SNP & TAD
Renee Matthews
2025-06-10
Last updated: 2025-07-09
Checks: 7 0
Knit directory: ATAC_learning/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20231016) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 0fa75c3. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .RData
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/H3K27ac_integration_noM.Rmd
Ignored: analysis/figure/
Ignored: data/ACresp_SNP_table.csv
Ignored: data/ARR_SNP_table.csv
Ignored: data/All_merged_peaks.tsv
Ignored: data/CAD_gwas_dataframe.RDS
Ignored: data/CTX_SNP_table.csv
Ignored: data/Collapsed_expressed_NG_peak_table.csv
Ignored: data/DEG_toplist_sep_n45.RDS
Ignored: data/FRiP_first_run.txt
Ignored: data/Final_four_data/
Ignored: data/Frip_1_reads.csv
Ignored: data/Frip_2_reads.csv
Ignored: data/Frip_3_reads.csv
Ignored: data/Frip_4_reads.csv
Ignored: data/Frip_5_reads.csv
Ignored: data/Frip_6_reads.csv
Ignored: data/GO_KEGG_analysis/
Ignored: data/HF_SNP_table.csv
Ignored: data/Ind1_75DA24h_dedup_peaks.csv
Ignored: data/Ind1_TSS_peaks.RDS
Ignored: data/Ind1_firstfragment_files.txt
Ignored: data/Ind1_fragment_files.txt
Ignored: data/Ind1_peaks_list.RDS
Ignored: data/Ind1_summary.txt
Ignored: data/Ind2_TSS_peaks.RDS
Ignored: data/Ind2_fragment_files.txt
Ignored: data/Ind2_peaks_list.RDS
Ignored: data/Ind2_summary.txt
Ignored: data/Ind3_TSS_peaks.RDS
Ignored: data/Ind3_fragment_files.txt
Ignored: data/Ind3_peaks_list.RDS
Ignored: data/Ind3_summary.txt
Ignored: data/Ind4_79B24h_dedup_peaks.csv
Ignored: data/Ind4_TSS_peaks.RDS
Ignored: data/Ind4_V24h_fraglength.txt
Ignored: data/Ind4_fragment_files.txt
Ignored: data/Ind4_fragment_filesN.txt
Ignored: data/Ind4_peaks_list.RDS
Ignored: data/Ind4_summary.txt
Ignored: data/Ind5_TSS_peaks.RDS
Ignored: data/Ind5_fragment_files.txt
Ignored: data/Ind5_fragment_filesN.txt
Ignored: data/Ind5_peaks_list.RDS
Ignored: data/Ind5_summary.txt
Ignored: data/Ind6_TSS_peaks.RDS
Ignored: data/Ind6_fragment_files.txt
Ignored: data/Ind6_peaks_list.RDS
Ignored: data/Ind6_summary.txt
Ignored: data/Knowles_4.RDS
Ignored: data/Knowles_5.RDS
Ignored: data/Knowles_6.RDS
Ignored: data/LiSiLTDNRe_TE_df.RDS
Ignored: data/MI_gwas.RDS
Ignored: data/SNP_GWAS_PEAK_MRC_id
Ignored: data/SNP_GWAS_PEAK_MRC_id.csv
Ignored: data/SNP_gene_cat_list.tsv
Ignored: data/SNP_supp_schneider.RDS
Ignored: data/TE_info/
Ignored: data/TFmapnames.RDS
Ignored: data/all_TSSE_scores.RDS
Ignored: data/all_four_filtered_counts.txt
Ignored: data/aln_run1_results.txt
Ignored: data/anno_ind1_DA24h.RDS
Ignored: data/anno_ind4_V24h.RDS
Ignored: data/annotated_gwas_SNPS.csv
Ignored: data/background_n45_he_peaks.RDS
Ignored: data/cardiac_muscle_FRIP.csv
Ignored: data/cardiomyocyte_FRIP.csv
Ignored: data/col_ng_peak.csv
Ignored: data/cormotif_full_4_run.RDS
Ignored: data/cormotif_full_4_run_he.RDS
Ignored: data/cormotif_full_6_run.RDS
Ignored: data/cormotif_full_6_run_he.RDS
Ignored: data/cormotif_probability_45_list.csv
Ignored: data/cormotif_probability_45_list_he.csv
Ignored: data/cormotif_probability_all_6_list.csv
Ignored: data/cormotif_probability_all_6_list_he.csv
Ignored: data/datasave.RDS
Ignored: data/embryo_heart_FRIP.csv
Ignored: data/enhancer_list_ENCFF126UHK.bed
Ignored: data/enhancerdata/
Ignored: data/filt_Peaks_efit2.RDS
Ignored: data/filt_Peaks_efit2_bl.RDS
Ignored: data/filt_Peaks_efit2_n45.RDS
Ignored: data/first_Peaksummarycounts.csv
Ignored: data/first_run_frag_counts.txt
Ignored: data/full_bedfiles/
Ignored: data/gene_ref.csv
Ignored: data/gwas_1_dataframe.RDS
Ignored: data/gwas_2_dataframe.RDS
Ignored: data/gwas_3_dataframe.RDS
Ignored: data/gwas_4_dataframe.RDS
Ignored: data/gwas_5_dataframe.RDS
Ignored: data/high_conf_peak_counts.csv
Ignored: data/high_conf_peak_counts.txt
Ignored: data/high_conf_peaks_bl_counts.txt
Ignored: data/high_conf_peaks_counts.txt
Ignored: data/hits_files/
Ignored: data/hyper_files/
Ignored: data/hypo_files/
Ignored: data/ind1_DA24hpeaks.RDS
Ignored: data/ind1_TSSE.RDS
Ignored: data/ind2_TSSE.RDS
Ignored: data/ind3_TSSE.RDS
Ignored: data/ind4_TSSE.RDS
Ignored: data/ind4_V24hpeaks.RDS
Ignored: data/ind5_TSSE.RDS
Ignored: data/ind6_TSSE.RDS
Ignored: data/initial_complete_stats_run1.txt
Ignored: data/left_ventricle_FRIP.csv
Ignored: data/median_24_lfc.RDS
Ignored: data/median_3_lfc.RDS
Ignored: data/mergedPeads.gff
Ignored: data/mergedPeaks.gff
Ignored: data/motif_list_full
Ignored: data/motif_list_n45
Ignored: data/motif_list_n45.RDS
Ignored: data/multiqc_fastqc_run1.txt
Ignored: data/multiqc_fastqc_run2.txt
Ignored: data/multiqc_genestat_run1.txt
Ignored: data/multiqc_genestat_run2.txt
Ignored: data/my_hc_filt_counts.RDS
Ignored: data/my_hc_filt_counts_n45.RDS
Ignored: data/n45_bedfiles/
Ignored: data/n45_files
Ignored: data/other_papers/
Ignored: data/peakAnnoList_1.RDS
Ignored: data/peakAnnoList_2.RDS
Ignored: data/peakAnnoList_24_full.RDS
Ignored: data/peakAnnoList_24_n45.RDS
Ignored: data/peakAnnoList_3.RDS
Ignored: data/peakAnnoList_3_full.RDS
Ignored: data/peakAnnoList_3_n45.RDS
Ignored: data/peakAnnoList_4.RDS
Ignored: data/peakAnnoList_5.RDS
Ignored: data/peakAnnoList_6.RDS
Ignored: data/peakAnnoList_Eight.RDS
Ignored: data/peakAnnoList_full_motif.RDS
Ignored: data/peakAnnoList_n45_motif.RDS
Ignored: data/siglist_full.RDS
Ignored: data/siglist_n45.RDS
Ignored: data/summarized_peaks_dataframe.txt
Ignored: data/summary_peakIDandReHeat.csv
Ignored: data/test.list.RDS
Ignored: data/testnames.txt
Ignored: data/toplist_6.RDS
Ignored: data/toplist_full.RDS
Ignored: data/toplist_full_DAR_6.RDS
Ignored: data/toplist_n45.RDS
Ignored: data/trimmed_seq_length.csv
Ignored: data/unclassified_full_set_peaks.RDS
Ignored: data/unclassified_n45_set_peaks.RDS
Ignored: data/xstreme/
Untracked files:
Untracked: RNA_seq_integration.Rmd
Untracked: Rplot.pdf
Untracked: Sig_meta
Untracked: analysis/.gitignore
Untracked: analysis/AF_HF_SNP_DAR.Rmd
Untracked: analysis/Cormotif_analysis_testing diff.Rmd
Untracked: analysis/Diagnosis-tmm.Rmd
Untracked: analysis/Expressed_RNA_associations.Rmd
Untracked: analysis/LFC_corr.Rmd
Untracked: analysis/SVA.Rmd
Untracked: analysis/Tan2020.Rmd
Untracked: analysis/Top2B_analysis.Rmd
Untracked: analysis/making_master_peaks_list.Rmd
Untracked: analysis/my_hc_filt_counts.csv
Untracked: code/Concatenations_for_export.R
Untracked: code/IGV_snapshot_code.R
Untracked: code/LongDARlist.R
Untracked: code/just_for_Fun.R
Untracked: my_plot.pdf
Untracked: my_plot.png
Untracked: output/cormotif_probability_45_list.csv
Untracked: output/cormotif_probability_all_6_list.csv
Untracked: setup.RData
Unstaged changes:
Modified: ATAC_learning.Rproj
Modified: analysis/AC_shared_analysis.Rmd
Modified: analysis/AF_HF_SNPs.Rmd
Modified: analysis/Cardiotox_SNPs.Rmd
Modified: analysis/Cormotif_analysis.Rmd
Modified: analysis/DEG_analysis.Rmd
Modified: analysis/GO_analysis_DAR.Rmd
Modified: analysis/H3K27ac_initial_QC.Rmd
Modified: analysis/H3K27ac_integration.Rmd
Modified: analysis/Jaspar_motif.Rmd
Modified: analysis/Jaspar_motif_DAR.Rmd
Modified: analysis/Jaspar_motif_ff.Rmd
Modified: analysis/TE_analysis_norm.Rmd
Modified: analysis/final_four_analysis.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/SNP_TAD_peaks.Rmd) and
HTML (docs/SNP_TAD_peaks.html) files. If you’ve configured
a remote Git repository (see ?wflow_git_remote), click on
the hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 0fa75c3 | reneeisnowhere | 2025-07-09 | wflow_publish("analysis/SNP_TAD_peaks.Rmd") |
library(tidyverse)
library(kableExtra)
library(broom)
library(RColorBrewer)
library(ChIPseeker)
library(ChIPpeakAnno)
library("TxDb.Hsapiens.UCSC.hg38.knownGene")
library("org.Hs.eg.db")
library(rtracklayer)
library(edgeR)
library(ggfortify)
library(limma)
library(readr)
library(BiocGenerics)
library(gridExtra)
library(VennDiagram)
library(scales)
library(BiocParallel)
library(ggpubr)
library(devtools)
library(biomaRt)
library(eulerr)
library(smplot2)
library(genomation)
library(ggsignif)
library(plyranges)
library(ggrepel)
library(epitools)
library(circlize)
library(readxl)
library(ComplexHeatmap)
library(gwascat)
library(liftOver)Loading data frames
### Pulling the all regions granges list from the motif list of lists
Motif_list_gr <- readRDS("data/Final_four_data/re_analysis/Motif_list_granges.RDS")
### no change motif_list_gr names so they do not overwrite the dataframes
names(Motif_list_gr) <- paste0(names(Motif_list_gr), "_gr")
list2env(Motif_list_gr[10],envir= .GlobalEnv)<environment: R_GlobalEnv>annotated_DARs<- readRDS("data/Final_four_data/re_analysis/DOX_DAR_annotated_peaks_chipannno.RDS")
Left_ventricle_TAD <- import(con = "C://Users/renee/Downloads/hg38.TADs/hg38/VentricleLeft_STL003_Leung_2015-raw_TADs.txt", format = "bed",genome="hg38")
mcols(Left_ventricle_TAD)$TAD_id <- paste0("TAD_", seq_along(Left_ventricle_TAD))
# mcols(Left_ventricle_TAD)$name <- Left_ventricle_TAD$TAD_id
##exporting the LEFt ventricle tad info for IGV visualization
# export(Left_ventricle_TAD, con = "data/Final_four_data/re_analysis/Other_bed_files/TAD_regions.bed", format="bed")
Schneider_all_SNPS <- read_delim("data/other_papers/Schneider_all_SNPS.txt",
delim = "\t", escape_double = FALSE,
trim_ws = TRUE)
Schneider_all_SNPS_df <- Schneider_all_SNPS %>%
dplyr::rename("RSID"="#Uploaded_variation") %>%
dplyr::select(RSID,Location,SYMBOL,Gene, SOURCE) %>%
distinct(RSID,Location,SYMBOL,.keep_all = TRUE) %>%
dplyr::rename("Close_SYMBOL"="SYMBOL") %>%
dplyr::filter(!str_starts(Location, "H")) %>%
separate_wider_delim(Location,delim=":",names=c("Chr","Coords")) %>%
separate_wider_delim(Coords,delim= "-", names= c("Start","End")) %>%
mutate(Chr=paste0("chr",Chr)) %>%
group_by(RSID) %>%
reframe(Chr=unique(Chr),
Start=unique(Start),
End=unique(End),
Close_SYMBOL=paste(unique(Close_SYMBOL),collapse=";"),
Gene=paste(Gene,collapse=";"),
SOURCE=paste(SOURCE,collapse=";")
) %>%
GRanges() %>% as.data.frame
schneider_gr <-Schneider_all_SNPS_df%>%
dplyr::select(seqnames,start,end,RSID:SOURCE) %>%
distinct() %>%
GRanges()
# export(schneider_gr, con = "data/Final_four_data/re_analysis/Other_bed_files/CardiotoxSNPs.bed", format="bed")
toptable_results <- readRDS("data/Final_four_data/re_analysis/Toptable_results.RDS")
all_results <- toptable_results %>%
imap(~ .x %>% tibble::rownames_to_column(var = "rowname") %>%
mutate(source = .y)) %>%
bind_rows()
all_results_pivot <- all_results %>%
dplyr::select(genes,logFC,source) %>%
pivot_wider(., id_cols = genes, names_from = source, values_from = logFC) %>%
dplyr::select(genes,DOX_3,EPI_3,DNR_3,MTX_3,TRZ_3,DOX_24,EPI_24,DNR_24,MTX_24,TRZ_24)
toplistall_RNA <- readRDS("data/other_papers/toplistall_RNA.RDS") %>%
mutate(logFC = logFC*(-1))
Assigned_genes_toPeak <- annotated_DARs$DOX_24 %>% as.data.frame() %>%
dplyr::select(mcols.genes,annotation, geneId, distanceToTSS) %>%
dplyr::rename("Peakid"=mcols.genes)
RNA_results <-
toplistall_RNA %>%
dplyr::select(time:logFC) %>%
tidyr::unite("sample",time, id) %>%
pivot_wider(., id_cols = c(ENTREZID,SYMBOL),names_from = sample, values_from = logFC) %>%
rename_with(~ str_replace(., "hours", "RNA"))
Peak_gene_RNA_LFC <- Assigned_genes_toPeak %>%
left_join(., RNA_results, by =c("geneId"="ENTREZID"))
entrez_ids <- Assigned_genes_toPeak$geneId
gene_info <- AnnotationDbi::select(
org.Hs.eg.db,
keys = entrez_ids,
columns = c("SYMBOL"),
keytype = "ENTREZID"
)
gene_info_collapsed <- gene_info %>%
group_by(ENTREZID) %>%
summarise(SYMBOL = paste(unique(SYMBOL), collapse = ","), .groups = "drop")
DOX_DAR_24hr_table <- annotated_DARs$DOX_24 %>%
as.data.frame()
# for_export <- DOX_24_DAR%>%
# # as.data.frame() %>%
# ### mark significance with color
# mutate(sig_24=if_else(mcols.adj.P.Val<0.05,"TRUE","FALSE")) %>%
# dplyr::select(seqnames:mcols.genes,sig_24) %>%
# GRanges
#
# ### add to the granges thingy for exporting
# mcols(for_export)$itemRgb <- ifelse(mcols(for_export)$sig_24,
# "255,0,0", # red for significant
# "190,190,190") # gray for not significant
# mcols(for_export)$sig_24 <- as.logical(mcols(for_export)$sig_24)
# mcols(for_export)$itemRgb <- as.character(mcols(for_export)$itemRgb)
#
# bed_df <- data.frame(
# seqnames = seqnames(for_export),
# start = start(for_export) - 1, # BED is 0-based
# end = end(for_export),
# strand = "*",
# thickStart = start(for_export) - 1,
# thickEnd = end(for_export),
# itemRgb = ifelse(mcols(for_export)$sig_24, "255,0,0", "190,190,190"),
# stringsAsFactors = FALSE)
# # )
#
# write.table(bed_df,
# file = "data/Final_four_data/re_analysis/Other_bed_files/DOX_24hour_sig_notsig_regions.bed",
# quote = FALSE, sep = "\t", row.names = FALSE, col.names = FALSE)Enrichment test of sig DAR and non-sig DAR of DOX within SNP-containing TADS
test_ol <- join_overlap_intersect(Left_ventricle_TAD, schneider_gr)
df <- as.data.frame(test_ol, row.names = NULL)
TAD_SNP_ol <- test_ol %>% as.data.frame() %>%
distinct(TAD_id, RSID)
peak_ol <- join_overlap_intersect(all_regions_gr, Left_ventricle_TAD)
TAD_SNP_Peak_ol <- peak_ol %>%
as.data.frame() %>%
dplyr::filter(TAD_id %in% TAD_SNP_ol$TAD_id)
snp_ol <- join_overlap_inner(schneider_gr, Left_ventricle_TAD)
TAD_peak_ol <- peak_ol %>%
as.data.frame() %>%
distinct(Peakid,.keep_all = TRUE)
left_ventricle_ol <- join_overlap_inner(all_regions_gr ,Left_ventricle_TAD) %>%
as.data.frame() %>%
distinct(Peakid,.keep_all = TRUE) %>%
dplyr::filter(TAD_id %in% TAD_SNP_ol$TAD_id)
peak_df <- as.data.frame(left_ventricle_ol)
SNP_df <- as.data.frame(snp_ol)
peak_snp_pairs <- inner_join(peak_df, SNP_df, by = "TAD_id", suffix = c(".peak", ".snp")) %>%
mutate(
peak_center = (start.peak + end.peak) / 2,
distance = abs(peak_center - start.snp) # or any metric you prefer
)reds <- colorRampPalette(brewer.pal(9, "Reds")[3:9])(12)
greens <- colorRampPalette(brewer.pal(9, "Greens")[3:9])(12)
blues <- colorRampPalette(brewer.pal(9, "Blues")[3:9])(12)
purples <- colorRampPalette(brewer.pal(9, "Purples")[3:9])(12)
oranges <- colorRampPalette(brewer.pal(9, "Oranges")[3:9])(12)
tads <- unique(peak_snp_pairs$TAD_id)
num_tads <- length(tads)
color_spectrum <- c(reds, greens, blues, purples, oranges)[1:num_tads]
if (num_tads > length(color_spectrum)) {
stop("Not enough colors for TADs. Add more palettes.")
