GTEx_genes
ERM
2023-06-15
Last updated: 2023-06-15
Checks: 7 0
Knit directory: Cardiotoxicity/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230109) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version d547877. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .RData
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/41588_2018_171_MOESM3_ESMeQTL_ST2_for paper.csv
Ignored: data/Arr_GWAS.txt
Ignored: data/Arr_geneset.RDS
Ignored: data/BC_cell_lines.csv
Ignored: data/CADGWASgene_table.csv
Ignored: data/CAD_geneset.RDS
Ignored: data/Clamp_Summary.csv
Ignored: data/Cormotif_24_k1-5_raw.RDS
Ignored: data/DAgostres24.RDS
Ignored: data/DAtable1.csv
Ignored: data/DDEMresp_list.csv
Ignored: data/DDE_reQTL.txt
Ignored: data/DDEresp_list.csv
Ignored: data/DEG-GO/
Ignored: data/DEG_cormotif.RDS
Ignored: data/DF_Plate_Peak.csv
Ignored: data/DRC48hoursdata.csv
Ignored: data/Da24counts.txt
Ignored: data/Dx24counts.txt
Ignored: data/Dx_reQTL_specific.txt
Ignored: data/Ep24counts.txt
Ignored: data/GOIsig.csv
Ignored: data/GOplots.R
Ignored: data/GTEX_setsimple.csv
Ignored: data/GTEx_gene_list.csv
Ignored: data/HFGWASgene_table.csv
Ignored: data/HF_geneset.RDS
Ignored: data/Heart_Left_Ventricle.v8.egenes.txt
Ignored: data/Hf_GWAS.txt
Ignored: data/K_cluster
Ignored: data/K_cluster_kisthree.csv
Ignored: data/K_cluster_kistwo.csv
Ignored: data/LDH48hoursdata.csv
Ignored: data/Mt24counts.txt
Ignored: data/RINsamplelist.txt
Ignored: data/Seonane2019supp1.txt
Ignored: data/TOP2Bi-24hoursGO_analysis.csv
Ignored: data/TR24counts.txt
Ignored: data/Top2biresp_cluster24h.csv
Ignored: data/Viabilitylistfull.csv
Ignored: data/allexpressedgenes.txt
Ignored: data/allgenes.txt
Ignored: data/allmatrix.RDS
Ignored: data/avgLD50.RDS
Ignored: data/backGL.txt
Ignored: data/cormotif_3hk1-8.RDS
Ignored: data/cormotif_initalK5.RDS
Ignored: data/cormotif_initialK5.RDS
Ignored: data/cormotif_initialall.RDS
Ignored: data/counts24hours.RDS
Ignored: data/cpmnorm_counts.csv
Ignored: data/crispr_genes.csv
Ignored: data/cvd_GWAS.txt
Ignored: data/dat_cpm.RDS
Ignored: data/data_outline.txt
Ignored: data/efit2.RDS
Ignored: data/efit2results.RDS
Ignored: data/ensembl_backup.RDS
Ignored: data/ensgtotal.txt
Ignored: data/filenameonly.txt
Ignored: data/filtered_cpm_counts.csv
Ignored: data/filtered_raw_counts.csv
Ignored: data/filtermatrix_x.RDS
Ignored: data/folder_05top/
Ignored: data/gene_corr_frame.RDS
Ignored: data/gene_prob_tran3h.RDS
Ignored: data/gene_probabilityk5.RDS
Ignored: data/gostresTop2bi_ER.RDS
Ignored: data/gostresTop2bi_LR
Ignored: data/gostresTop2bi_LR.RDS
Ignored: data/gostresTop2bi_TI.RDS
Ignored: data/gostrescoNR
Ignored: data/gtex/
Ignored: data/heartgenes.csv
Ignored: data/individualDRCfile.RDS
Ignored: data/individual_DRC48.RDS
Ignored: data/individual_LDH48.RDS
Ignored: data/knowfig4.csv
Ignored: data/knowfig5.csv
Ignored: data/knowles56.GMT
Ignored: data/knowlesGMT.GMT
Ignored: data/mymatrix.RDS
Ignored: data/nonresponse_cluster24h.csv
Ignored: data/norm_LDH.csv
Ignored: data/norm_counts.csv
Ignored: data/old_sets/
Ignored: data/plan2plot.png
Ignored: data/raw_counts.csv
Ignored: data/response_cluster24h.csv
Ignored: data/sigVDA24.txt
