Merge samples
Mechthild Lütge
14 May 2020
Last updated: 2022-07-19
Checks: 6 1
Knit directory: humanCardiacFibroblasts/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R
Markdown file created these results, you’ll want to first commit it to
the Git repo. If you’re still working on the analysis, you can ignore
this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20210903) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 3e98bf3. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/figure/
Ignored: data/GSEA/
Ignored: data/humanFibroblast/
Untracked files:
Untracked: analysis/DEgenesGZplusSG_Groups.Rmd
Untracked: analysis/DEgenesGZplusSG_SubsetLT.Rmd
Untracked: figure/DEgenesGZplusSG_Groups.Rmd/
Untracked: figure/DEgenesGZplusSG_SubsetLT.Rmd/
Unstaged changes:
Modified: analysis/integrateAcrossPatientsGZplusSG.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with
wflow_publish() to start tracking its development.
load packages
suppressPackageStartupMessages({
library(SingleCellExperiment)
library(tidyverse)
library(Seurat)
library(magrittr)
library(dplyr)
library(purrr)
library(ggplot2)
library(here)
library(runSeurat3)
library(ggsci)
library(ggpubr)
library(pheatmap)
library(viridis)
library(sctransform)
library(fgsea)
library(grid)
library(gridExtra)
library(clusterProfiler)
library(org.Hs.eg.db)
library(DOSE)
library(enrichplot)
library(msigdbr)
})sign plot funct
## random plotting order
shuf <- function(df){
return(df[sample(1:dim(df)[1], dim(df)[1]),])
}
## adapted from CellMixS
visGroup_adapt <- function (sce,group,dim_red = "TSNE",col_group=pal_nejm()(8))
{
if (!is(sce, "SingleCellExperiment")) {
stop("Error:'sce' must be a 'SingleCellExperiment' object.")
}
if (!group %in% names(colData(sce))) {
stop("Error: 'group' variable must be in 'colData(sce)'")
}
cell_names <- colnames(sce)
if (!dim_red %in% "TSNE") {
if (!dim_red %in% reducedDimNames(sce)) {
stop("Please provide a dim_red method listed in reducedDims of sce")
}
red_dim <- as.data.frame(reducedDim(sce, dim_red))
}
else {
if (!"TSNE" %in% reducedDimNames(sce)) {
if ("logcounts" %in% names(assays(sce))) {
sce <- runTSNE(sce)
}
else {
sce <- runTSNE(sce, exprs_values = "counts")
}
}
red_dim <- as.data.frame(reducedDim(sce, "TSNE"))
}
colnames(red_dim) <- c("red_dim1", "red_dim2")
df <- data.frame(sample_id = cell_names, group_var = colData(sce)[,
group], red_Dim1 = red_dim$red_dim1, red_Dim2 = red_dim$red_dim2)
t <- ggplot(shuf(df), aes_string(x = "red_Dim1", y = "red_Dim2")) +
xlab(paste0(dim_red, "_1")) + ylab(paste0(dim_red, "_2")) +
theme_void() + theme(aspect.ratio = 1,
panel.grid.minor = element_blank(),
panel.grid.major = element_line(color = "grey", size = 0.3))
t_group <- t + geom_point(size = 1, alpha = 0.7,
aes_string(color = "group_var")) +
guides(color = guide_legend(override.aes = list(size = 1),
title = group)) + ggtitle(group)
if (is.numeric(df$group_var)) {
t_group <- t_group + scale_color_viridis(option = "D")
}
else {
t_group <- t_group + scale_color_manual(values = col_group)
}
t_group
}integrate data
basedir <- here()
seurat <- readRDS(file = paste0(basedir,
"/data/humanHeartsPlusGraz_intPatients_merged",
"labeled_seurat.rds"))
myoGrp <- c("GZ1","GZ4","GZ6","SG29","SG32")
myoLTGrp <- c("GZ2","GZ3","GZ5","GZ7","SG31")
CtrlGrp <- c("GZ8","GZ10","GZ11","GZ12")
seurat$cond2 <- "HH"
