Genome Browser 2022
Marie Saitou
4/23/2022
Last updated: 2022-04-25
Checks: 7 0
Knit directory: Bio326/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20210128) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 173a1b4. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .RData
Ignored: .Rhistory
Ignored: analysis/.DS_Store
Ignored: analysis/popgen.simu.nb.html
Untracked files:
Untracked: BIO326 URL genome annotatin computer lab_24_MAR_2021.docx
Untracked: BIO326-121VGenomesequencingBIO326-121VGenomsekvensering;verktøyoganalyser-BIO326-121VGenomesequencing_PhillipByronPope.pdf
Untracked: BIO326-RNAseq.pptx
Untracked: BIO326-genome/
Untracked: BIO326.MS.10th_FEB_2021function.pptx
Untracked: BIO326_Introduction to sequence technology and protocols_3rd_FEB_2021.pdf
Untracked: BIO326_Introduction to sequence technology and protocols_3rd_FEB_2021.pptx
Untracked: BIO326_RNAseq_5th_FEB_2021.pptx
Untracked: BIO326_SQK-RAD004 DNA challenge.docx
Untracked: BIO326_visual_30_APR_2021.pptx
Untracked: Bio326.2022.1.Rmd
Untracked: Bio326.genome.html
Untracked: Nanopore_SumStatQC_Tutorial.Rmd
Untracked: PCRdemo.R
Untracked: Pig_mutation_hist.csv
Untracked: PopGenBio326.322/
Untracked: RNAseq.Rplot.pdf
Untracked: Untitled.R
Untracked: [eng]BIO326-121VGenomesequencingBIO326-121VGenomsekvensering;verktøyoganalyser-BIO326-121VGenomesequencing_PhillipByronPope.mht
Untracked: [eng]BIO326-121VGenomesequencingBIO326-121VGenomsekvensering;verktøyoganalyser-BIO326-121VGenomesequencing_PhillipByronPope.pdf
Untracked: analysis/AnimalGenomics.Rmd
Untracked: analysis/AnimalGenomicsVariant2022.Rmd
Untracked: analysis/RNAseq_for_lab.Rmd
Untracked: prepare.txt
Untracked: samples.xlsx
Untracked: test/
Untracked: trial/
Untracked: vis.xlsx
Untracked: workflowR.bio326.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/genomebrowser_for_lab.Rmd) and HTML (docs/genomebrowser_for_lab.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 173a1b4 | mariesaitou | 2022-04-25 | wflow_publish(c(“analysis/genomebrowser_for_lab.Rmd”)) |
| html | 366d293 | mariesaitou | 2022-04-25 | Build site. |
| Rmd | 45cacf3 | mariesaitou | 2022-04-25 | wflow_publish(c(“analysis/genomebrowser_for_lab.Rmd”)) |
| html | f5f4065 | mariesaitou | 2022-04-25 | Build site. |
| Rmd | 8452478 | mariesaitou | 2022-04-25 | image data set |
| html | 8452478 | mariesaitou | 2022-04-25 | image data set |
| html | ef2a13c | mariesaitou | 2022-04-25 | Build site. |
| Rmd | 6f44b4b | mariesaitou | 2022-04-25 | wflow_publish(c(“analysis/genomebrowser_for_lab.Rmd”)) |
| html | f12c39f | mariesaitou | 2022-04-25 | Build site. |
| Rmd | c7517b0 | mariesaitou | 2022-04-25 | Add my first analysis |
0 Introduction
National Center for Biotechnology Information. Good for primer design and litereture search.
The grafical use intereface is light and helpful. Most non-model species genomes are not up-to-date, but you can send them a request.
Most non-model species friendly. Good for evolutionary and functional genetics analysis.
1 Primer Design
Mission: You want to investigate if TP53 gene is expressed in a particular tissue of pig and want to design a primer set for its cDNA.
1-1 Search for gene info at NCBI
Go to:NCBI and search for “Sus scrofa TP53”.