}
tad_colors <- color_spectrum[1:num_tads]
names(tad_colors) <- tads # Assign color names to TAD IDs
#ha <- HeatmapAnnotation(TAD = df$TAD_id, col = list(TAD = tad_colors))DOX_24_DAR <- as.data.frame(annotated_DARs$DOX_24)
EPI_24_DAR <- as.data.frame(annotated_DARs$EPI_24)
DNR_24_DAR <- as.data.frame(annotated_DARs$DNR_24)
MTX_24_DAR <- as.data.frame(annotated_DARs$MTX_24)
TAD_count_df <- DOX_24_DAR %>%
dplyr::select(mcols.genes, mcols.adj.P.Val,annotation:distanceToTSS) %>%
mutate(sig_24=if_else(mcols.adj.P.Val<0.05,"sig","not_sig")) %>%
mutate(sig_24=factor(sig_24, levels = c("sig","not_sig"))) %>%
mutate(TAD_all_status=if_else(mcols.genes %in% peak_ol$Peakid,"TAD_peak","not_TAD_peak")) %>%
mutate(SNP_TAD_status= if_else(mcols.genes %in% TAD_SNP_Peak_ol$Peakid,"SNP_TAD","not_SNP_TAD"))
TAD_count_df %>% #dplyr::filter(TAD_all_status=="TAD_peak") %>%
group_by(sig_24,SNP_TAD_status,TAD_all_status) %>%
tally #%>% # A tibble: 6 × 4
# Groups: sig_24, SNP_TAD_status [4]
sig_24 SNP_TAD_status TAD_all_status n
<fct> <chr> <chr> <int>
1 sig SNP_TAD TAD_peak 2047
2 sig not_SNP_TAD TAD_peak 56865
3 sig not_SNP_TAD not_TAD_peak 5908
4 not_sig SNP_TAD TAD_peak 3111
5 not_sig not_SNP_TAD TAD_peak 78627
6 not_sig not_SNP_TAD not_TAD_peak 8999 # pivot_wider(., id_cols = sig_24, names_from = SNP_TAD_status, values_from = n)
# print("Odds ratio of SNP_TADs across DOXall regions, regardless of TAD_status")
# TAD_count_df %>% #dplyr::filter(TAD_all_status=="TAD_peak") %>%
# group_by(sig_24,SNP_TAD_status) %>%
# tally %>%
# pivot_wider(., id_cols = sig_24, names_from = SNP_TAD_status, values_from = n) %>%
# column_to_rownames( "sig_24") %>% as.matrix() %>%
# # chisq.test()
# epitools::oddsratio()
print("Odds ratio testing proportion SNP-containing TADs of sig-DOX DARs vs non-sig DARs at 24 hours")[1] "Odds ratio testing proportion SNP-containing TADs of sig-DOX DARs vs non-sig DARs at 24 hours"TAD_count_df %>% dplyr::filter(TAD_all_status=="TAD_peak") %>%
group_by(sig_24,SNP_TAD_status) %>%
tally %>%
pivot_wider(., id_cols = sig_24, names_from = SNP_TAD_status, values_from = n) %>%
column_to_rownames( "sig_24") %>% as.matrix() %>%
# chisq.test()
epitools::oddsratio()$data
SNP_TAD not_SNP_TAD Total
sig 2047 56865 58912
not_sig 3111 78627 81738
Total 5158 135492 140650
$measure
NA
odds ratio with 95% C.I. estimate lower upper
sig 1.000000 NA NA
not_sig 0.909844 0.8594795 0.9629201
$p.value
NA
two-sided midp.exact fisher.exact chi.square
sig NA NA NA
not_sig 0.00107761 0.001098901 0.001105101
$correction
[1] FALSE
attr(,"method")
[1] "median-unbiased estimate & mid-p exact CI"TAD_count_df %>%
dplyr::filter(TAD_all_status=="TAD_peak") %>%
group_by(sig_24,SNP_TAD_status) %>%
tally ()%>%
mutate(sig_24=factor(sig_24, levels = c("sig","not_sig"))) %>%
ggplot(.,aes(x=sig_24, y= n,fill=SNP_TAD_status))+
geom_col(position="fill")+
theme_bw()+
ggtitle("Proportion of significant regions by 24 hours")+
ylab("proportion")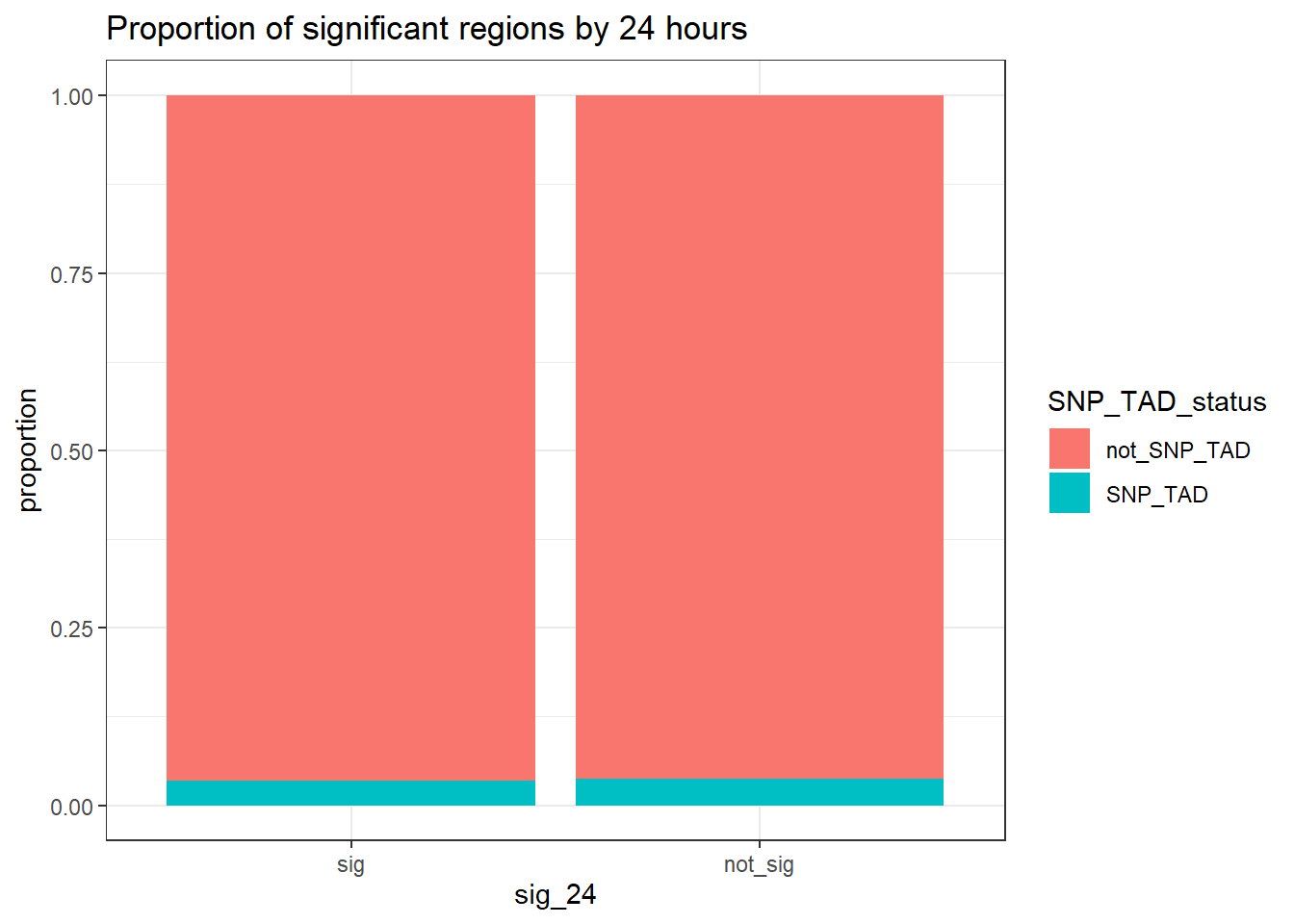
# TAD_count_df %>%
# dplyr::filter(TAD_all_status=="TAD_peak") %>%
#
# group_by(sig_24,SNP_TAD_status) %>%
# tally ()%>%
# mutate(sig_24=factor(sig_24, levels = c("sig","not_sig"))) %>%
# ggplot(.,aes(x=SNP_TAD_status, y= n,fill=sig_24))+
# geom_col(position="fill")+
# theme_bw()+
# ggtitle("Proportion of significant regions by 24 hours")+
# ylab("proportion")Calculating Distance to TAD-SNP from peak
DOX 24 hours
