*rich.cond + x["b_log1pPtoidt1_log1pBRBR_t:temp"]*3,
x["b_log1pPtoidt1_log1pLYER_t"],
0),
ncol = 3, byrow = TRUE)
# constrain intraspecific BRBR effect to a max of zero
if(tmp.mat[1,1] > 0){
tmp.mat[1,1] <- 0
}
# constrain BRBR-Ptoid interaction to a min of zero
if(tmp.mat[3,1] < 0){
tmp.mat[3,1] <- 0
}
tmp.r = matrix(c(x["b_log1pBRBRt1_intercept"] + x["b_log1pBRBRt1_temp"]*3,
x["b_log1pLYERt1_intercept"] + x["b_log1pLYERt1_rich"]*rich.cond,
x["b_log1pPtoidt1_intercept"] + x["b_log1pPtoidt1_rich"]*rich.cond + x["b_log1pPtoidt1_temp"]*3),
ncol = 1)
# constrain BRBR intrinsic growth rate to positive values
if(tmp.r[1] < 0){
tmp.r[1] <- 0.1 # small positive value
}
# constrain Ptoid intrinsic growth rate to negative values
if(tmp.r[3] > 0){
tmp.r[3] <- -0.1 # small negative value
}
Feasibility = as.numeric(FeasibilityBoundary(tmp.mat, tmp.r)["feasibility"])
FeasibilityBoundary23 = as.numeric(FeasibilityBoundary(tmp.mat, tmp.r)["alpha.A23"])
Extinctions = paste0(which(-1*inv(tmp.mat) %*% tmp.r < 0), collapse = ",")
Resilience = max(Re(eigen((tmp.mat + diag(3)))$values))
list(tmp.mat = tmp.mat,
tmp.r = tmp.r,
Feasibility = Feasibility,
FeasibilityBoundary23 = FeasibilityBoundary23,
Extinctions = Extinctions,
Resilience = Resilience)
})
temp23C_stability.df <- data.frame(
temp = "23 C",
posterior_sample = 1:nrow(pp.temp_model),
Feasibility = unlist(lapply(temp23C_stability, FUN = function(x) x$Feasibility)),
FeasibilityBoundary23 = unlist(lapply(temp23C_stability, FUN = function(x) x$FeasibilityBoundary23)),
Extinctions = unlist(lapply(temp23C_stability, FUN = function(x) x$Extinctions)),
Resilience = unlist(lapply(temp23C_stability, FUN = function(x) x$Resilience))
)
all.temp_stability.df <- bind_rows(temp20C_stability.df, temp23C_stability.df) Reproduce Fig. S2
# Boundary determined by LYER-Ptoid (23)
all.temp_stability.df %>%