Now you can see the gene information, exon-intron structure and gene/progein IDs. 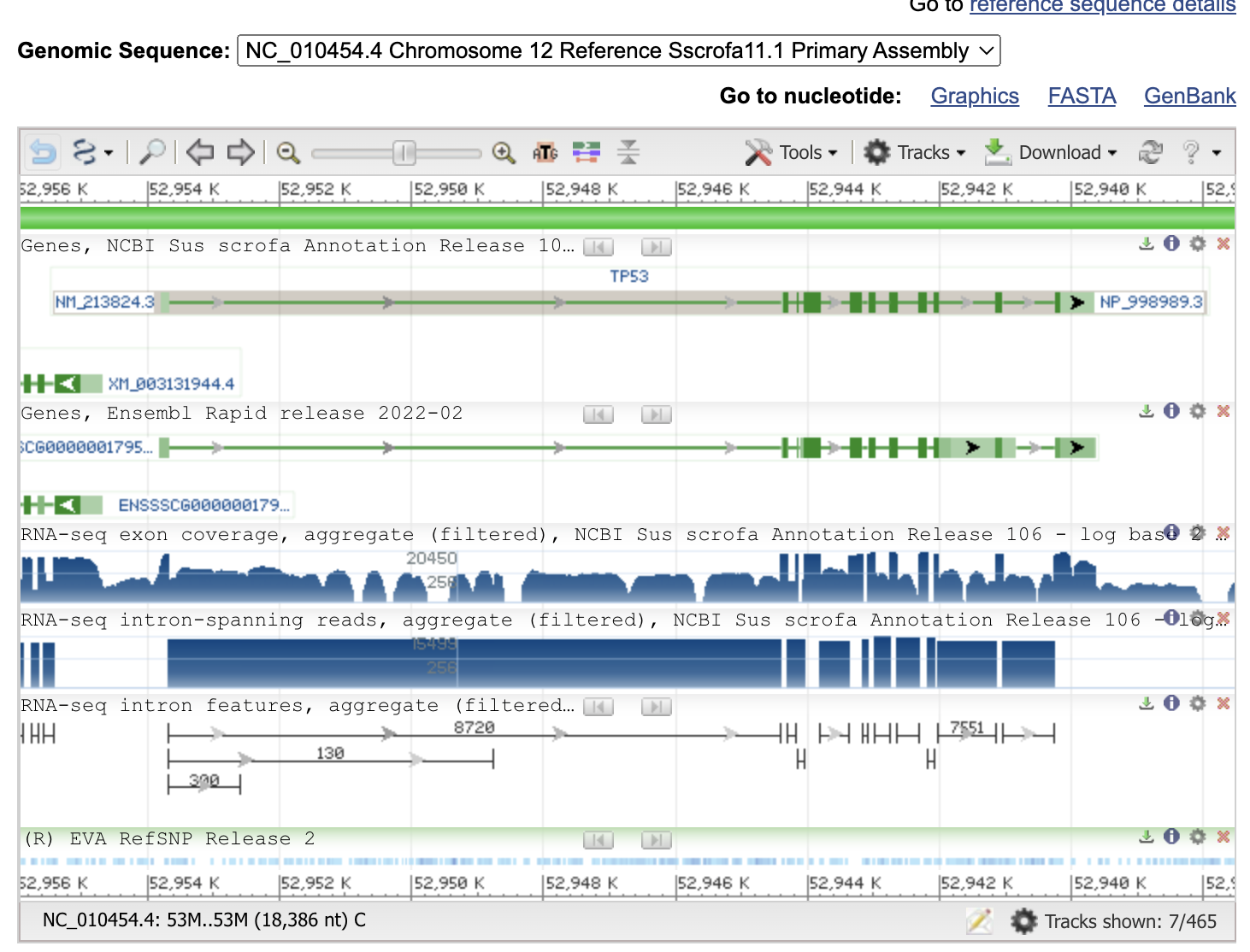
Thin green lines are the introns, thick green boxes are exons.