DOX_DAR_sig <- DOX_24_DAR %>%
dplyr::filter(mcols.adj.P.Val<0.05) %>%
distinct (mcols.genes) %>%
dplyr::rename("Peakid"="mcols.genes")
EPI_DAR_sig <- EPI_24_DAR %>%
dplyr::filter(mcols.adj.P.Val<0.05) %>%
distinct (mcols.genes) %>%
dplyr::rename("Peakid"="mcols.genes")
DNR_DAR_sig <- DNR_24_DAR %>%
dplyr::filter(mcols.adj.P.Val<0.05) %>%
distinct (mcols.genes) %>%
dplyr::rename("Peakid"="mcols.genes")
MTX_DAR_sig <- MTX_24_DAR %>%
dplyr::filter(mcols.adj.P.Val<0.05) %>%
distinct (mcols.genes) %>%
dplyr::rename("Peakid"="mcols.genes")
MCF7_DAR_snp_pairs_dist <- readRDS("data/Final_four_data/re_analysis/MCF7_DAR_snp_pairs_dist.RDS") %>%
dplyr::rename("Peakid"=names) %>%
mutate(sig_24="MCF7_DAR")
snp_tad_df <-
join_overlap_inner(schneider_gr, Left_ventricle_TAD) %>%
as_tibble() %>%
dplyr::select(RSID, snp_start = start, snp_chr = seqnames, TAD_id)
peak_tad_df <-
join_overlap_inner(all_regions_gr, Left_ventricle_TAD) %>%
as_tibble() %>%
dplyr::select(Peakid, peak_start = start, peak_chr = seqnames, TAD_id)
peak_snp_pairs <- peak_tad_df %>%
inner_join(snp_tad_df, by = "TAD_id")
peak_snp_pairs_dist <- peak_snp_pairs %>%
mutate(distance = abs(peak_start - snp_start)) %>%
mutate(sig_24= if_else(Peakid %in% DOX_DAR_sig$Peakid, "sig","not_sig"))
peak_snp_pairs_dist %>%
mutate(sig_24=factor(sig_24, levels= c("sig","not_sig"))) %>%
ggplot(., aes(x= sig_24, y=distance))+
geom_boxplot()+
theme_bw()+
geom_signif(comparisons = list(c("sig", "not_sig")),
map_signif_level = FALSE, test = "wilcox.test")+
ggtitle("DOX 24 hour distances of DAR-SNP pairs and non-DAR-SNP pairs")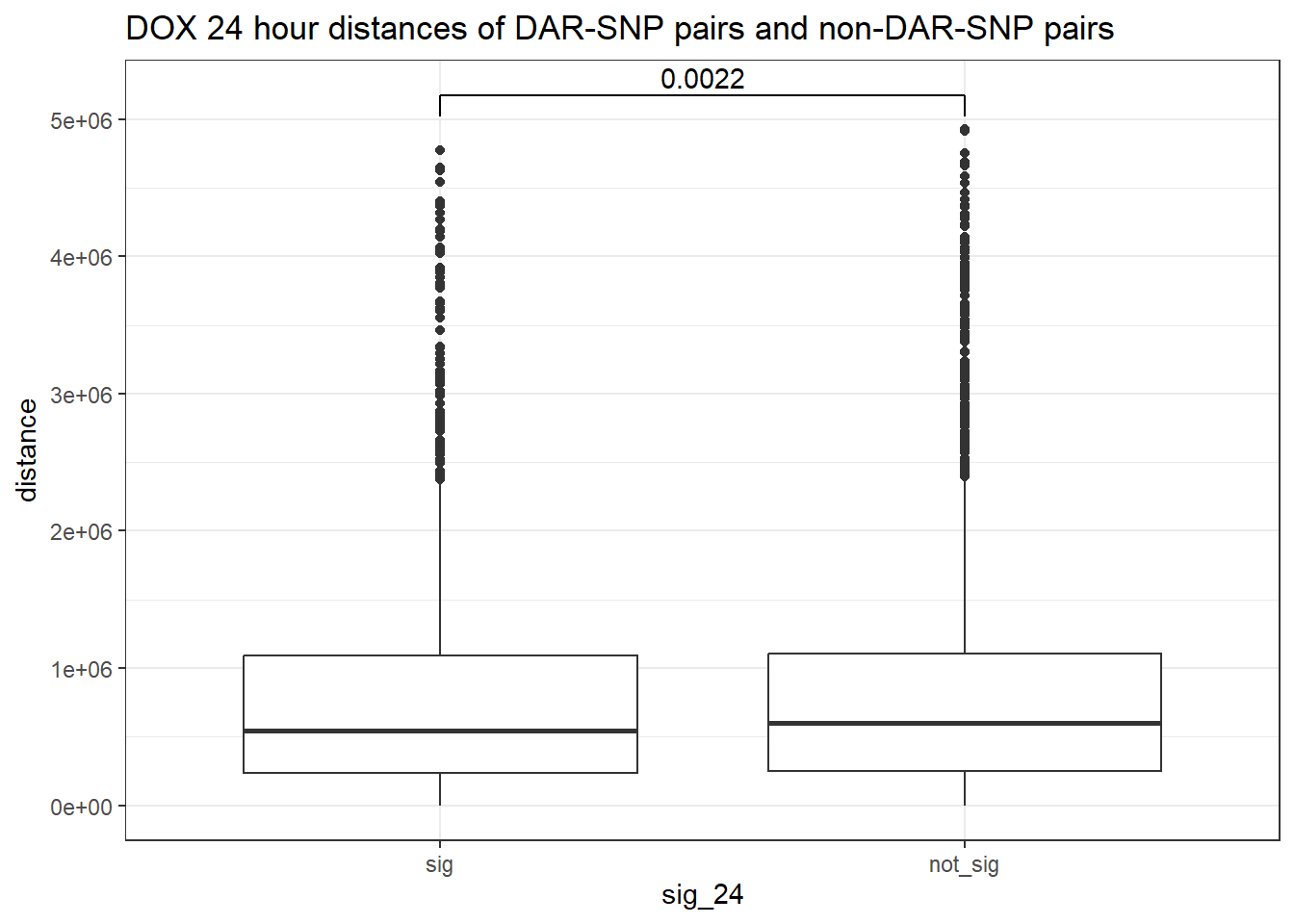
wilcox.test(distance ~ sig_24, data = peak_snp_pairs_dist)
Wilcoxon rank sum test with continuity correction
data: distance by sig_24
W = 9463083, p-value = 0.002185
alternative hypothesis: true location shift is not equal to 0Cardiotox_gwas_df <- peak_snp_pairs_dist %>%
dplyr::filter(sig_24=="sig") %>%
group_by(TAD_id,RSID) %>%
slice_min(order_by = distance, with_ties = FALSE) %>%
ungroup() %>%
arrange(snp_chr,snp_start) %>%
left_join(., all_results_pivot, by=c("Peakid"="genes")) %>%
tidyr::unite(., name,Peakid,RSID)
Cardiotox_gwas_collaped_df <-
peak_snp_pairs_dist %>%
dplyr::filter(sig_24=="sig") %>%
group_by(TAD_id,RSID) %>%
slice_min(order_by = distance, with_ties = FALSE) %>%
ungroup() %>%
arrange(snp_chr,snp_start) %>%
group_by(Peakid, peak_chr, peak_start, TAD_id, sig_24) %>%
summarise(
min_distance = min(distance),
mean_distance = mean(distance),
snp_list = paste(unique(RSID), collapse = ","),
.groups = "drop"
) %>%
left_join(., all_results_pivot, by=c("Peakid"="genes")) %>%
left_join(., Peak_gene_RNA_LFC, by=c("Peakid"="Peakid")) %>%
left_join(.,gene_info_collapsed, by=c("geneId"="ENTREZID")) %>%
mutate(SYMBOL=if_else(is.na(SYMBOL.x),SYMBOL.y,if_else(SYMBOL.x==SYMBOL.y, SYMBOL.x,paste0(SYMBOL.x,"_",SYMBOL.y)))) %>%
tidyr::unite(., name,Peakid,SYMBOL,snp_list) %>%
mutate(snp_dist=case_when(min_distance <2000 ~"2kb",
min_distance > 2000 & min_distance<20000 ~ "20kb",
min_distance >20000 ~">20kb"))DOX_MCF7 added