seurat$cond2[which(seurat$ID %in% myoGrp)] <- "MyocarditisHT"
seurat$cond2[which(seurat$ID %in% myoLTGrp)] <- "MyocarditisLT"color vectors
colPal <- pal_igv()(length(levels(seurat)))
colTec <- pal_jama()(length(unique(seurat$technique)))
colSmp <- c(pal_uchicago()(8), pal_npg()(8), pal_aaas()(10))[1:length(unique(seurat$dataset))]
colCond <- pal_npg()(length(unique(seurat$cond2)))
colID <- c(pal_jco()(10), pal_npg()(10))[1:length(unique(seurat$ID))]
colOrig <- pal_aaas()(length(unique(seurat$origin)))
colIso <- pal_nejm()(length(unique(seurat$isolation)))
colProc <- pal_aaas()(length(unique(seurat$processing)))
colLab <- pal_futurama()(length(unique(seurat$label)))
names(colPal) <- levels(seurat)
names(colTec) <- unique(seurat$technique)
names(colSmp) <- unique(seurat$dataset)
names(colCond) <- unique(seurat$cond2)
names(colID) <- unique(seurat$ID)
names(colOrig) <- unique(seurat$origin)
names(colIso) <- unique(seurat$isolation)
names(colProc) <- unique(seurat$processing)
names(colLab) <- unique(seurat$label)vis data
clusters
DimPlot(seurat, reduction = "umap", cols=colPal)+
theme_bw() +
theme(axis.text = element_blank(), axis.ticks = element_blank(),
panel.grid.minor = element_blank()) +
xlab("UMAP1") +
ylab("UMAP2")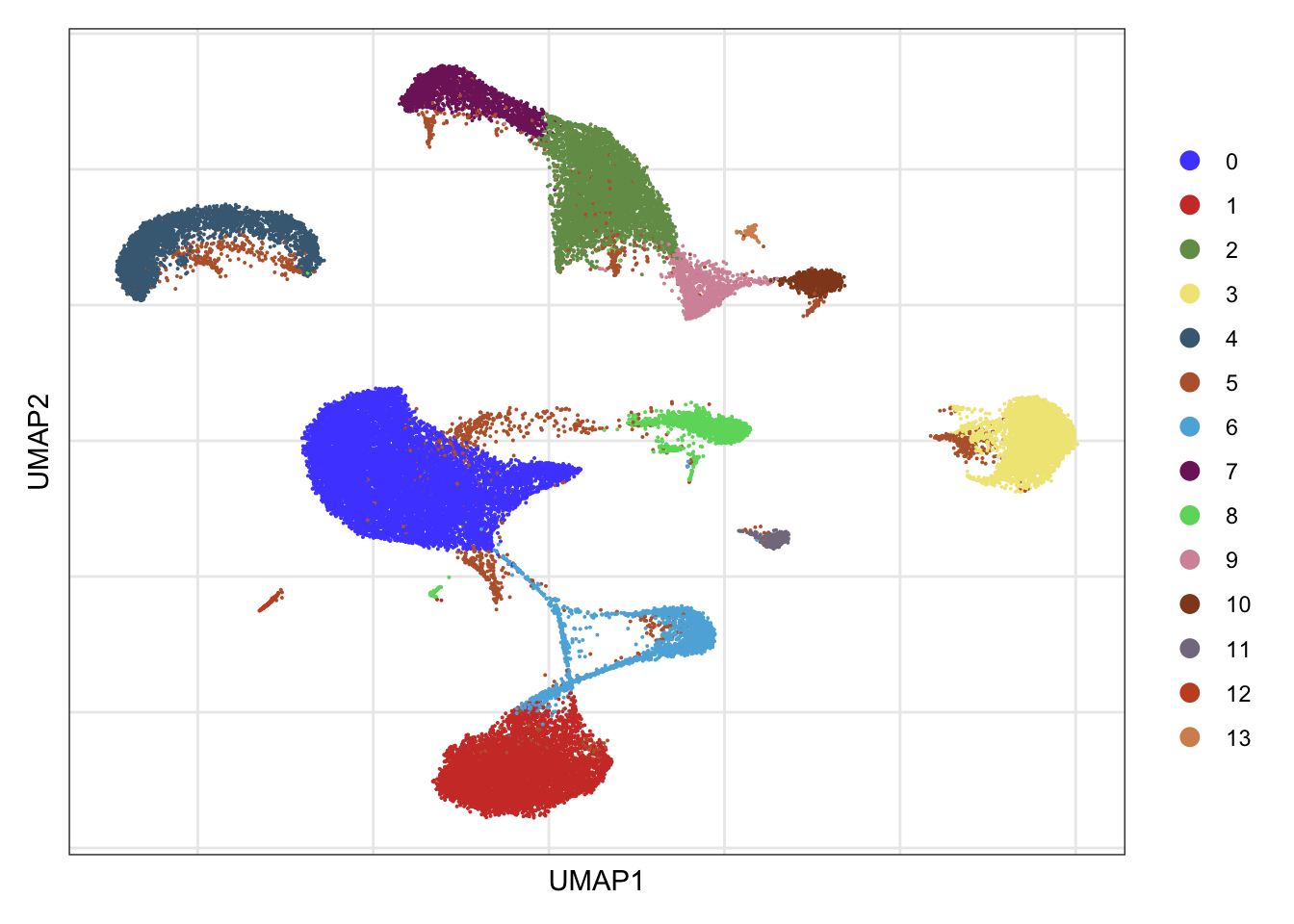
label
DimPlot(seurat, reduction = "umap", group.by = "label", cols=colLab)+
theme_bw() +
theme(axis.text = element_blank(), axis.ticks = element_blank(),
panel.grid.minor = element_blank()) +
xlab("UMAP1") +
ylab("UMAP2")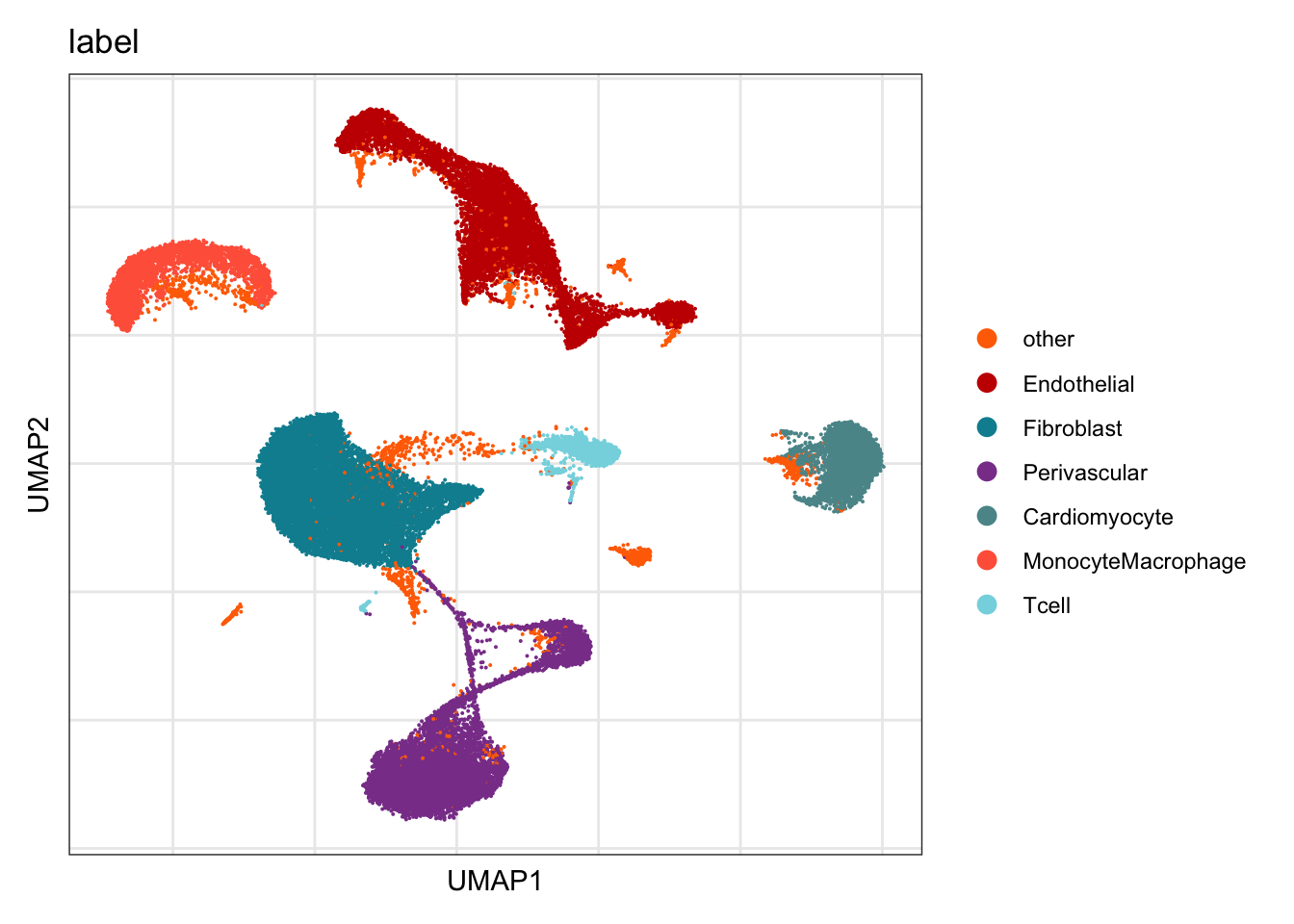
technique
DimPlot(seurat, reduction = "umap", group.by = "technique", cols=colTec)+
theme_bw() +
theme(axis.text = element_blank(), axis.ticks = element_blank(),
panel.grid.minor = element_blank()) +
xlab("UMAP1") +
ylab("UMAP2")
Sample
DimPlot(seurat, reduction = "umap", group.by = "dataset", cols=colSmp)+
theme_bw() +
theme(axis.text = element_blank(), axis.ticks = element_blank(),
panel.grid.minor = element_blank()) +
xlab("UMAP1") +
ylab("UMAP2")
ID
DimPlot(seurat, reduction = "umap", group.by = "ID", cols=colID)+
theme_bw() +
theme(axis.text = element_blank(), axis.ticks = element_blank(),
panel.grid.minor = element_blank()) +
xlab("UMAP1") +
ylab("UMAP2")
Origin
DimPlot(seurat, reduction = "umap", group.by = "origin", cols=colOrig)+
theme_bw() +
theme(axis.text = element_blank(), axis.ticks = element_blank(),
panel.grid.minor = element_blank()) +
xlab("UMAP1") +
ylab("UMAP2")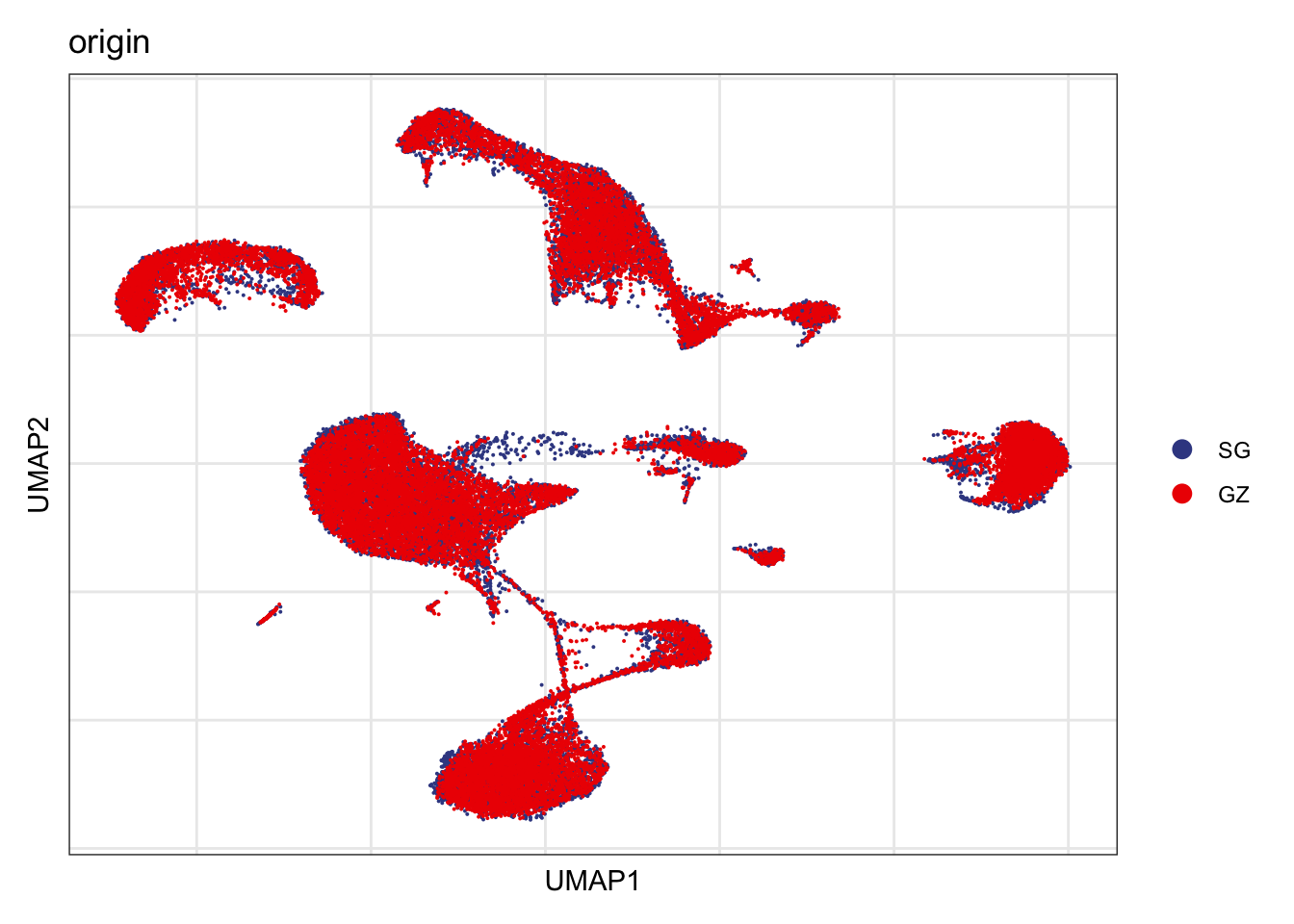
isolation
DimPlot(seurat, reduction = "umap", group.by = "isolation", cols=colIso)+
theme_bw() +
theme(axis.text = element_blank(), axis.ticks = element_blank(),
panel.grid.minor = element_blank()) +
xlab("UMAP1") +
ylab("UMAP2")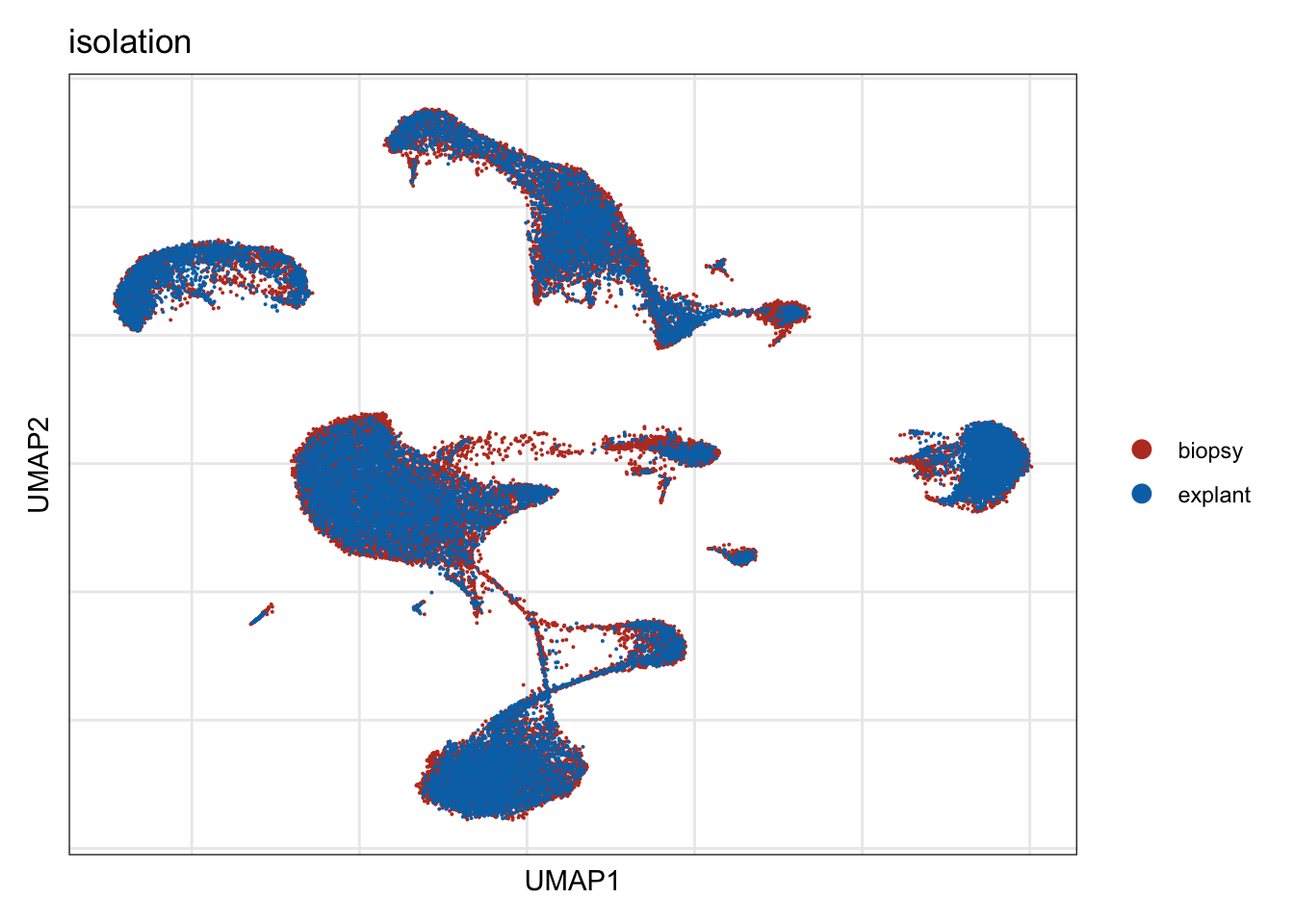
cond
DimPlot(seurat, reduction = "umap", group.by = "cond2", cols=colCond)+
theme_bw() +
theme(axis.text = element_blank(), axis.ticks = element_blank(),
panel.grid.minor = element_blank()) +
xlab("UMAP1") +
ylab("UMAP2")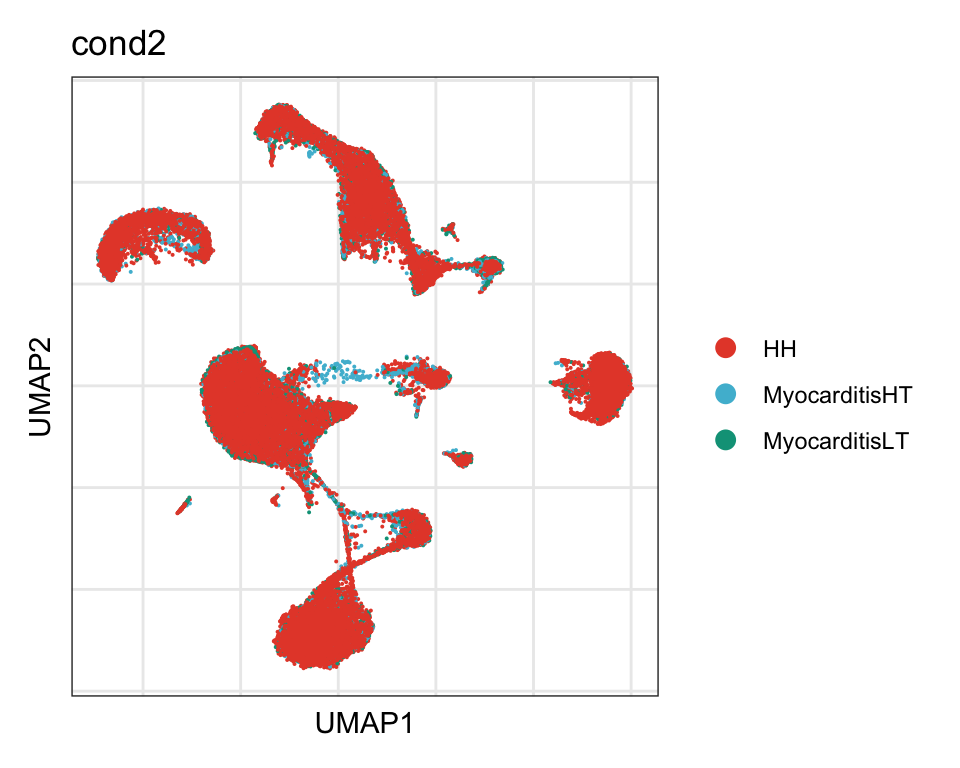
processing
DimPlot(seurat, reduction = "umap", group.by = "processing", cols=colProc)+
theme_bw() +
theme(axis.text = element_blank(), axis.ticks = element_blank(),
panel.grid.minor = element_blank()) +
xlab("UMAP1") +
ylab("UMAP2")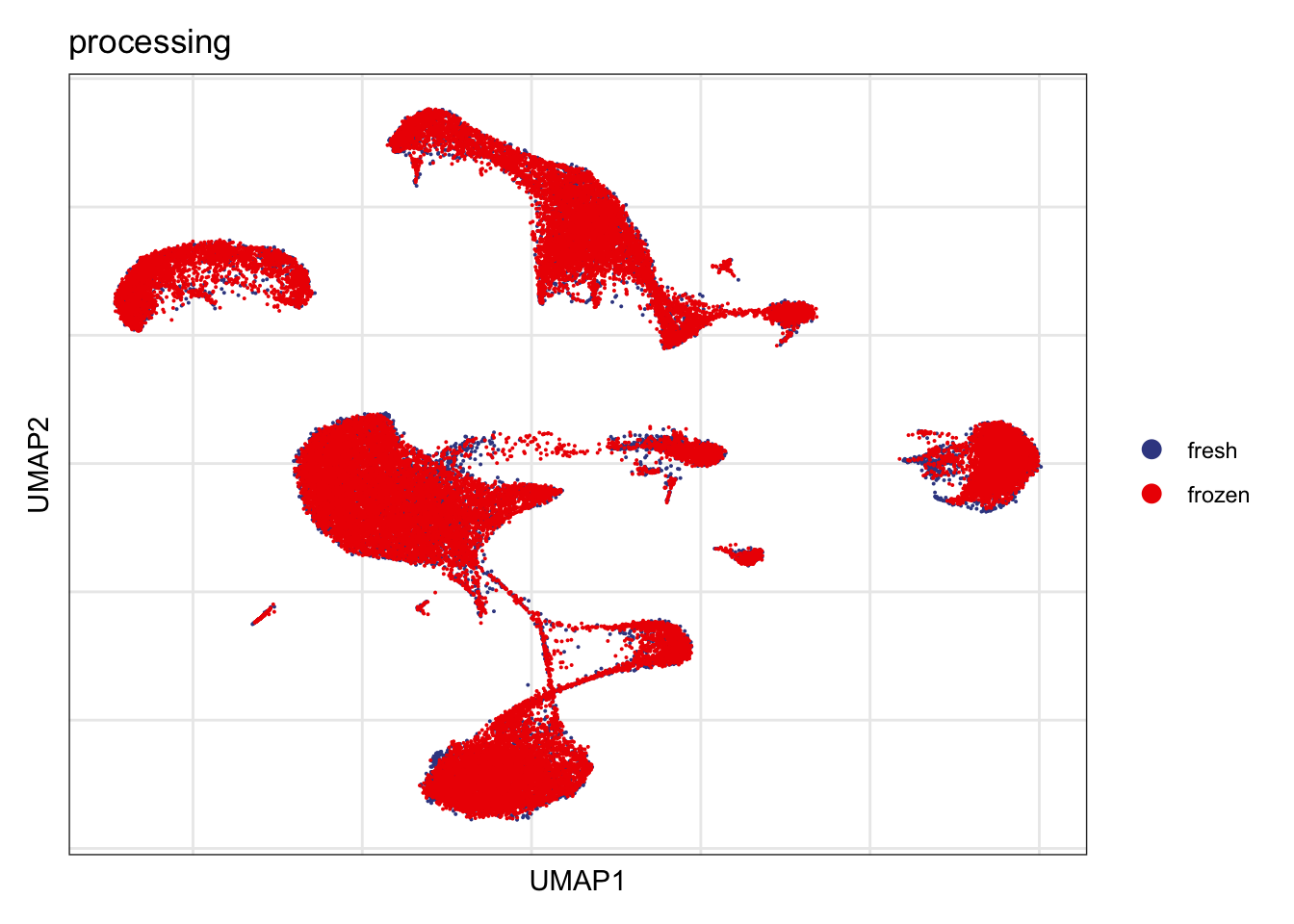
overall DE genes HH vs MyocarditisLT
seurat <- subset(seurat, cond2 == "MyocarditisHT", invert=T)