Also when you scroll it down, you can see the expression pattern of this gene in various tissues. 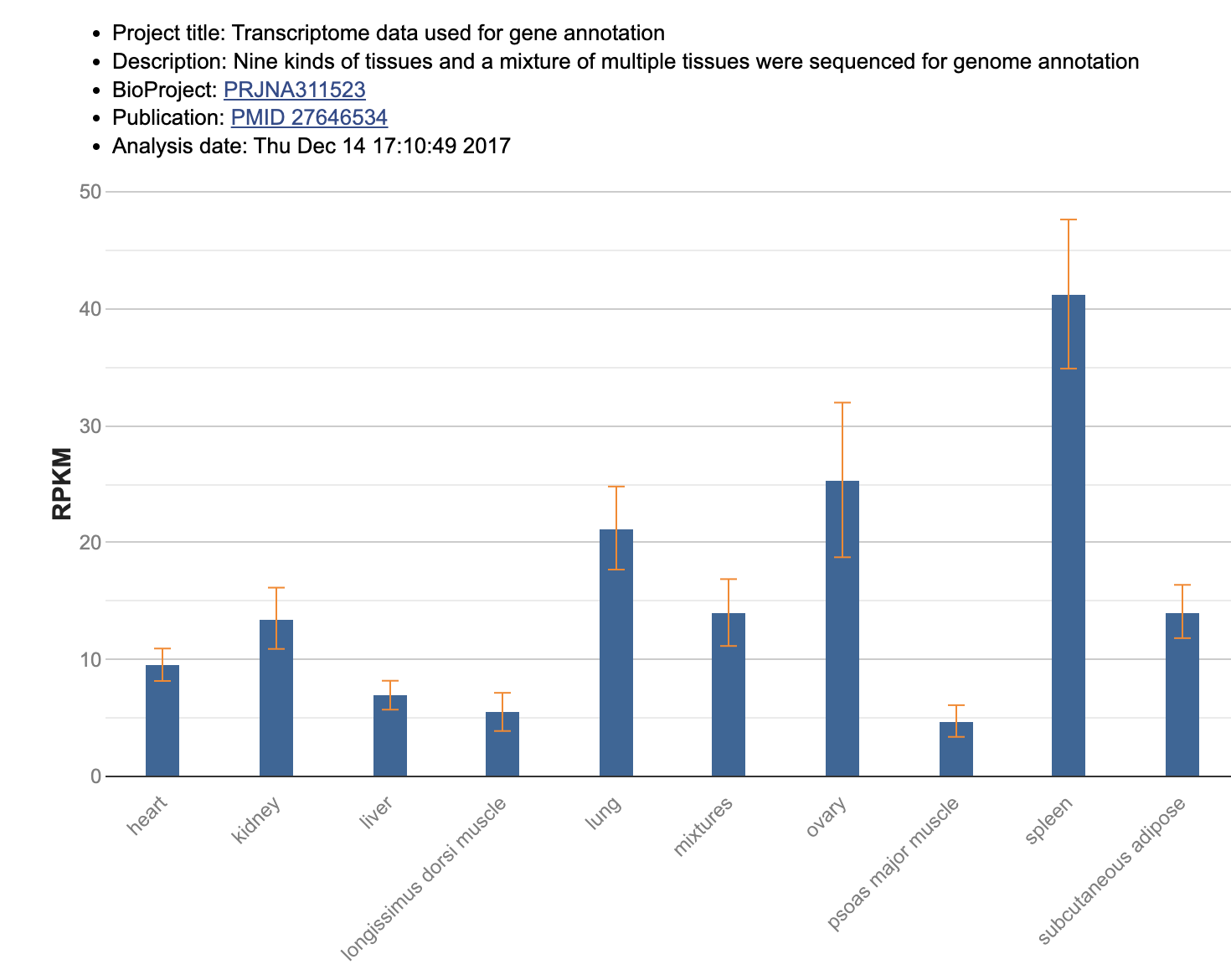
RPKM is a unit for gene expression
Furthere below, you can see relevant publications and expected function of this gene.
1-2 Primer blast - primer design
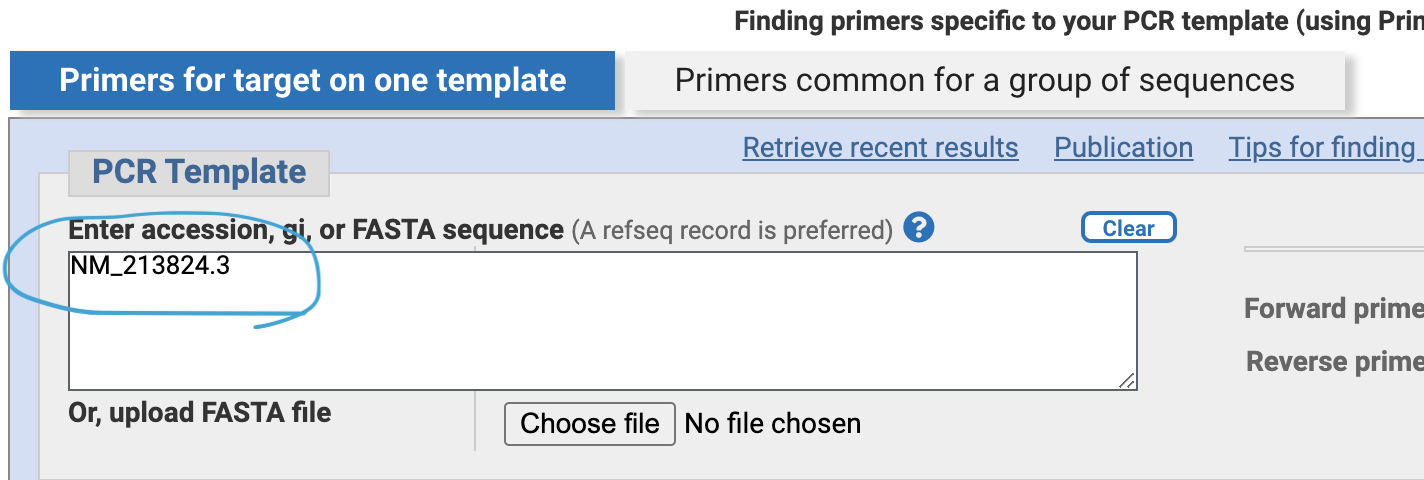
Now let’s design a primer set to amplify the cDNA. Put the mouse cursor on the gene image, and copy and note the refseq mRNA ID (NM_21824.3)
And go to:NCBI primer blast
Put the transcript ID “NM_21824.3” in the PCR template box.