bind_rows(MCF7_DAR_snp_pairs_dist,peak_snp_pairs_dist) %>%
mutate(sig_24=factor(sig_24, levels= c("sig","not_sig", "MCF7_DAR"))) %>% ggplot(., aes(x= sig_24, y=distance))+
geom_boxplot()+
theme_bw()+
geom_signif(comparisons = list(c("sig", "not_sig"),
c("sig","MCF7_DAR"),
c("not_sig","MCF7_DAR")),
step_increase = 0.1,
map_signif_level = FALSE,
test = "wilcox.test")+
ggtitle("DOX 24 hour distances of DAR-SNP pairs\n and non-DAR-SNP pairs with MCF7 DARs")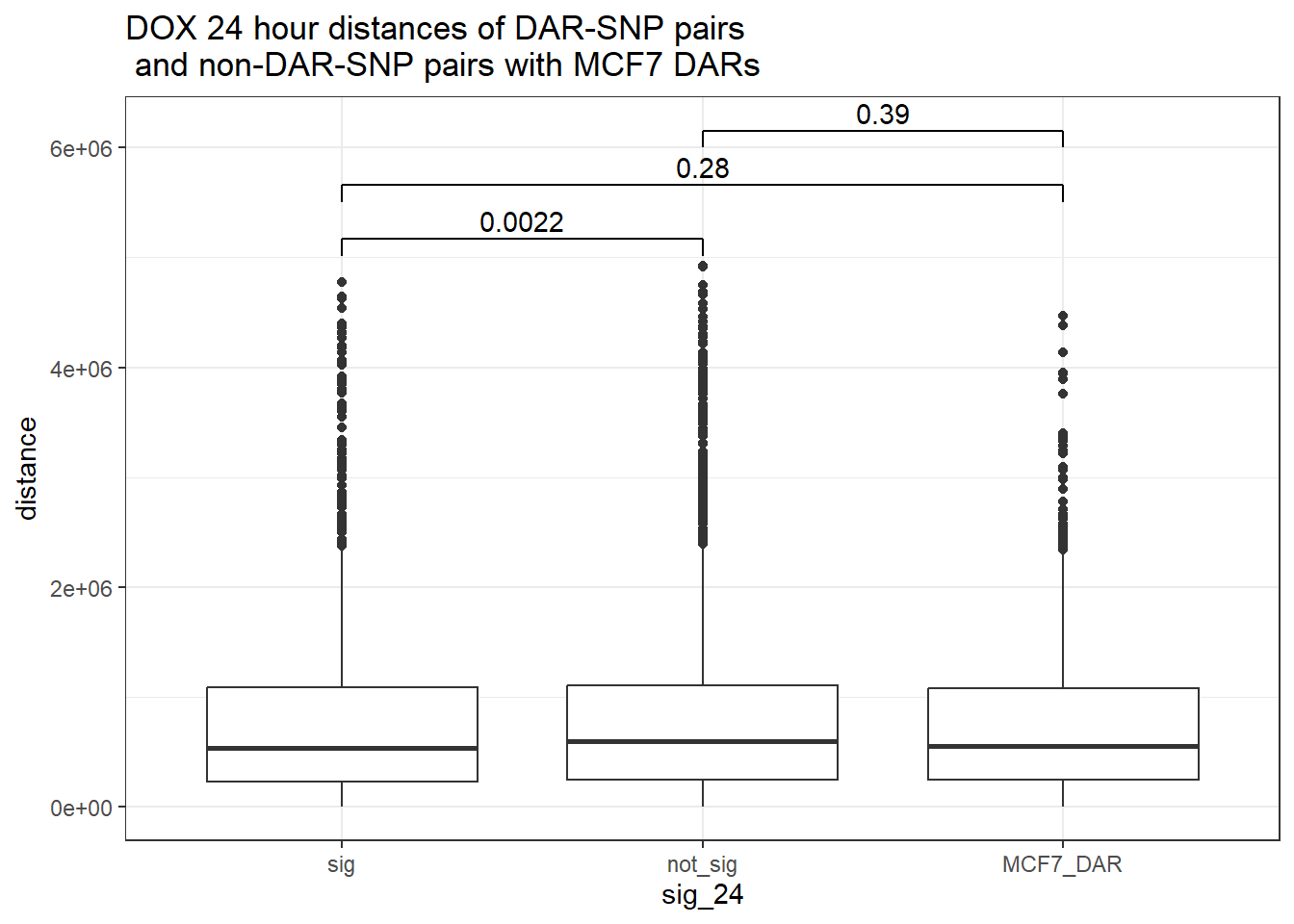
EPI 24 hours
peak_snp_pairs_dist_EPI <- peak_snp_pairs %>%
mutate(distance = abs(peak_start - snp_start)) %>%
mutate(sig_24= if_else(Peakid %in% EPI_DAR_sig$Peakid, "sig","not_sig"))
peak_snp_pairs_dist_EPI %>%
mutate(sig_24=factor(sig_24, levels= c("sig","not_sig"))) %>%
ggplot(., aes(x= sig_24, y=distance))+
geom_boxplot()+
theme_bw()+
geom_signif(comparisons = list(c("sig", "not_sig")),
map_signif_level = FALSE, test = "wilcox.test")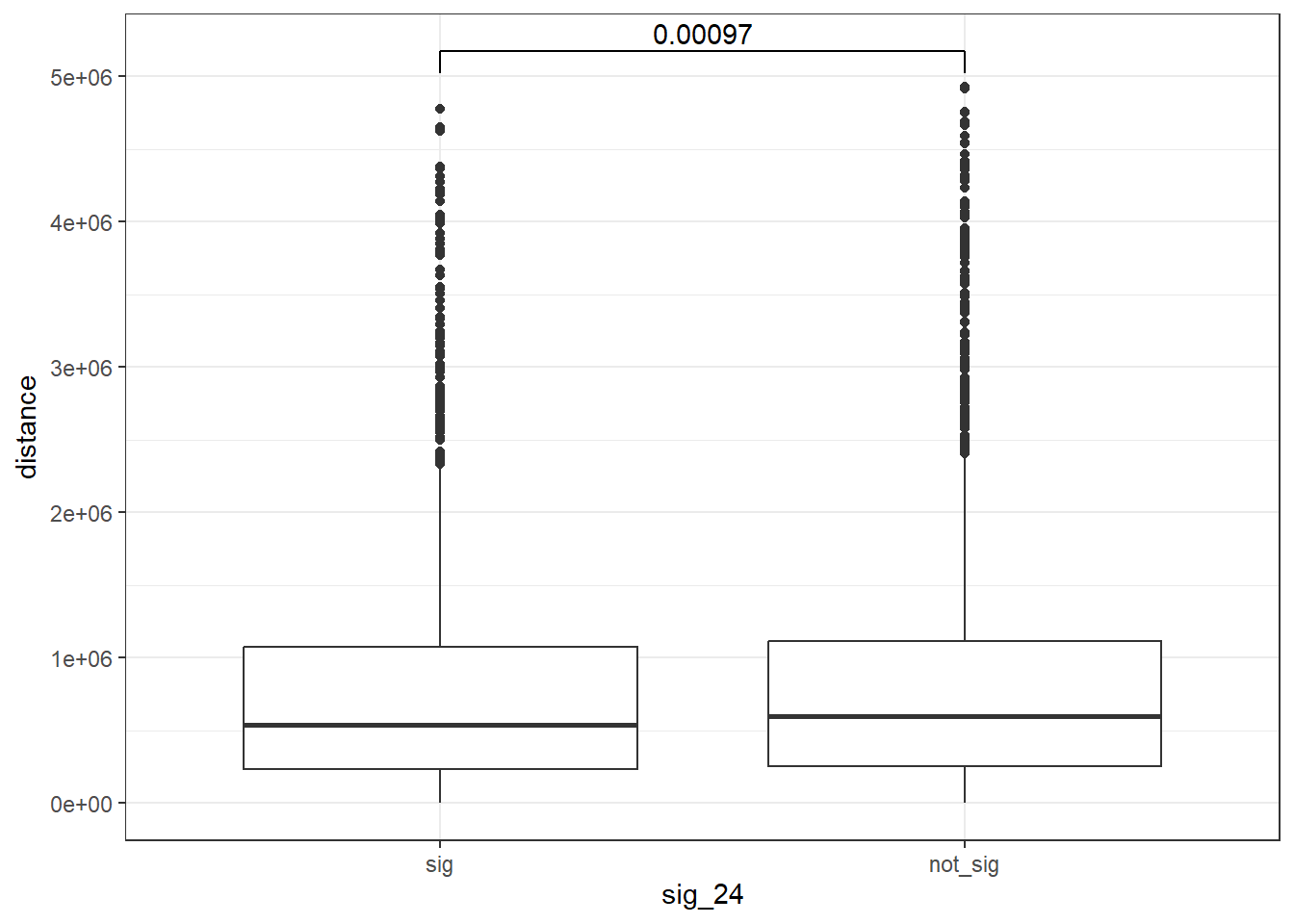
wilcox.test(distance ~ sig_24, data = peak_snp_pairs_dist)
Wilcoxon rank sum test with continuity correction
data: distance by sig_24
W = 9463083, p-value = 0.002185
alternative hypothesis: true location shift is not equal to 0Cardiotox_gwas_EPI <- peak_snp_pairs_dist_EPI %>%
dplyr::filter(sig_24=="sig") %>%
group_by(TAD_id,RSID) %>%
slice_min(order_by = distance, with_ties = FALSE) %>%
ungroup() %>%
arrange(snp_chr,snp_start) %>%
left_join(., all_results_pivot, by=c("Peakid"="genes")) %>%
tidyr::unite(., name,Peakid,RSID)EPI_MCF7 added
bind_rows(MCF7_DAR_snp_pairs_dist,peak_snp_pairs_dist_EPI) %>%
mutate(sig_24=factor(sig_24, levels= c("sig","not_sig", "MCF7_DAR"))) %>% ggplot(., aes(x= sig_24, y=distance))+
geom_boxplot()+
theme_bw()+
geom_signif(comparisons = list(c("sig", "not_sig"),
c("sig","MCF7_DAR"),
c("not_sig","MCF7_DAR")),
step_increase = 0.1,
map_signif_level = FALSE,
test = "wilcox.test")+
ggtitle("EPI 24 hour distances of DAR-SNP pairs\n and non-DAR-SNP pairs with MCF7 DARs")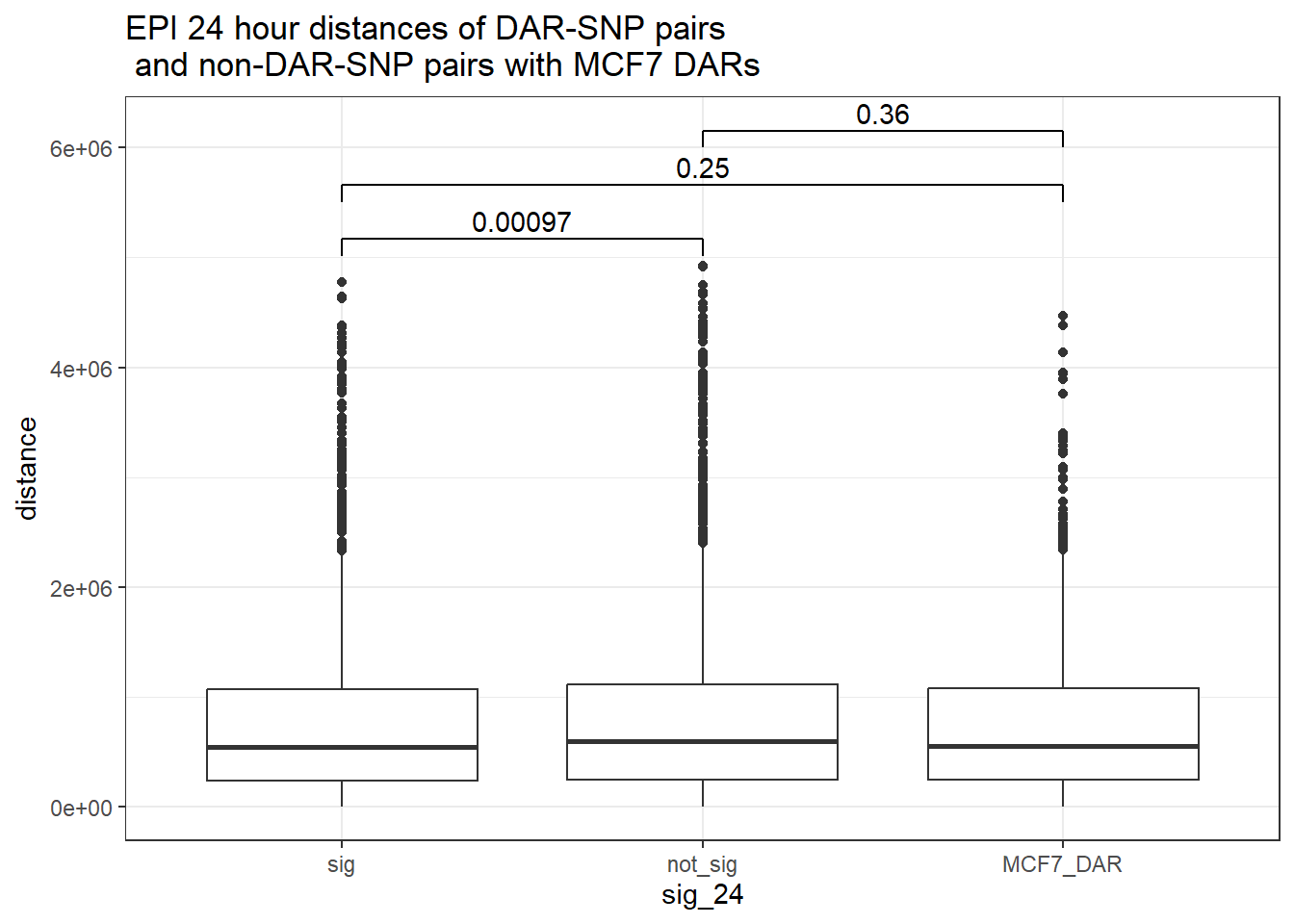
DNR 24 hours
peak_snp_pairs_dist_DNR <- peak_snp_pairs %>%
mutate(distance = abs(peak_start - snp_start)) %>%
mutate(sig_24= if_else(Peakid %in% DNR_DAR_sig$Peakid, "sig","not_sig"))
peak_snp_pairs_dist_DNR %>%
mutate(sig_24=factor(sig_24, levels= c("sig","not_sig"))) %>%
ggplot(., aes(x= sig_24, y=distance))+
geom_boxplot()+
theme_bw()+
geom_signif(comparisons = list(c("sig", "not_sig")),
map_signif_level = FALSE, test = "wilcox.test")+
ggtitle("DNR 24 hour distances of DAR-SNP pairs and non-DAR-SNP pairs")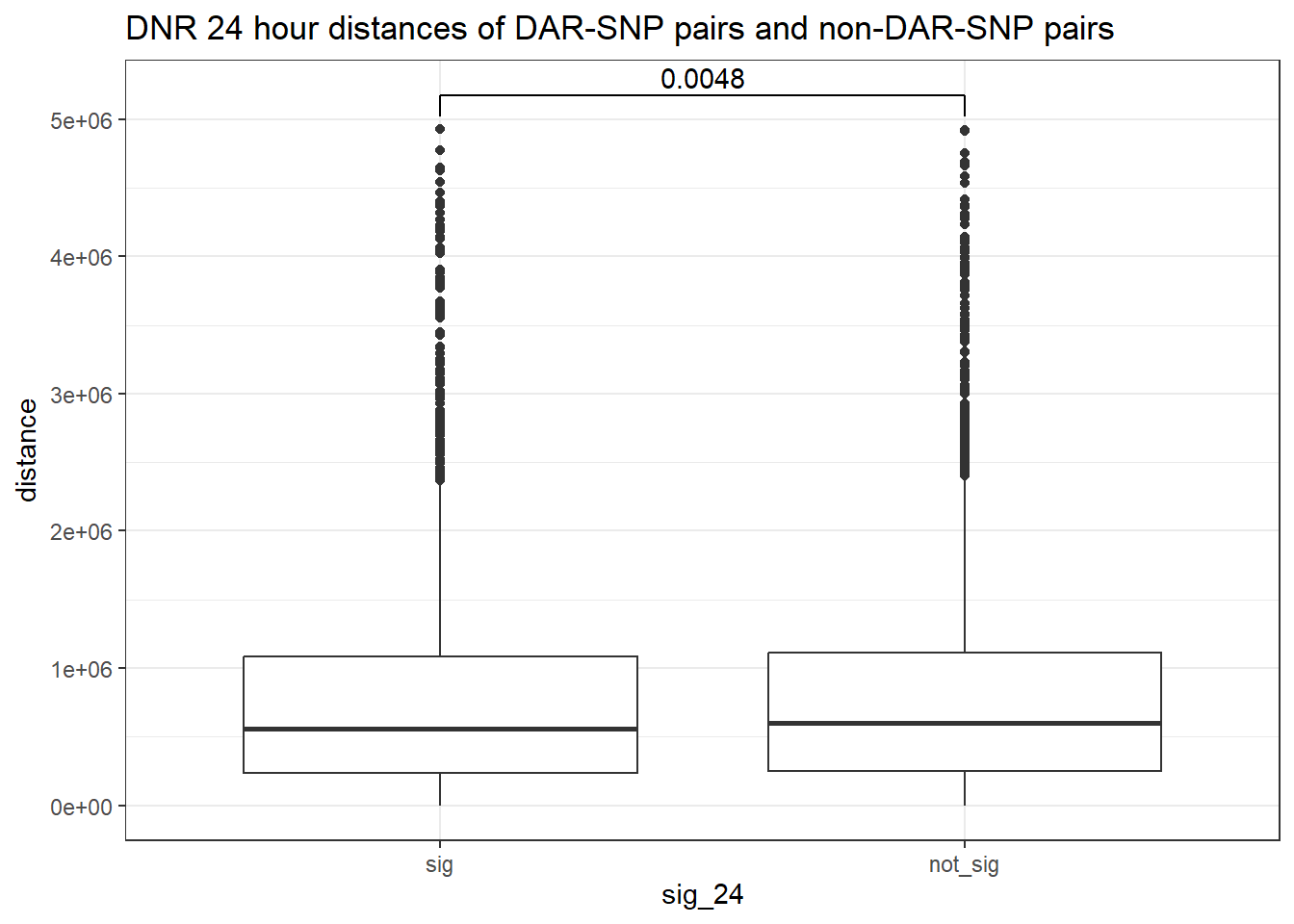
wilcox.test(distance ~ sig_24, data = peak_snp_pairs_dist)
Wilcoxon rank sum test with continuity correction
data: distance by sig_24
W = 9463083, p-value = 0.002185
alternative hypothesis: true location shift is not equal to 0Cardiotox_gwas_DNR <- peak_snp_pairs_dist_DNR %>%
dplyr::filter(sig_24=="sig") %>%
group_by(TAD_id,RSID) %>%
slice_min(order_by = distance, with_ties = FALSE) %>%
ungroup() %>%
arrange(snp_chr,snp_start) %>%
left_join(., all_results_pivot, by=c("Peakid"="genes")) %>%
tidyr::unite(., name,Peakid,RSID)DNR_MCF7 added
bind_rows(MCF7_DAR_snp_pairs_dist,peak_snp_pairs_dist_DNR) %>%