Idents(seurat) <- seurat$cond2
DEgenes <-FindAllMarkers(seurat, only.pos=T, logfc.threshold = 0.1,
min.pct = 0.01)
clVec <- unique(seurat$cond2)
GOcons <- lapply(clVec, function(cl){
clustDE_DatSub <- DEgenes[which(DEgenes$cluster == cl),] %>%
mutate(ENS=gsub("\\..*$", "", gene)) #%>%
#slice_min(., max_pval, n=200)
egoSS <- enrichGO(gene = unique(clustDE_DatSub$ENS),
OrgDb = org.Hs.eg.db,
keyType = 'ENSEMBL',
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
qvalueCutoff = 0.05)
egoSS <- setReadable(egoSS, OrgDb = org.Hs.eg.db)
egoSSres <- egoSS@result %>% filter(p.adjust < 0.05) %>%
mutate(subset=cl)
})
names(GOcons) <- clVec
## table to select pathways
GOconsDat <- do.call("rbind", GOcons)
write.table(GOconsDat, quote=F, row.names = T, col.names = T, sep= "\t",
file = paste0(basedir,"/data/humanHeartsPlusGraz_intPatients_",
"merged_SubsetLT_overallDEGO.txt"))cw DE genes
Idents(seurat) <- seurat$cond2
grpVec <- unique(seurat$label)
clustDE <- lapply(grpVec, function(grp){
grpSub <- unique(seurat$label)[which(
unique(seurat$label)==grp)]
seuratSub <- subset(seurat, label == grpSub)
DEgenes <-FindAllMarkers(seuratSub, only.pos=T, logfc.threshold = 0.1,
min.pct = 0.01)
if(nrow(DEgenes)>1){
DEgenes <- DEgenes %>% filter(p_val_adj<0.01) %>%
mutate(group=paste0(grp, "_", cluster)) %>%
mutate(geneID=gsub(".*\\.", "", gene)) %>%
filter(nchar(geneID)>1)
}
})
names(clustDE) <- grpVec
clustDE_Dat <- data.frame(do.call("rbind", clustDE))
write.table(clustDE_Dat,
file=paste0(basedir,
"/data/humanHeartsPlusGraz_intPatients_",
"merged_SubsetLT_cwDEGenes.txt"),
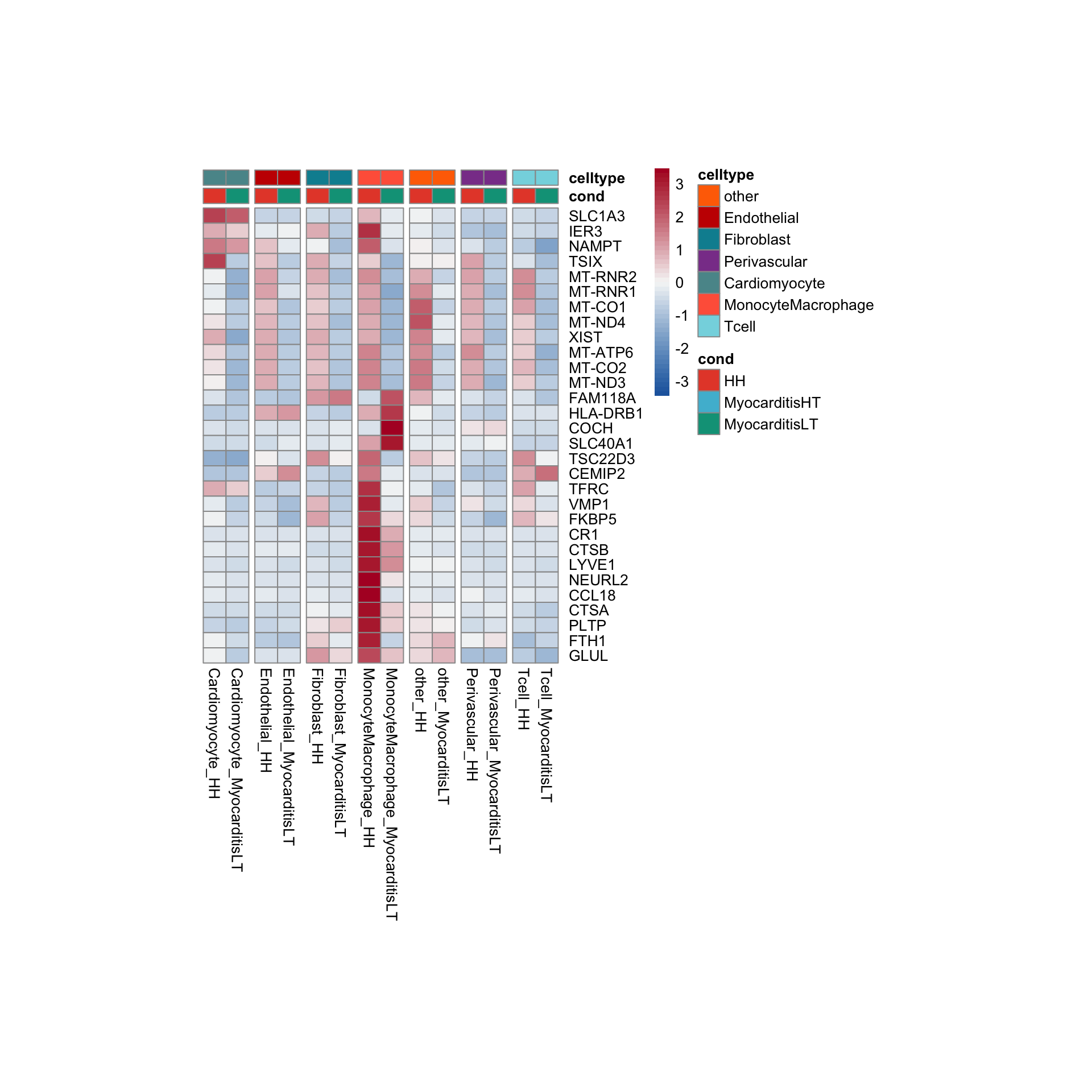
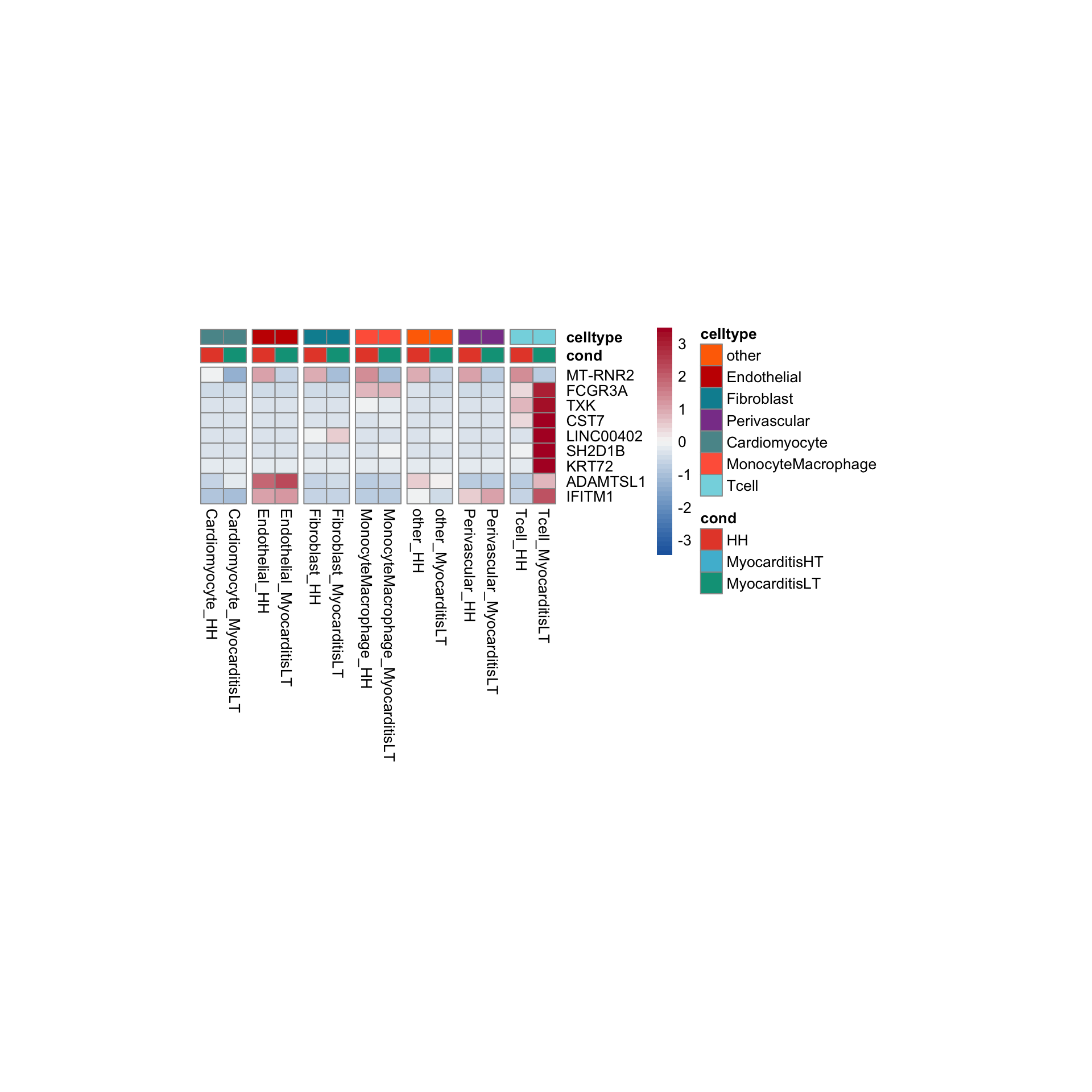
row.names = FALSE, col.names = TRUE, quote = FALSE, sep = "\t")avg Heatmap top cwDE genes
other
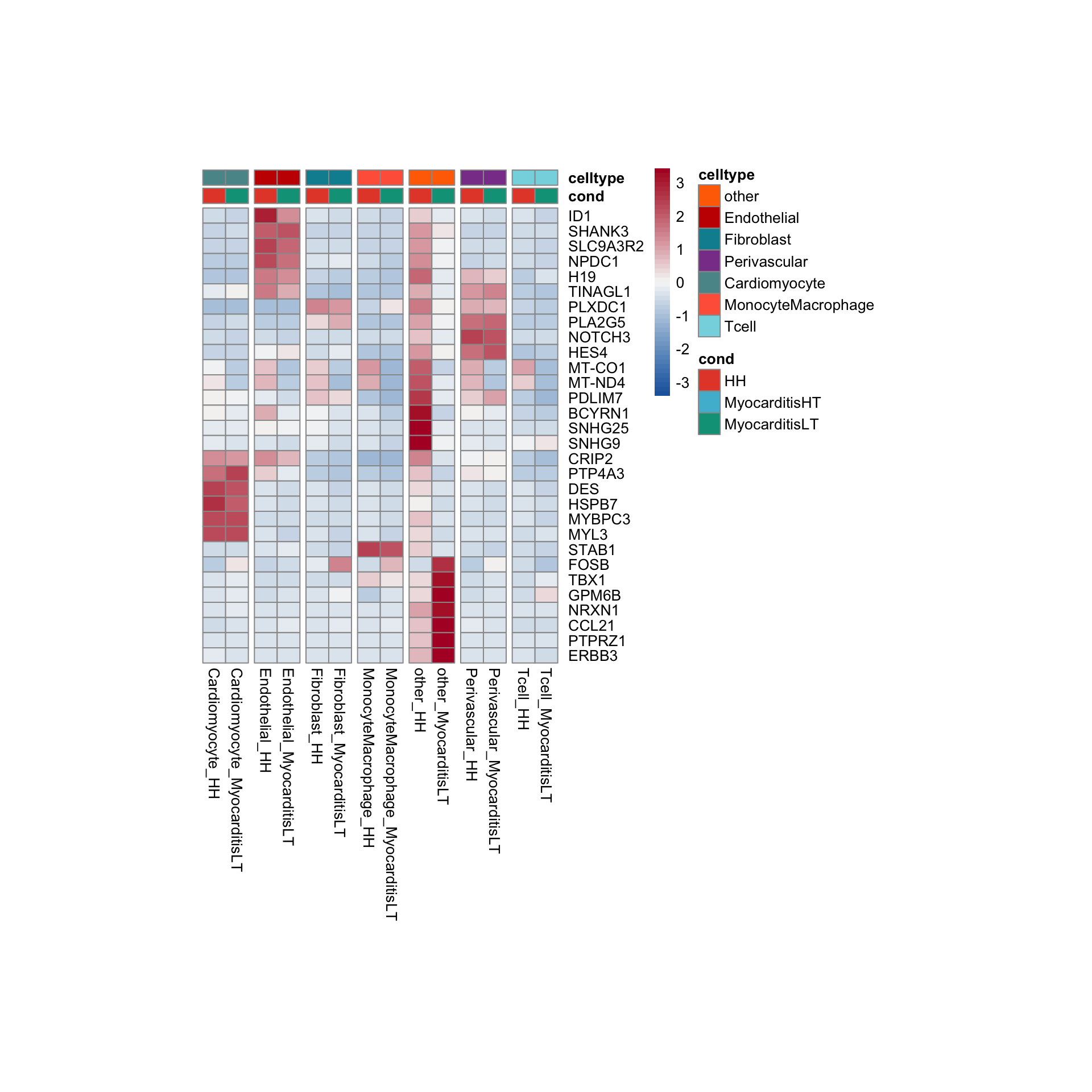
Endothelial
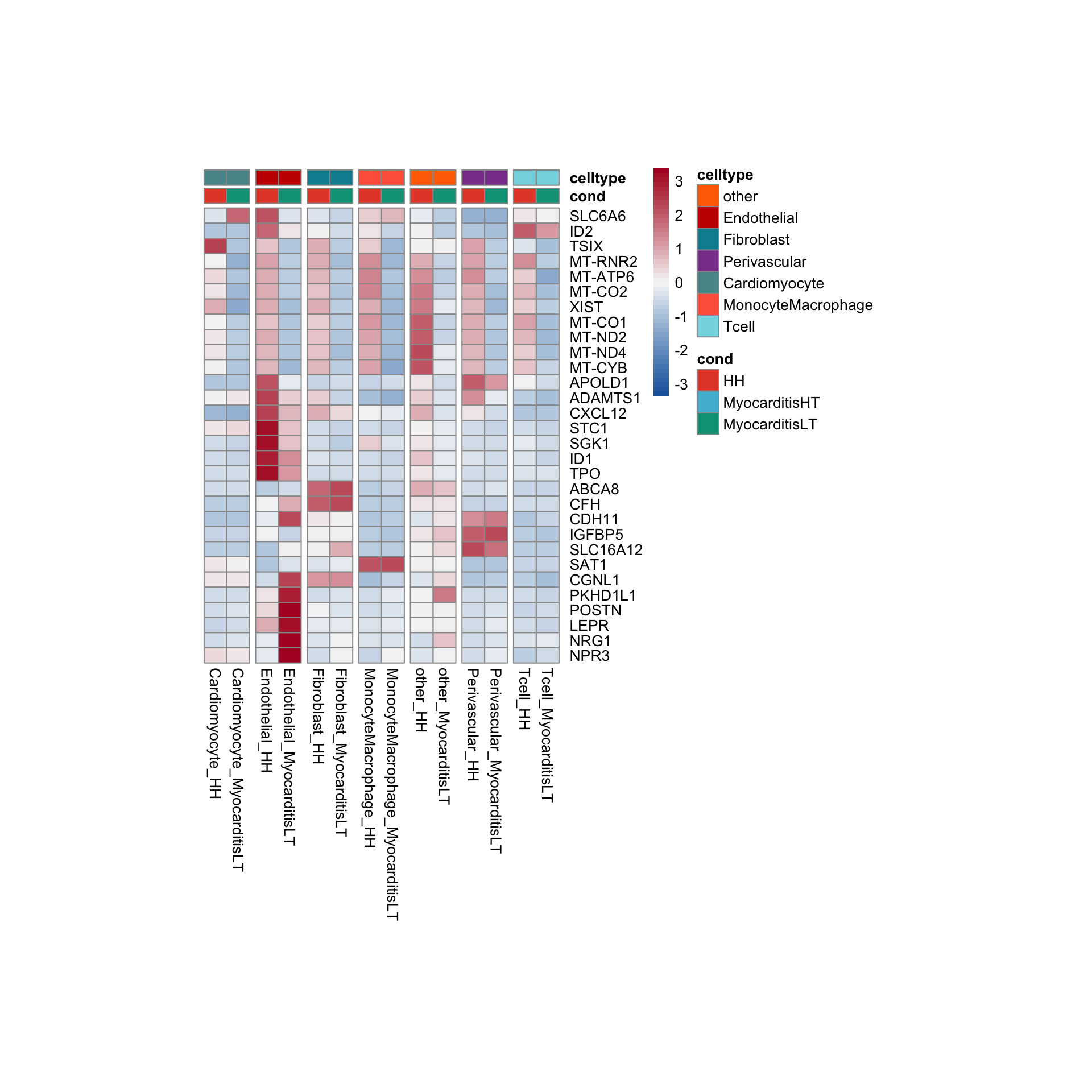
Fibroblast
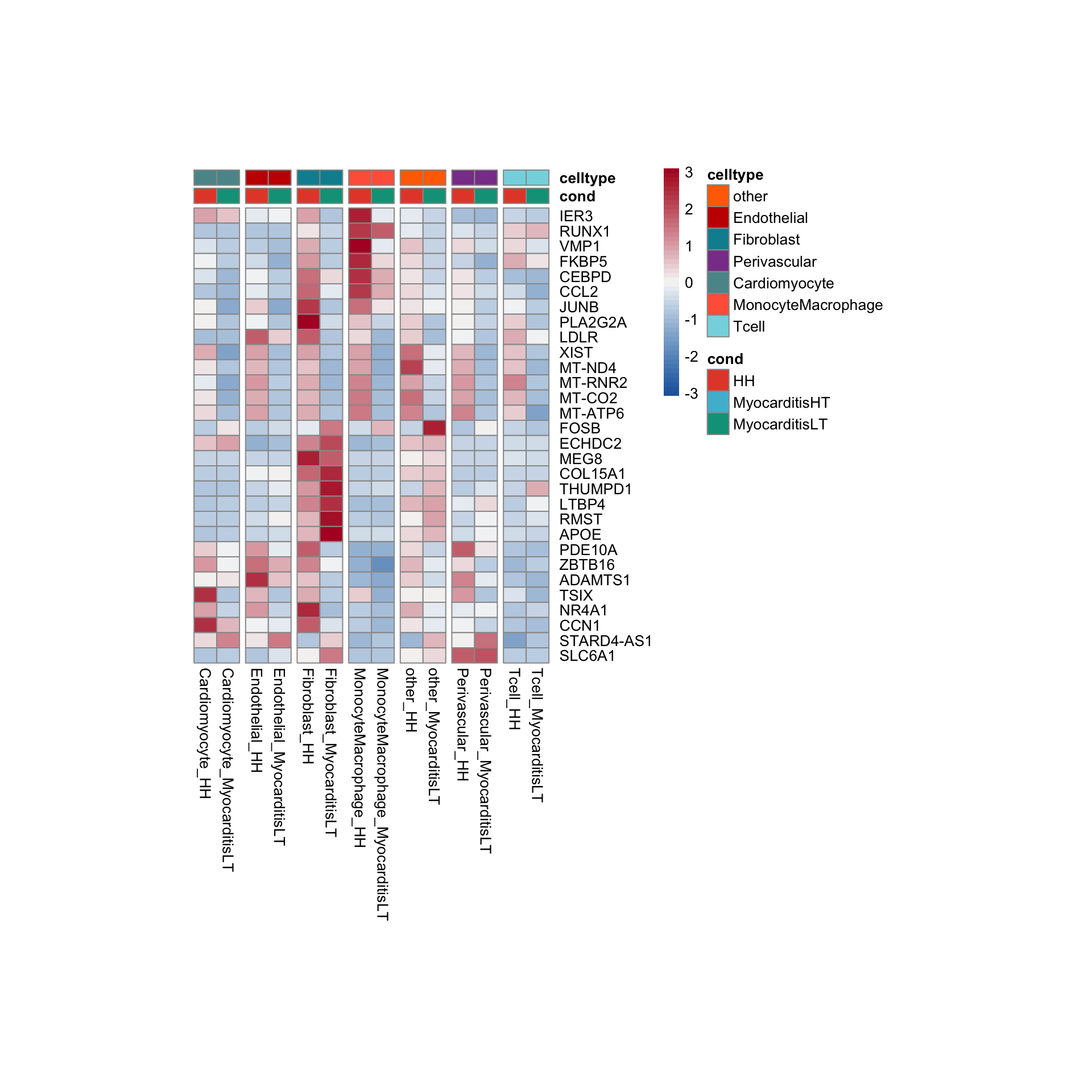
Perivascular
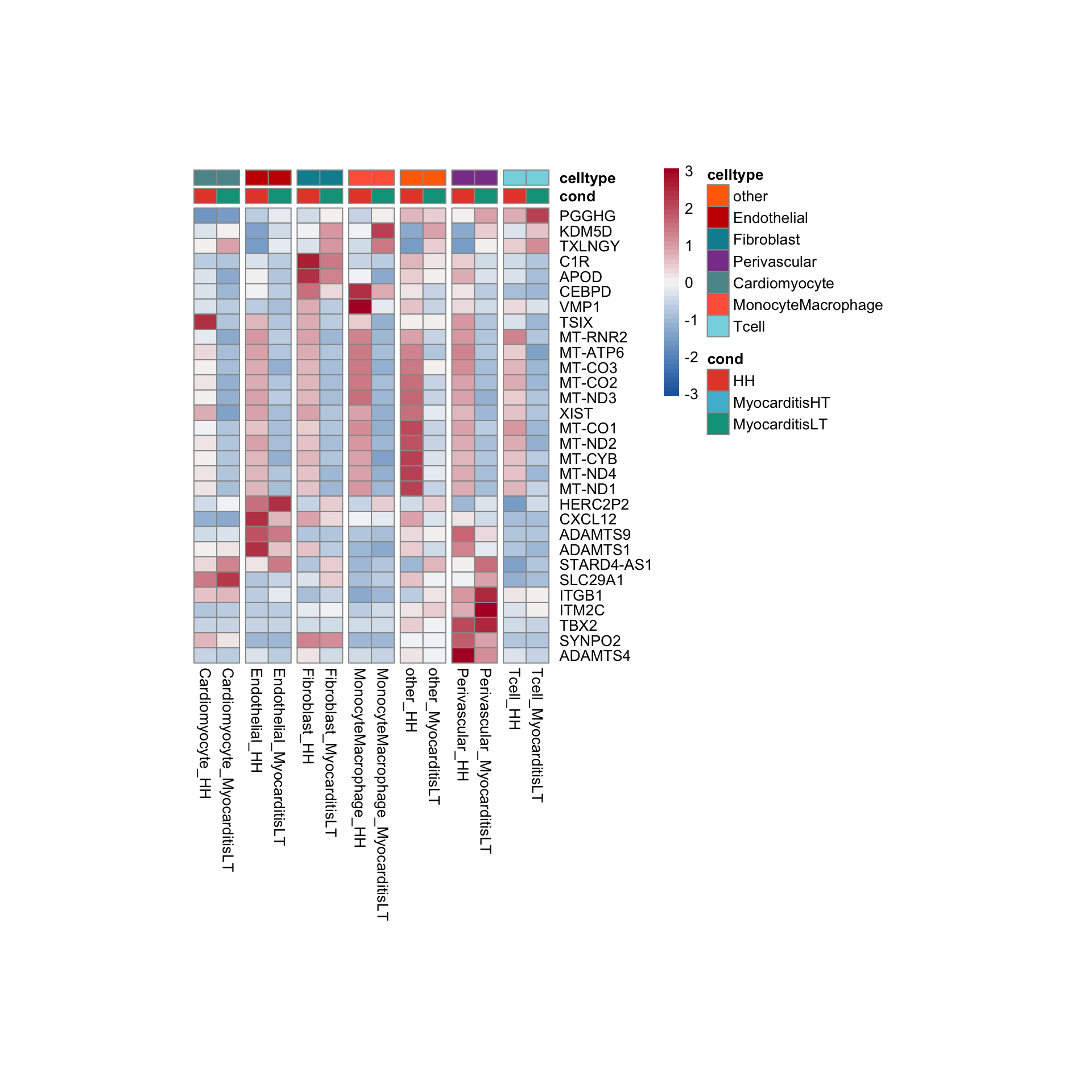
Cardiomyocyte
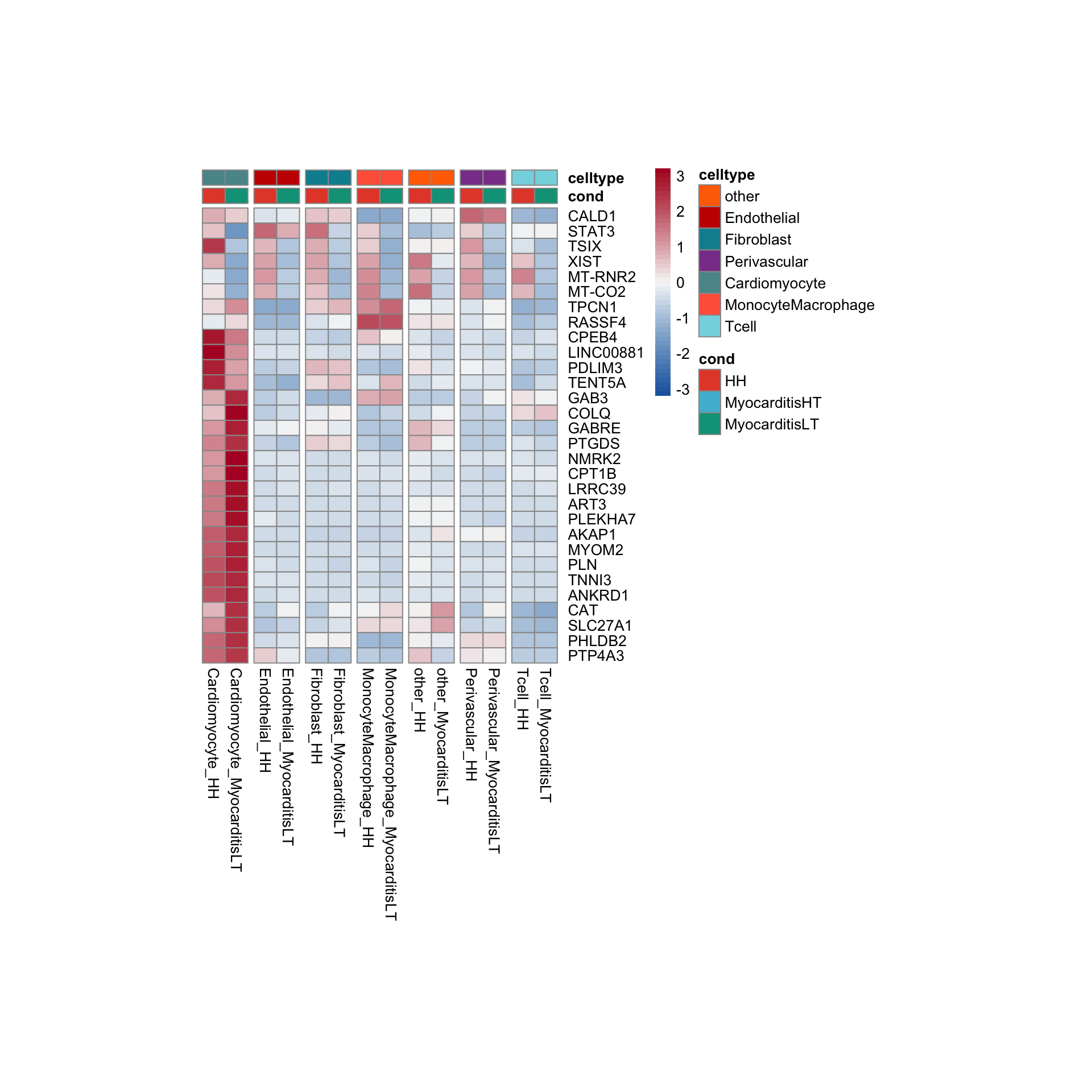
MonocyteMacrophage
Tcell
GSEA clusterProfiler cw DEgenes
#clVec <- levels(seurat$label_plus_cond2)
## only include cluster with >1 DE gene
clInclude <- data.frame(table(clustDE_Dat$group)) %>% filter(Freq>1)
clVec <- clInclude$Var1
GOcons <- lapply(clVec, function(cl){
clustDE_DatSub <- clustDE_Dat[which(clustDE_Dat$group == cl),] %>%
mutate(ENS=gsub("\\..*$", "", gene)) #%>%
#slice_min(., max_pval, n=200)
egoSS <- enrichGO(gene = unique(clustDE_DatSub$ENS),
OrgDb = org.Hs.eg.db,
keyType = 'ENSEMBL',
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
qvalueCutoff = 0.05)
egoSS <- setReadable(egoSS, OrgDb = org.Hs.eg.db)
egoSSres <- egoSS@result %>% filter(p.adjust < 0.05) %>%
mutate(subset=cl)
})
names(GOcons) <- clVec
## table to select pathways
GOconsDat <- do.call("rbind", GOcons)
write.table(GOconsDat, quote=F, row.names = T, col.names = T, sep= "\t",
file = paste0(basedir,"/data/humanHeartsPlusGraz_intPatients_",
"merged_SubsetLT_cwDEGO.txt"))vis sel GO Fibroblasts
selGO <- read_tsv(paste0(basedir,"/data/GSEA/selGO_Fibroblasts.txt"))
GOconsDatSel <- GOconsDat %>% filter(ID %in% selGO$GOterm) %>%
mutate(cond2 = gsub(".*_", "", subset)) %>%
mutate(label= gsub("_.*", "", subset)) %>%
filter(label == "Fibroblast")
grpVec <- unique(selGO$cond2)
lapply(grpVec, function(grp){
selGODat <- GOconsDatSel %>% filter(cond2 == grp)