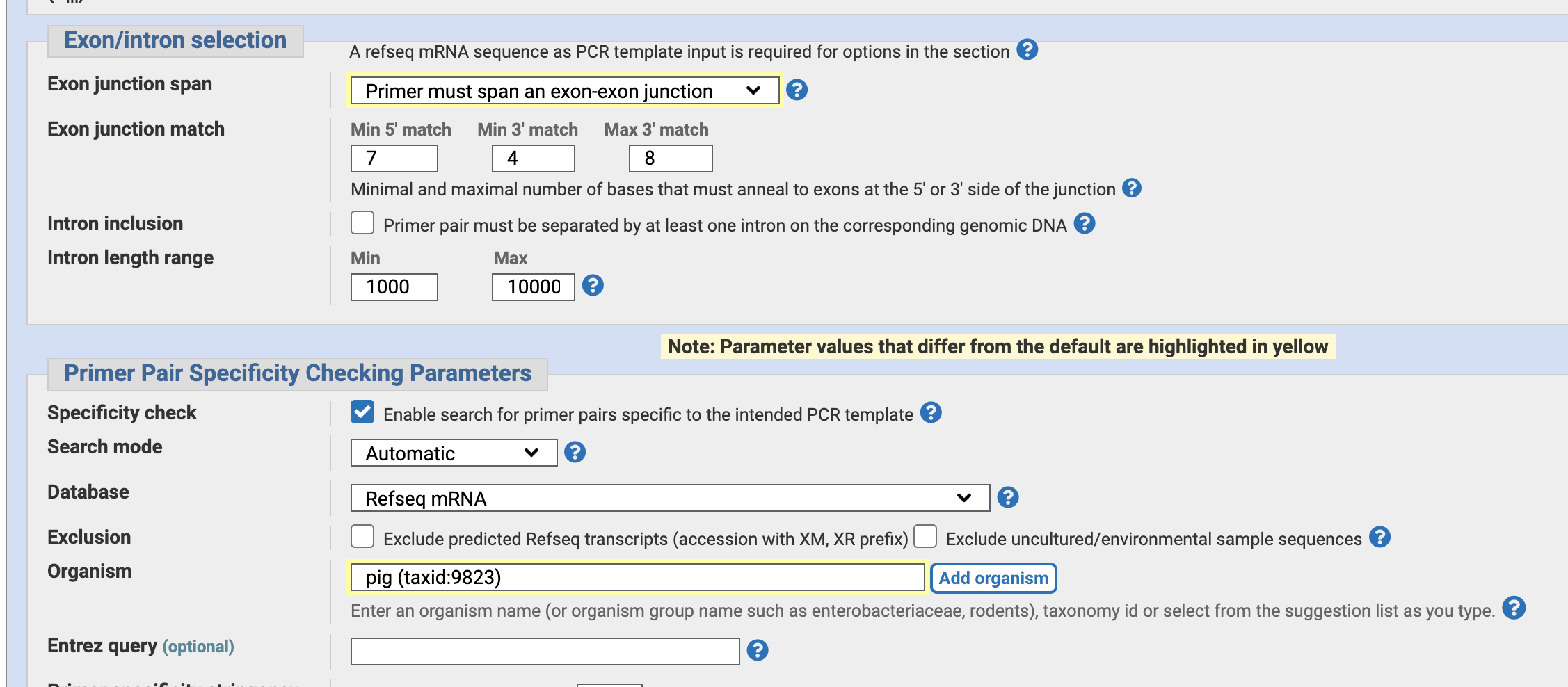
Specify “Primer mast span an exon-exon junction”. — to avoid false positive PCR amplification of genomic DNA and only observe mRNA by PCR.