mutate(sig_24=factor(sig_24, levels= c("sig","not_sig", "MCF7_DAR"))) %>% ggplot(., aes(x= sig_24, y=distance))+
geom_boxplot()+
theme_bw()+
geom_signif(comparisons = list(c("sig", "not_sig"),
c("sig","MCF7_DAR"),
c("not_sig","MCF7_DAR")),
step_increase = 0.1,
map_signif_level = FALSE,
test = "wilcox.test")+
ggtitle("DNR 24 hour distances of DAR-SNP pairs\n and non-DAR-SNP pairs with MCF7 DARs")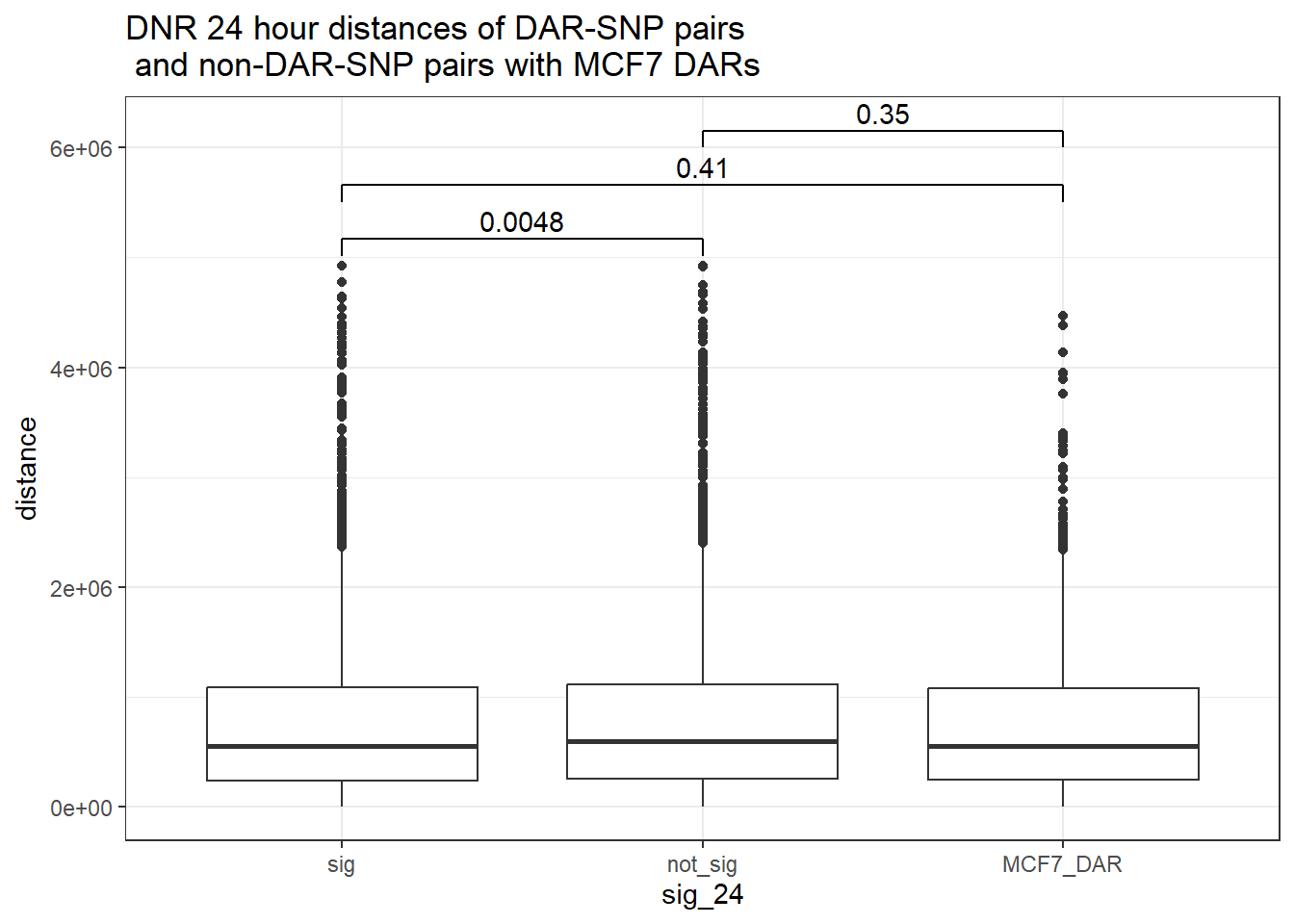
MTX 24 hours
peak_snp_pairs_dist_MTX <- peak_snp_pairs %>%
mutate(distance = abs(peak_start - snp_start)) %>%
mutate(sig_24= if_else(Peakid %in% MTX_DAR_sig$Peakid, "sig","not_sig"))
peak_snp_pairs_dist_MTX %>%
mutate(sig_24=factor(sig_24, levels= c("sig","not_sig"))) %>%
ggplot(., aes(x= sig_24, y=distance))+
geom_boxplot()+
theme_bw()+
geom_signif(comparisons = list(c("sig", "not_sig")),
map_signif_level = FALSE, test = "wilcox.test")+
ggtitle("MTX 24 hour distances of DAR-SNP pairs and non-DAR-SNP pairs")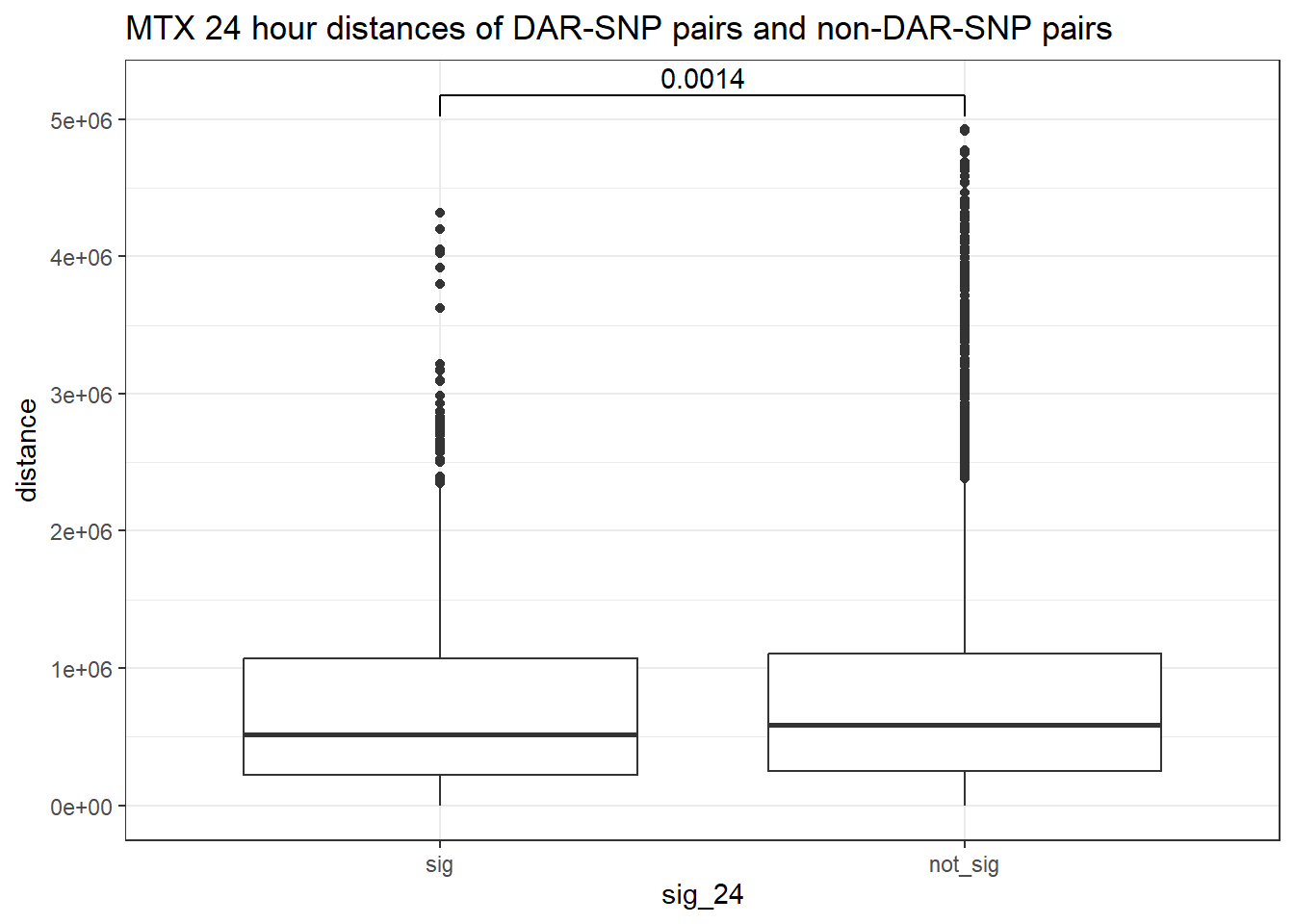
wilcox.test(distance ~ sig_24, data = peak_snp_pairs_dist)
Wilcoxon rank sum test with continuity correction
data: distance by sig_24
W = 9463083, p-value = 0.002185
alternative hypothesis: true location shift is not equal to 0Cardiotox_gwas_MTX <- peak_snp_pairs_dist_MTX %>%
dplyr::filter(sig_24=="sig") %>%
group_by(TAD_id,RSID) %>%
slice_min(order_by = distance, with_ties = FALSE) %>%
ungroup() %>%
arrange(snp_chr,snp_start) %>%
left_join(., all_results_pivot, by=c("Peakid"="genes")) %>%
tidyr::unite(., name,Peakid,RSID)MTX_MCF7 added
bind_rows(MCF7_DAR_snp_pairs_dist,peak_snp_pairs_dist_MTX) %>%
mutate(sig_24=factor(sig_24, levels= c("sig","not_sig", "MCF7_DAR"))) %>% ggplot(., aes(x= sig_24, y=distance))+
geom_boxplot()+
theme_bw()+
geom_signif(comparisons = list(c("sig", "not_sig"),
c("sig","MCF7_DAR"),
c("not_sig","MCF7_DAR")),
step_increase = 0.1,
map_signif_level = FALSE,
test = "wilcox.test")+
ggtitle("MTX 24 hour distances of DAR-SNP pairs\n and non-DAR-SNP pairs with MCF7 DARs")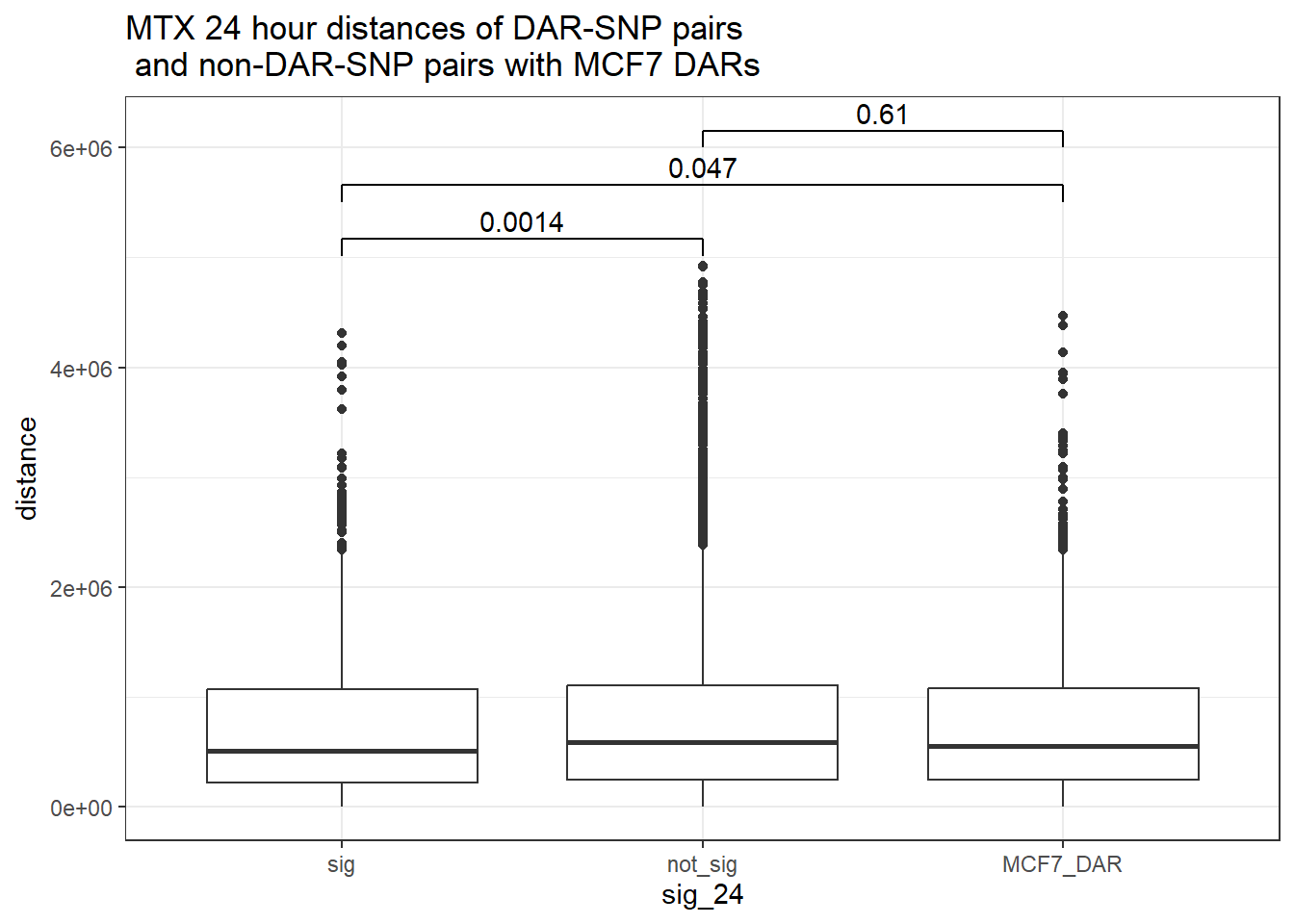 ### Creating SNP_TAD distance DF
### Creating SNP_TAD distance DF
For combining the above 24 hour trt-distance to SNP data frames for box-plots
SNP_TAD_dist_DF <- bind_rows((peak_snp_pairs_dist_MTX %>%
mutate(trt="MTX")),
(peak_snp_pairs_dist %>%
mutate(trt="DOX"))) %>%
bind_rows(.,(peak_snp_pairs_dist_EPI %>%
mutate(trt="EPI"))) %>%
bind_rows(.,(peak_snp_pairs_dist_DNR %>%
mutate(trt="DNR"))) %>%
mutate(trt=factor(trt,levels=c("DOX","EPI","DNR","MTX"))) %>%
mutate(sig_24=factor(sig_24, levels= c("sig","not_sig")))
SNP_TAD_dist_DF%>%
ggplot(., aes(x= interaction(sig_24,trt), y=distance))+
geom_boxplot(aes(fill=trt))+
theme_bw()+
geom_signif(comparisons = list(c("sig.DOX", "not_sig.DOX"),
c("sig.EPI","not_sig.EPI"),
c("sig.DNR", "not_sig.DNR"),
c("sig.MTX", "not_sig.MTX")),
step_increase = 0.1,
map_signif_level = FALSE,
test = "wilcox.test")+
ggtitle("ALL dist")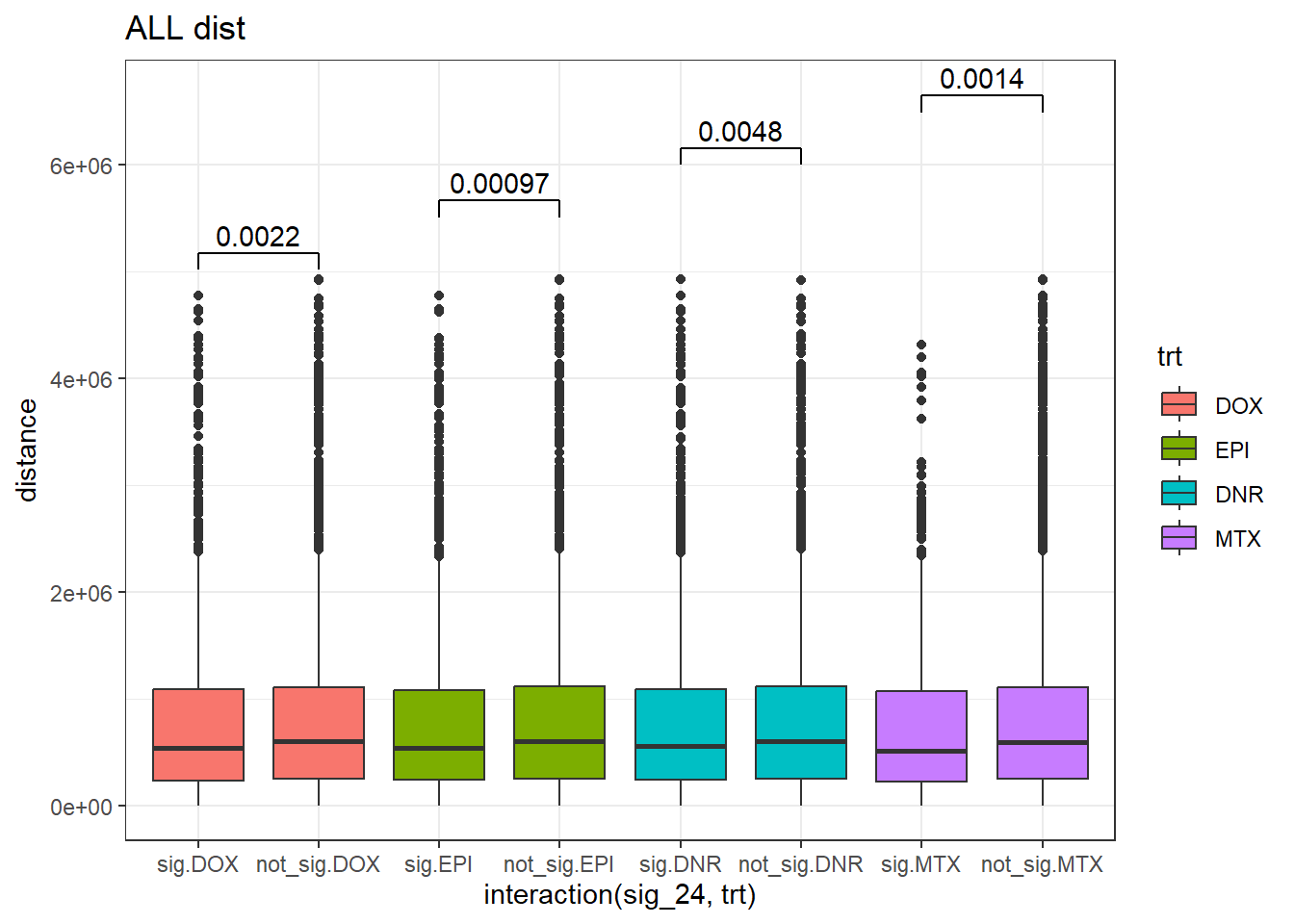 ### MCF7 tissue specific, non-tissue specific.
### MCF7 tissue specific, non-tissue specific.
Here I am overlapping my data with the MCF7 DAR data. This will create DARS for each treatment that overlap MCF7 DARS (tissue-shared regions) and DARS that do not overlap MCF7 DARS (tissue-specific regions). I will then calculate the distance between the SNPs in the shared vs specific for each treatment.
