alkati_cell_line_tae684_response
Haider Inam
3/6/2019
Last updated: 2019-03-06
workflowr checks: (Click a bullet for more information)-
✔ R Markdown file: up-to-date
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
-
✔ Environment: empty
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
-
✔ Seed:
set.seed(20190211)The command
set.seed(20190211)was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible. -
✔ Session information: recorded
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
-
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.✔ Repository version: 6f57396
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can usewflow_publishorwflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.Ignored files: Ignored: .Rhistory Ignored: .Rproj.user/ Untracked files: Untracked: code/alldata_compiler.R Untracked: code/contab_maker.R Untracked: code/mut_excl_genes_datapoints.R Untracked: code/mut_excl_genes_generator.R Untracked: code/quadratic_solver.R Untracked: code/simresults_generator.R Untracked: data/ALKATI_ccle.csv Untracked: data/All_Data_V2.csv Untracked: data/CCLE_NP24.2009_Drug_data_2015.02.24.csv Untracked: data/alkati_growthcurvedata.csv Untracked: data/alkati_growthcurvedata_popdoublings.csv Untracked: data/alkati_melanoma_vemurafenib_figure_data.csv Untracked: data/all_data.csv Untracked: data/tcga_luad_expression/ Untracked: data/tcga_skcm_expression/ Untracked: docs/figure/Filteranalysis.Rmd/ Untracked: output/alkati_ccle_tae684_plot.pdf Untracked: output/alkati_filtercutoff_allfilters.csv Untracked: output/alkati_luad_exonimbalance.pdf Untracked: output/alkati_mtn_pval_fig2B.pdf Untracked: output/alkati_skcm_exonimbalance.pdf Untracked: output/all_data_luad.csv Untracked: output/all_data_luad_egfr.csv Untracked: output/all_data_skcm.csv Untracked: output/baf3_alkati_figure_deltaadjusted_doublings.pdf Untracked: output/baf3_barplot.pdf Untracked: output/baf3_elisa_barplot.pdf Untracked: output/egfr_luad_exonimbalance.pdf Untracked: output/fig2b2_filtercutoff_atinras_totalalk.pdf Untracked: output/fig2b_filtercutoff_atibraf.pdf Untracked: output/fig2b_filtercutoff_atinras.pdf Untracked: output/luad_alk_exon_expression.csv Untracked: output/luad_egfr_exon_expression.csv Untracked: output/melanoma_vemurafenib_fig.pdf Untracked: output/skcm_alk_exon_expression.csv Unstaged changes: Modified: analysis/ALKATI_Filter_Cutoff_Analysis.Rmd Modified: analysis/baf3_alkati_transformations.Rmd
Expand here to see past versions:
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 6f57396 | haiderinam | 2019-03-06 | Added CCLE analyses on cancer cell line TAE684 response. This includes a logistic |
Here, we combine data from 3 sources: TCGA SKCM ALK expressiossion, CCLE TAE684 IC50, and ALK expression for some of the cell lines on CCLE
The inputs here are 3 .csv files.
- alldata.csv contains SKCM mutation data for BRAF,NRAS and expression data for ALK
- CCLE_NP24.2009_Drug_data_2015.02.24.csv contains SKCM dose responses for different drugs for many cell lines. Of these drugs, only TAE684 targets ALK. There are 504 cell lines with dose repsonses for drug. We had expression data for only 54 of these 504 cell lines.
- ALKATI_ccle.csv contains ALK exon expression RPKMs for 54 cell lines. Obtained from crownbio
ccle_drug=read.csv("data/CCLE_NP24.2009_Drug_data_2015.02.24.csv",sep=",",header=T,stringsAsFactors=F)
ccle_alk=ccle_drug[ccle_drug$Target=="ALK",]
rm(ccle_drug)
ccle_rpkm=read.csv("data/ALKATI_ccle.csv", sep=",",header=T, stringsAsFactors=F)
ccleRpkmT=t(ccle_rpkm)
#Extracting our desired cell lines
data_mat=data.frame(ccleRpkmT[5:57,])
colnames(data_mat)[1:28]=ccleRpkmT[4,2:29]
data_mat_rpkm=data.frame(cbind(rownames(data_mat),data_mat))
rownames(data_mat_rpkm)=NULL
data_mat_rpkm$rownames.data_mat.=sub("^G[0-9]{5}.([A-Za-z0-9._]*).[0-9].bam","\\1",data_mat_rpkm$rownames.data_mat.)