Ignored: data/sigVDA3.txt
Ignored: data/sigVDX24.txt
Ignored: data/sigVDX3.txt
Ignored: data/sigVEP24.txt
Ignored: data/sigVEP3.txt
Ignored: data/sigVMT24.txt
Ignored: data/sigVMT3.txt
Ignored: data/sigVTR24.txt
Ignored: data/sigVTR3.txt
Ignored: data/siglist.RDS
Ignored: data/table3a.omar
Ignored: data/toplistall.RDS
Ignored: data/tvl24hour.txt
Ignored: data/tvl24hourw.txt
Ignored: data/venn_code.R
Untracked files:
Untracked: .RDataTmp
Untracked: .RDataTmp1
Untracked: .RDataTmp2
Untracked: analysis/Seoane_chrom.Rmd
Untracked: cormotif_probability_genelist.csv
Untracked: individual-legenddark2.png
Untracked: installed_old.rda
Untracked: motif_ER.txt
Untracked: motif_LR.txt
Untracked: motif_NR.txt
Untracked: motif_TI.txt
Untracked: output/DNRmeQTLs.RDS
Untracked: output/DNRmeSNPs.RDS
Untracked: output/DNRreQTLs.RDS
Untracked: output/DOXmeQTLs.RDS
Untracked: output/DOXmeSNPs.RDS
Untracked: output/DOXreQTLs.RDS
Untracked: output/EPImeQTLs.RDS
Untracked: output/EPImeSNPs.RDS
Untracked: output/EPIreQTLs.RDS
Untracked: output/Fig_summary1.png
Untracked: output/GWAS_goi.csv
Untracked: output/MTXmeQTLs.RDS
Untracked: output/MTXmeSNPs.RDS
Untracked: output/MTXreQTLs.RDS
Untracked: output/Sup4seoane.csv
Untracked: output/egenes.csv
Untracked: output/egenes_hgnc.csv
Untracked: output/egenes_set.csv
Untracked: output/figure_1.Rmd
Untracked: output/heart_gtex.csv
Untracked: output/knowles4.RDS
Untracked: output/knowles5.RDS
Untracked: output/not_eqtls_GTEX.csv
Untracked: output/output-old/
Untracked: output/plan2plot.png
Untracked: output/plan48ldh.png
Untracked: reneebasecode.R
Unstaged changes:
Modified: analysis/DEG-GO_analysis.Rmd
Modified: analysis/Figure1.Rmd
Modified: analysis/Knowles2019.Rmd
Modified: code/eQTLcodes.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/GTEx_genes.Rmd) and HTML
(docs/GTEx_genes.html) files. If you’ve configured a remote
Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | d547877 | reneeisnowhere | 2023-06-15 | update on GTEx |
| html | 7600128 | reneeisnowhere | 2023-06-14 | Build site. |
| Rmd | ff02989 | reneeisnowhere | 2023-06-14 | update for website |
| html | ae073be | reneeisnowhere | 2023-06-14 | Build site. |
| Rmd | e9f1a70 | reneeisnowhere | 2023-06-14 | update for website |
| Rmd | 7fc7ec7 | reneeisnowhere | 2023-06-14 | updating code |
GTEx egenes and analysis
library(ComplexHeatmap)
library(tidyverse)
library(ggsignif)
library(biomaRt)
library(RColorBrewer)
library(cowplot)
library(scales)
library(sjmisc)
library(kableExtra)
library(broom)
library(ggstats)Data import
toplistall<- read.csv("output/toplistall.csv", row.names = 1)
my_exp_genes <- read.csv("data/backGL.txt")
egenes_set <- read.csv("output/egenes_set.csv",row.names = 1)
egenes_hgnc <- read.csv("output/egenes_hgnc.csv",row.names = 1)
GTEx_genes <- read.csv("data/GTEx_gene_list.csv",row.names = 1)
not_eqtls <- read.csv("output/not_eqtls_GTEX.csv",row.names = 1)
heart_gtex <- read.csv("output/heart_gtex.csv",row.names = 1)
egenes <- read.csv("output/egenes.csv",row.names = 1)Obtaining the GTEx data set
I downloaded the GTEx_Analysis_v8.metasoft.txt.gz files from the Consortium at https://www.gtexportal.org/home/datasets .