MCF7_DARs_hyper <- read_excel("C:/Users/renee/Downloads/MCF7-doxATAC/Table 4.XLSX",
sheet = "hyper") %>% GRanges()
MCF7_DARs_hypo <- read_excel("C:/Users/renee/Downloads/MCF7-doxATAC/Table 4.XLSX",
sheet = "hypo") %>% GRanges()
# MCF7_ARsmcf7_1 <- read_excel("C:/Users/renee/Downloads/MCF7-doxATAC/Table 3.XLSX") %>%
# GRanges()
MCF7_DARs_hyper$names <- paste0("hyper_", seq_along(seqnames(MCF7_DARs_hyper)))
MCF7_DARs_hypo$names <- paste0("hypo_", seq_along(seqnames(MCF7_DARs_hypo)))
MCF7_DAR_all <- c(MCF7_DARs_hyper,MCF7_DARs_hypo)
ch = import.chain("C:/Users/renee/ATAC_folder/liftOver_genome/hg19ToHg38.over.chain")
# MCF7_ARsmcf7_1_LO <- as.data.frame(liftOver(MCF7_ARsmcf7_1,ch)) %>%
# GRanges()
MCF7_DARs_hyper_LO <- as.data.frame(liftOver(MCF7_DARs_hyper,ch)) %>%
GRanges()
MCF7_DARs_hypo_LO <- as.data.frame(liftOver(MCF7_DARs_hypo,ch)) %>%
GRanges()
MCF7_DAR_all_LO <- c(MCF7_DARs_hyper_LO,MCF7_DARs_hypo_LO)
MCF7DAR_AR_ol <- join_overlap_intersect(all_regions_gr, MCF7_DAR_all_LO)
DAR_AR_overlap_df <-MCF7DAR_AR_ol %>%
as.data.frame() %>%
distinct(Peakid)Attempting with DOX
peak_snp_pairs_dist %>%
dplyr::filter(sig_24 =="sig") %>%
mutate(specific=if_else(Peakid %in%DAR_AR_overlap_df$Peakid,"specific","not_specific")) %>%
mutate(specific=factor(specific,levels= c("specific","not_specific"))) %>%
ggplot(.,aes(x=specific, y=distance))+
geom_boxplot()+
theme_bw()+
geom_signif(comparisons = list(c("specific", "not_specific")),
step_increase = 0.1,
map_signif_level = FALSE,
test = "wilcox.test")+
ggtitle("DOX 24 hour distances of DAR-SNP pairs\n that are specific for iPSC-CMs or not-specific")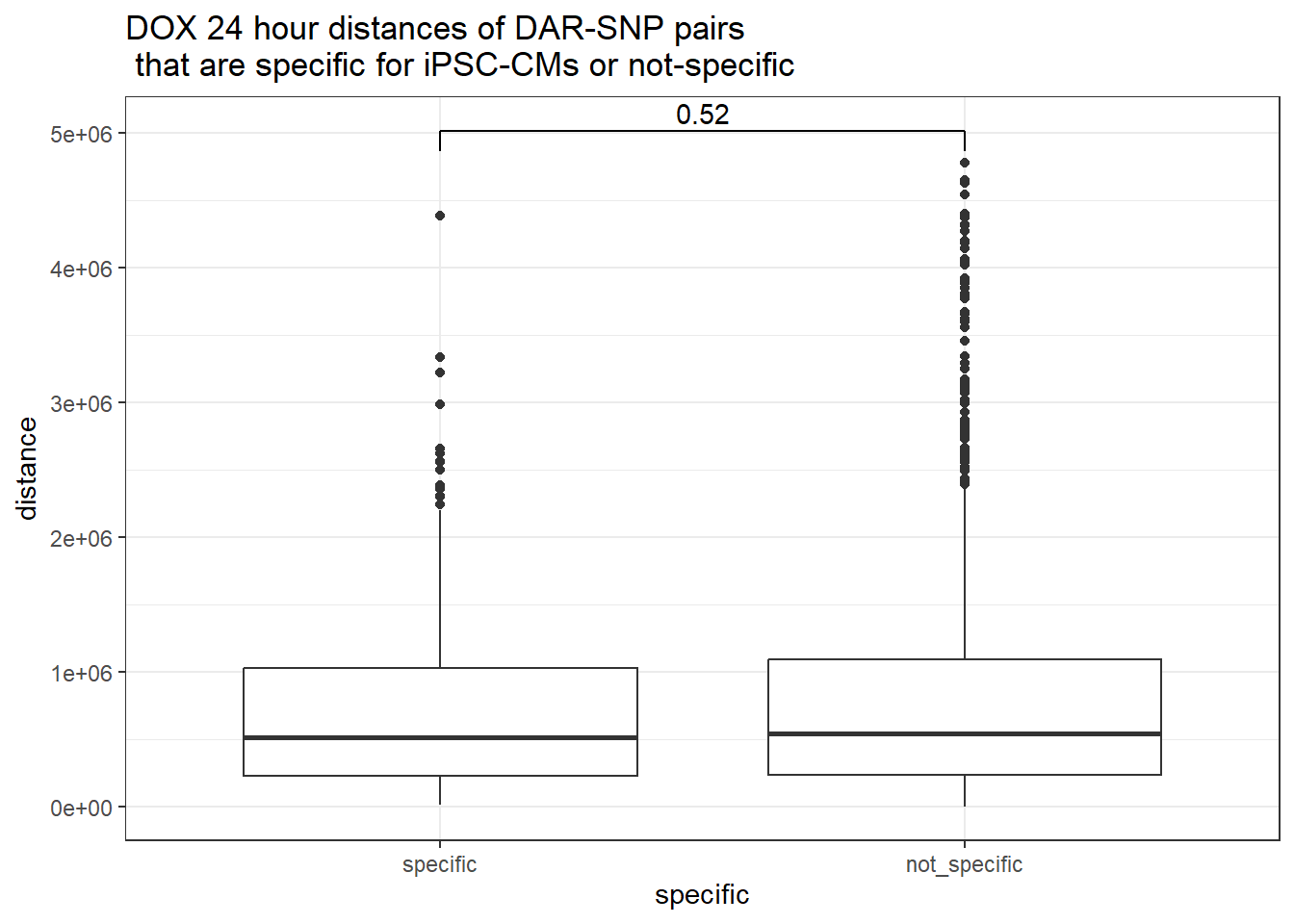
peak_snp_pairs_dist_EPI %>%
dplyr::filter(sig_24 =="sig") %>%
mutate(specific=if_else(Peakid %in%DAR_AR_overlap_df$Peakid,"specific","not_specific")) %>%
mutate(specific=factor(specific,levels= c("specific","not_specific"))) %>%
ggplot(.,aes(x=specific, y=distance))+
geom_boxplot()+
theme_bw()+
geom_signif(comparisons = list(c("specific", "not_specific")),
step_increase = 0.1,
map_signif_level = FALSE,
test = "wilcox.test")+
ggtitle("EPI 24 hour distances of DAR-SNP pairs\n that are specific for iPSC-CMs or not-specific")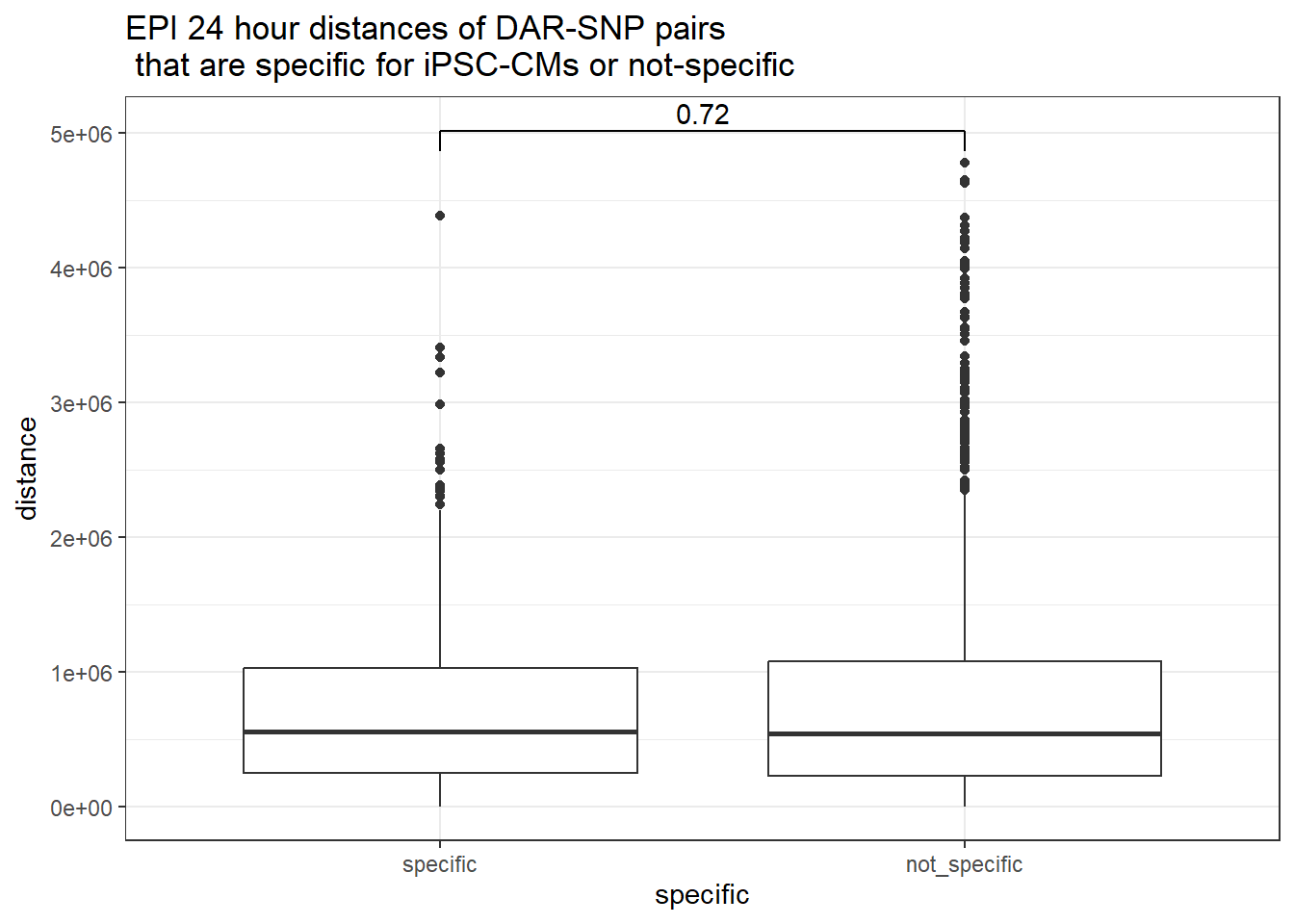
peak_snp_pairs_dist_DNR %>%
dplyr::filter(sig_24 =="sig") %>%
mutate(specific=if_else(Peakid %in%DAR_AR_overlap_df$Peakid,"specific","not_specific")) %>%
mutate(specific=factor(specific,levels= c("specific","not_specific"))) %>%
ggplot(.,aes(x=specific, y=distance))+
geom_boxplot()+
theme_bw()+
geom_signif(comparisons = list(c("specific", "not_specific")),
step_increase = 0.1,
map_signif_level = FALSE,
test = "wilcox.test")+
ggtitle("DNR 24 hour distances of DAR-SNP pairs\n that are specific for iPSC-CMs or not-specific")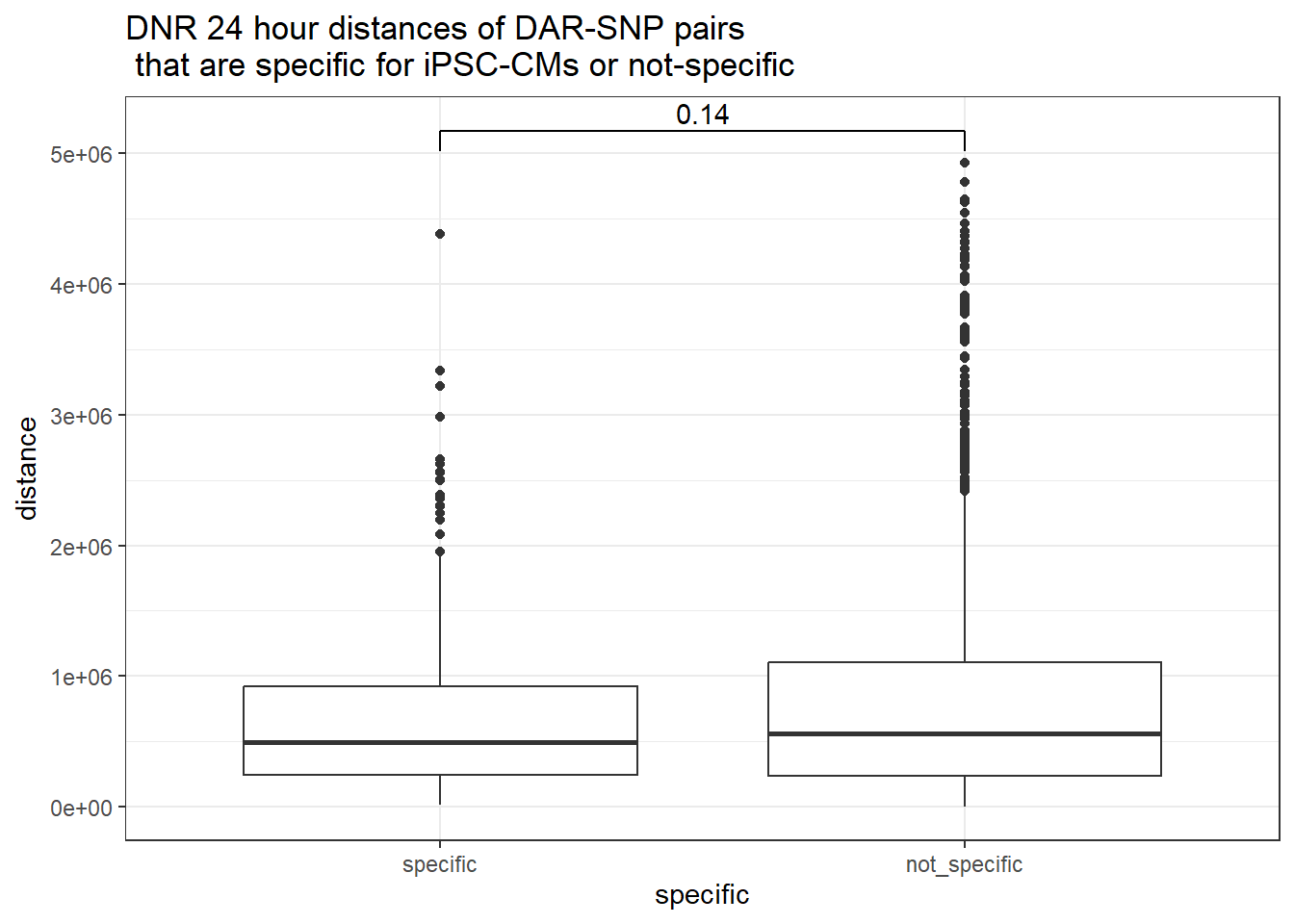
peak_snp_pairs_dist_MTX %>%
dplyr::filter(sig_24 =="sig") %>%
mutate(specific=if_else(Peakid %in%DAR_AR_overlap_df$Peakid,"specific","not_specific")) %>%
mutate(specific=factor(specific,levels= c("specific","not_specific"))) %>%
ggplot(.,aes(x=specific, y=distance))+
geom_boxplot()+
theme_bw()+
geom_signif(comparisons = list(c("specific", "not_specific")),