Also, Specify tne organism “pig”.
Wait for a while…
Then the algorism gaves us the candidate primer sets. 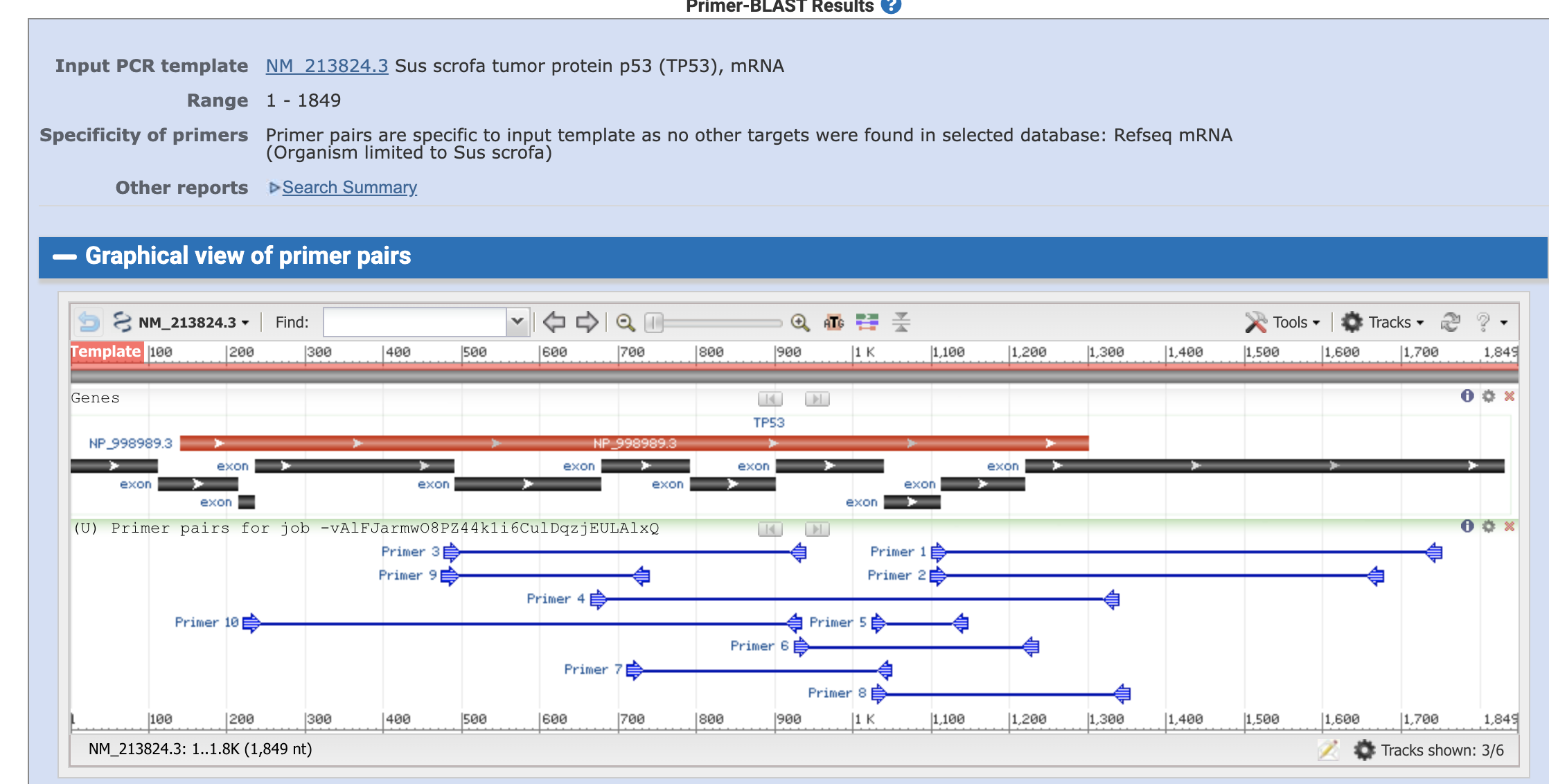
black bars are the exons and the blue thin lines are the primers and planned amplified region.
We can take a close look on each primer pair by clicking the primers.

We can now see the primer sequences, locations, melting tempreture, GC content, self complementarity, and the product length. It also tells us that the forward primer spans a exon-exon junction (location 1112+1113 th of the nucleotide)
1-3 Primer blast - primer reuse
Now you want to know if the primer sets can be used for other species. To do so, we can extract the target template sequence from the pig transcriptome data and compare it for those of other species.