I the extracted the Heart_Left_Ventricle.v8.egenes.txt file and uploaded into R under the data folder.
heart_gtex <-
readr::read_delim("data/Heart_Left_Ventricle.v8.egenes.txt",
delim = "\t",
escape_double = FALSE,
trim_ws = TRUE)
egenes <- heart_gtex %>%
dplyr::select(gene_id, gene_name, qval) %>%
filter(qval<0.05) %>%
separate(gene_id, into =c('ensembl_gene_id', 'gene_version'))
not_eqtl <- heart_gtex %>%
dplyr::select(gene_id, gene_name, qval) %>%
filter(qval>0.05) %>%
separate(gene_id, into =c('ensembl_gene_id', 'gene_version'))
egenes_set <- getBM(attributes=my_attributes,
filters ='ensembl_gene_id',
values =egenes$ensembl_gene_id,
mart = ensembl)
egenes_hgnc <- getBM(attributes=my_attributes,
filters ='hgnc_symbol',
values =egenes$gene_name,
mart = ensembl)
not_eqtl_set <- getBM(attributes=my_attributes,
filters ='ensembl_gene_id',
values =not_eqtl$ensembl_gene_id,
mart = ensembl)
not_eqtls <- not_eqtl_set %>%
distinct(entrezgene_id,.keep_all = TRUE) %>%
filter(entrezgene_id %in% my_exp_genes$ENTREZID)
##6711 not_eqtls
GTEx_genes <- egenes_set %>%
distinct(entrezgene_id,.keep_all = TRUE)This file contains several columns gene_id, gene_name, gene_chr, gene_start, gene_end, strand, num_var, beta_shape1, beta_shape2, true_df, pval_true_df, variant_id, tss_distance, chr, variant_pos, ref, alt, num_alt_per_site, rs_id_dbSNP151_GRCh38p7, minor_allele_samples, minor_allele_count, maf, ref_factor, pval_nominal, slope, slope_se, pval_perm, pval_beta, qval, pval_nominal_threshold, log2_aFC, log2_aFC_lower, log2_aFC_upper.
I then chose the the ‘gene_id’,‘gene_name’, and ‘qval’ columns. This
left me with 21353 genes. Next I filtered the tissue specific expressed
genes using a the ‘qval < 0.05’ for a total of 9642. I then took the
gene_name column and used biomart to convert to ‘entrezgene_id’.
Because results vary by which way I look up genes in BioMart, I tested
both egenes using ensemble_gene_id and hgnc_symbol columns. I found 7813
for the ensemble_gene_ set and 7271 for the hgnc_symbol set.
I went with using ensemble_gene_id because I found ~600 more genes
overall than using the hgnc_symbol filter.
GTEx <- intersect(GTEx_genes$entrezgene_id,my_exp_genes$ENTREZID)
nQTL <- not_eqtls
# '%!in%' <- function(x,y)!('%in%'(x,y))
nQTLmy <- my_exp_genes %>%
dplyr:: filter(!ENTREZID %in%GTEx)
# testset <- toplistall %>%
# filter(adj.P.Val<0.05) %>%
# select(ENTREZID) %>% distinct(ENTREZID) %>%
# dplyr:: filter(ENTREZID %in%GTEx_genes$entrezgene_id)To find out how many genes from the gtex egenes were expressed in my data, I intersected my expressed genes list of 14084 genes with the GTEx_genes and found 6261 genes were shared between them. I called this set ‘GTEx’. Using the other eGenes from GTEx, I made another set intersected with my expressed gene set called ‘nQTL’. This nQTL set contains 7823 genes. Next, I then took my DEG top list and filtered out genes with an adj.P.value < 0.05.
drug_palspc <- c("#8B006D","#DF707E","#8B006D","#DF707E")The next step is to wrangle the data so that I can test the difference between the proportions of significantly DE genes found in the GTEx and nQTLs.