selGODat <- selGODat %>% mutate(qscore=-log(p.adjust, base=10))
p <- ggbarplot(selGODat, x = "Description", y = "qscore",
fill = "cond2",
color = "cond2",
palette = colCond,
sort.val = "asc",
sort.by.groups = TRUE
#x.text.angle = 90
) +
rotate()
p
})[[1]]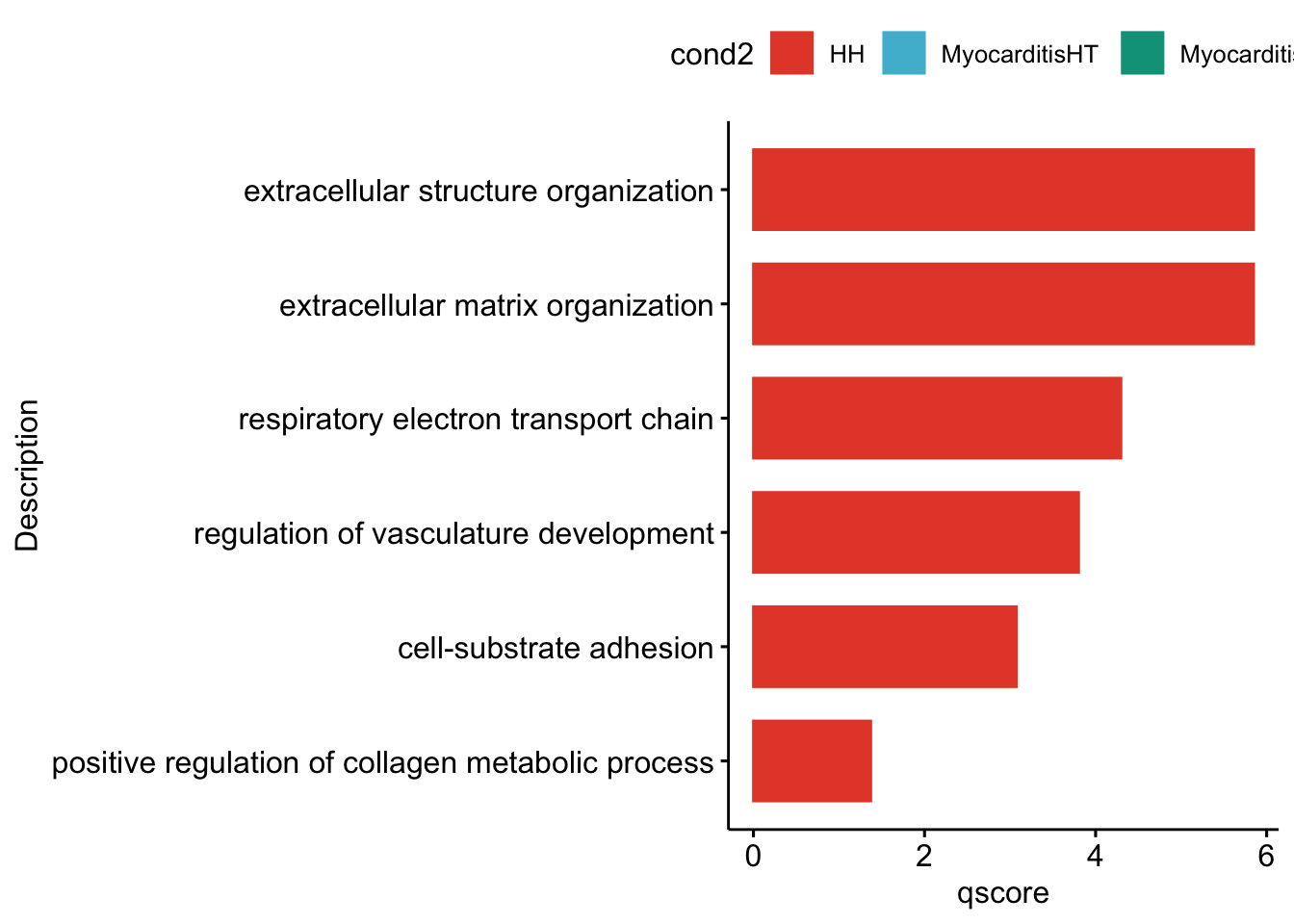
[[2]]
vis sel GO T cells
selGO <- read_tsv(paste0(basedir,"/data/GSEA/selGO_Tcell.txt"))
GOconsDatSel <- GOconsDat %>% filter(ID %in% selGO$GOterm) %>%
mutate(cond2 = gsub(".*_", "", subset)) %>%
mutate(label= gsub("_.*", "", subset)) %>%
filter(label == "Tcell")
grpVec <- unique(selGO$cond2)
lapply(grpVec, function(grp){
selGODat <- GOconsDatSel %>% filter(cond2 == grp)
selGODat <- selGODat %>% mutate(qscore=-log(p.adjust, base=10))
p <- ggbarplot(selGODat, x = "Description", y = "qscore",
fill = "cond2",
color = "cond2",
palette = colCond,
sort.val = "asc",
sort.by.groups = TRUE
#x.text.angle = 90
) +
rotate()
p
})[[1]]
signatures viridis
split by grp
signDat <- read_delim(file = paste0(basedir,
"/data/SelSignaturesTreat2.txt"),
delim = "\t")
genes <- data.frame(geneID=rownames(seurat)) %>%
mutate(gene=gsub("^.*\\.", "", geneID))
signDat <- signDat %>% left_join(.,genes, by="gene")
allSign <- unique(signDat$signature)
DefaultAssay(object = seurat) <- "integrated"
sce2 <- as.SingleCellExperiment(seurat)
DefaultAssay(object = seurat) <- "RNA"
sce <- as.SingleCellExperiment(seurat)
reducedDims(sce) <- list(PCA=reducedDim(sce2, "PCA"),
TSNE=reducedDim(sce2, "TSNE"),
UMAP=reducedDim(sce2, "UMAP"))
treatGrps <- unique(sce$cond2)
cutOff <- 3
pal = viridis(100)
sc <- scale_colour_gradientn(colours = pal, limits=c(0, cutOff))
lapply(unique(signDat$signature), function(sign){
signGenes <- signDat %>% dplyr::filter(signature == sign)
sceSub <- sce[which(rownames(sce) %in% signGenes$geneID),]
cntMat <- rowSums(t(as.matrix(sceSub@assays@data$logcounts)))/nrow(signGenes)
sceSub$sign <- cntMat
sceSub$sign[which(sceSub$sign > cutOff)] <- cutOff
sceSub$sign[which(sceSub$sign < 0)] <- 0
lapply(treatGrps, function(treat){
sceSubT <- sceSub[, which(sceSub$cond2 == treat)]
p <- visGroup_adapt(sceSubT, 'sign', dim_red = 'UMAP') +
sc +
guides(colour = guide_colourbar(title = '')) +
ggtitle(paste0(sign, ' signature - ', treat)) +
theme_classic() +
theme(axis.text = element_blank(),
axis.ticks = element_blank()) +
labs(x='Dimension 1', y='Dimension 2')
p
})
})[[1]]
[[1]][[1]]
[[1]][[2]]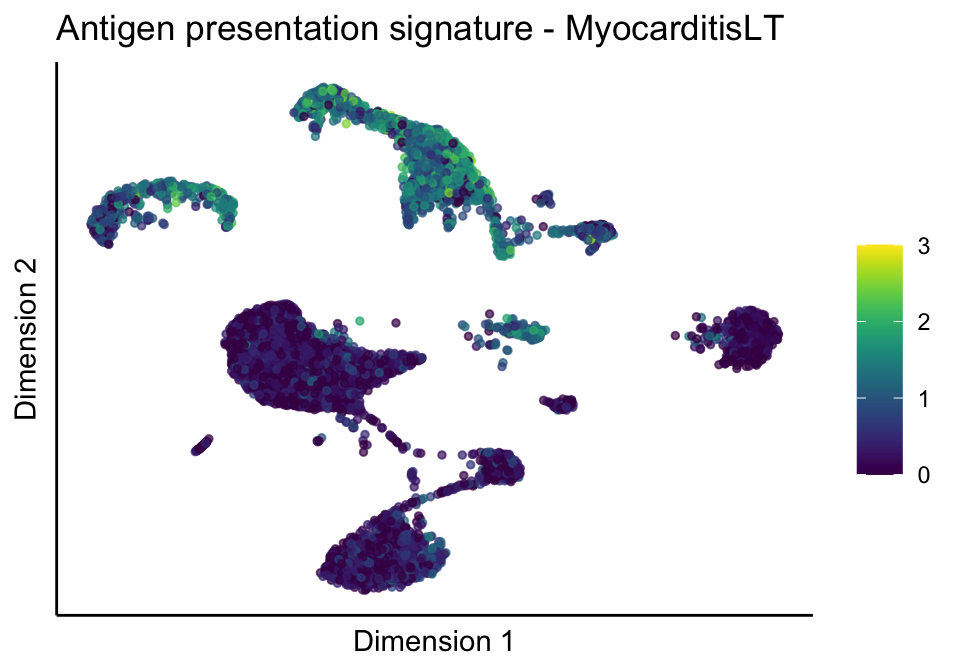
[[2]]
[[2]][[1]]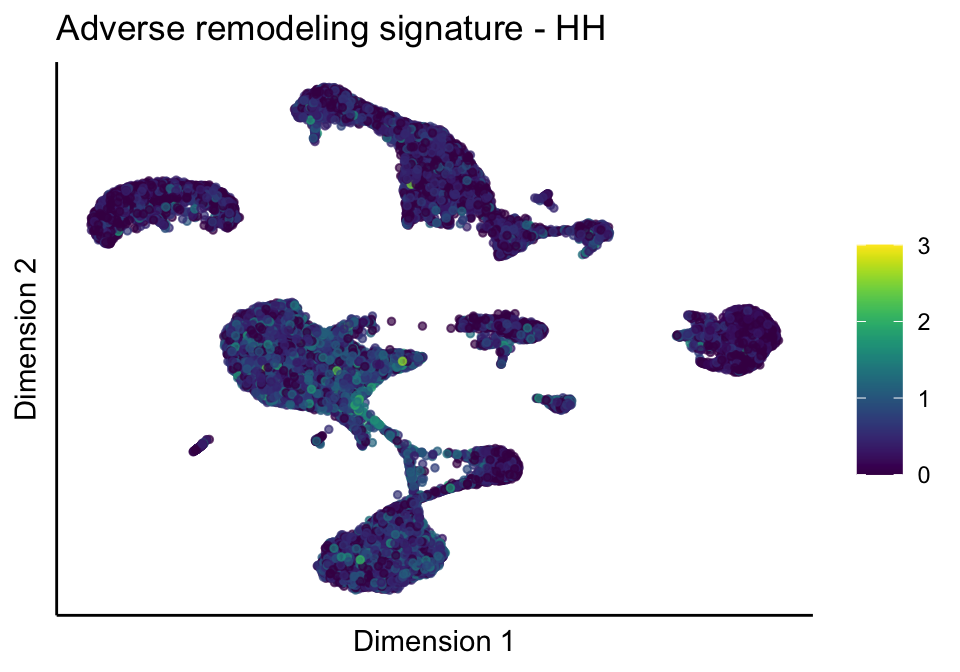
[[2]][[2]]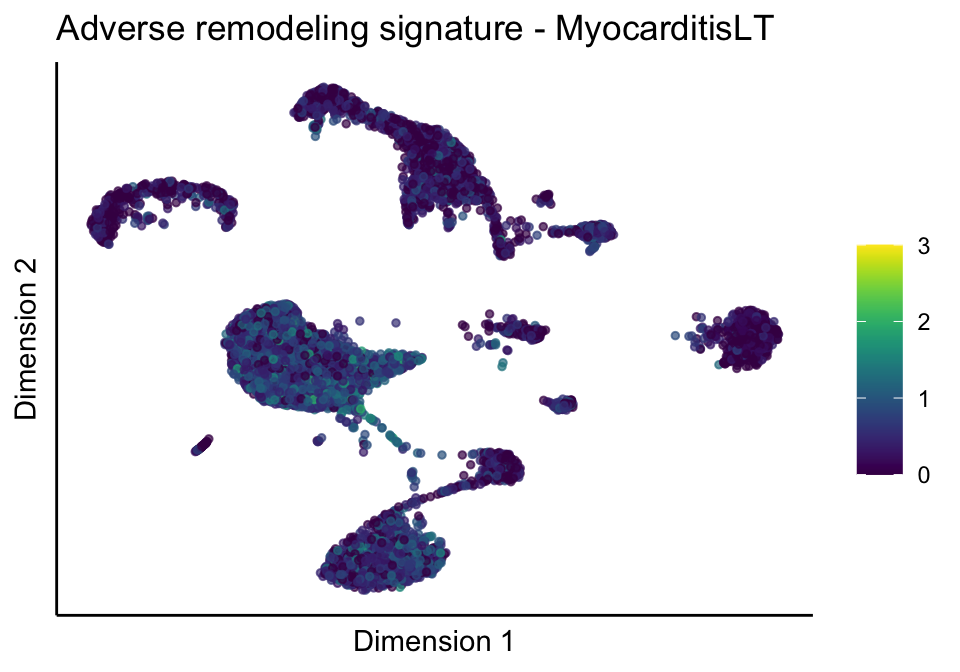
[[3]]
[[3]][[1]]
[[3]][[2]]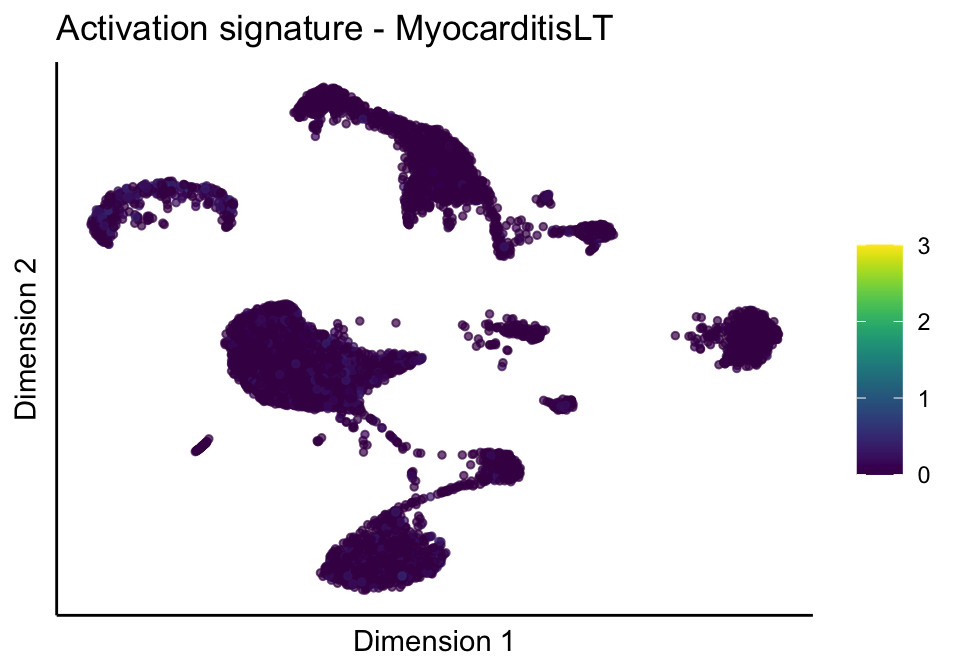
cutOff <- 2
pal = viridis(100)
sc <- scale_colour_gradientn(colours = pal, limits=c(0, cutOff))
lapply(unique(signDat$signature), function(sign){
signGenes <- signDat %>% dplyr::filter(signature == sign)
sceSub <- sce[which(rownames(sce) %in% signGenes$geneID),]
cntMat <- rowSums(t(as.matrix(sceSub@assays@data$logcounts)))/nrow(signGenes)
sceSub$sign <- cntMat
sceSub$sign[which(sceSub$sign > cutOff)] <- cutOff
sceSub$sign[which(sceSub$sign < 0)] <- 0
lapply(treatGrps, function(treat){
sceSubT <- sceSub[, which(sceSub$cond2 == treat)]
p <- visGroup_adapt(sceSubT, 'sign', dim_red = 'UMAP') +
sc +
guides(colour = guide_colourbar(title = '')) +
ggtitle(paste0(sign, ' signature - ', treat)) +
theme_classic() +
theme(axis.text = element_blank(),