filter(posterior_sample %in% rsamp) %>% # plot subset of posterior for easier visualization
mutate(n.temp = as.numeric(as.factor(temp))) %>%
ggplot(., aes(x = n.temp, y = FeasibilityBoundary23)) +
geom_line(aes(group = posterior_sample), alpha = 0.1) +
stat_summary(fun.y = mean, geom = "line", color = "red", size = 1) +
stat_summary(fun.y = mean, geom = "point", color = "red", size = 1.5) +
theme_minimal_hgrid() +
scale_x_continuous(name = "Temperature", breaks = c(1,2), labels = c("20 C", "23 C"), expand = c(0.1,0.1)) +
ylab("Critical boundary (L-P)")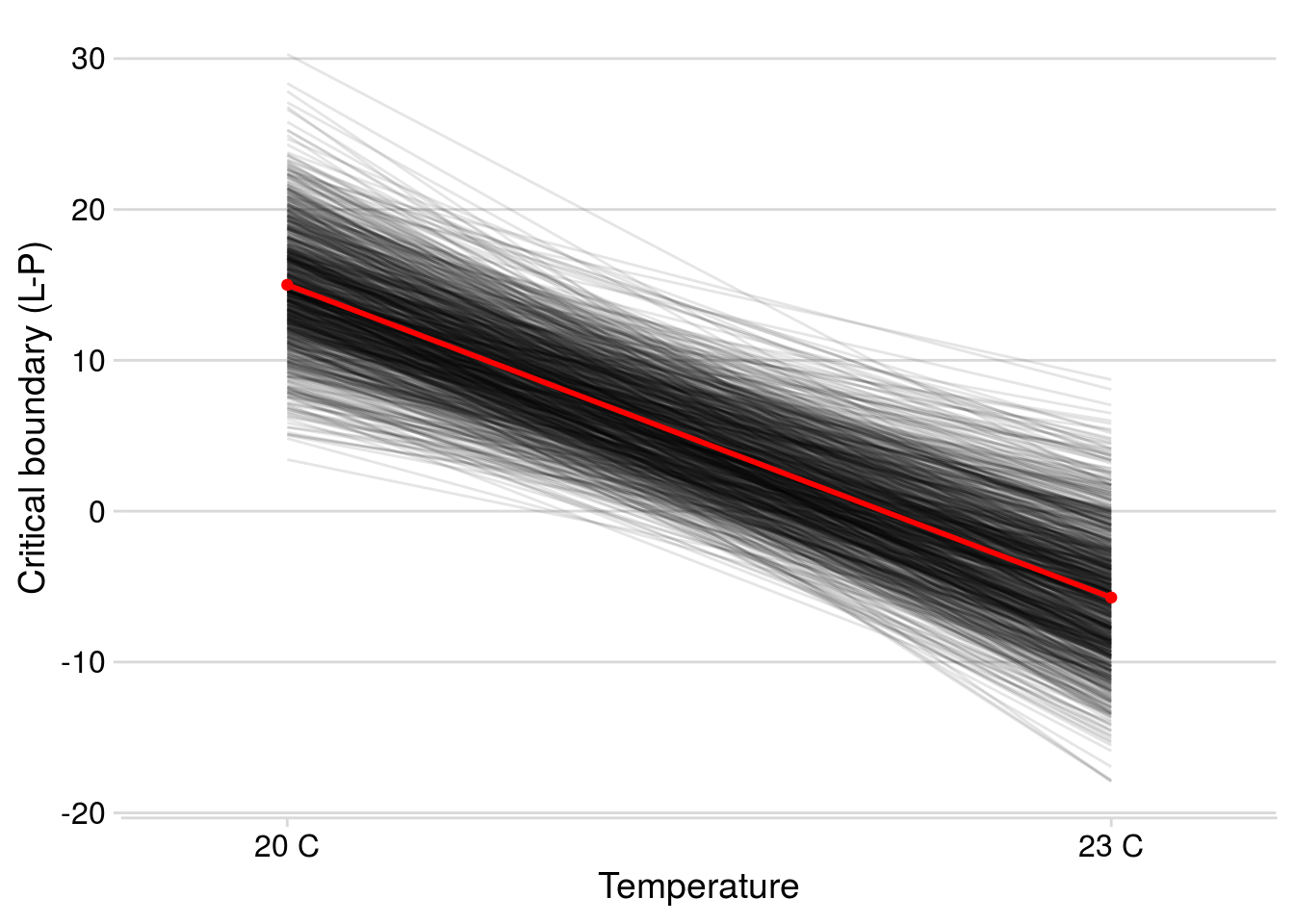
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
## calculate percentage of posterior estimates where temperature decreases structural stability
# organize data
FeasibilityBoundary23_df <- all.temp_stability.df %>%
select(temp, posterior_sample, FeasibilityBoundary23) %>%
spread(key = temp, value = FeasibilityBoundary23) %>%
mutate(Diff = `23 C` - `20 C`)
# calculate percentage
mean(FeasibilityBoundary23_df$Diff < 0)[1] 1Non-equilibrium simulation (Fig. S3)
noneq_temp20 <- SimulateCommunityDynamics(IGR.Vector = temp20.IGR, Interaction.Matrix = (temp20.mat + diag(3)), Duration = 9) %>% mutate(temp = "20 C")
noneq_temp23 <- SimulateCommunityDynamics(IGR.Vector = temp23.IGR, Interaction.Matrix = (temp23.mat + diag(3)), Duration = 7) %>% mutate(temp = "23 C")
bind_rows(noneq_temp20, noneq_temp23) %>%
gather(Species, value = "Abundance", -Week, - temp) %>%
ggplot(., aes(x = Week, y = Abundance, color = Species)) +
geom_hline(yintercept = 0, linetype = "dotted") +
geom_line() +
geom_point() +
facet_wrap(~temp, ncol = 1) +
theme_minimal_hgrid() +
ylab("Abundance (log scale)") +
scale_color_viridis_d(labels = c("B. brassicae","L. erysimi","D. rapae"))
| Version | Author | Date |
|---|---|---|
| 86116c8 | mabarbour | 2020-06-23 |
Save analysis
Write out an .RData file to use for the Supplementary Online Materiak.