nQTLsum <- toplistall %>%
mutate(id = as.factor(id)) %>%
mutate(time=factor(time, levels=c("3_hours","24_hours"))) %>%
dplyr::filter(adj.P.Val <0.05) %>%
mutate(nQTL=if_else(ENTREZID %in% nQTLmy$ENTREZID,'nQTL_y','nQTL_no')) %>%
group_by(id,time,nQTL) %>%
tally() %>%
separate(nQTL, into=c('set', 'group')) %>%
mutate(total=length(nQTLmy$ENTREZID) - n) %>%
dplyr::filter(group=="y")
GTExsum <- toplistall %>%
mutate(id = as.factor(id)) %>%
mutate(time=factor(time, levels=c("3_hours","24_hours"))) %>%
dplyr::filter(adj.P.Val <0.05) %>%
mutate(GTEx=if_else(ENTREZID %in%GTEx,"GTEx_y","GTEx_no")) %>%
group_by(id,time,GTEx) %>%
tally() %>%
separate(GTEx, into=c('set', 'group')) %>%
mutate(total=length(GTEx) - n) %>%
dplyr::filter(group=="y")
GTEXcr8z <- GTExsum %>%
rbind(., nQTLsum) %>%
dplyr::select(id,time,set, n,total) %>%
pivot_longer(cols=n:total, names_to="group",values_to="total")
GTEXcr8z %>%
ggplot(., aes(x=set,y=total, fill=group))+
geom_col(position='fill')+
facet_wrap(time~id,nrow=2,ncol=4)+
theme_classic()+
# scale_color_manual(values=drug_palNoVeh[c(1,2)])+
scale_fill_manual(values=drug_palspc)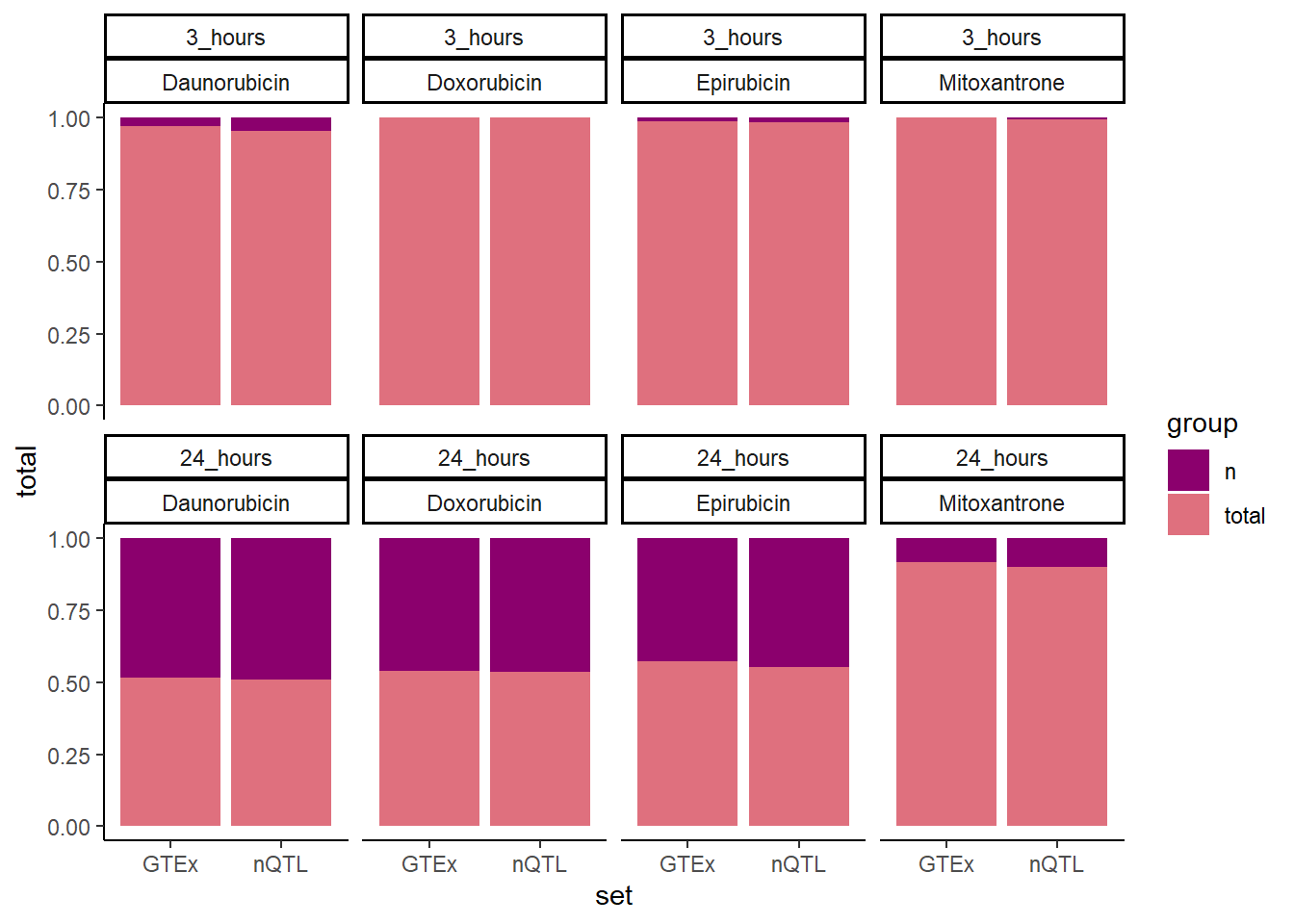
GTExsum %>%
rbind(., nQTLsum) %>%
dplyr::select(id,time,set, n, total) %>%
mutate(time = case_match(time, '3_hours'~'3 hrs',
'24_hours'~'24 hrs',.default = time)) %>%
mutate(id =case_match( id,
'Daunorubicin'~'DNR',
'Doxorubicin'~'DOX' ,
'Epirubicin'~'EPI' ,
'Mitoxantrone' ~ 'MTX', .default = id)) #%>% # A tibble: 16 × 5
# Groups: id, time [8]
id time set n total
<chr> <chr> <chr> <int> <int>
1 DNR 3 hrs GTEx 190 6071
2 DNR 24 hrs GTEx 3032 3229
3 DOX 3 hrs GTEx 2 6259
4 DOX 24 hrs GTEx 2877 3384
5 EPI 3 hrs GTEx 77 6184
6 EPI 24 hrs GTEx 2686 3575
7 MTX 3 hrs GTEx 13 6248
8 MTX 24 hrs GTEx 534 5727
9 DNR 3 hrs nQTL 365 7458
10 DNR 24 hrs nQTL 3832 3991
11 DOX 3 hrs nQTL 14 7809
12 DOX 24 hrs nQTL 3639 4184
13 EPI 3 hrs nQTL 143 7680
14 EPI 24 hrs nQTL 3516 4307
15 MTX 3 hrs nQTL 45 7778
16 MTX 24 hrs nQTL 793 7030testDNR3chix <- matrix(c(365,190,7458, 6071), nrow=2, ncol=2, byrow=FALSE,
dimnames=list(c("nogtex","gtex"),c( "y", "n")))
DNR_3chix <- chisq.test(testDNR3chix,correct=TRUE)$p.value
testDNR24chix <- matrix(c(3832,3032,3991,3229), nrow=2, ncol=2, byrow=FALSE,