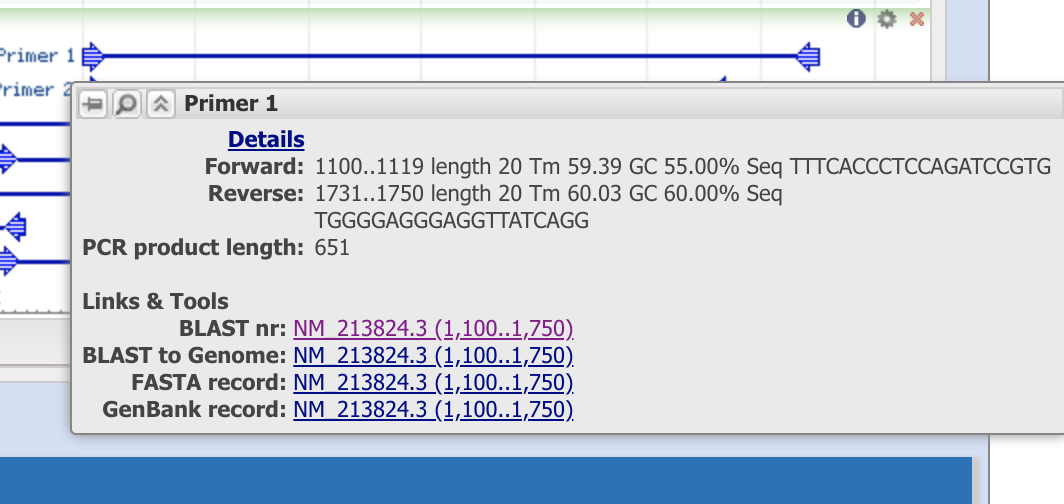
When you put the cursor on primer1, it shows that this primer set will amplity the “1,100 - 1,750 th nucleotide” of the sequence from mRNA, refseq ID (NM_21824.3). Note the info so see if the PCR template can be observed in other species.
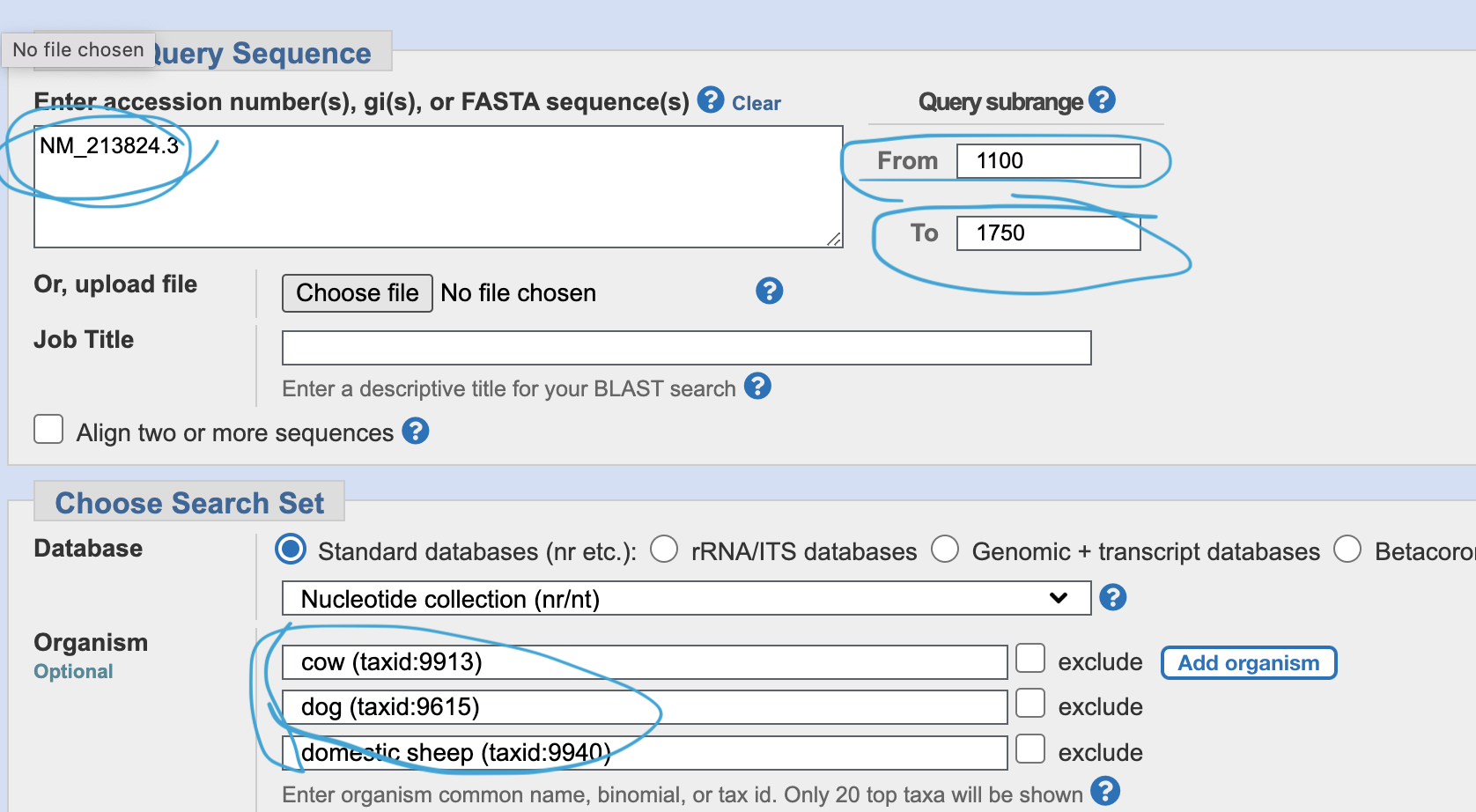
Go to primer blast, and input NM_21824.3, and range “from 1100 - to 1750”. Add your favorite species in the “organism” space.
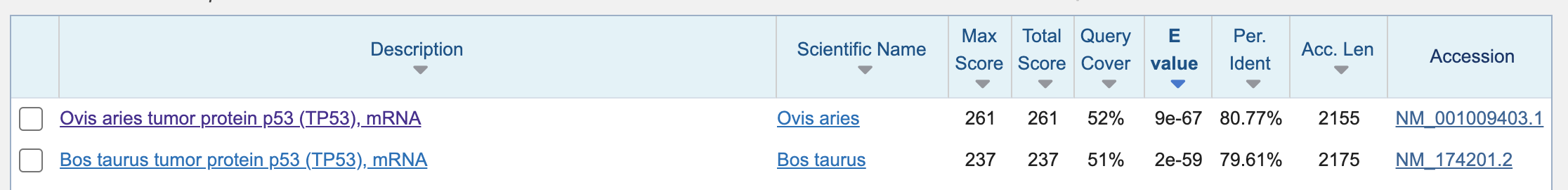
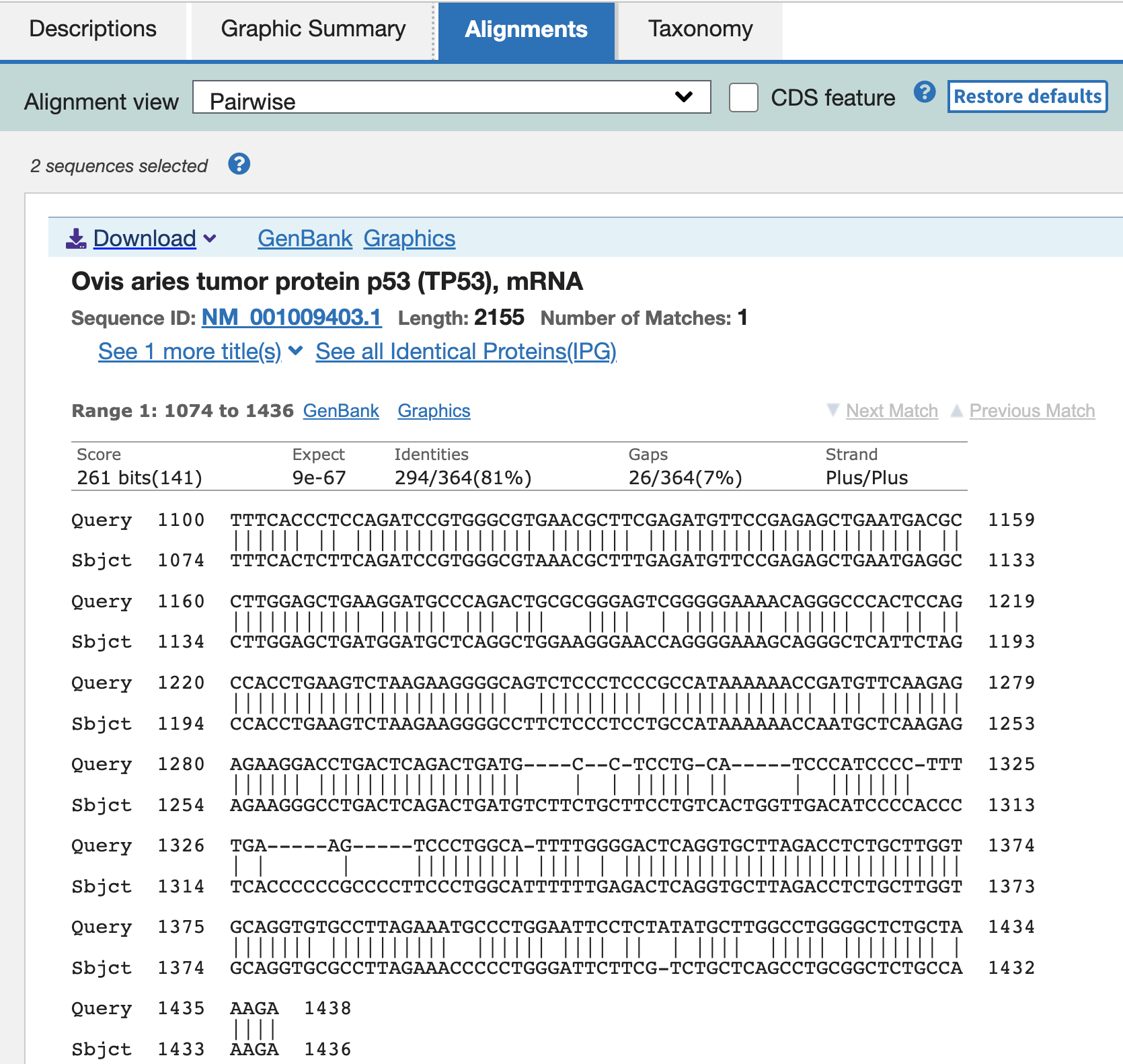
So probably we have to design new primer sets for these species…
2 The effect of genetic variants
Mission: You found a genetic variant in your sequenced individual. You want to investigate the potential effect of the variants.
2-2 Variant effect predictor
Assume that you found the following deletion polymorphism at in a rabbit genome.