step_increase = 0.1,
map_signif_level = FALSE,
test = "wilcox.test")+
ggtitle("MTX 24 hour distances of DAR-SNP pairs\n that are specific for iPSC-CMs or not-specific")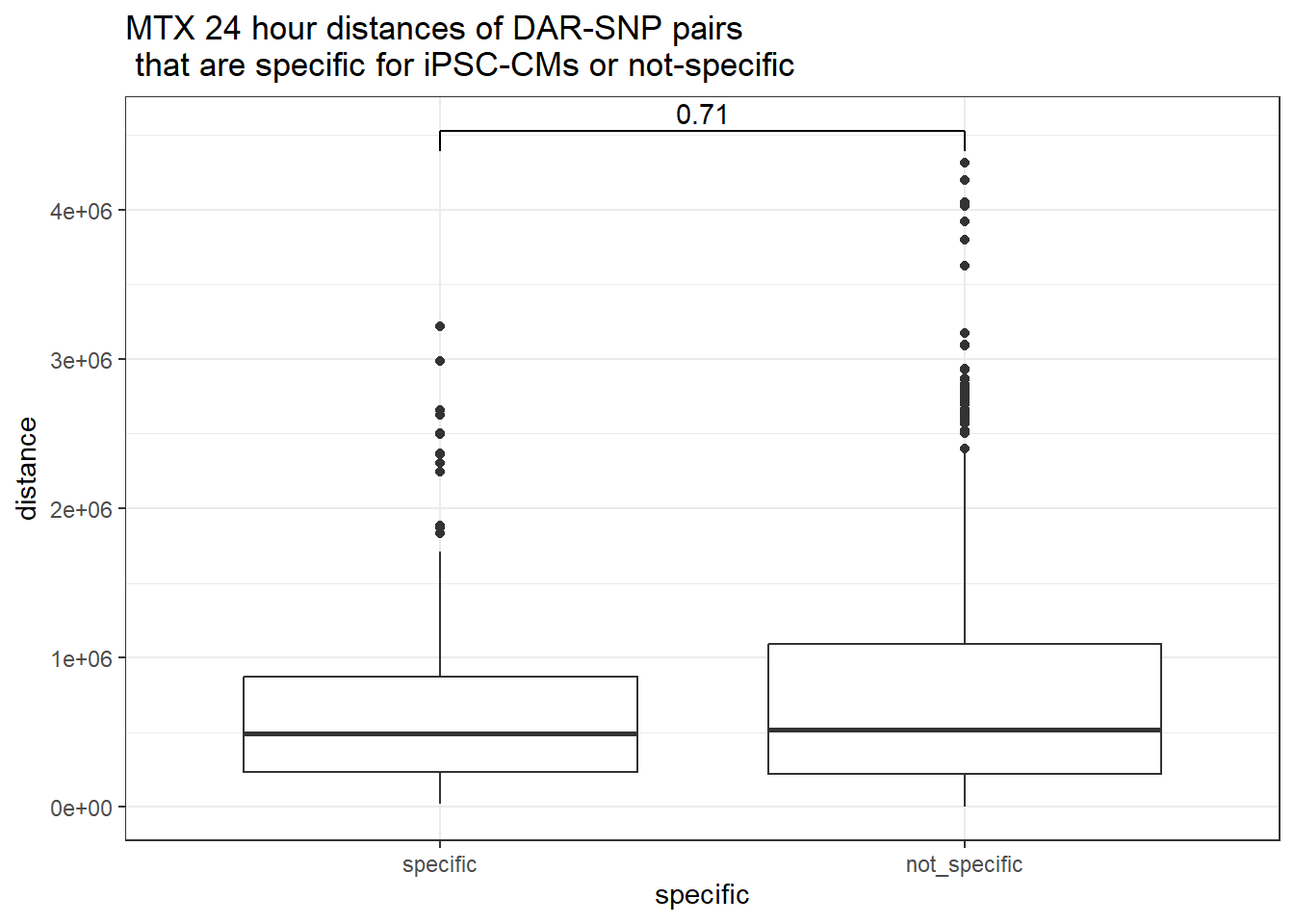
peak_snp_pairs_dist %>%
dplyr::filter(!Peakid %in% DAR_AR_overlap_df$Peakid) %>%
mutate(sig_24=factor(sig_24,levels = c ("sig","not_sig"))) %>%
ggplot(.,aes(x=sig_24, y=distance))+
geom_boxplot()+
theme_bw()+
geom_signif(comparisons = list(c("sig", "not_sig")),
step_increase = 0.1,
map_signif_level = FALSE,
test = "wilcox.test")+
ggtitle("DOX 24 hour distances of DAR-SNP pairs\n that are specific for iPSC-CMs")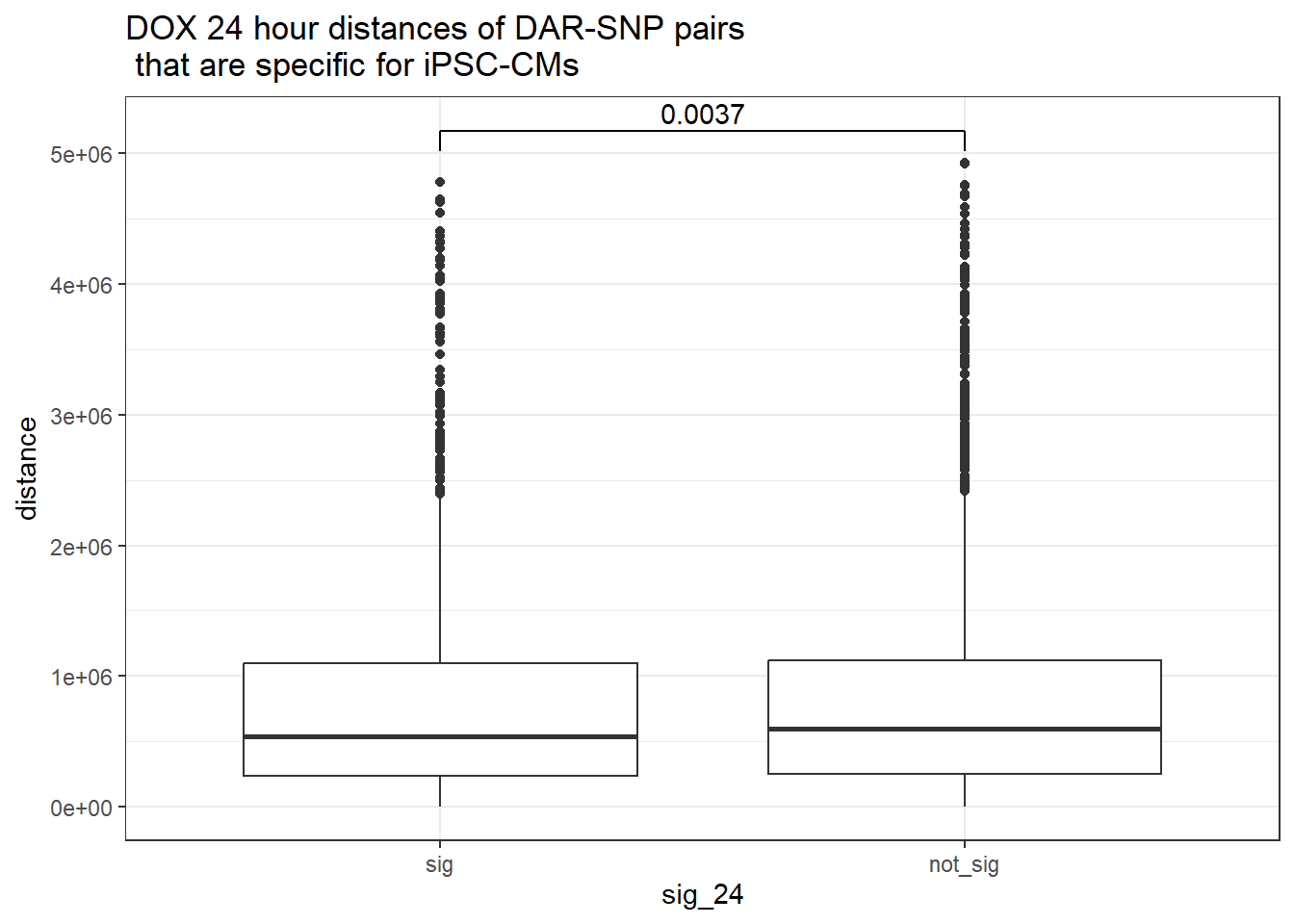
peak_snp_pairs_dist_EPI %>%
dplyr::filter(!Peakid %in% DAR_AR_overlap_df$Peakid) %>%
mutate(sig_24=factor(sig_24,levels = c ("sig","not_sig"))) %>%
ggplot(.,aes(x=sig_24, y=distance))+
geom_boxplot()+
theme_bw()+
geom_signif(comparisons = list(c("sig", "not_sig")),
step_increase = 0.1,
map_signif_level = FALSE,
test = "wilcox.test")+
ggtitle("EPI 24 hour distances of DAR-SNP pairs\n that are specific for iPSC-CMs")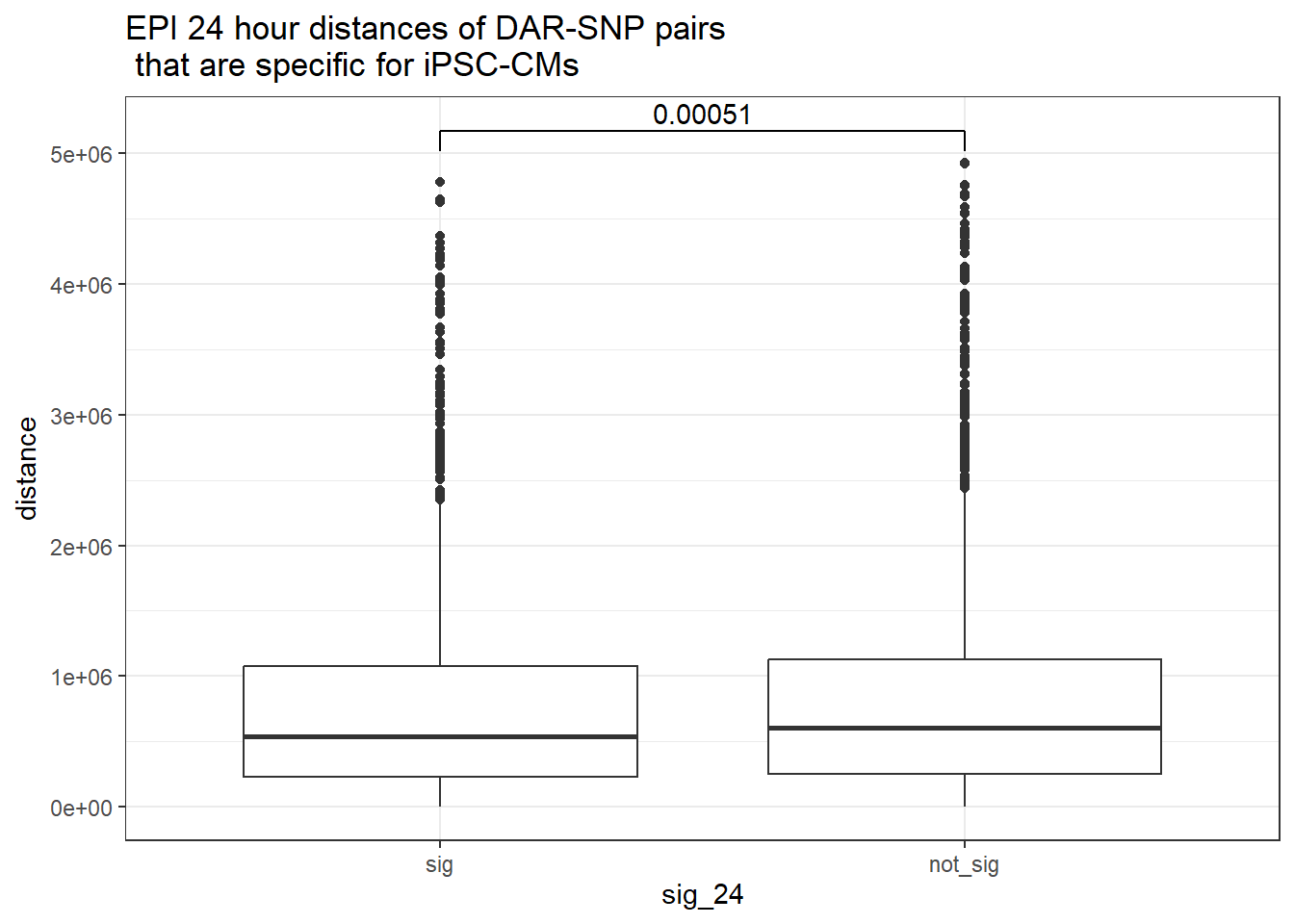
peak_snp_pairs_dist_DNR %>%
dplyr::filter(!Peakid %in% DAR_AR_overlap_df$Peakid) %>%
mutate(sig_24=factor(sig_24,levels = c ("sig","not_sig"))) %>%
ggplot(.,aes(x=sig_24, y=distance))+
geom_boxplot()+
theme_bw()+
geom_signif(comparisons = list(c("sig", "not_sig")),
step_increase = 0.1,
map_signif_level = FALSE,
test = "wilcox.test")+
ggtitle("DNR 24 hour distances of DAR-SNP pairs\n that are specific for iPSC-CMs")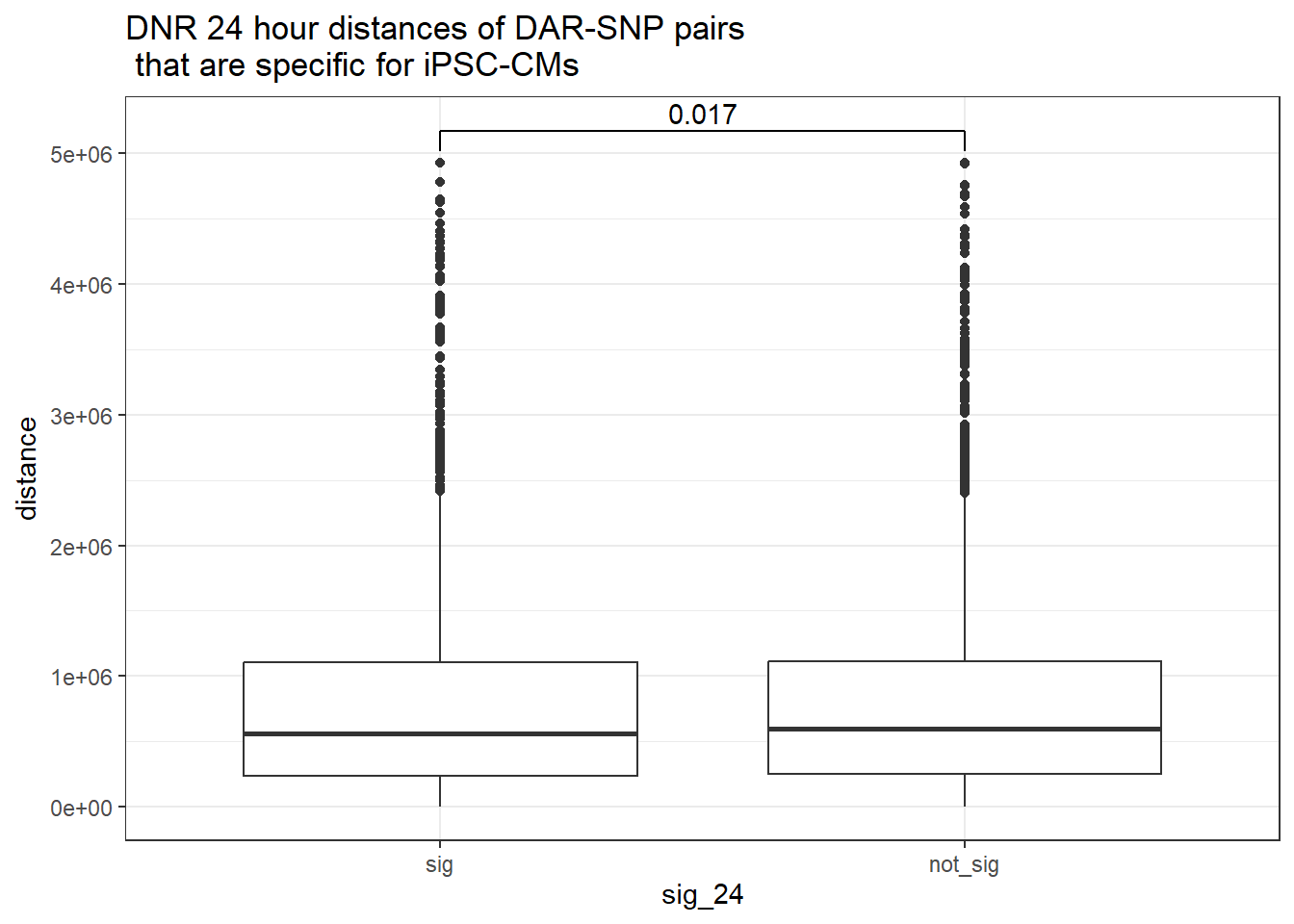
peak_snp_pairs_dist_MTX %>%
dplyr::filter(!Peakid %in% DAR_AR_overlap_df$Peakid) %>%
mutate(sig_24=factor(sig_24,levels = c ("sig","not_sig"))) %>%
ggplot(.,aes(x=sig_24, y=distance))+
geom_boxplot()+
theme_bw()+
geom_signif(comparisons = list(c("sig", "not_sig")),
step_increase = 0.1,
map_signif_level = FALSE,
test = "wilcox.test")+
ggtitle("MTX 24 hour distances of DAR-SNP pairs\n that are specific for iPSC-CMs")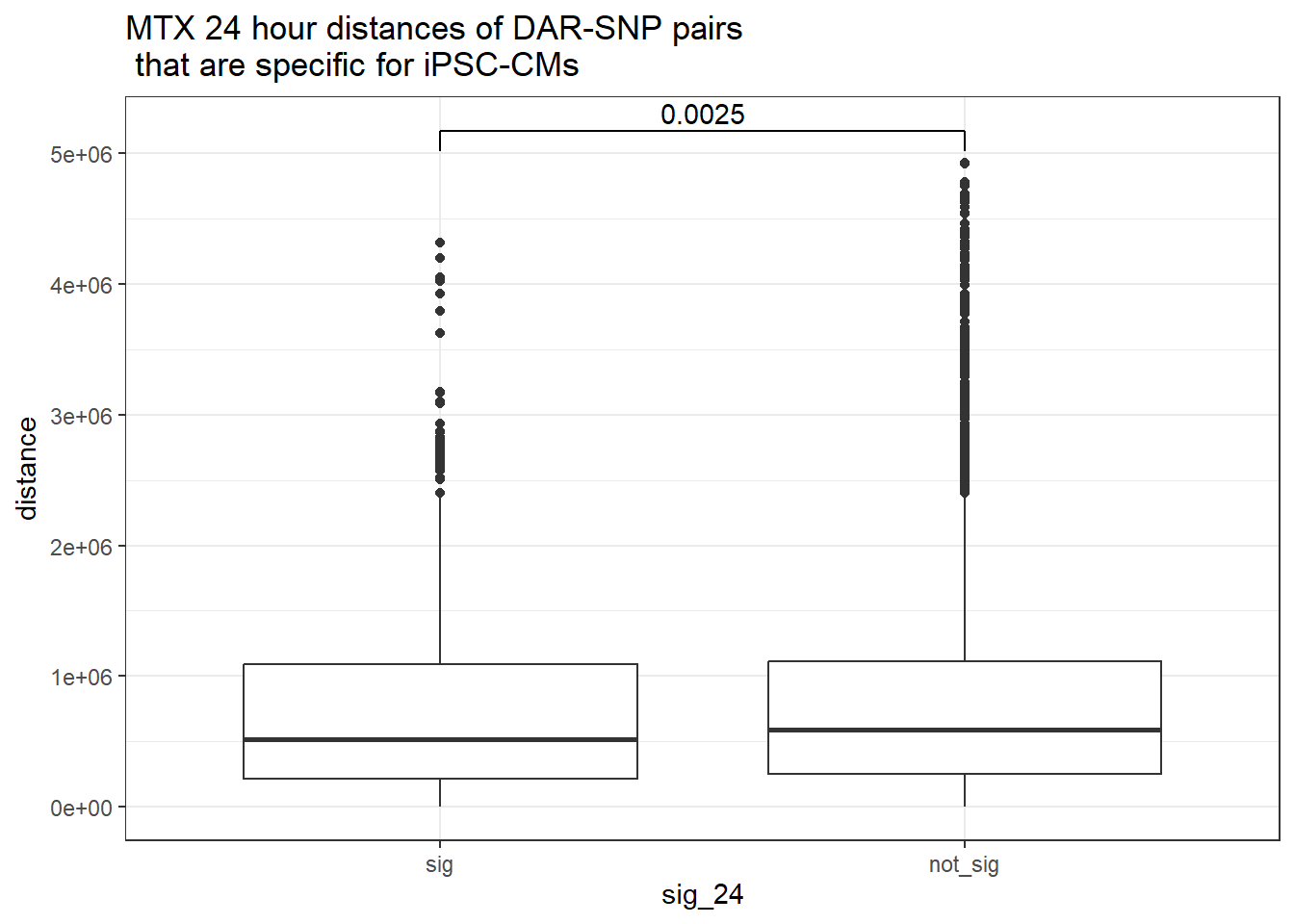
Attempt at heatmap with RNA expression, decided not to use or plot here.
Cardotox_mat <- Cardiotox_gwas_collaped_df %>%
dplyr::select(name,DOX_3:TRZ_24,`3_RNA_DOX`,`3_RNA_EPI`,`3_RNA_DNR`,`3_RNA_MTX`,`3_RNA_TRZ`,`24_RNA_DOX`,`24_RNA_EPI`,`24_RNA_DNR`,`24_RNA_MTX`,`24_RNA_TRZ`) %>%
column_to_rownames("name") %>%
as.matrix()
annot_map_df <- Cardiotox_gwas_collaped_df %>%
dplyr::select(name,snp_dist) %>%
column_to_rownames("name")
annot_map <-
rowAnnotation(
snp_dist=Cardiotox_gwas_collaped_df$snp_dist,
TAD_id=Cardiotox_gwas_collaped_df$TAD_id,
col= list(snp_dist=c("2kb"="goldenrod4",
"20kb"="pink",
">20kb"="tan2"),
TAD_id=tad_colors))
simply_map_lfc <- ComplexHeatmap::Heatmap(Cardotox_mat,
# col = col_fun,
left_annotation = annot_map,
show_row_names = TRUE,
row_names_max_width= ComplexHeatmap::max_text_width(rownames(Cardotox_mat), gp=gpar(fontsize=14)),
heatmap_legend_param = list(direction = "horizontal"),
show_column_names = TRUE,
cluster_rows = FALSE,
cluster_columns = FALSE)
ComplexHeatmap::draw(simply_map_lfc,
merge_legend = TRUE,
heatmap_legend_side = "left",
annotation_legend_side = "left")Alternative of final graph that we may use.