axis.ticks = element_blank()) +
labs(x='Dimension 1', y='Dimension 2')
p
})
})[[1]]
[[1]][[1]]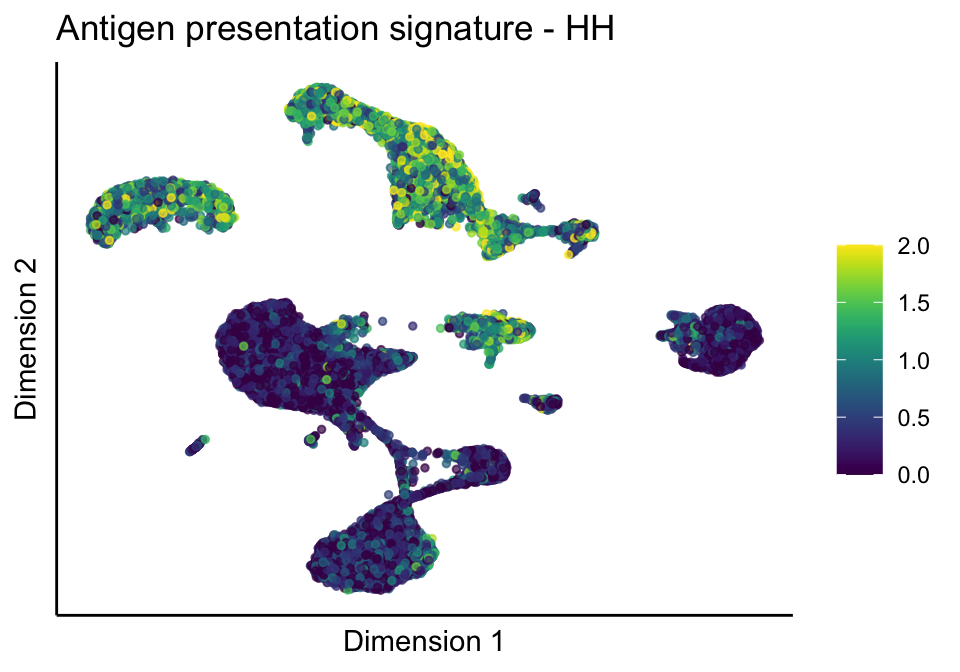
[[1]][[2]]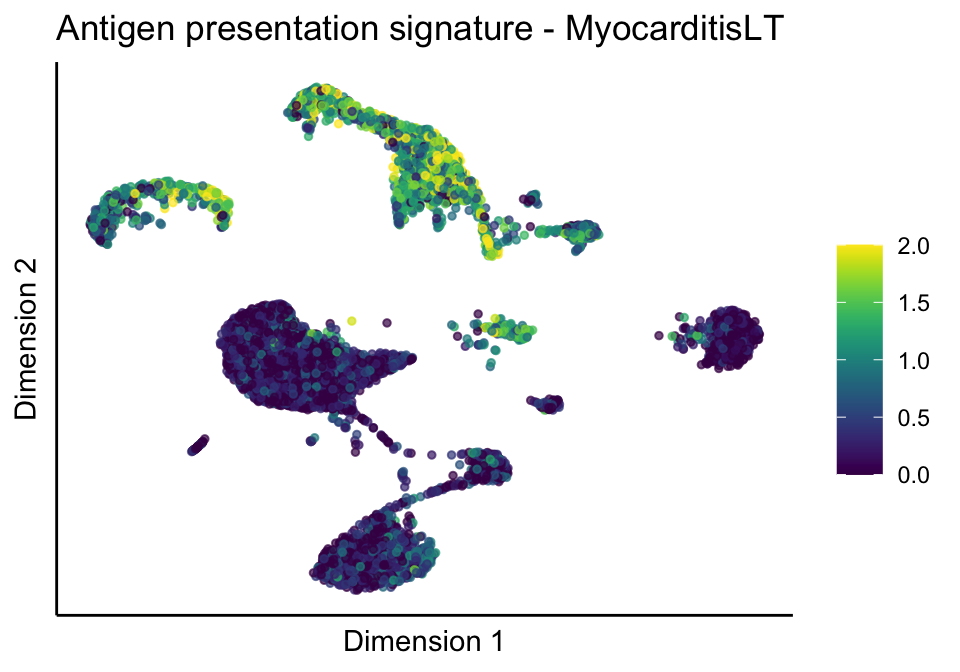
[[2]]
[[2]][[1]]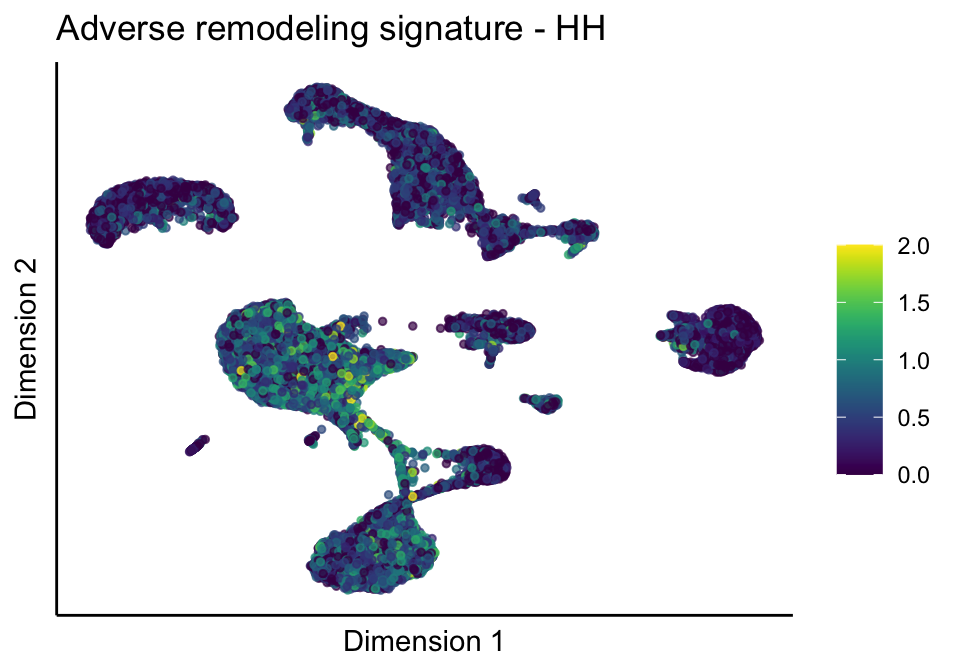
[[2]][[2]]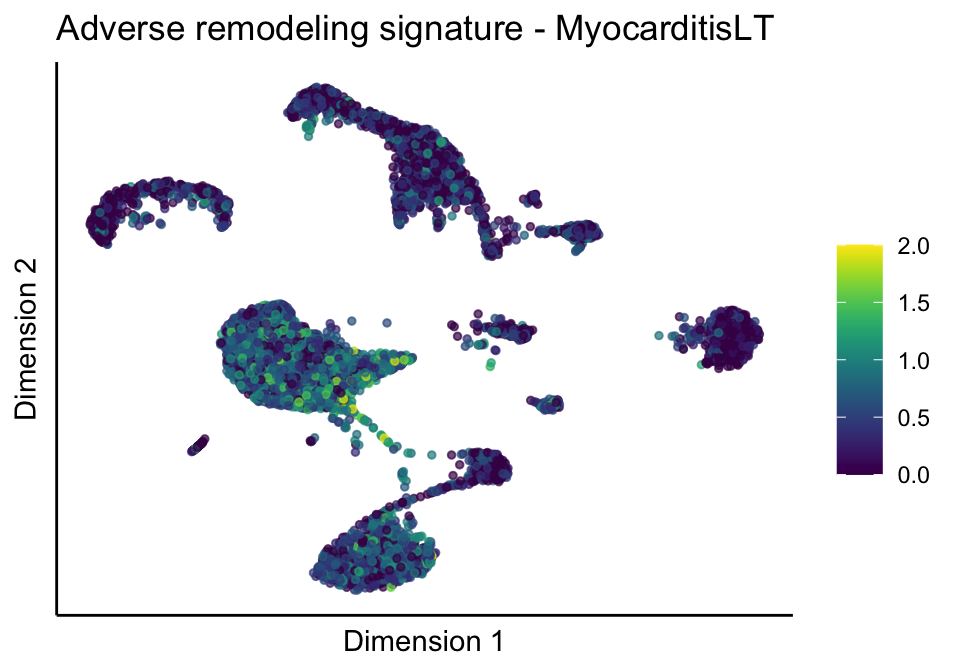
[[3]]
[[3]][[1]]
[[3]][[2]]
cutOff <- 1.5
pal = viridis(100)
sc <- scale_colour_gradientn(colours = pal, limits=c(0, cutOff))
lapply(unique(signDat$signature), function(sign){
signGenes <- signDat %>% dplyr::filter(signature == sign)
sceSub <- sce[which(rownames(sce) %in% signGenes$geneID),]
cntMat <- rowSums(t(as.matrix(sceSub@assays@data$logcounts)))/nrow(signGenes)
sceSub$sign <- cntMat
sceSub$sign[which(sceSub$sign > cutOff)] <- cutOff
sceSub$sign[which(sceSub$sign < 0)] <- 0
lapply(treatGrps, function(treat){
sceSubT <- sceSub[, which(sceSub$cond2 == treat)]
p <- visGroup_adapt(sceSubT, 'sign', dim_red = 'UMAP') +
sc +
guides(colour = guide_colourbar(title = '')) +
ggtitle(paste0(sign, ' signature - ', treat)) +
theme_classic() +
theme(axis.text = element_blank(),
axis.ticks = element_blank()) +
labs(x='Dimension 1', y='Dimension 2')
p
})
})[[1]]
[[1]][[1]]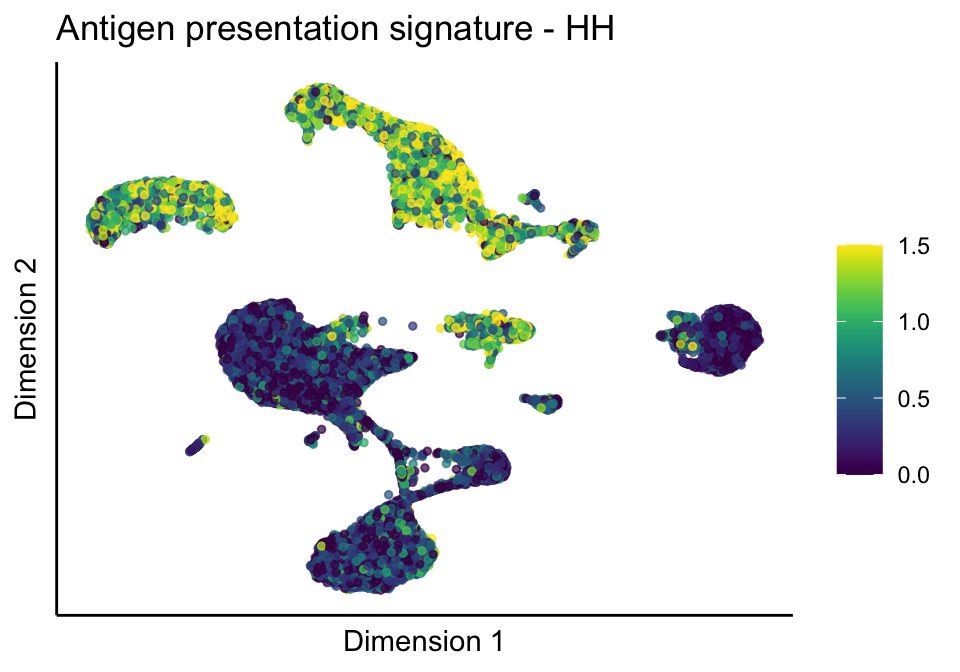
[[1]][[2]]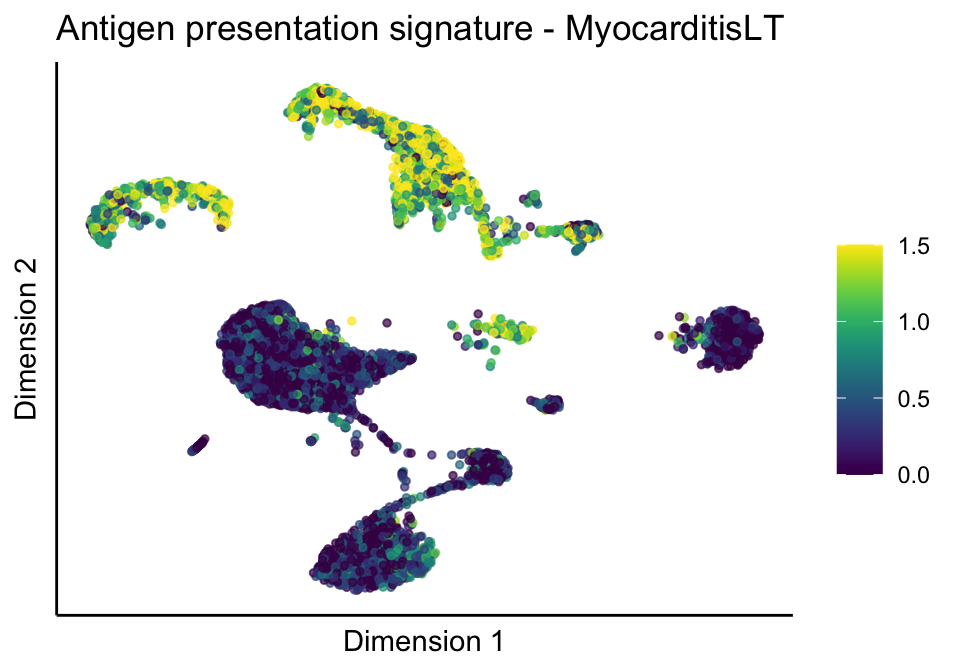
[[2]]
[[2]][[1]]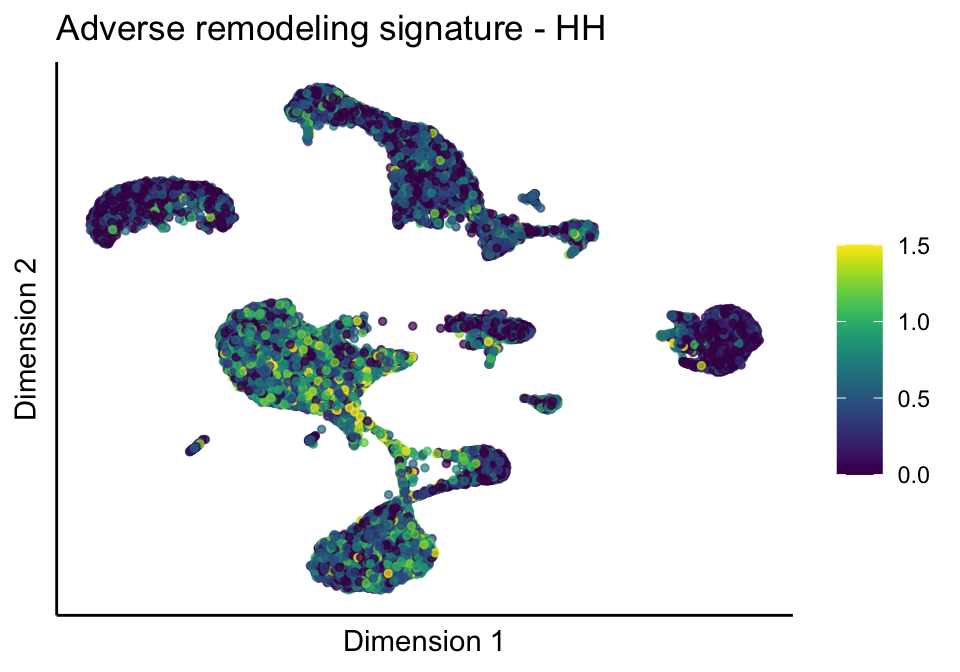
[[2]][[2]]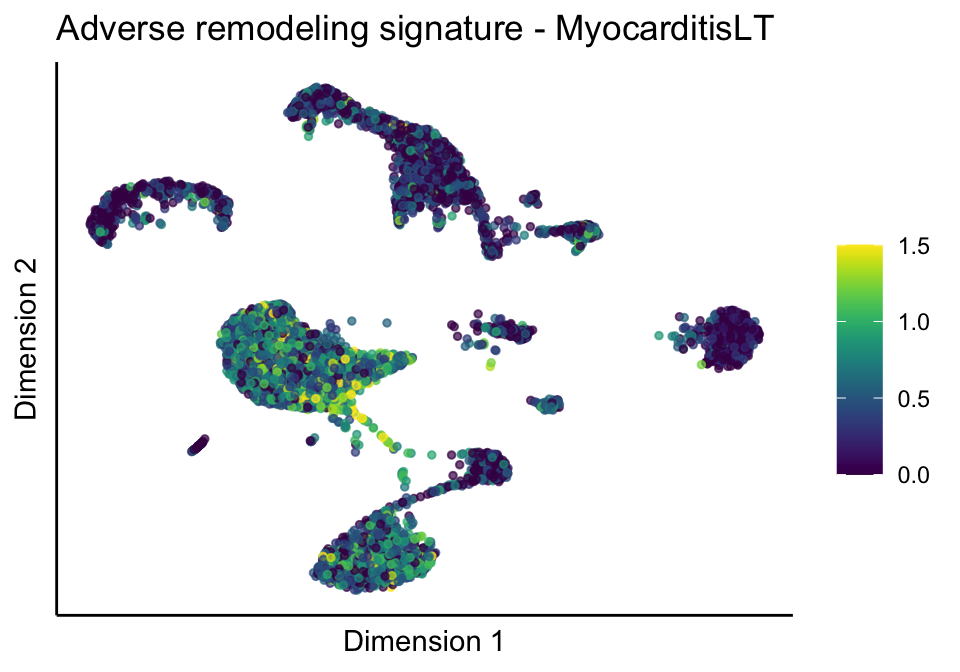
[[3]]
[[3]][[1]]
[[3]][[2]]
signatures red blue
split by grp
cutOff <- 3
pal = colorRampPalette(c("#053061", "#f7f7f7","#85122d"))(100)
sc <- scale_colour_gradientn(colours = pal, limits=c(0, cutOff))
lapply(unique(signDat$signature), function(sign){
signGenes <- signDat %>% dplyr::filter(signature == sign)
sceSub <- sce[which(rownames(sce) %in% signGenes$geneID),]
cntMat <- rowSums(t(as.matrix(sceSub@assays@data$logcounts)))/nrow(signGenes)
sceSub$sign <- cntMat
sceSub$sign[which(sceSub$sign > cutOff)] <- cutOff
sceSub$sign[which(sceSub$sign < 0)] <- 0
lapply(treatGrps, function(treat){
sceSubT <- sceSub[, which(sceSub$cond2 == treat)]
p <- visGroup_adapt(sceSubT, 'sign', dim_red = 'UMAP') +
sc +
guides(colour = guide_colourbar(title = '')) +
ggtitle(paste0(sign, ' signature - ', treat)) +
theme_classic() +
theme(axis.text = element_blank(),
axis.ticks = element_blank()) +
labs(x='Dimension 1', y='Dimension 2')
p
})
})[[1]]
[[1]][[1]]
[[1]][[2]]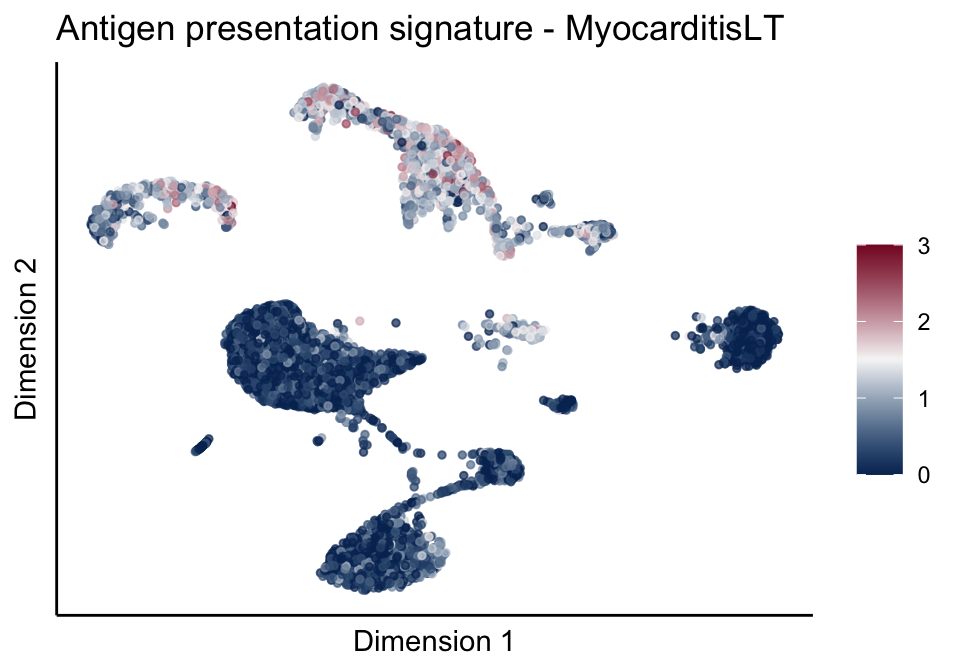
[[2]]
[[2]][[1]]
[[2]][[2]]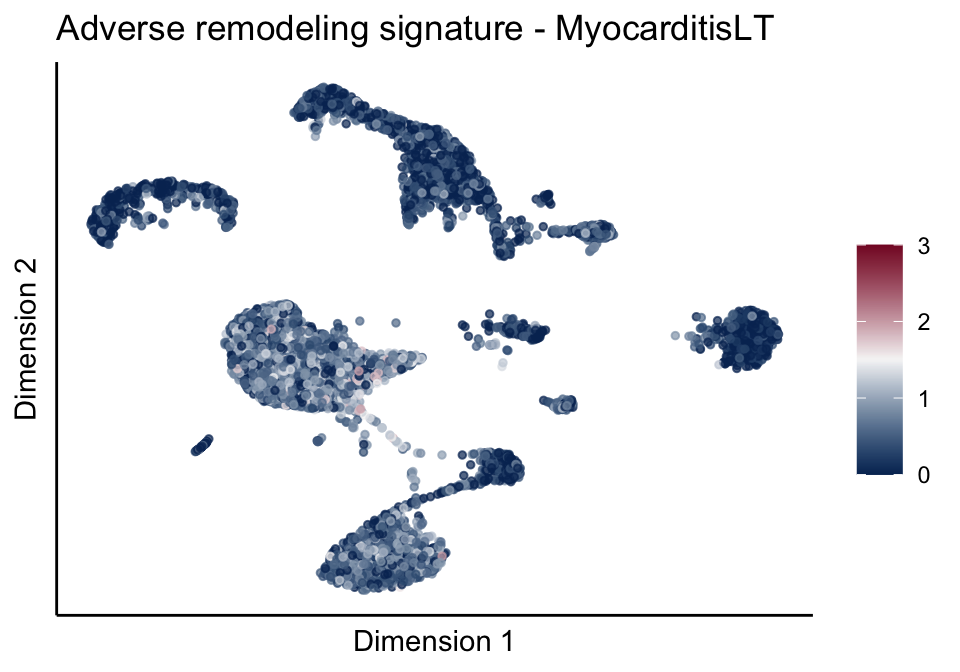
[[3]]
[[3]][[1]]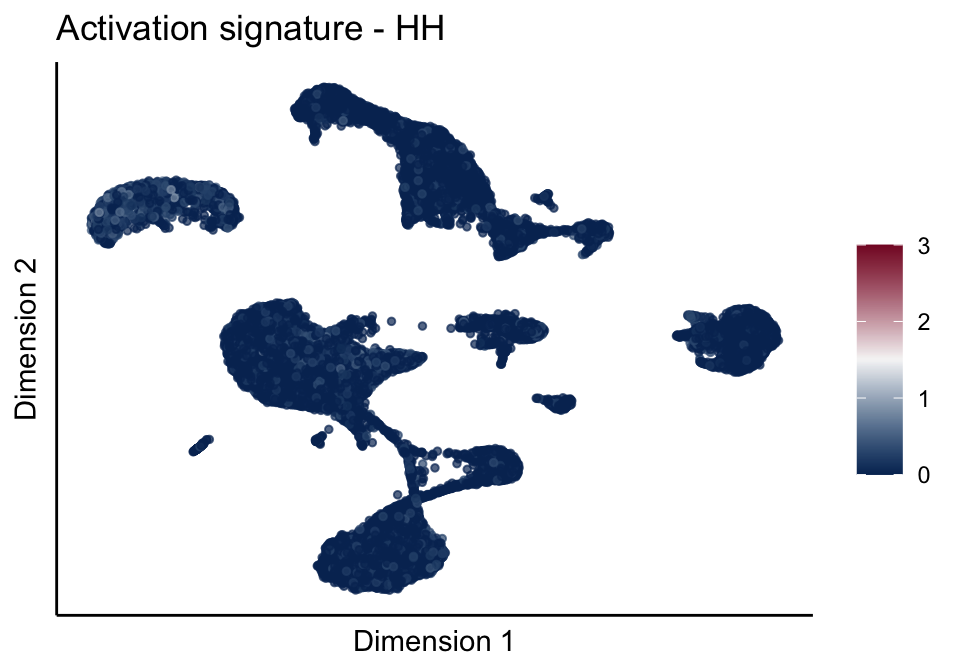
[[3]][[2]]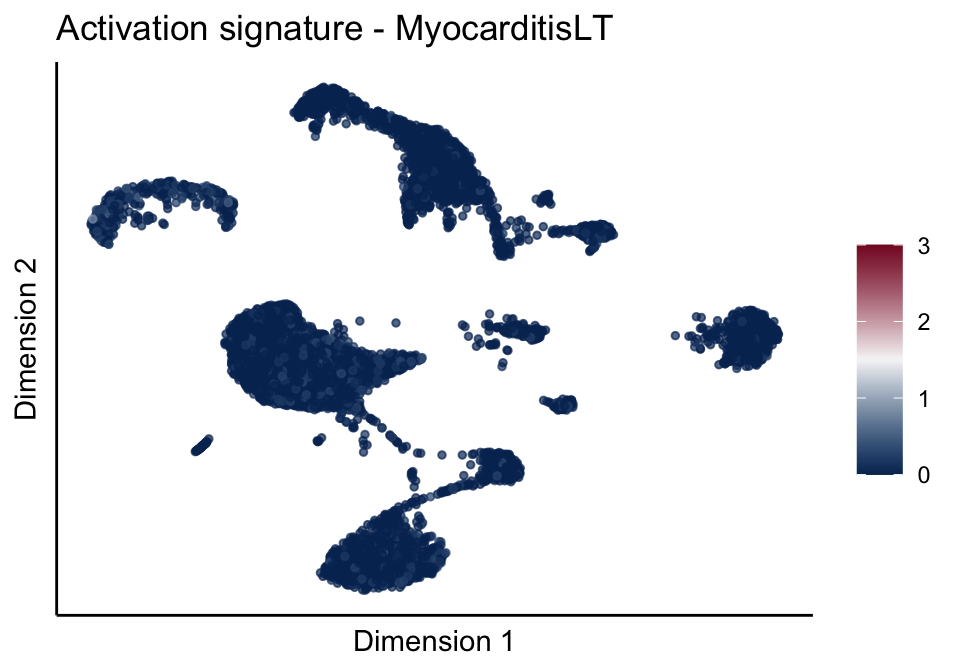
cutOff <- 2
pal = colorRampPalette(c("#053061", "#f7f7f7","#85122d"))(100)
sc <- scale_colour_gradientn(colours = pal, limits=c(0, cutOff))
lapply(unique(signDat$signature), function(sign){
signGenes <- signDat %>% dplyr::filter(signature == sign)
sceSub <- sce[which(rownames(sce) %in% signGenes$geneID),]
cntMat <- rowSums(t(as.matrix(sceSub@assays@data$logcounts)))/nrow(signGenes)
sceSub$sign <- cntMat
sceSub$sign[which(sceSub$sign > cutOff)] <- cutOff
sceSub$sign[which(sceSub$sign < 0)] <- 0
lapply(treatGrps, function(treat){
sceSubT <- sceSub[, which(sceSub$cond2 == treat)]
p <- visGroup_adapt(sceSubT, 'sign', dim_red = 'UMAP') +
sc +
guides(colour = guide_colourbar(title = '')) +
ggtitle(paste0(sign, ' signature - ', treat)) +
theme_classic() +
theme(axis.text = element_blank(),
axis.ticks = element_blank()) +