12: 107,236,296-107,236,969
Variant Effect Predictor And input:
Species - rabbit (Oryctolagus_cuniculus)
Variant - 12 107236296 107236969 DEL + deletion1
the variant format, left to rignt … chromosome, starting point, ending point, kind, strand, variant ID (you can name it as you like) Variants.
You can also find various acceptable input formats here
2-2 What is this gene doing?
2-3 Comparative Analysis
Rabbit and Alpaca
3 the genomic architecture
3-1
3-2
genomic landscape genetic variants
UCSC - check
chr6:43,426,669-43,433,274
Repeats.
"" RepeatMasker Information
Name: (GGAT)n
>danRer11_rmsk_(GGAT)n range=chr6:43428241-43428346 5'pad=0 3'pad=0 strand=+ repeatMasking=none
GGATGGATGGATGGATGGATGGATGGATGGATGGATGGATGGATGGATGG
ATGGATGGATCGATGGAAGGATGGATGGATGGAAGGATGGATGGACAGAT
GGATGGVariant.
sessionInfo()R version 4.1.2 (2021-11-01)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur 10.16
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] workflowr_1.7.0
loaded via a namespace (and not attached):
[1] Rcpp_1.0.8.2 bslib_0.3.1 compiler_4.1.2 pillar_1.7.0
[5] later_1.3.0 git2r_0.29.0 jquerylib_0.1.4 tools_4.1.2
[9] getPass_0.2-2 digest_0.6.29 jsonlite_1.8.0 evaluate_0.15
[13] tibble_3.1.6 lifecycle_1.0.1 pkgconfig_2.0.3 rlang_1.0.2
[17] cli_3.2.0 rstudioapi_0.13 yaml_2.3.5 xfun_0.30
[21] fastmap_1.1.0 httr_1.4.2 stringr_1.4.0 knitr_1.37
[25] sass_0.4.0 fs_1.5.2 vctrs_0.3.8 rprojroot_2.0.2
[29] glue_1.6.2 R6_2.5.1 processx_3.5.2 fansi_1.0.2
[33] rmarkdown_2.13 callr_3.7.0 magrittr_2.0.2 whisker_0.4
[37] ps_1.6.0 promises_1.2.0.1 htmltools_0.5.2 ellipsis_0.3.2
[41] httpuv_1.6.5 utf8_1.2.2 stringi_1.7.6 crayon_1.5.0