dimnames=list(c("nogtex","gtex"),c( "y", "n")))
DNR_24chix <- chisq.test(testDNR24chix,correct=TRUE)$p.value
testDOX3chix <- matrix(c(14,2,7809,6259), nrow=2, ncol=2, byrow=FALSE,
dimnames=list(c("nogtex","gtex"),c( "y", "n")))
DOX_3chix <- chisq.test(testDOX3chix,correct=TRUE)$p.value
testDOX24chix <- matrix(c(3639,2877,4184,3384), nrow=2, ncol=2, byrow=FALSE,
dimnames=list(c("nogtex","gtex"),c( "y", "n")))
DOX_24chix <- chisq.test(testDOX24chix,correct=TRUE)$p.value
testEPI3chix <- matrix(c(143,77,7680,6184), nrow=2, ncol=2, byrow=FALSE,
dimnames=list(c("nogtex","gtex"),c( "y", "n")))
EPI_3chix <- chisq.test(testEPI3chix)$p.value
testEPI24chix <- matrix(c(3516,2686,4307,3575), nrow=2, ncol=2, byrow=FALSE,
dimnames=list(c("nogtex","gtex"),c( "y", "n")))
EPI_24chix <- chisq.test(testEPI24chix)$p.value
testMTX3chix <- matrix(c(45,13,7778,6248), nrow=2, ncol=2, byrow=FALSE,
dimnames=list(c("nogtex","nogtex"),c( "y", "n")))
MTX_3chix <- chisq.test(testMTX3chix)$p.value
testMTX24chix <- matrix(c(793,534,7030,5727), nrow=2, ncol=2, byrow=FALSE,
dimnames=list(c("nogtex","nogtex"),c( "y", "n")))
MTX_24chix <- chisq.test(testMTX24chix,correct=TRUE)$p.value
GTEX_table_chix <- data.frame(treatment=c('DNR_3','DNR_24','DOX_3','DOX_24','EPI_3','EPI_24','MTX_3','MTX_24'), chi_p.value=c(DNR_3chix,DNR_24chix,DOX_3chix,DOX_24chix,EPI_3chix,EPI_24chix,MTX_3chix,MTX_24chix))
GTEX_table_chix %>%
separate(treatment, into= c('Drug','time')) %>%
pivot_wider(id_cols = Drug, names_from = time, values_from = chi_p.value) %>%
kable(., caption= "Chi Square p. values from chi-square test between proportions of sig-DE meQTLs and reQTLS by time and treatment") %>%
kable_paper("striped", full_width = TRUE) %>%
kable_styling(full_width = FALSE, font_size = 16) %>%
scroll_box( height = "500px")| Drug | 3 | 24 |
|---|---|---|
| DNR | 0.0000010 | 0.5220483 |
| DOX | 0.0202323 | 0.5144642 |
| EPI | 0.0055023 | 0.0159127 |
| MTX | 0.0011436 | 0.0012975 |
sessionInfo()R version 4.2.2 (2022-10-31 ucrt)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 19045)
Matrix products: default
locale:
[1] LC_COLLATE=English_United States.utf8
[2] LC_CTYPE=English_United States.utf8