labs(x='Dimension 1', y='Dimension 2')
p
})
})[[1]]
[[1]][[1]]
[[1]][[2]]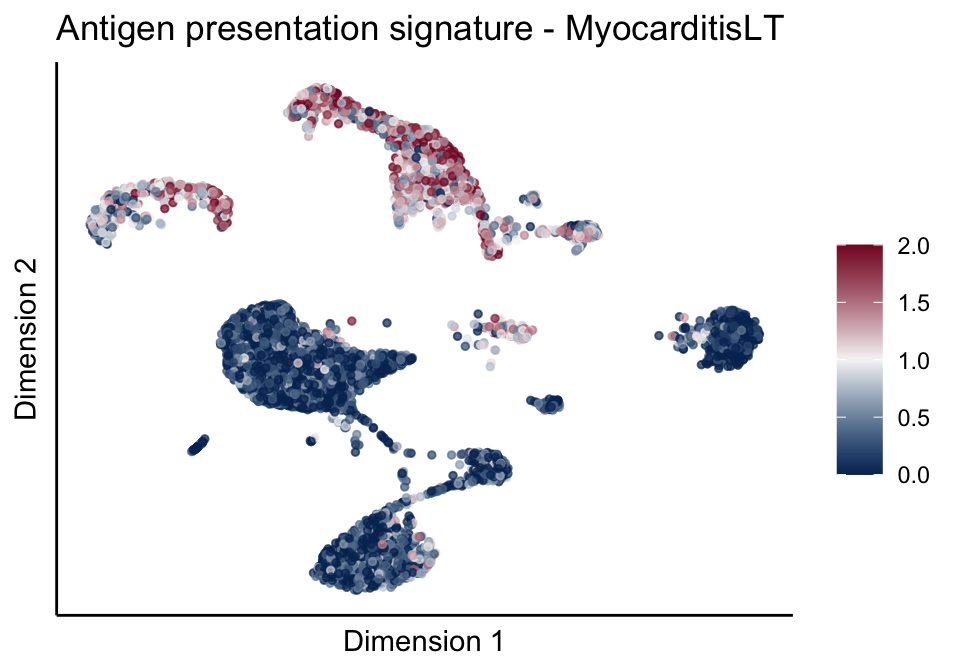
[[2]]
[[2]][[1]]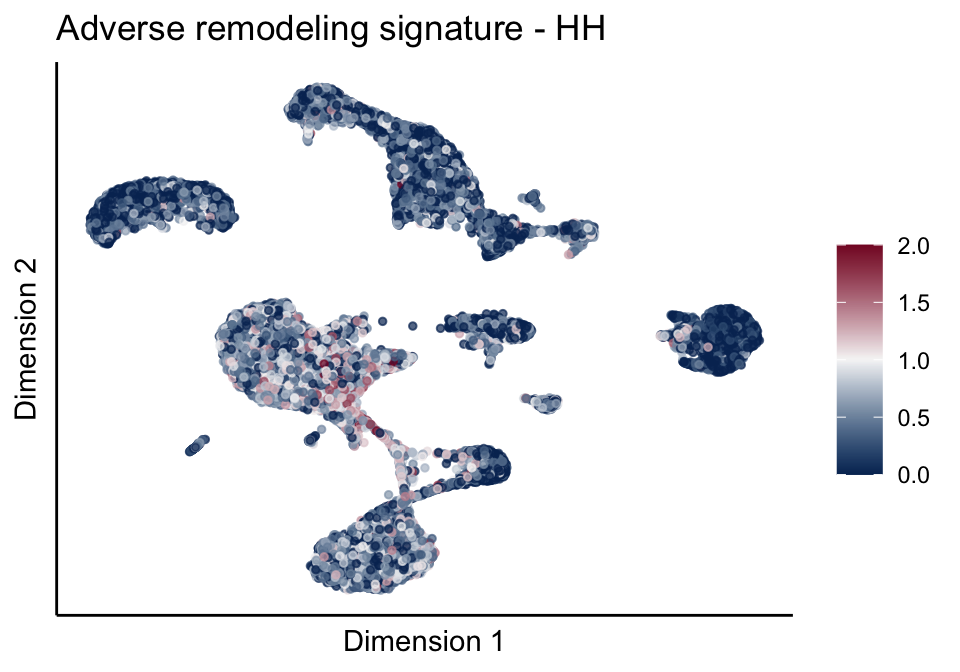
[[2]][[2]]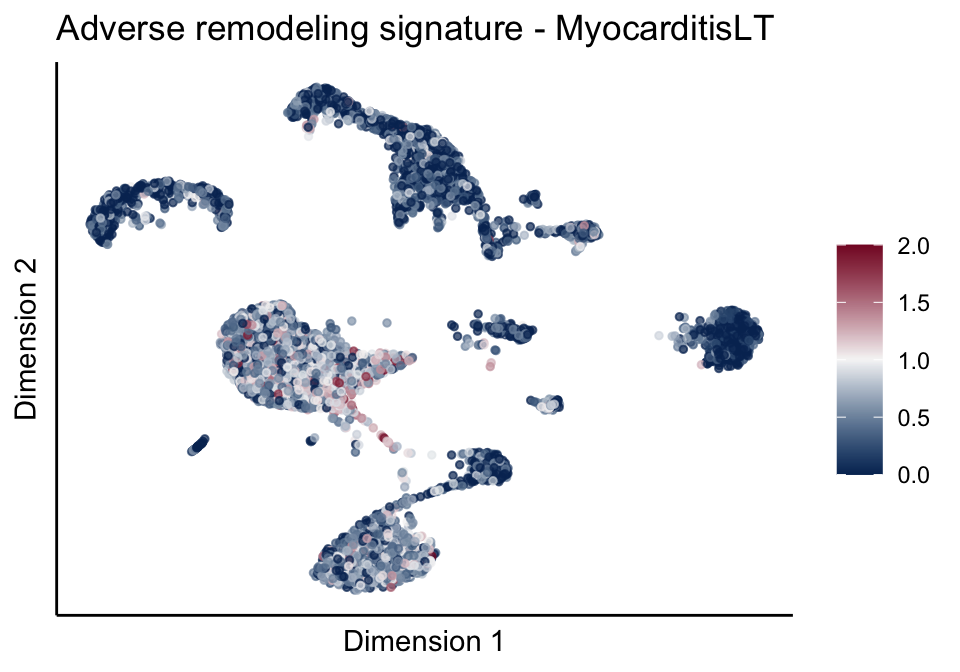
[[3]]
[[3]][[1]]
[[3]][[2]]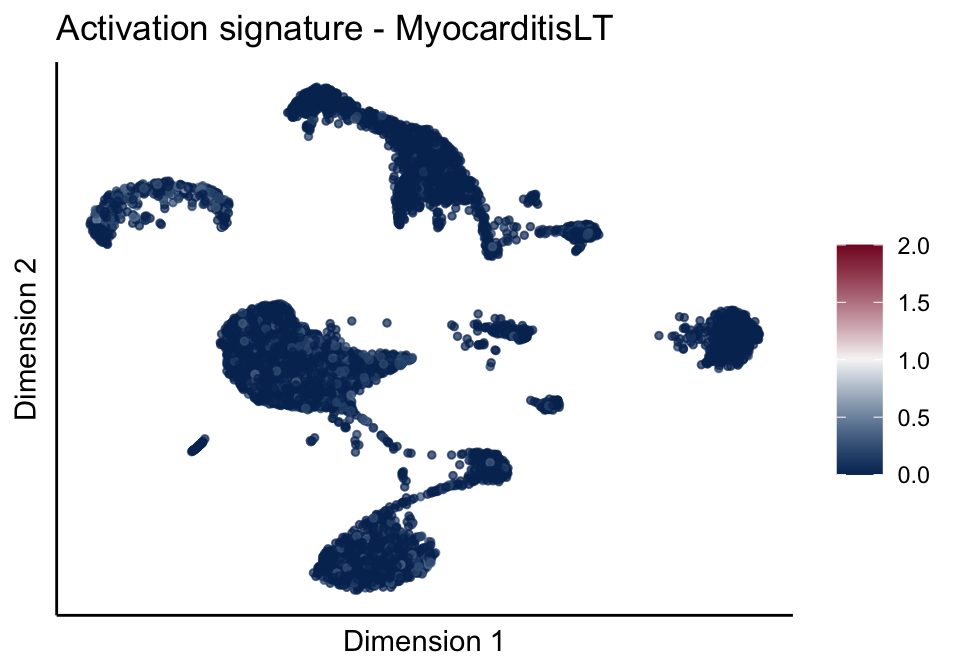
cutOff <- 1.5
pal = colorRampPalette(c("#053061", "#f7f7f7","#85122d"))(100)
sc <- scale_colour_gradientn(colours = pal, limits=c(0, cutOff))
lapply(unique(signDat$signature), function(sign){
signGenes <- signDat %>% dplyr::filter(signature == sign)
sceSub <- sce[which(rownames(sce) %in% signGenes$geneID),]
cntMat <- rowSums(t(as.matrix(sceSub@assays@data$logcounts)))/nrow(signGenes)
sceSub$sign <- cntMat
sceSub$sign[which(sceSub$sign > cutOff)] <- cutOff
sceSub$sign[which(sceSub$sign < 0)] <- 0
lapply(treatGrps, function(treat){
sceSubT <- sceSub[, which(sceSub$cond2 == treat)]
p <- visGroup_adapt(sceSubT, 'sign', dim_red = 'UMAP') +
sc +
guides(colour = guide_colourbar(title = '')) +
ggtitle(paste0(sign, ' signature - ', treat)) +
theme_classic() +
theme(axis.text = element_blank(),
axis.ticks = element_blank()) +
labs(x='Dimension 1', y='Dimension 2')
p
})
})[[1]]
[[1]][[1]]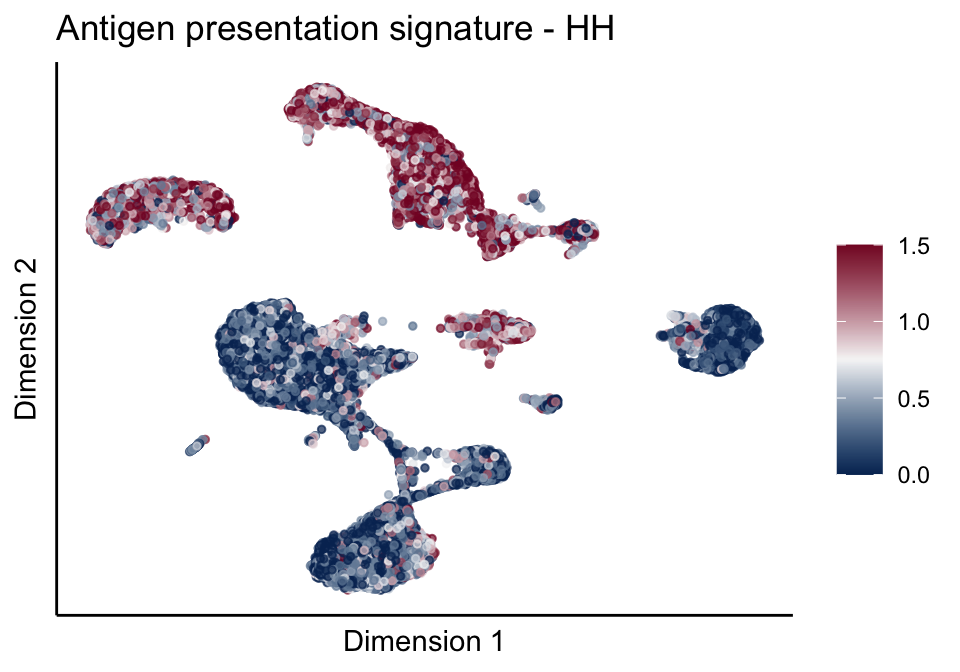
[[1]][[2]]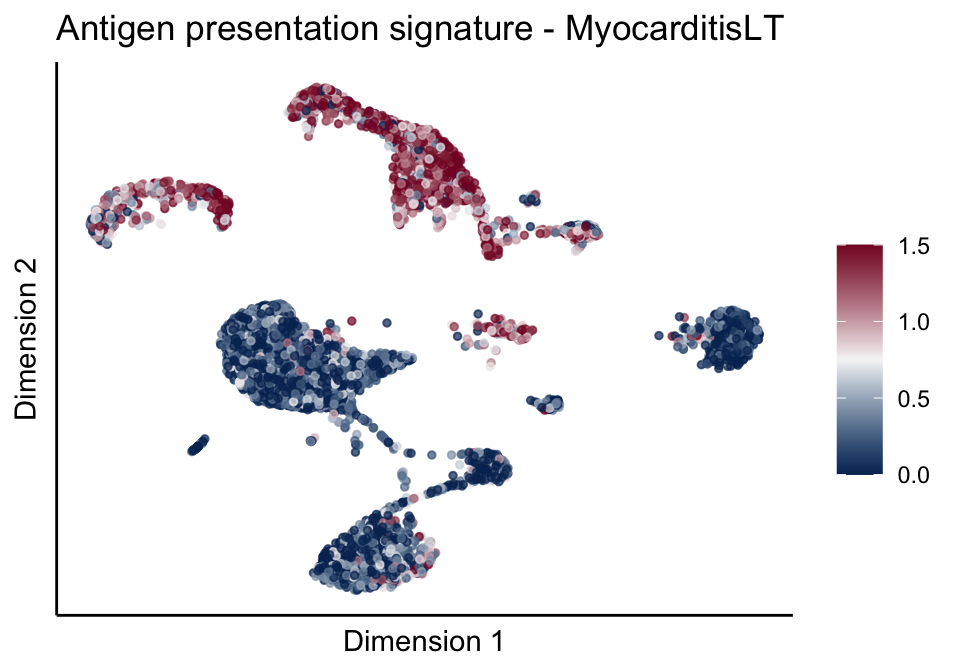
[[2]]
[[2]][[1]]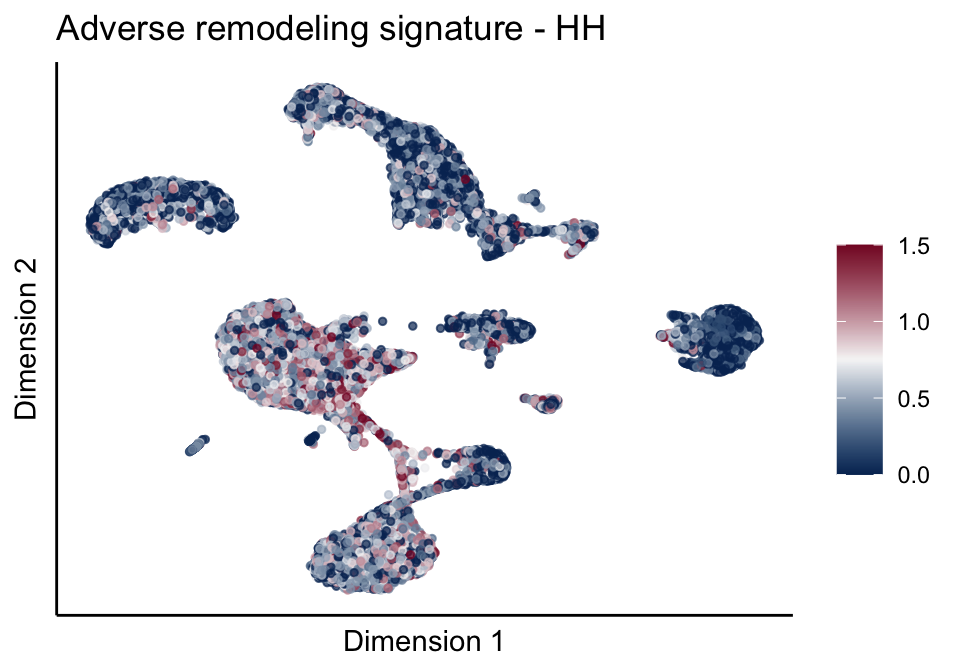
[[2]][[2]]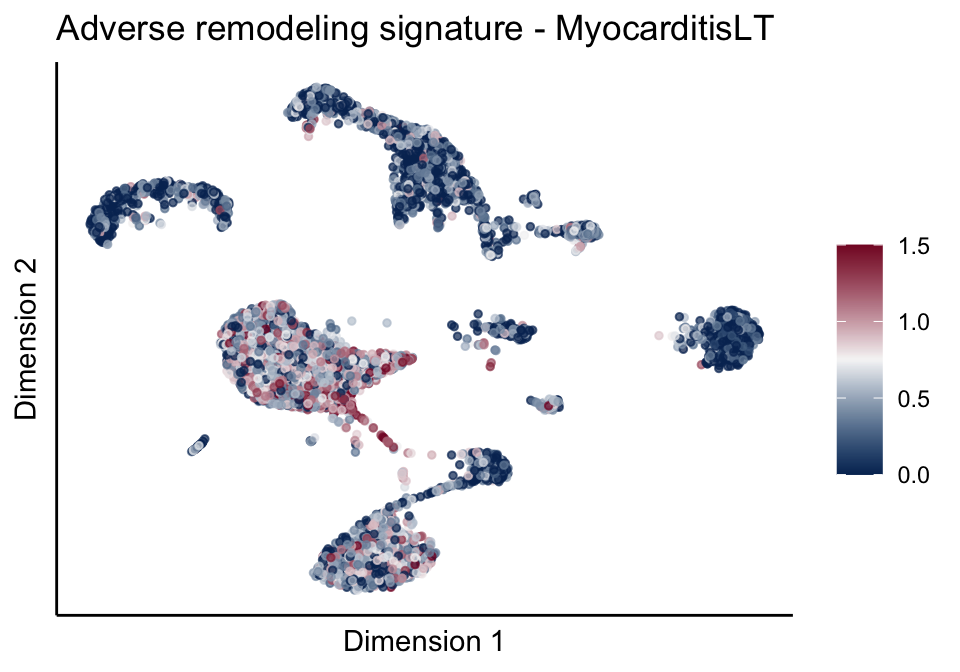
[[3]]
[[3]][[1]]
[[3]][[2]]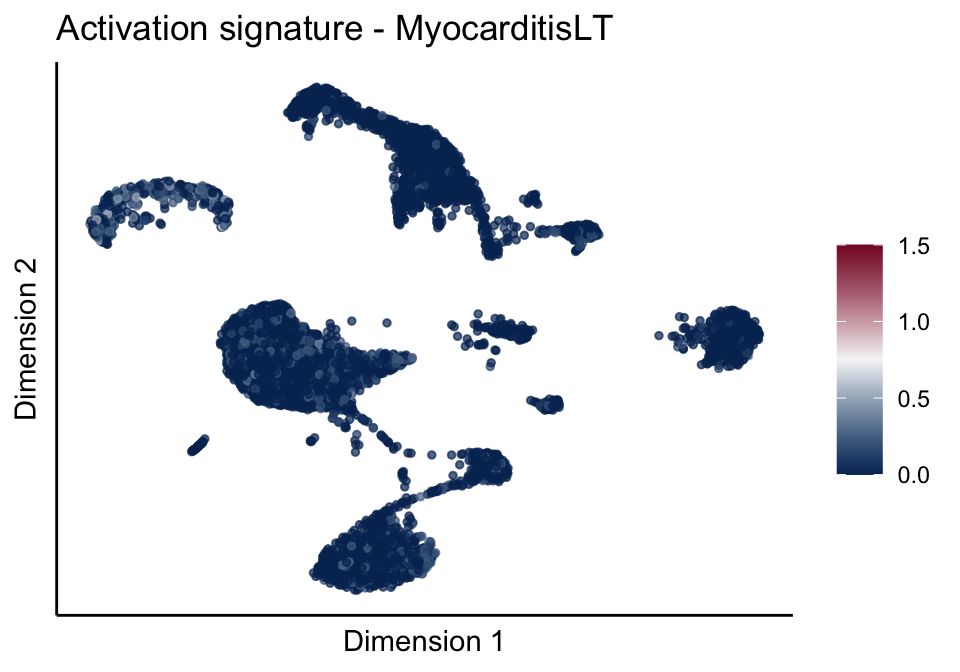
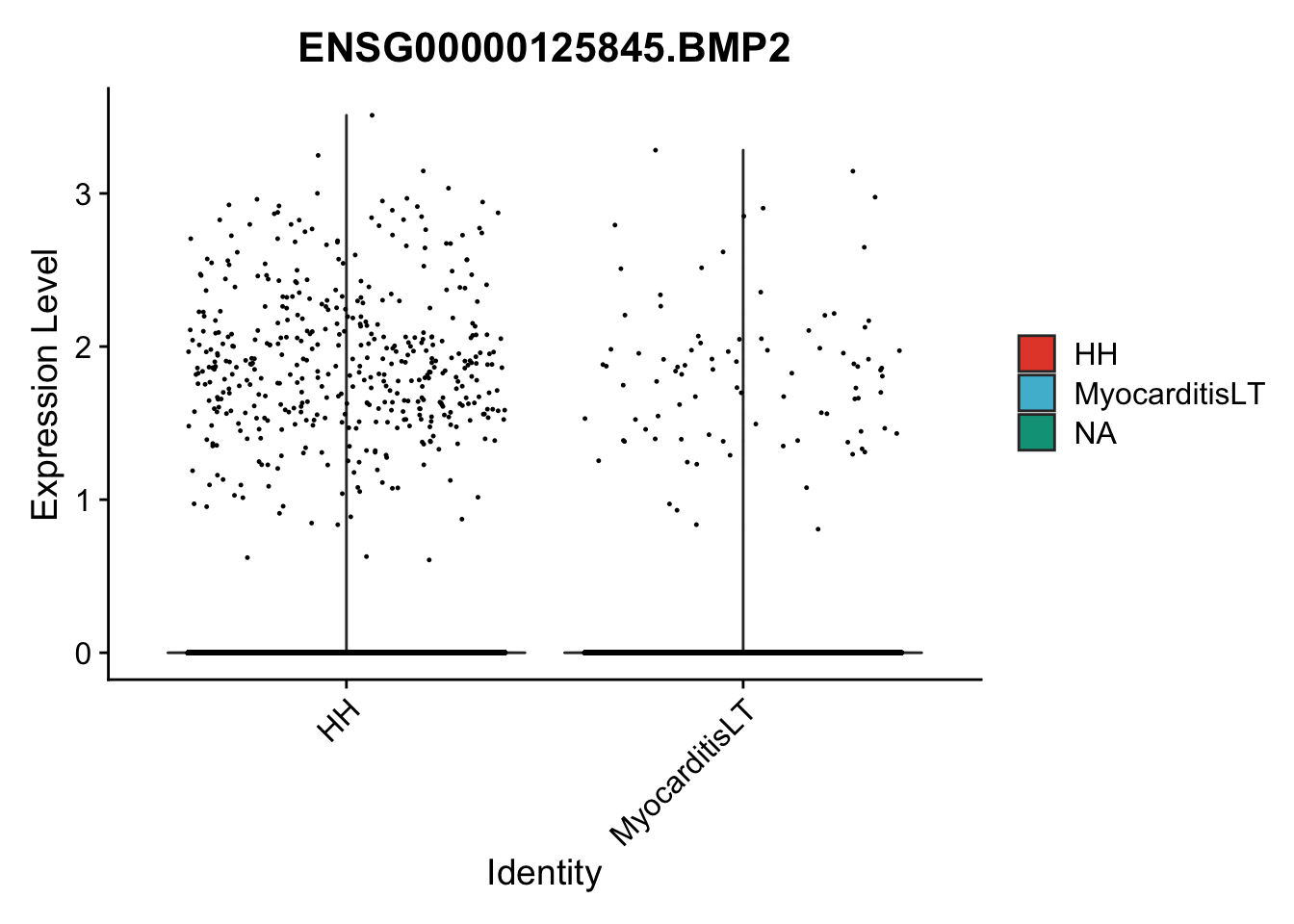
vis sel genes violin
genesDat <- data.frame(EnsID=rownames(seurat)) %>%
mutate(gene=gsub(".*\\.", "", EnsID))
selGenes <- data.frame(gene=c("BMP2", "BMP4", "BMPR1A", "BMPR2")) %>%
left_join(., genesDat, by="gene")
## subsample to equal number
seuratSub <- subset(seurat, downsample = min(table(seurat$cond2)))
pList <- sapply(selGenes$EnsID, function(x){
p <- VlnPlot(object = seuratSub, features = x,
group.by = "cond2",
cols = colCond, pt.size = 0.2
)
plot(p)
})
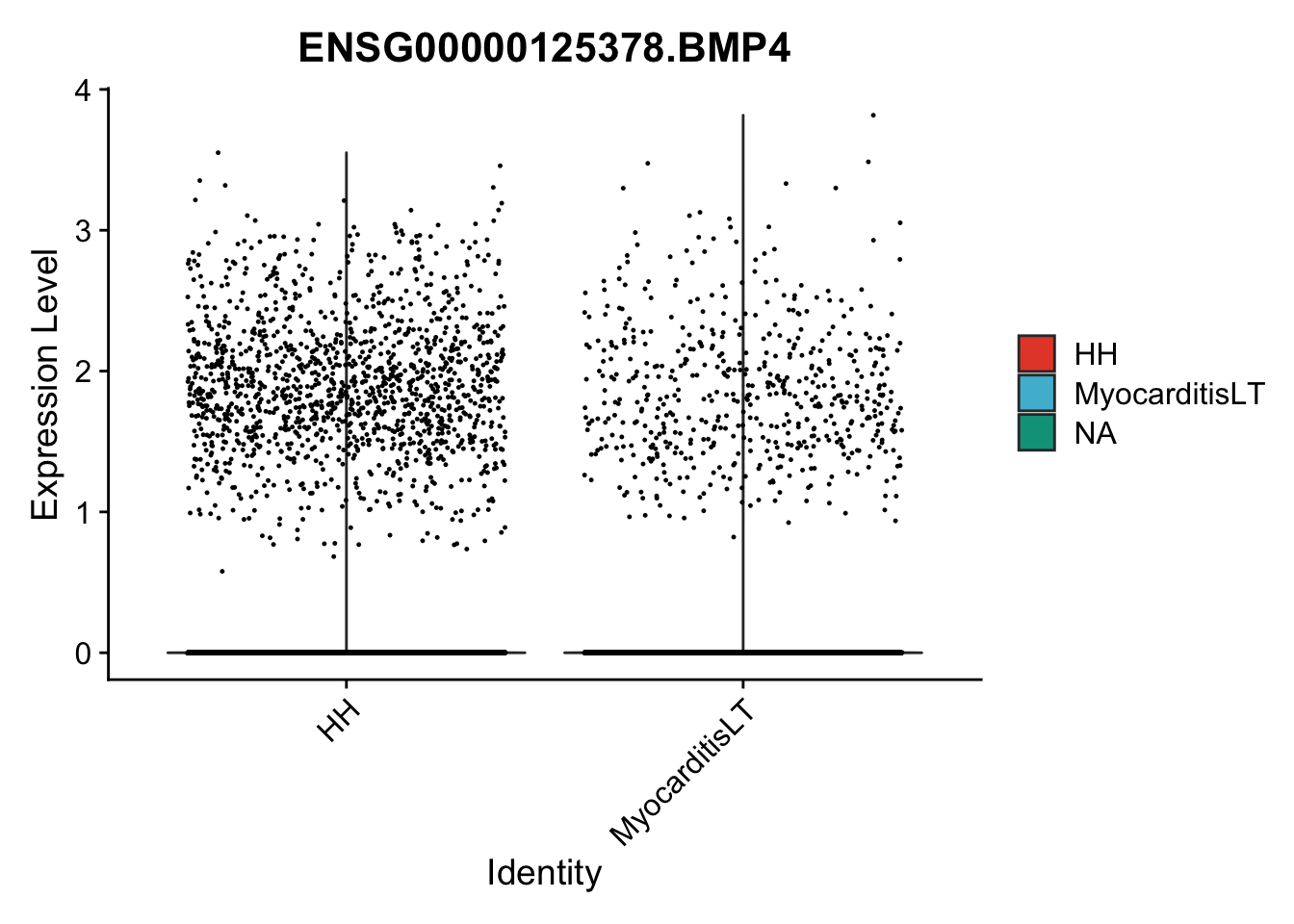
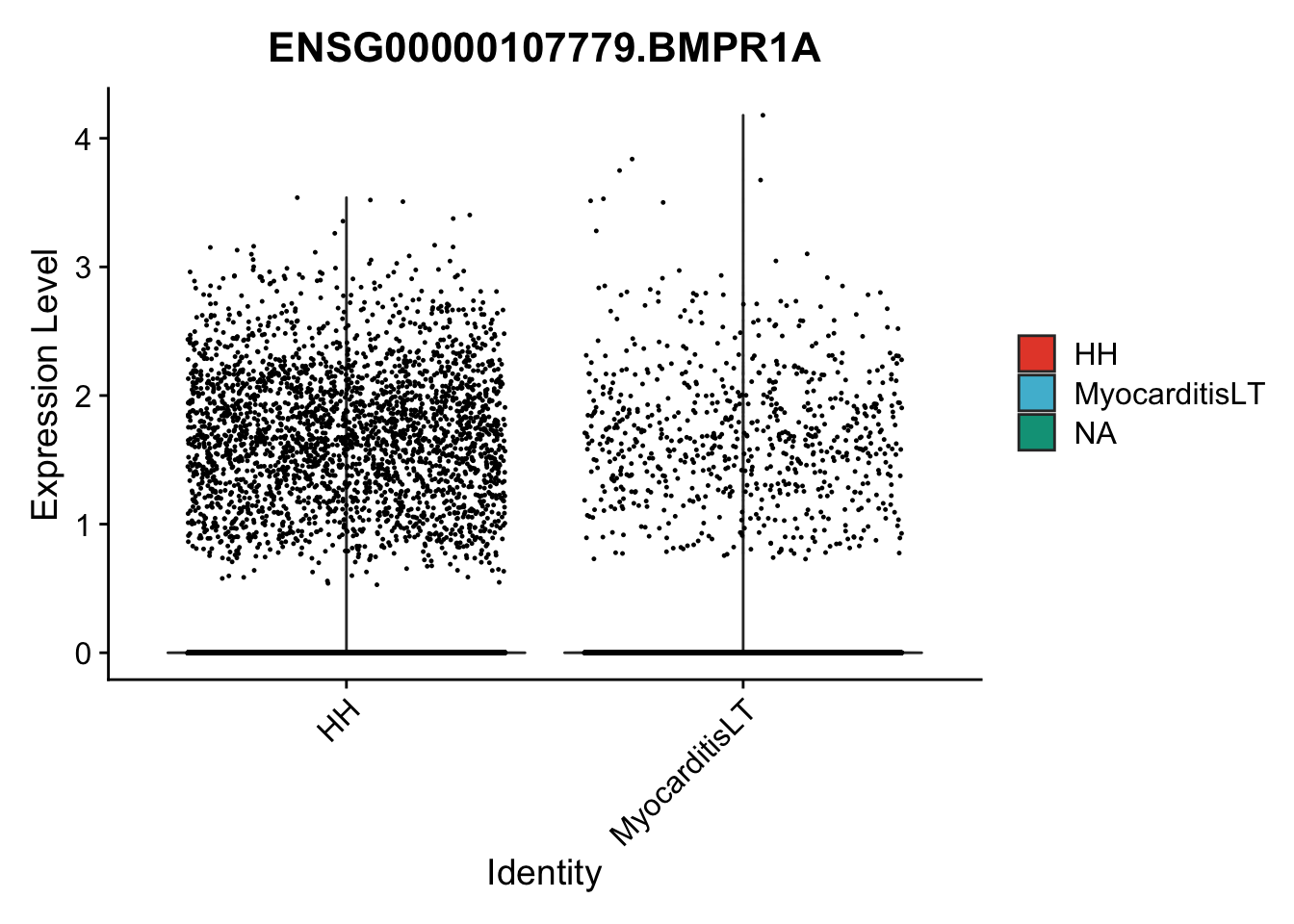
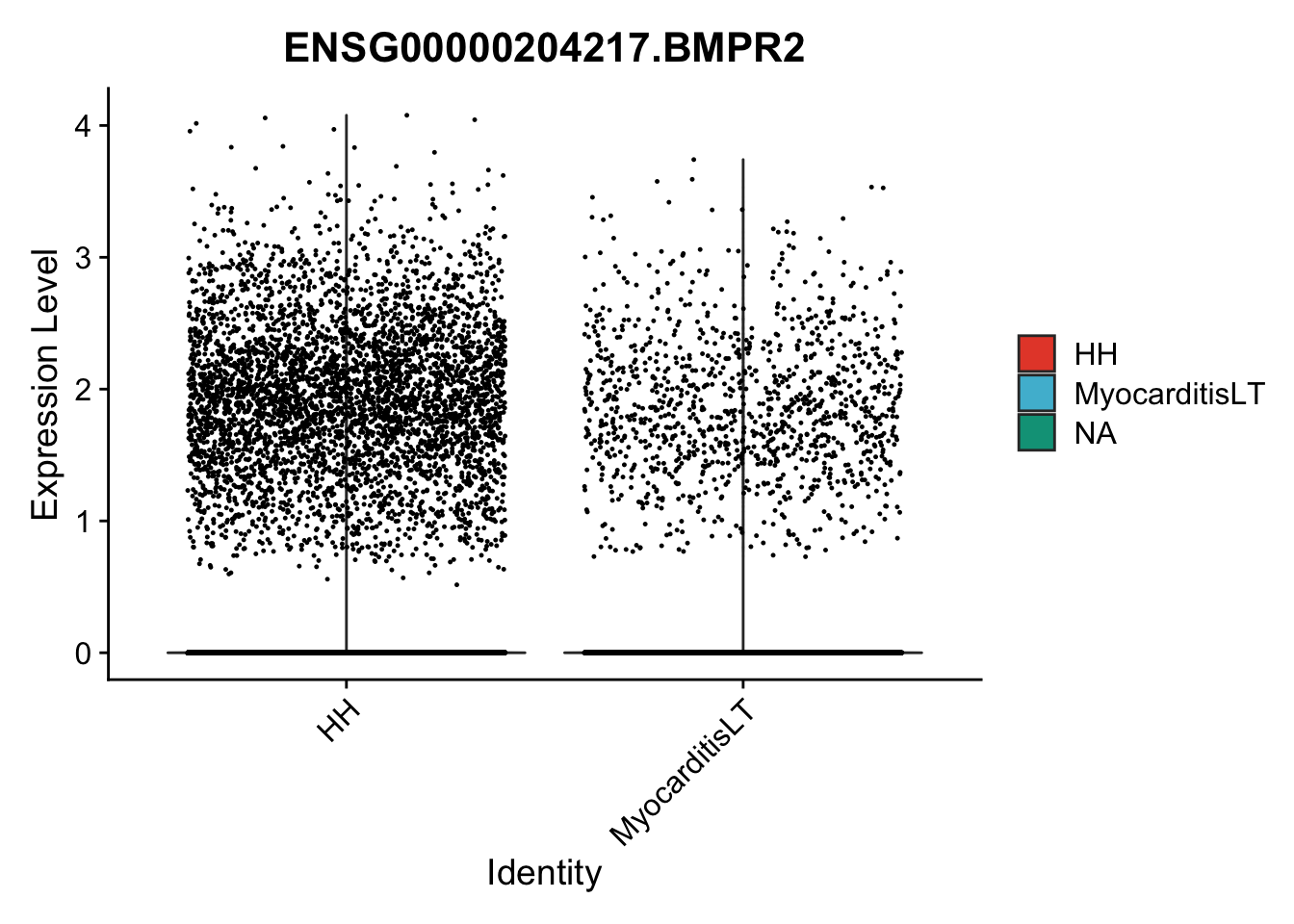
vis sel genes violin Fibroblasts
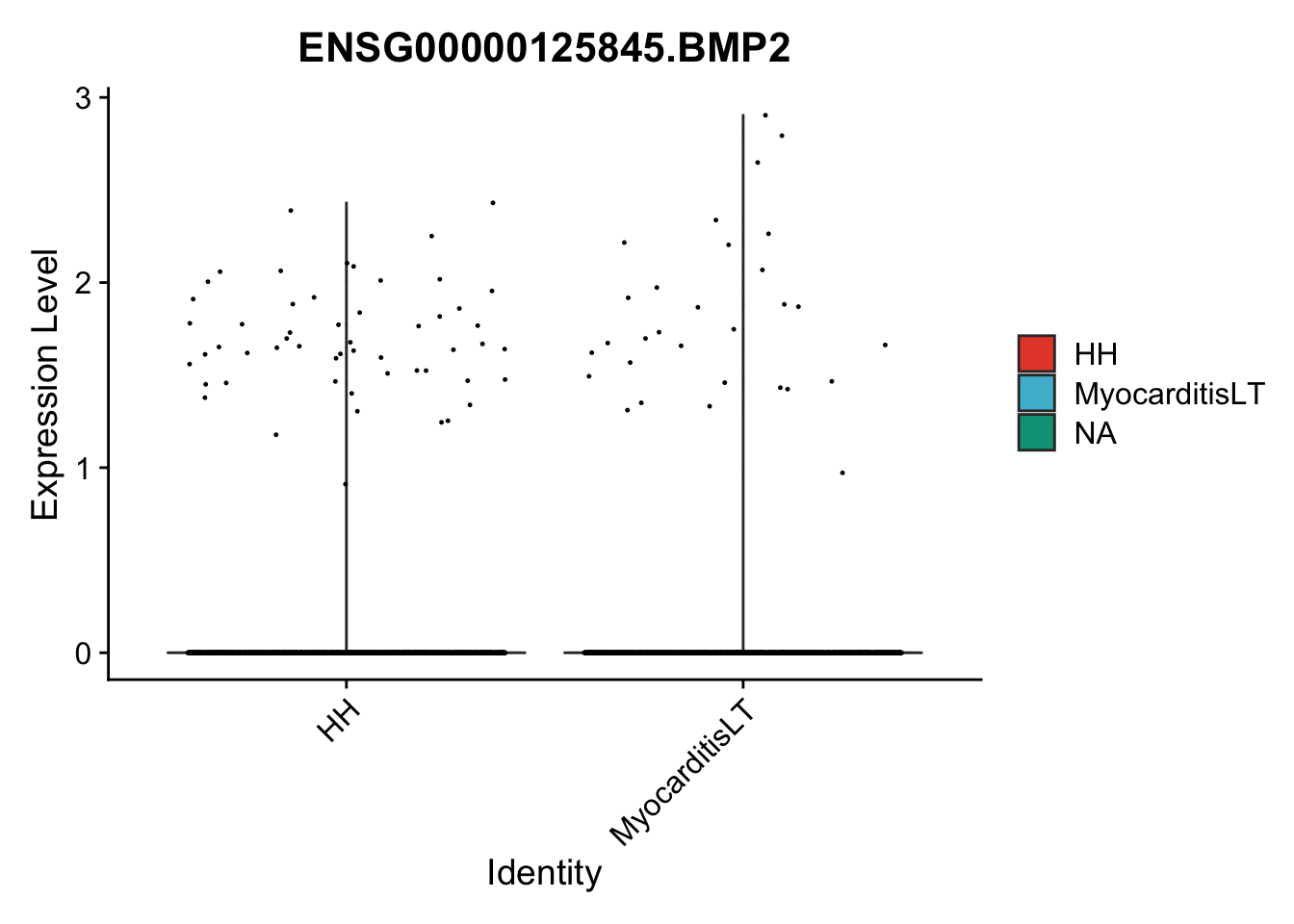
seuratSub <- subset(seurat, label == "Fibroblast")
## subsample to equal number
seuratSub <- subset(seuratSub, downsample = min(table(seuratSub$cond2)))
pList <- sapply(selGenes$EnsID, function(x){
p <- VlnPlot(object = seuratSub, features = x,
group.by = "cond2",
cols = colCond, pt.size = 0.2
)
plot(p)
})
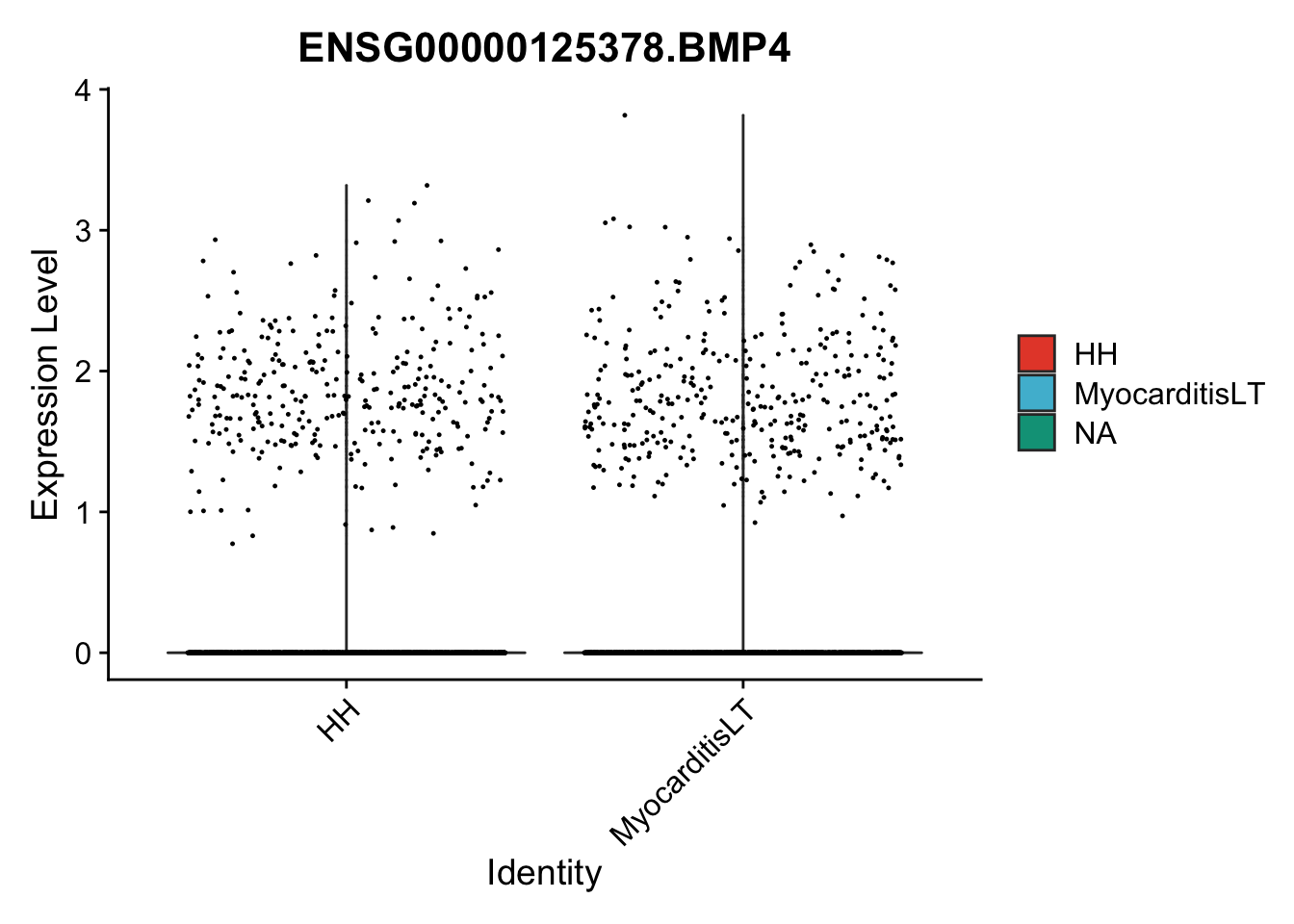
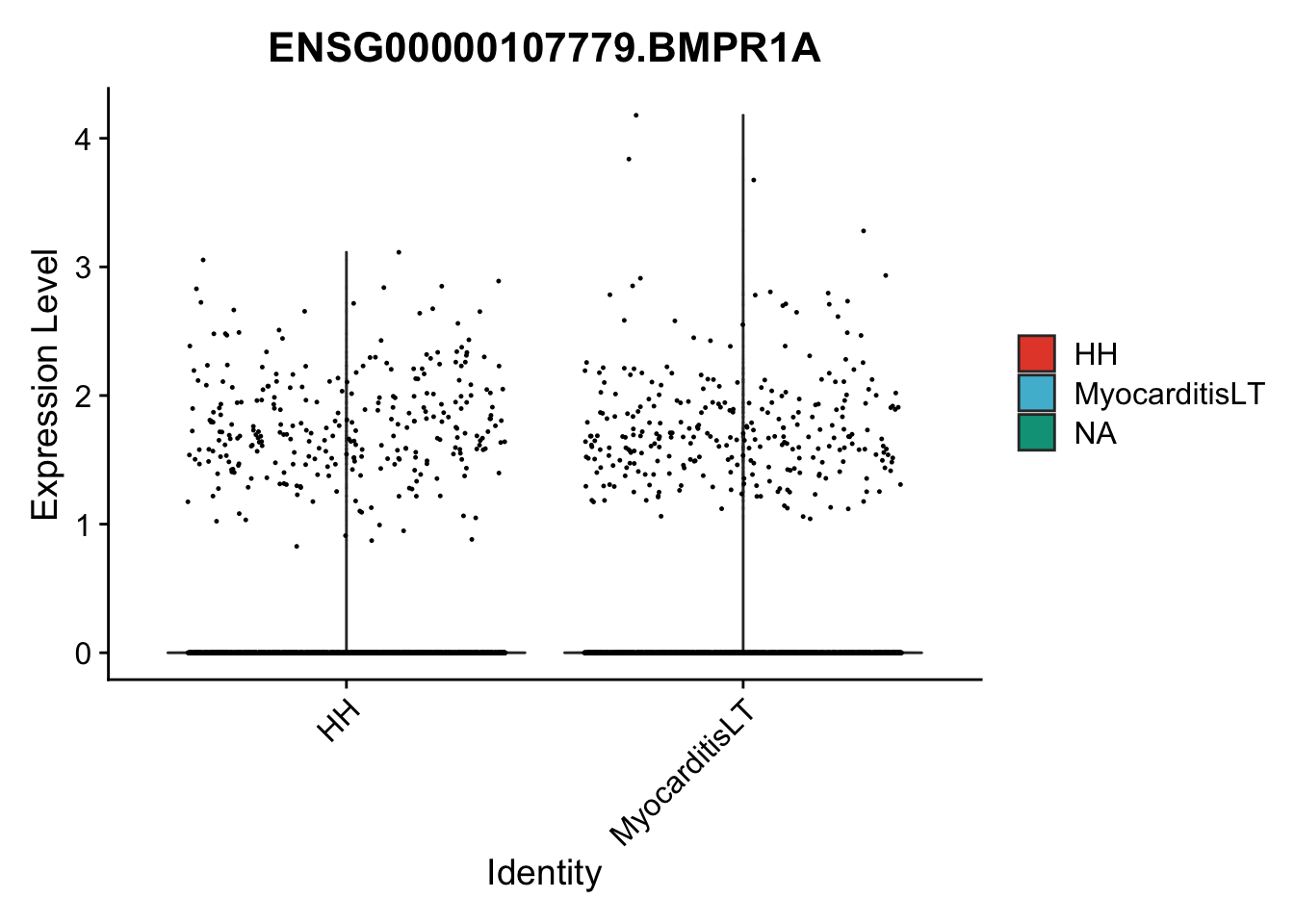
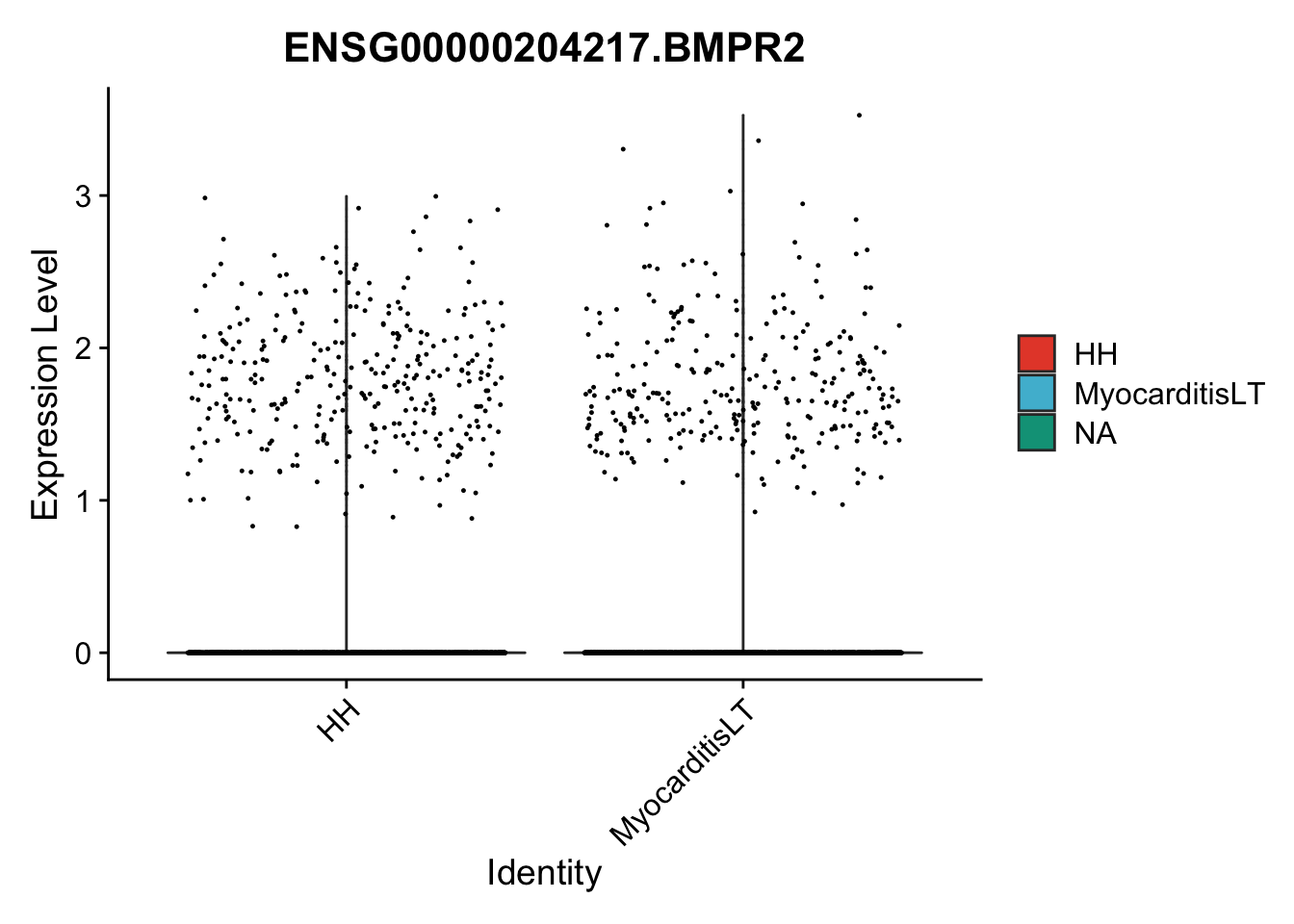
session info
sessionInfo()R version 4.1.0 (2021-05-18)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur 10.16
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRblas.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] grid parallel stats4 stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] msigdbr_7.5.1 enrichplot_1.12.3
[3] DOSE_3.18.3 org.Hs.eg.db_3.13.0
[5] AnnotationDbi_1.54.1 clusterProfiler_4.0.5
[7] gridExtra_2.3 fgsea_1.18.0
[9] sctransform_0.3.3 viridis_0.6.2
[11] viridisLite_0.4.0 pheatmap_1.0.12
[13] ggpubr_0.4.0 ggsci_2.9
[15] runSeurat3_0.1.0 here_1.0.1
[17] magrittr_2.0.3 sp_1.4-7
[19] SeuratObject_4.1.0 Seurat_4.1.1
[21] forcats_0.5.1 stringr_1.4.0
[23] dplyr_1.0.9 purrr_0.3.4
[25] readr_2.1.2 tidyr_1.2.0
[27] tibble_3.1.7 ggplot2_3.3.6