############################################################################################################
ccle_alk$CCLE.Cell.Line.Name=sub("^([0-9A-Z]*)_[A-Za-z0-9_]*","\\1",ccle_alk$CCLE.Cell.Line.Name)
data_mat_rpkm$rownames.data_mat.=gsub("[_]","",data_mat_rpkm$rownames.data_mat.)
data_mat_rpkm$rownames.data_mat.=gsub("[.]","",data_mat_rpkm$rownames.data_mat.)
data_mat_rpkm$rownames.data_mat.=toupper(data_mat_rpkm$rownames.data_mat.)
#they were character because of auto import
for (i in 2:ncol(data_mat_rpkm)){
data_mat_rpkm[,i]=as.numeric(as.character(data_mat_rpkm[,i]))
}
for (i in 2:nrow(data_mat_rpkm)){
data_mat_rpkm[i,31]=sum(data_mat_rpkm[i,2:29])
data_mat_rpkm[i,32]=sum(data_mat_rpkm[i,2:19])/19
data_mat_rpkm[i,33]=sum(data_mat_rpkm[i,20:29])/10
}
colnames(data_mat_rpkm)[31]="SumRPKM"
colnames(data_mat_rpkm)[32]="Avg1_19RPKM"
colnames(data_mat_rpkm)[33]="Avg20_29RPKM"
alldata=merge(data_mat_rpkm,ccle_alk, by.x="rownames.data_mat.", by.y="CCLE.Cell.Line.Name")
alldata=data.frame(cbind(alldata,alldata$Avg20_29RPKM/alldata$Avg1_19RPKM))
data_mat_rpkm=data.frame(cbind(data_mat_rpkm,data_mat_rpkm$Avg20_29RPKM/data_mat_rpkm$Avg1_19RPKM))
#####Makingderivative columns for RPKM ratio and Sum RPKM#########
skcm=read.csv("data/all_data.csv",sep=",",header=T,stringsAsFactors=F)#Downloaded from firehose 02-xx-2016,compiled with mutation data, 340 patients with RNAseq and Muts
skcm_comp=data.frame(cbind(skcm,skcm$mean_RPKM_20.29/skcm$mean_RPKM_1.19))
dim(skcm_comp)[1] 351 11skcm_comp[12]=skcm_comp[5]
skcm_comp[13]=data.frame(20*(skcm_comp$mean_RPKM_1.19)+10*(skcm_comp$mean_RPKM_20.29))
colnames(skcm_comp)[13]="Total_RPKM"