[3] LC_MONETARY=English_United States.utf8
[4] LC_NUMERIC=C
[5] LC_TIME=English_United States.utf8
attached base packages:
[1] grid stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] ggstats_0.3.0 broom_1.0.5 kableExtra_1.3.4
[4] sjmisc_2.8.9 scales_1.2.1 cowplot_1.1.1
[7] RColorBrewer_1.1-3 biomaRt_2.52.0 ggsignif_0.6.4
[10] lubridate_1.9.2 forcats_1.0.0 stringr_1.5.0
[13] dplyr_1.1.2 purrr_1.0.1 readr_2.1.4
[16] tidyr_1.3.0 tibble_3.2.1 ggplot2_3.4.2
[19] tidyverse_2.0.0 ComplexHeatmap_2.12.1 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] colorspace_2.1-0 rjson_0.2.21 sjlabelled_1.2.0
[4] rprojroot_2.0.3 circlize_0.4.15 XVector_0.36.0
[7] GlobalOptions_0.1.2 fs_1.6.2 clue_0.3-64
[10] rstudioapi_0.14 farver_2.1.1 bit64_4.0.5
[13] AnnotationDbi_1.58.0 fansi_1.0.4 xml2_1.3.4
[16] codetools_0.2-19 doParallel_1.0.17 cachem_1.0.8
[19] knitr_1.43 jsonlite_1.8.5 cluster_2.1.4
[22] dbplyr_2.3.2 png_0.1-8 compiler_4.2.2
[25] httr_1.4.6 backports_1.4.1 fastmap_1.1.1
[28] cli_3.6.1 later_1.3.1 htmltools_0.5.5
[31] prettyunits_1.1.1 tools_4.2.2 gtable_0.3.3
[34] glue_1.6.2 GenomeInfoDbData_1.2.8 rappdirs_0.3.3
[37] Rcpp_1.0.10 Biobase_2.56.0 jquerylib_0.1.4
[40] vctrs_0.6.2 Biostrings_2.64.1 svglite_2.1.1
[43] iterators_1.0.14 insight_0.19.2 xfun_0.39
[46] ps_1.7.5 rvest_1.0.3 timechange_0.2.0
[49] lifecycle_1.0.3 XML_3.99-0.14 getPass_0.2-2
[52] zlibbioc_1.42.0 hms_1.1.3 promises_1.2.0.1
[55] parallel_4.2.2 yaml_2.3.7 curl_5.0.1
[58] memoise_2.0.1 sass_0.4.6 stringi_1.7.12
[61] RSQLite_2.3.1 highr_0.10 S4Vectors_0.34.0
[64] foreach_1.5.2 BiocGenerics_0.42.0 filelock_1.0.2
[67] shape_1.4.6 GenomeInfoDb_1.32.4 rlang_1.1.1
[70] pkgconfig_2.0.3 systemfonts_1.0.4 matrixStats_1.0.0
[73] bitops_1.0-7 evaluate_0.21 labeling_0.4.2
[76] bit_4.0.5 processx_3.8.1 tidyselect_1.2.0
[79] magrittr_2.0.3 R6_2.5.1 IRanges_2.30.1
[82] generics_0.1.3 DBI_1.1.3 pillar_1.9.0
[85] whisker_0.4.1 withr_2.5.0 KEGGREST_1.36.3
[88] RCurl_1.98-1.12 crayon_1.5.2 utf8_1.2.3
[91] BiocFileCache_2.4.0 tzdb_0.4.0 rmarkdown_2.22
[94] GetoptLong_1.0.5 progress_1.2.2 blob_1.2.4
[97] callr_3.7.3 git2r_0.32.0 digest_0.6.31
[100] webshot_0.5.4 httpuv_1.6.11 stats4_4.2.2
[103] munsell_0.5.0 viridisLite_0.4.2 bslib_0.5.0