[29] tidyverse_1.3.1 SingleCellExperiment_1.14.1
[31] SummarizedExperiment_1.22.0 Biobase_2.52.0
[33] GenomicRanges_1.44.0 GenomeInfoDb_1.28.4
[35] IRanges_2.26.0 S4Vectors_0.30.2
[37] BiocGenerics_0.38.0 MatrixGenerics_1.4.3
[39] matrixStats_0.62.0
loaded via a namespace (and not attached):
[1] scattermore_0.8 bit64_4.0.5 knitr_1.39
[4] irlba_2.3.5 DelayedArray_0.18.0 data.table_1.14.2
[7] rpart_4.1.16 KEGGREST_1.32.0 RCurl_1.98-1.6
[10] generics_0.1.2 cowplot_1.1.1 RSQLite_2.2.14
[13] shadowtext_0.1.2 RANN_2.6.1 future_1.25.0
[16] bit_4.0.4 tzdb_0.3.0 spatstat.data_2.2-0
[19] xml2_1.3.3 lubridate_1.8.0 httpuv_1.6.5
[22] assertthat_0.2.1 xfun_0.30 hms_1.1.1
[25] jquerylib_0.1.4 babelgene_22.3 evaluate_0.15
[28] promises_1.2.0.1 fansi_1.0.3 dbplyr_2.1.1
[31] readxl_1.4.0 igraph_1.3.1 DBI_1.1.2
[34] htmlwidgets_1.5.4 spatstat.geom_2.4-0 ellipsis_0.3.2
[37] backports_1.4.1 deldir_1.0-6 vctrs_0.4.1
[40] ROCR_1.0-11 abind_1.4-5 cachem_1.0.6
[43] withr_2.5.0 ggforce_0.3.3 progressr_0.10.0
[46] vroom_1.5.7 treeio_1.16.2 goftest_1.2-3
[49] cluster_2.1.3 ape_5.6-2 lazyeval_0.2.2
[52] crayon_1.5.1 pkgconfig_2.0.3 labeling_0.4.2
[55] tweenr_1.0.2 nlme_3.1-157 rlang_1.0.2
[58] globals_0.15.0 lifecycle_1.0.1 miniUI_0.1.1.1
[61] downloader_0.4 modelr_0.1.8 cellranger_1.1.0
[64] rprojroot_2.0.3 polyclip_1.10-0 lmtest_0.9-40
[67] Matrix_1.4-1 aplot_0.1.4 carData_3.0-5
[70] zoo_1.8-10 reprex_2.0.1 ggridges_0.5.3
[73] png_0.1-7 bitops_1.0-7 KernSmooth_2.23-20
[76] Biostrings_2.60.2 blob_1.2.3 workflowr_1.7.0
[79] qvalue_2.24.0 parallelly_1.31.1 spatstat.random_2.2-0
[82] rstatix_0.7.0 gridGraphics_0.5-1 ggsignif_0.6.3
[85] scales_1.2.0 memoise_2.0.1 plyr_1.8.7
[88] ica_1.0-2 zlibbioc_1.38.0 compiler_4.1.0
[91] scatterpie_0.1.7 RColorBrewer_1.1-3 fitdistrplus_1.1-8
[94] cli_3.3.0 XVector_0.32.0 listenv_0.8.0
[97] patchwork_1.1.1 pbapply_1.5-0 MASS_7.3-57
[100] mgcv_1.8-40 tidyselect_1.1.2 stringi_1.7.6
[103] highr_0.9 yaml_2.3.5 GOSemSim_2.18.1
[106] ggrepel_0.9.1 sass_0.4.1 fastmatch_1.1-3
[109] tools_4.1.0 future.apply_1.9.0 rstudioapi_0.13
[112] git2r_0.30.1 farver_2.1.0 Rtsne_0.16
[115] ggraph_2.0.5 digest_0.6.29 rgeos_0.5-9
[118] shiny_1.7.1 Rcpp_1.0.8.3 car_3.0-13
[121] broom_0.8.0 later_1.3.0 RcppAnnoy_0.0.19
[124] httr_1.4.3 colorspace_2.0-3 rvest_1.0.2
[127] fs_1.5.2 tensor_1.5 reticulate_1.24
[130] splines_4.1.0 uwot_0.1.11 yulab.utils_0.0.4
[133] tidytree_0.3.9 spatstat.utils_2.3-1 graphlayouts_0.8.0
[136] ggplotify_0.1.0 plotly_4.10.0 xtable_1.8-4
[139] jsonlite_1.8.0 ggtree_3.0.4 tidygraph_1.2.1
[142] ggfun_0.0.6 R6_2.5.1 pillar_1.7.0
[145] htmltools_0.5.2 mime_0.12 glue_1.6.2
[148] fastmap_1.1.0 BiocParallel_1.26.2 codetools_0.2-18
[151] utf8_1.2.2 lattice_0.20-45 bslib_0.3.1
[154] spatstat.sparse_2.1-1 leiden_0.3.10 GO.db_3.13.0
[157] limma_3.48.3 survival_3.3-1 rmarkdown_2.14
[160] munsell_0.5.0 DO.db_2.9 GenomeInfoDbData_1.2.6
[163] haven_2.5.0 reshape2_1.4.4 gtable_0.3.0
[166] spatstat.core_2.4-2 date()[1] "Tue Jul 19 15:03:36 2022"
sessionInfo()R version 4.1.0 (2021-05-18)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur 10.16
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRblas.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] grid parallel stats4 stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] msigdbr_7.5.1 enrichplot_1.12.3
[3] DOSE_3.18.3 org.Hs.eg.db_3.13.0
