H3K27ac and TE overlap
Renee Matthews
2026-02-05
Last updated: 2026-02-05
Checks: 7 0
Knit directory: DXR_continue/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20250701) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version dbe48c5. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/Bed_exports/
Ignored: data/Cormotif_data/
Ignored: data/DER_data/
Ignored: data/Other_paper_data/
Ignored: data/RDS_files/
Ignored: data/TE_annotation/
Ignored: data/alignment_summary.txt
Ignored: data/all_peak_final_dataframe.txt
Ignored: data/cell_line_info_.tsv
Ignored: data/full_summary_QC_metrics.txt
Ignored: data/motif_lists/
Ignored: data/number_frag_peaks_summary.txt
Untracked files:
Untracked: H3K27ac_all_regions_test.bed
Untracked: H3K27ac_consensus_clusters_test.bed
Untracked: analysis/ATAC_integration_data1.Rmd
Untracked: analysis/GREAT_H3K27ac.Rmd
Untracked: analysis/H3K27ac_ChromHMM_FC.Rmd
Untracked: analysis/H3K27ac_cisRE.Rmd
Untracked: analysis/H3K27me3_TE_investigation.Rmd
Untracked: analysis/H3K36me3_TE_investigation.Rmd
Untracked: analysis/Top2a_Top2b_expression.Rmd
Untracked: analysis/maps_and_plots.Rmd
Untracked: analysis/proteomics.Rmd
Untracked: code/For_john.R
Untracked: other_analysis/
Unstaged changes:
Modified: analysis/H3K27ac_RNA_integration.Rmd
Modified: analysis/H3K27ac_summit_processing.Rmd
Modified: analysis/dual_histone_TE_investigation.Rmd
Modified: analysis/final_analysis.Rmd
Modified: analysis/summit_files_processing.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/H3K27_TE_overlap.Rmd) and
HTML (docs/H3K27_TE_overlap.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | dbe48c5 | reneeisnowhere | 2026-02-05 | adding in proportion plots |
| html | 26f72a7 | reneeisnowhere | 2026-02-05 | Build site. |
| Rmd | f3fb1ca | reneeisnowhere | 2026-02-05 | wflow_publish("analysis/H3K27_TE_overlap.Rmd") |
| html | 0606abf | reneeisnowhere | 2026-01-27 | Build site. |
| Rmd | 6ec7f8f | reneeisnowhere | 2026-01-27 | wflow_publish("analysis/H3K27_TE_overlap.Rmd") |
| html | 8f7ef07 | reneeisnowhere | 2026-01-26 | Build site. |
| Rmd | a44db38 | reneeisnowhere | 2026-01-26 | new overlaps autosomal only |
| html | 177cb71 | reneeisnowhere | 2026-01-23 | Build site. |
| Rmd | e8e2585 | reneeisnowhere | 2026-01-23 | image update |
| html | adde0e9 | reneeisnowhere | 2026-01-15 | Build site. |
| Rmd | 26ea501 | reneeisnowhere | 2026-01-15 | wflow_publish("analysis/H3K27_TE_overlap.Rmd") |
| html | 2944d3e | reneeisnowhere | 2026-01-15 | Build site. |
| Rmd | a8dd5cd | reneeisnowhere | 2026-01-15 | wflow_publish("analysis/H3K27_TE_overlap.Rmd") |
| html | 6d24630 | reneeisnowhere | 2026-01-15 | Build site. |
| html | dac3bca | reneeisnowhere | 2026-01-13 | Build site. |
| Rmd | 75f31af | reneeisnowhere | 2026-01-13 | updates |
| html | d19b31e | reneeisnowhere | 2026-01-12 | Build site. |
| html | dee0fe2 | reneeisnowhere | 2026-01-08 | Build site. |
| Rmd | ceecc02 | reneeisnowhere | 2026-01-08 | wflow_publish("analysis/H3K27_TE_overlap.Rmd") |
| html | 95452bd | reneeisnowhere | 2026-01-08 | Build site. |
| Rmd | f549ad9 | reneeisnowhere | 2026-01-08 | wflow_publish("analysis/H3K27_TE_overlap.Rmd") |
library(tidyverse)
library(GenomicRanges)
library(plyranges)
library(genomation)
library(readr)
library(rtracklayer)
library(stringr)
library(ggrepel)
library(DT)First steps: breakdown repeatmasker into groups and pull out the ones by each class I am interested in.