save.image(file = "output/structural-stability.RData")
sessionInfo()R version 3.6.3 (2020-02-29)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 16.04.6 LTS
Matrix products: default
BLAS: /usr/lib/libblas/libblas.so.3.6.0
LAPACK: /usr/lib/lapack/liblapack.so.3.6.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] rgl_0.100.54 knitr_1.26 tidybayes_2.0.1 matlib_0.9.2
[5] cowplot_1.0.0 forcats_0.4.0 stringr_1.4.0 dplyr_0.8.3
[9] purrr_0.3.3 readr_1.3.1 tidyr_1.0.2 tibble_2.1.3
[13] ggplot2_3.2.1 tidyverse_1.3.0 brms_2.12.7 Rcpp_1.0.2
[17] RCurl_1.95-4.12 bitops_1.0-6
loaded via a namespace (and not attached):
[1] colorspace_1.4-1 ellipsis_0.3.0 rio_0.5.16
[4] ggridges_0.5.1 rsconnect_0.8.13 rprojroot_1.3-2
[7] markdown_1.1 base64enc_0.1-3 fs_1.3.1
[10] rstudioapi_0.10 rstan_2.19.2 svUnit_0.7-12
[13] DT_0.6 mvtnorm_1.0-10 lubridate_1.7.4
[16] xml2_1.2.2 bridgesampling_0.6-0 shinythemes_1.1.2
[19] bayesplot_1.7.0 jsonlite_1.6 workflowr_1.6.0
[22] broom_0.5.2 dbplyr_1.4.2 shiny_1.3.2
[25] compiler_3.6.3 httr_1.4.1 backports_1.1.4
[28] assertthat_0.2.1 Matrix_1.2-17 lazyeval_0.2.2
[31] cli_1.1.0 later_1.0.0 htmltools_0.3.6
[34] prettyunits_1.0.2 tools_3.6.3 igraph_1.2.4.1
[37] coda_0.19-2 gtable_0.3.0 glue_1.3.1
[40] reshape2_1.4.3 carData_3.0-2 cellranger_1.1.0
[43] vctrs_0.2.2 nlme_3.1-140 crosstalk_1.0.0
[46] xfun_0.9 ps_1.3.0 openxlsx_4.1.0.1
[49] rvest_0.3.5 mime_0.7 miniUI_0.1.1.1
[52] lifecycle_0.1.0 gtools_3.8.1 MASS_7.3-51.4
[55] zoo_1.8-6 scales_1.0.0 colourpicker_1.0
[58] hms_0.5.3 promises_1.0.1 Brobdingnag_1.2-6
[61] parallel_3.6.3 inline_0.3.15 shinystan_2.5.0
[64] curl_4.0 yaml_2.2.0 gridExtra_2.3
[67] loo_2.2.0 StanHeaders_2.19.0 stringi_1.4.3
[70] dygraphs_1.1.1.6 zip_2.0.3 pkgbuild_1.0.3
[73] manipulateWidget_0.10.0 rlang_0.4.4 pkgconfig_2.0.2
[76] matrixStats_0.54.0 evaluate_0.14 lattice_0.20-38
[79] labeling_0.3 rstantools_2.0.0 htmlwidgets_1.3
[82] processx_3.3.1 tidyselect_0.2.5 plyr_1.8.4
[85] magrittr_1.5 R6_2.4.0 generics_0.0.2
[88] DBI_1.0.0 foreign_0.8-71 pillar_1.4.2
[91] haven_2.2.0 whisker_0.3-2 withr_2.1.2
[94] xts_0.11-2 abind_1.4-5 car_3.0-3
[97] modelr_0.1.5 crayon_1.3.4 arrayhelpers_1.1-0
[100] rmarkdown_2.0 viridis_0.5.1 grid_3.6.3
[103] readxl_1.3.1 data.table_1.12.2 callr_3.2.0
[106] git2r_0.26.1 threejs_0.3.1 reprex_0.3.0
[109] digest_0.6.20 webshot_0.5.1 xtable_1.8-4
[112] httpuv_1.5.1 stats4_3.6.3 munsell_0.5.0
[115] viridisLite_0.3.0 shinyjs_1.0