AR_Cardiotox_gwas_collaped_df <-
peak_snp_pairs_dist %>%
# dplyr::filter(sig_24=="sig") %>%
group_by(TAD_id,RSID) %>%
slice_min(order_by = distance, with_ties = FALSE) %>%
ungroup() %>%
arrange(snp_chr,snp_start) %>%
group_by(Peakid, peak_chr, peak_start, TAD_id, sig_24) %>%
summarise(
min_distance = min(distance),
mean_distance = mean(distance),
snp_list = paste(unique(RSID), collapse = ","),
.groups = "drop"
) %>%
left_join(., all_results_pivot, by=c("Peakid"="genes")) %>%
# left_join(., Peak_gene_RNA_LFC, by=c("Peakid"="Peakid")) %>%
# left_join(.,gene_info_collapsed, by=c("geneId"="ENTREZID")) %>%
# mutate(SYMBOL=if_else(is.na(SYMBOL.x),SYMBOL.y,if_else(SYMBOL.x==SYMBOL.y, SYMBOL.x,paste0(SYMBOL.x,"_",SYMBOL.y)))) %>%
tidyr::unite(., name,Peakid,snp_list) %>%
mutate(snp_dist=case_when(min_distance <2000 ~"2kb",
min_distance > 2000 & min_distance<20000 ~ "20kb",
min_distance >20000 ~">20kb"))
Cardotox_mat_2 <- AR_Cardiotox_gwas_collaped_df %>%
dplyr::select(name,DOX_3:TRZ_24) %>%
column_to_rownames("name") %>%
as.matrix()
annot_map_df_2 <- AR_Cardiotox_gwas_collaped_df %>%
dplyr::select(name,snp_dist,sig_24) %>%
column_to_rownames("name")
annot_map_2 <-
ComplexHeatmap::rowAnnotation(
snp_dist=AR_Cardiotox_gwas_collaped_df$snp_dist,
TAD_id=AR_Cardiotox_gwas_collaped_df$TAD_id,
DOX_24hr_DAR=AR_Cardiotox_gwas_collaped_df$sig_24,
col= list(snp_dist=c("2kb"="goldenrod4",
"20kb"="pink",
">20kb"="tan2"),
TAD_id=tad_colors))
simply_map_lfc_2 <- ComplexHeatmap::Heatmap(Cardotox_mat_2,
# col = col_fun,
left_annotation = annot_map_2,
show_row_names = TRUE,
row_names_max_width= ComplexHeatmap::max_text_width(rownames(Cardotox_mat_2), gp=gpar(fontsize=14)),
heatmap_legend_param = list(direction = "horizontal"),
show_column_names = TRUE,
cluster_rows = FALSE,
cluster_columns = FALSE)
ComplexHeatmap::draw(simply_map_lfc_2,
merge_legend = TRUE,
heatmap_legend_side = "left",
annotation_legend_side = "left") ### adding in Park data:
### adding in Park data:
ParkSNPs <- readRDS("data/other_papers/ParkSNPs_pull_VEF.RDS")
ParkSNP_table <-
ParkSNPs %>%
dplyr::select(1:2) %>%
distinct() %>%
separate_wider_delim(.,Location,delim=":",names=c("chr","position"), cols_remove=FALSE) %>%
separate_wider_delim(.,position,delim="-",names=c("begin","term")) %>%
mutate(chr=paste0("chr",chr))
ParkSNP_gr <- ParkSNP_table %>%
mutate("start" = begin, "end"=term) %>%
GRanges()
Park_snp_tad_df <- join_overlap_inner(ParkSNP_gr, Left_ventricle_TAD) %>%
as_tibble() %>%
dplyr::rename("RSID"=X.Uploaded_variation) %>%
dplyr::select(RSID, snp_start = start, snp_chr = seqnames, TAD_id)
Park_snp_pairs <- peak_tad_df %>%
inner_join(Park_snp_tad_df, by = "TAD_id")
Park_snp_pairs %>%
distinct(RSID)# A tibble: 7 × 1
RSID
<chr>
1 rs147631684
2 rs2113374
3 rs11894115
4 rs6804462
5 rs17530621
6 rs58328254
7 rs117299725Park_snp_pairs_dist <- Park_snp_pairs %>%
mutate(distance = abs(peak_start - snp_start)) %>%
mutate(sig_24= if_else(Peakid %in% DOX_DAR_sig$Peakid, "sig","not_sig"))
Park_snp_pairs_dist %>%
mutate(sig_24=factor(sig_24, levels= c("sig","not_sig"))) %>%
ggplot(., aes(x= sig_24, y=distance))+
geom_boxplot()+
theme_bw()+
geom_signif(comparisons = list(c("sig", "not_sig")),
map_signif_level = FALSE, test = "wilcox.test")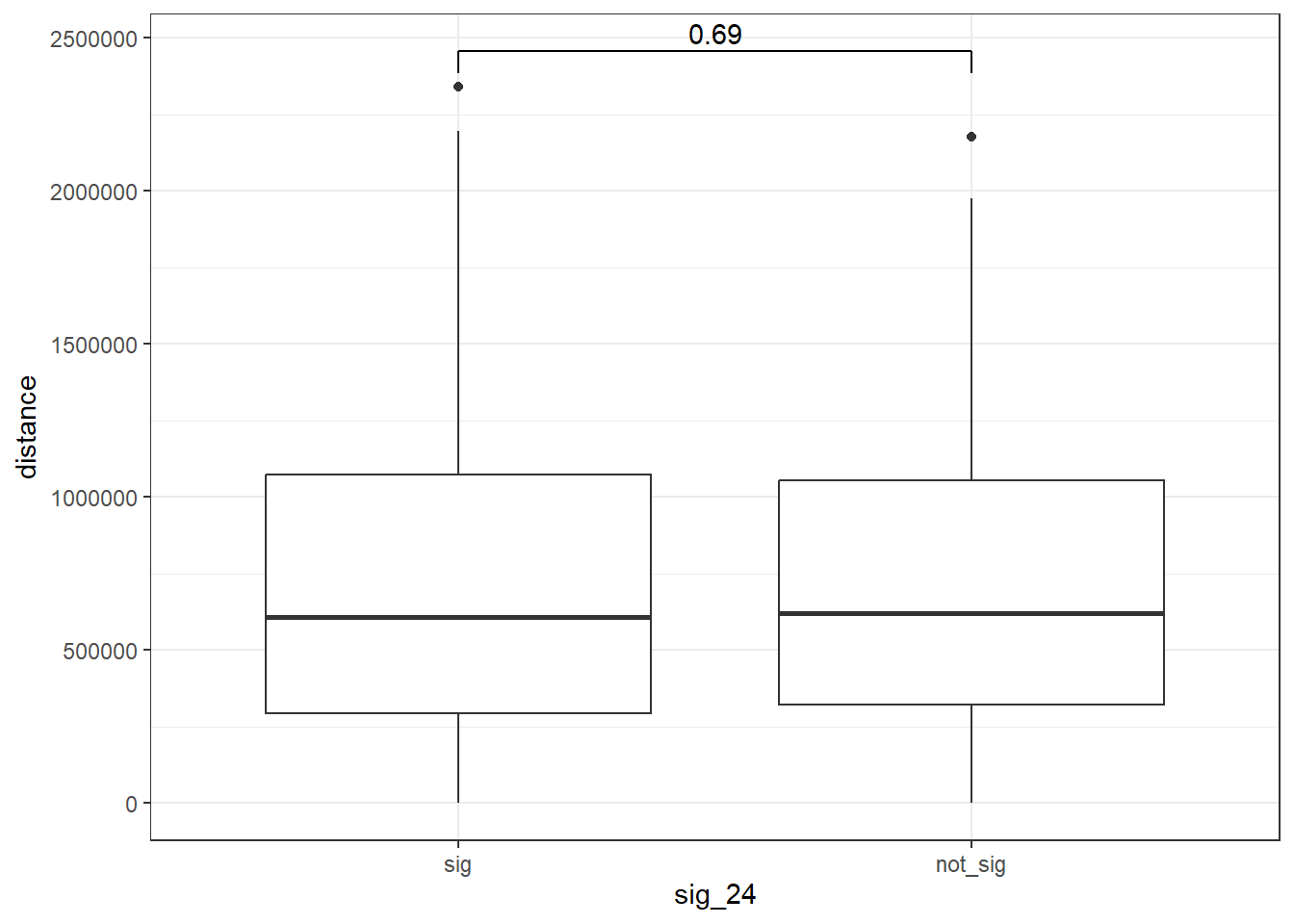
wilcox.test(distance ~ sig_24, data = Park_snp_pairs_dist)
Wilcoxon rank sum test with continuity correction
data: distance by sig_24
W = 71794, p-value = 0.6862
alternative hypothesis: true location shift is not equal to 0Park_Cardiotox_gwas_df <- Park_snp_pairs_dist %>%
dplyr::filter(sig_24=="sig") %>%
group_by(TAD_id,RSID) %>%
slice_min(order_by = distance, with_ties = FALSE) %>%
ungroup() %>%
arrange(snp_chr,snp_start) %>%
left_join(., all_results_pivot, by=c("Peakid"="genes")) %>%
tidyr::unite(., name,Peakid,RSID)Park_Cardiotox_gwas_collaped_df <-
Park_snp_pairs_dist %>%
# dplyr::filter(sig_24=="sig") %>%
group_by(TAD_id,RSID) %>%
slice_min(order_by = distance, with_ties = FALSE) %>%
ungroup() %>%
arrange(snp_chr,snp_start) %>%
group_by(Peakid, peak_chr, peak_start, TAD_id, sig_24) %>%
summarise(
min_distance = min(distance),
mean_distance = mean(distance),
snp_list = paste(unique(RSID), collapse = ","),
.groups = "drop"
) %>%
left_join(., all_results_pivot, by=c("Peakid"="genes")) %>%
# left_join(., Peak_gene_RNA_LFC, by=c("Peakid"="Peakid")) %>%
# left_join(.,gene_info_collapsed, by=c("geneId"="ENTREZID")) %>%
# mutate(SYMBOL=if_else(is.na(SYMBOL.x),SYMBOL.y,if_else(SYMBOL.x==SYMBOL.y, SYMBOL.x,paste0(SYMBOL.x,"_",SYMBOL.y)))) %>%
tidyr::unite(., name,Peakid,snp_list) %>%
mutate(snp_dist=case_when(min_distance <2000 ~"2kb",
min_distance > 2000 & min_distance<20000 ~ "20kb",
min_distance >20000 ~">20kb"))
Cardotox_mat_park <- Park_Cardiotox_gwas_collaped_df %>%
dplyr::select(name,DOX_3:TRZ_24) %>%
column_to_rownames("name") %>%
as.matrix()
annot_map_df_park <- Park_Cardiotox_gwas_collaped_df %>%
dplyr::select(name,snp_dist,sig_24) %>%
column_to_rownames("name")
annot_map_park <-
ComplexHeatmap::rowAnnotation(
snp_dist=Park_Cardiotox_gwas_collaped_df$snp_dist,
TAD_id=Park_Cardiotox_gwas_collaped_df$TAD_id,
DOX_24hr_DAR=Park_Cardiotox_gwas_collaped_df$sig_24,
col= list(snp_dist=c("2kb"="goldenrod4",
"20kb"="pink",
">20kb"="tan2")))
simply_map_lfc_park <- ComplexHeatmap::Heatmap(Cardotox_mat_park,
# col = col_fun,
left_annotation = annot_map_park,
show_row_names = TRUE,
row_names_max_width= ComplexHeatmap::max_text_width(rownames(Cardotox_mat_park), gp=gpar(fontsize=14)),
heatmap_legend_param = list(direction = "horizontal"),
show_column_names = TRUE,
cluster_rows = FALSE,
cluster_columns = FALSE)
ComplexHeatmap::draw(simply_map_lfc_park,
merge_legend = TRUE,
heatmap_legend_side = "left",
annotation_legend_side = "left")
sessionInfo()R version 4.4.2 (2024-10-31 ucrt)
Platform: x86_64-w64-mingw32/x64
Running under: Windows 11 x64 (build 26100)
Matrix products: default
locale:
[1] LC_COLLATE=English_United States.utf8
[2] LC_CTYPE=English_United States.utf8
[3] LC_MONETARY=English_United States.utf8
[4] LC_NUMERIC=C
[5] LC_TIME=English_United States.utf8
time zone: America/Chicago
tzcode source: internal
attached base packages:
[1] grid stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] BSgenome.Hsapiens.UCSC.hg38_1.4.5
[2] BSgenome_1.74.0
[3] BiocIO_1.16.0
[4] Biostrings_2.74.1
[5] XVector_0.46.0
[6] liftOver_1.30.0
[7] Homo.sapiens_1.3.1
[8] TxDb.Hsapiens.UCSC.hg19.knownGene_3.2.2
[9] GO.db_3.20.0
[10] OrganismDbi_1.48.0
[11] gwascat_2.38.0
[12] ComplexHeatmap_2.22.0
[13] readxl_1.4.5
[14] circlize_0.4.16
[15] epitools_0.5-10.1
[16] ggrepel_0.9.6
[17] plyranges_1.26.0
[18] ggsignif_0.6.4
[19] genomation_1.38.0
[20] smplot2_0.2.5
[21] eulerr_7.0.2
[22] biomaRt_2.62.1
[23] devtools_2.4.5
[24] usethis_3.1.0
[25] ggpubr_0.6.1
[26] BiocParallel_1.40.2
[27] scales_1.4.0
[28] VennDiagram_1.7.3
[29] futile.logger_1.4.3
[30] gridExtra_2.3
[31] ggfortify_0.4.18
[32] edgeR_4.4.2
[33] limma_3.62.2
[34] rtracklayer_1.66.0
[35] org.Hs.eg.db_3.20.0
[36] TxDb.Hsapiens.UCSC.hg38.knownGene_3.20.0
[37] GenomicFeatures_1.58.0
[38] AnnotationDbi_1.68.0
[39] Biobase_2.66.0
[40] ChIPpeakAnno_3.40.0
[41] GenomicRanges_1.58.0
[42] GenomeInfoDb_1.42.3
[43] IRanges_2.40.1
[44] S4Vectors_0.44.0
[45] BiocGenerics_0.52.0
[46] ChIPseeker_1.42.1
[47] RColorBrewer_1.1-3
[48] broom_1.0.8
[49] kableExtra_1.4.0
[50] lubridate_1.9.4
[51] forcats_1.0.0
[52] stringr_1.5.1
[53] dplyr_1.1.4
[54] purrr_1.0.4
[55] readr_2.1.5
[56] tidyr_1.3.1
[57] tibble_3.3.0
[58] ggplot2_3.5.2
[59] tidyverse_2.0.0
[60] workflowr_1.7.1
loaded via a namespace (and not attached):
[1] R.methodsS3_1.8.2 dichromat_2.0-0.1
[3] vroom_1.6.5 progress_1.2.3
[5] urlchecker_1.0.1 nnet_7.3-20
[7] vctrs_0.6.5 ggtangle_0.0.7
[9] digest_0.6.37 png_0.1-8
[11] shape_1.4.6.1 git2r_0.36.2
[13] magick_2.8.7 MASS_7.3-65
[15] reshape2_1.4.4 foreach_1.5.2
[17] httpuv_1.6.16 qvalue_2.38.0
[19] withr_3.0.2 xfun_0.52
[21] ggfun_0.1.9 ellipsis_0.3.2
[23] survival_3.8-3 memoise_2.0.1
[25] profvis_0.4.0 systemfonts_1.2.3
[27] tidytree_0.4.6 zoo_1.8-14
[29] GlobalOptions_0.1.2 gtools_3.9.5
[31] R.oo_1.27.1 Formula_1.2-5
[33] prettyunits_1.2.0 KEGGREST_1.46.0
[35] promises_1.3.3 httr_1.4.7
[37] rstatix_0.7.2 restfulr_0.0.16
[39] ps_1.9.1 rstudioapi_0.17.1
[41] UCSC.utils_1.2.0 miniUI_0.1.2
[43] generics_0.1.4 DOSE_4.0.1
[45] base64enc_0.1-3 processx_3.8.6
[47] curl_6.4.0 zlibbioc_1.52.0
[49] GenomeInfoDbData_1.2.13 SparseArray_1.6.2
[51] RBGL_1.82.0 xtable_1.8-4
[53] doParallel_1.0.17 evaluate_1.0.4
[55] S4Arrays_1.6.0 BiocFileCache_2.14.0
[57] hms_1.1.3 colorspace_2.1-1
[59] filelock_1.0.3 magrittr_2.0.3
[61] later_1.4.2 ggtree_3.14.0
[63] lattice_0.22-7 getPass_0.2-4
[65] XML_3.99-0.18 cowplot_1.1.3
[67] matrixStats_1.5.0 Hmisc_5.2-3
[69] pillar_1.11.0 nlme_3.1-168
[71] iterators_1.0.14 pwalign_1.2.0
[73] gridBase_0.4-7 caTools_1.18.3
[75] compiler_4.4.2 stringi_1.8.7
[77] SummarizedExperiment_1.36.0 GenomicAlignments_1.42.0
[79] plyr_1.8.9 crayon_1.5.3
[81] abind_1.4-8 gridGraphics_0.5-1
[83] locfit_1.5-9.12 bit_4.6.0
[85] fastmatch_1.1-6 whisker_0.4.1
[87] codetools_0.2-20 textshaping_1.0.1
[89] bslib_0.9.0 GetoptLong_1.0.5
[91] multtest_2.62.0 mime_0.13
[93] splines_4.4.2 Rcpp_1.1.0
[95] dbplyr_2.5.0 cellranger_1.1.0
[97] utf8_1.2.6 knitr_1.50
[99] blob_1.2.4 clue_0.3-66
[101] AnnotationFilter_1.30.0 fs_1.6.6
[103] checkmate_2.3.2 pkgbuild_1.4.8
[105] ggplotify_0.1.2 Matrix_1.7-3
[107] callr_3.7.6 statmod_1.5.0
[109] tzdb_0.5.0 svglite_2.2.1
[111] pkgconfig_2.0.3 tools_4.4.2
[113] cachem_1.1.0 RSQLite_2.4.1
[115] viridisLite_0.4.2 DBI_1.2.3
[117] impute_1.80.0 fastmap_1.2.0
[119] rmarkdown_2.29 Rsamtools_2.22.0
[121] sass_0.4.10 patchwork_1.3.1
[123] BiocManager_1.30.26 VariantAnnotation_1.52.0
[125] graph_1.84.1 carData_3.0-5
[127] rpart_4.1.24 farver_2.1.2
[129] yaml_2.3.10 MatrixGenerics_1.18.1
[131] foreign_0.8-90 cli_3.6.5
[133] txdbmaker_1.2.1 lifecycle_1.0.4
[135] lambda.r_1.2.4 sessioninfo_1.2.3
[137] backports_1.5.0 timechange_0.3.0
[139] gtable_0.3.6 rjson_0.2.23
[141] parallel_4.4.2 ape_5.8-1
[143] jsonlite_2.0.0 bitops_1.0-9
[145] bit64_4.6.0-1 pwr_1.3-0
[147] yulab.utils_0.2.0 futile.options_1.0.1
[149] jquerylib_0.1.4 GOSemSim_2.32.0
[151] R.utils_2.13.0 snpStats_1.56.0
[153] lazyeval_0.2.2 shiny_1.11.1
[155] htmltools_0.5.8.1 enrichplot_1.26.6
[157] rappdirs_0.3.3 formatR_1.14
[159] ensembldb_2.30.0 glue_1.8.0
[161] httr2_1.1.2 RCurl_1.98-1.17
[163] InteractionSet_1.34.0 rprojroot_2.0.4
[165] treeio_1.30.0 boot_1.3-31
[167] universalmotif_1.24.2 igraph_2.1.4
[169] R6_2.6.1 gplots_3.2.0
[171] labeling_0.4.3 cluster_2.1.8.1
[173] pkgload_1.4.0 regioneR_1.38.0
[175] aplot_0.2.8 DelayedArray_0.32.0
[177] tidyselect_1.2.1 plotrix_3.8-4
[179] ProtGenerics_1.38.0 htmlTable_2.4.3
[181] xml2_1.3.8 car_3.1-3
[183] seqPattern_1.38.0 KernSmooth_2.23-26
[185] data.table_1.17.6 htmlwidgets_1.6.4
[187] fgsea_1.32.4 rlang_1.1.6
[189] remotes_2.5.0 Cairo_1.6-2