colnames(skcm_comp)[12]="Ratio_ALK_Exons20_29vsExons1_19"
skcm_comp[14]=data.frame(skcm_comp$BRAF!=NaN)
colnames(skcm_comp)[14]="is.BRAF"
skcm_comp[15]=data.frame(grepl("V600",skcm_comp$BRAF),"BRAF")Warning in `[<-.data.frame`(`*tmp*`, 15, value =
structure(list(grepl..V600...skcm_comp.BRAF. = c(TRUE, : provided 2
variables to replace 1 variablescolnames(skcm_comp)[15]="is.V600"
skcm_comp[16]=data.frame(skcm_comp$NRAS!=NaN)
colnames(skcm_comp)[16]="is.NRAS"
skcm_comp[17]=data.frame(grepl("Q61",skcm_comp$NRAS),"NRAS")Warning in `[<-.data.frame`(`*tmp*`, 17, value =
structure(list(grepl..Q61...skcm_comp.NRAS. = c(FALSE, : provided 2
variables to replace 1 variablescolnames(skcm_comp)[17]="is.Q61"
skcm_comp[skcm_comp$BRAF==NaN & skcm_comp$NRAS==NaN, 18]= "DoubleNegative"
colnames(skcm_comp)[18]="is.neitherBRAForNRAS"
skcm_comp[skcm_comp$BRAF!=NaN, 19]= "BRAF Mutant"
skcm_comp[skcm_comp$NRAS!=NaN, 19]= "NRAS Mutant"
skcm_comp[skcm_comp$BRAF==NaN & skcm_comp$NRAS==NaN, 19]= "DoubleNegative"
colnames(skcm_comp)[19]="is.mutant"
######Graphing########
ggplot()+geom_point(data=skcm_comp,aes(x=Ratio_ALK_Exons20_29vsExons1_19, y=Total_RPKM,color=is.mutant))+geom_point(data=alldata,aes(x=alldata.Avg20_29RPKM.alldata.Avg1_19RPKM,y=SumRPKM,size=IC50..uM.))+scale_x_log10()+scale_y_log10()Warning: Transformation introduced infinite values in continuous y-axisWarning: Removed 2 rows containing missing values (geom_point).
# ggsave("cellline,IC50.png",width=10,length=10)
# ggplot()+geom_point(data=skcm_comp,aes(x=Ratio_ALK_Exons20_29vsExons1_19, y=Total_RPKM,color=is.mutant))+geom_point(data=data_mat_rpkm,aes(x=alldata.Avg20_29RPKM.alldata.Avg1_19RPKM,y=SumRPKM,size=IC50..uM.))+scale_x_log10()+scale_y_log10()
# ggsave("cellline_clinical_all.png",width=10,length=10)
ggplot()+