[5] AnnotationDbi_1.54.1 clusterProfiler_4.0.5
[7] gridExtra_2.3 fgsea_1.18.0
[9] sctransform_0.3.3 viridis_0.6.2
[11] viridisLite_0.4.0 pheatmap_1.0.12
[13] ggpubr_0.4.0 ggsci_2.9
[15] runSeurat3_0.1.0 here_1.0.1
[17] magrittr_2.0.3 sp_1.4-7
[19] SeuratObject_4.1.0 Seurat_4.1.1
[21] forcats_0.5.1 stringr_1.4.0
[23] dplyr_1.0.9 purrr_0.3.4
[25] readr_2.1.2 tidyr_1.2.0
[27] tibble_3.1.7 ggplot2_3.3.6
[29] tidyverse_1.3.1 SingleCellExperiment_1.14.1
[31] SummarizedExperiment_1.22.0 Biobase_2.52.0
[33] GenomicRanges_1.44.0 GenomeInfoDb_1.28.4
[35] IRanges_2.26.0 S4Vectors_0.30.2
[37] BiocGenerics_0.38.0 MatrixGenerics_1.4.3
[39] matrixStats_0.62.0
loaded via a namespace (and not attached):
[1] scattermore_0.8 bit64_4.0.5 knitr_1.39
[4] irlba_2.3.5 DelayedArray_0.18.0 data.table_1.14.2
[7] rpart_4.1.16 KEGGREST_1.32.0 RCurl_1.98-1.6
[10] generics_0.1.2 cowplot_1.1.1 RSQLite_2.2.14
[13] shadowtext_0.1.2 RANN_2.6.1 future_1.25.0
[16] bit_4.0.4 tzdb_0.3.0 spatstat.data_2.2-0
[19] xml2_1.3.3 lubridate_1.8.0 httpuv_1.6.5
[22] assertthat_0.2.1 xfun_0.30 hms_1.1.1
[25] jquerylib_0.1.4 babelgene_22.3 evaluate_0.15
[28] promises_1.2.0.1 fansi_1.0.3 dbplyr_2.1.1
[31] readxl_1.4.0 igraph_1.3.1 DBI_1.1.2
[34] htmlwidgets_1.5.4 spatstat.geom_2.4-0 ellipsis_0.3.2
[37] backports_1.4.1 deldir_1.0-6 vctrs_0.4.1
[40] ROCR_1.0-11 abind_1.4-5 cachem_1.0.6
[43] withr_2.5.0 ggforce_0.3.3 progressr_0.10.0
[46] vroom_1.5.7 treeio_1.16.2 goftest_1.2-3
[49] cluster_2.1.3 ape_5.6-2 lazyeval_0.2.2
[52] crayon_1.5.1 pkgconfig_2.0.3 labeling_0.4.2
[55] tweenr_1.0.2 nlme_3.1-157 rlang_1.0.2
[58] globals_0.15.0 lifecycle_1.0.1 miniUI_0.1.1.1
[61] downloader_0.4 modelr_0.1.8 cellranger_1.1.0
[64] rprojroot_2.0.3 polyclip_1.10-0 lmtest_0.9-40
[67] Matrix_1.4-1 aplot_0.1.4 carData_3.0-5
[70] zoo_1.8-10 reprex_2.0.1 ggridges_0.5.3
[73] png_0.1-7 bitops_1.0-7 KernSmooth_2.23-20
[76] Biostrings_2.60.2 blob_1.2.3 workflowr_1.7.0
[79] qvalue_2.24.0 parallelly_1.31.1 spatstat.random_2.2-0
[82] rstatix_0.7.0 gridGraphics_0.5-1 ggsignif_0.6.3
[85] scales_1.2.0 memoise_2.0.1 plyr_1.8.7
[88] ica_1.0-2 zlibbioc_1.38.0 compiler_4.1.0
[91] scatterpie_0.1.7 RColorBrewer_1.1-3 fitdistrplus_1.1-8
[94] cli_3.3.0 XVector_0.32.0 listenv_0.8.0
[97] patchwork_1.1.1 pbapply_1.5-0 MASS_7.3-57
[100] mgcv_1.8-40 tidyselect_1.1.2 stringi_1.7.6
[103] highr_0.9 yaml_2.3.5 GOSemSim_2.18.1
[106] ggrepel_0.9.1 sass_0.4.1 fastmatch_1.1-3
[109] tools_4.1.0 future.apply_1.9.0 rstudioapi_0.13
[112] git2r_0.30.1 farver_2.1.0 Rtsne_0.16
[115] ggraph_2.0.5 digest_0.6.29 rgeos_0.5-9
[118] shiny_1.7.1 Rcpp_1.0.8.3 car_3.0-13
[121] broom_0.8.0 later_1.3.0 RcppAnnoy_0.0.19
[124] httr_1.4.3 colorspace_2.0-3 rvest_1.0.2
[127] fs_1.5.2 tensor_1.5 reticulate_1.24
[130] splines_4.1.0 uwot_0.1.11 yulab.utils_0.0.4
[133] tidytree_0.3.9 spatstat.utils_2.3-1 graphlayouts_0.8.0
[136] ggplotify_0.1.0 plotly_4.10.0 xtable_1.8-4
[139] jsonlite_1.8.0 ggtree_3.0.4 tidygraph_1.2.1
[142] ggfun_0.0.6 R6_2.5.1 pillar_1.7.0
[145] htmltools_0.5.2 mime_0.12 glue_1.6.2
[148] fastmap_1.1.0 BiocParallel_1.26.2 codetools_0.2-18
[151] utf8_1.2.2 lattice_0.20-45 bslib_0.3.1
[154] spatstat.sparse_2.1-1 leiden_0.3.10 GO.db_3.13.0
[157] limma_3.48.3 survival_3.3-1 rmarkdown_2.14
[160] munsell_0.5.0 DO.db_2.9 GenomeInfoDbData_1.2.6
[163] haven_2.5.0 reshape2_1.4.4 gtable_0.3.0
[166] spatstat.core_2.4-2