geom_point(data=skcm_comp,aes(x=Ratio_ALK_Exons20_29vsExons1_19, y=Total_RPKM,color=factor(skcm_comp$is.mutant,levels = c("DoubleNegative","BRAF Mutant","NRAS Mutant")),size=2.5))+
geom_point(data=alldata,aes(x=alldata.Avg20_29RPKM.alldata.Avg1_19RPKM,y=SumRPKM,size=IC50..uM.))+
annotate("rect", xmin = 10, xmax = Inf, ymin = 10, ymax = Inf,fill="#66C2A5",alpha = .4)+
scale_x_log10()+
scale_y_log10()+
cleanup+
# scale_color_brewer(palette="Set2",name="Mutation")+
scale_color_manual(values =c("#FFD92F","#E78AC3","#8DA0CB"),name="Mutation")+
scale_size_continuous("Cell Line IC50[uM]")+
xlab("Ratio ALK Ex20-29 to Ex1-19")+
ylab("Total RPKM")+
theme(plot.title = element_text(hjust=.5),
text = element_text(size=26,face="bold"),
axis.title = element_text(face="bold",size="26",color="black"),
axis.text=element_text(face="bold",size="24",color="black"))Warning: Transformation introduced infinite values in continuous y-axis
Warning: Removed 2 rows containing missing values (geom_point).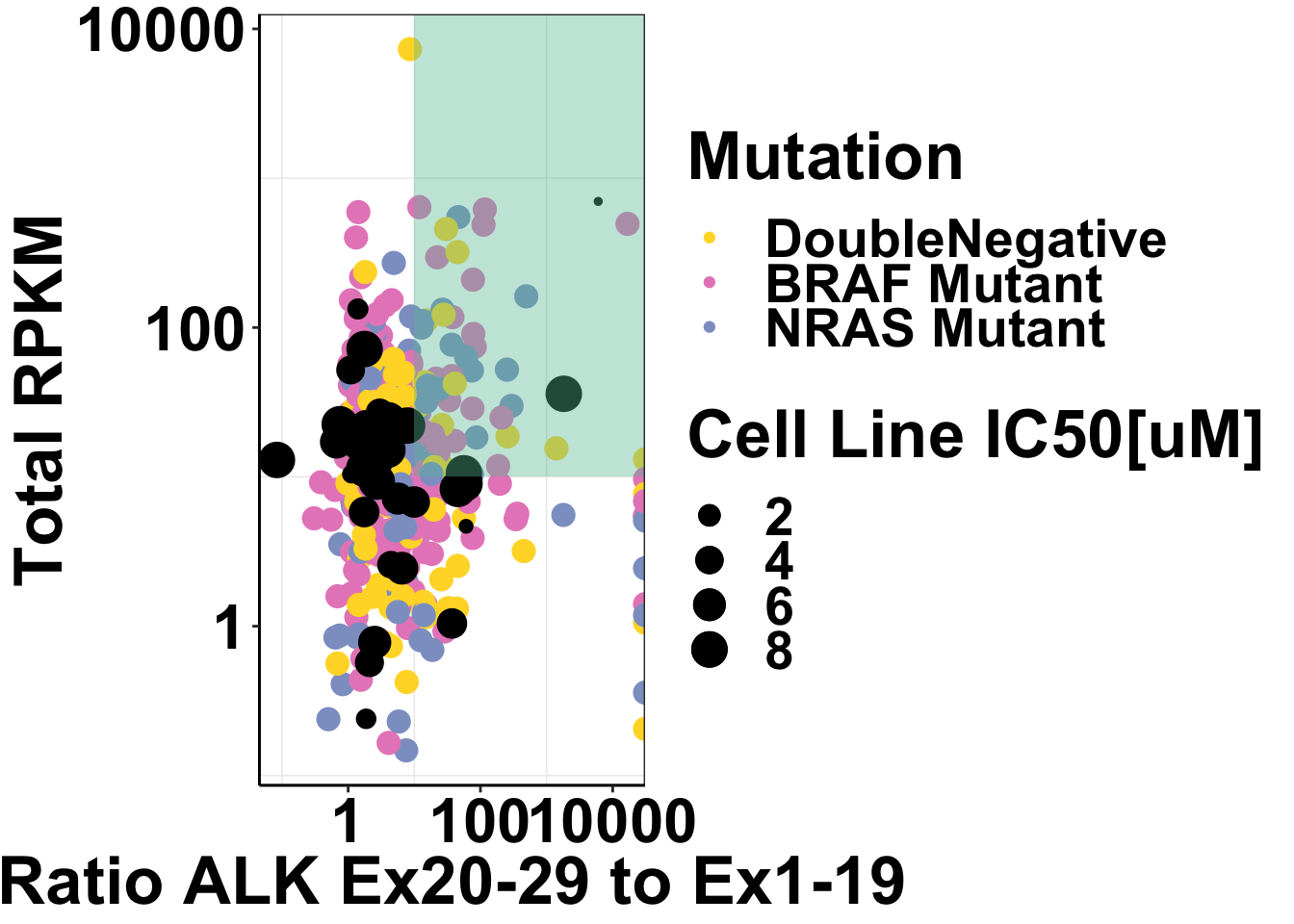
ggsave("output/alkati_ccle_tae684_plot.pdf",height=8,width=12, useDingbats=FALSE)Warning: Transformation introduced infinite values in continuous y-axis
Warning: Removed 2 rows containing missing values (geom_point).Logistic regression to see if IC50 can predict ALKATI or Overexpression
#Notice that we are keeping the EML4ALK translocation from the supm2 lung cancer cell line. These improving IC50's predictive power, if anything.
#Since we only have two ALK
#We are going to look at how well IC50 predicts whether a hit is ALKATI.
#Checking for overexpression. overexpression taken as 1.5x expression in kinase & RPKM of at least 50
#Detecting ALKATI
alldata$alkati=0
alldata$alkati[alldata$alldata.Avg20_29RPKM.alldata.Avg1_19RPKM>5&alldata$SumRPKM>50]=1
#Predictive power of IC50 in a logistic regression model
logistic=glm(alkati~IC50..uM.,data = alldata,family = "binomial")Warning: glm.fit: algorithm did not convergeWarning: glm.fit: fitted probabilities numerically 0 or 1 occurredsummary(logistic)
Call:
glm(formula = alkati ~ IC50..uM., family = "binomial", data = alldata)
Deviance Residuals:
Min 1Q Median 3Q Max
-1.548e-04 -2.100e-08 -2.100e-08 -2.100e-08 1.237e-04
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) 28.6 8921.1 0.003 0.997
IC50..uM. -106.0 25472.7 -0.004 0.997
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 9.0818e+00 on 34 degrees of freedom
Residual deviance: 3.9275e-08 on 33 degrees of freedom
AIC: 4
Number of Fisher Scoring iterations: 25#Checking for overexpression. overexpression taken as twice the expression in the kinase
#Detecting ALKATI
alldata$alkati=as.numeric(alldata$alldata.Avg20_29RPKM.alldata.Avg1_19RPKM>2,na.rm=T)
alldata$alkati[is.na(alldata$alkati)]=0
alldata$alkati=as.factor(alldata$alkati)
#Predictive power of IC50 in a logistic regression model
logistic=glm(alkati~IC50..uM.,data = alldata,family = "binomial")
summary(logistic)
Call:
glm(formula = alkati ~ IC50..uM., family = "binomial", data = alldata)
Deviance Residuals:
Min 1Q Median 3Q Max
-1.315 -1.245 1.045 1.098 1.247
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -0.16801 0.81338 -0.207 0.836
IC50..uM. 0.06082 0.13242 0.459 0.646
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 48.263 on 34 degrees of freedom
Residual deviance: 48.051 on 33 degrees of freedom
AIC: 52.051
Number of Fisher Scoring iterations: 4Session information
sessionInfo()R version 3.5.2 (2018-12-20)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Mojave 10.14.3
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] parallel grid stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] ggsignif_0.4.0 usethis_1.4.0 devtools_2.0.1
[4] RColorBrewer_1.1-2 reshape2_1.4.3 ggplot2_3.1.0
[7] doParallel_1.0.14 iterators_1.0.10 foreach_1.4.4
[10] dplyr_0.7.8 VennDiagram_1.6.20 futile.logger_1.4.3
[13] workflowr_1.1.1 tictoc_1.0 knitr_1.21
loaded via a namespace (and not attached):
[1] tidyselect_0.2.5 xfun_0.4 remotes_2.0.2
[4] purrr_0.3.0 colorspace_1.4-0 htmltools_0.3.6
[7] yaml_2.2.0 rlang_0.3.1 pkgbuild_1.0.2
[10] R.oo_1.22.0 pillar_1.3.1 glue_1.3.0
[13] withr_2.1.2 R.utils_2.7.0 sessioninfo_1.1.1
[16] lambda.r_1.2.3 bindrcpp_0.2.2 bindr_0.1.1
[19] plyr_1.8.4 stringr_1.3.1 munsell_0.5.0
[22] gtable_0.2.0 R.methodsS3_1.7.1 codetools_0.2-16
[25] evaluate_0.12 memoise_1.1.0 labeling_0.3
[28] callr_3.1.1 ps_1.3.0 Rcpp_1.0.0
[31] backports_1.1.3 scales_1.0.0 formatR_1.5
[34] desc_1.2.0 pkgload_1.0.2 fs_1.2.6
[37] digest_0.6.18 stringi_1.2.4 processx_3.2.1
[40] rprojroot_1.3-2 cli_1.0.1 tools_3.5.2
[43] magrittr_1.5 lazyeval_0.2.1 tibble_2.0.1
[46] futile.options_1.0.1 crayon_1.3.4 whisker_0.3-2
[49] pkgconfig_2.0.2 prettyunits_1.0.2 assertthat_0.2.0
[52] rmarkdown_1.11 R6_2.3.0 git2r_0.24.0
[55] compiler_3.5.2 This reproducible R Markdown analysis was created with workflowr 1.1.1