repeatmasker <- read_delim("data/Other_paper_data/repeatmasker_20250911.txt",
delim = "\t", escape_double = FALSE,
trim_ws = TRUE)
colnames(repeatmasker) [1] "#bin" "swScore" "milliDiv" "milliDel" "milliIns" "genoName"
[7] "genoStart" "genoEnd" "genoLeft" "strand" "repName" "repClass"
[13] "repFamily" "repStart" "repEnd" "repLeft" "id" autosomes <- paste0("chr", 1:22)
repeatmasker_clean <- repeatmasker %>% mutate(
strand = ifelse(strand == "C", "-", "+")
) %>%
mutate(
start = genoStart + 1,
end = genoEnd)%>%
mutate(repFamily= str_remove(repFamily, "\\?$")) %>%
dplyr::filter(genoName %in% autosomes)
rpt_split <- split(repeatmasker_clean, repeatmasker_clean$repClass)
rpt_split_gr_list <- lapply(rpt_split, function(df) {
GRanges(
seqnames = df$genoName,
ranges = IRanges(start = df$start, end = df$end),
strand = df$strand,
repName = df$repName,
repClass = df$repClass,
repFamily = df$repFamily,
swScore = df$swScore,
milliDiv = df$milliDiv,
id = df$id
)
})SINE_gr <- rpt_split_gr_list$SINE
LINE_gr <- rpt_split_gr_list$LINE
LTR_gr <- rpt_split_gr_list$LTR
SVA_gr <- rpt_split_gr_list$Retroposon
DNA_gr <- rpt_split_gr_list$DNAH3K27ac_summit_gr <- readRDS("data/RDS_files/H3K27ac_complete_summit_gr.RDS")
peakAnnoList_H3K27ac <- readRDS("data/motif_lists/H3K27ac_annotated_peaks.RDS")
H3K27ac_lookup <- imap_dfr(peakAnnoList_H3K27ac[1:3], ~
tibble(Peakid = .x@anno$Peakid, cluster = .y)
)
H3K27ac_sets_gr <- lapply(peakAnnoList_H3K27ac, function(df) {
as_granges(df)
})
##assigning Peakid as name of summit region
mcols(H3K27ac_summit_gr)$name <- mcols(H3K27ac_summit_gr)$Peakid
comparisons <- tibble(
cluster2 = c("Set_2", "Set_3"),
cluster1 = c("Set_1", "Set_1")
)# Generic pairwise Fisher test
test_pair_TE_generic <- function(df_long, te_name, cluster1, cluster2) {
sub_df <- df_long %>%
filter(TE_type == te_name) %>%
complete(
cluster = c(cluster1, cluster2),
status = c("TE", "not_TE"),
fill = list(count = 0))
# enforce fixed order
status_levels <- c("TE", "not_TE")
# assume "status" column has TE vs wnot_TE automatically
statuses <- unique(sub_df$status)
if(length(statuses) != 2) {
# ensure we have exactly two categories, fill missing with 0
sub_df <- sub_df %>%
complete(cluster, status, fill = list(count = 0))
statuses <- unique(sub_df$status)
}
# extract counts for cluster1
c1_counts <- sub_df %>%
filter(cluster == cluster1) %>%
arrange(factor(status, levels = status_levels)) %>% # ensure same order
pull(count)
# extract counts for cluster2
c2_counts <- sub_df %>%
filter(cluster == cluster2) %>%
arrange(factor(status, levels = status_levels)) %>%
pull(count)
# build 2x2 matrix
mat <- matrix(
c(c2_counts, c1_counts),
nrow = 2,
byrow = TRUE,
dimnames = list(
cluster = c(cluster2, cluster1),
category = status_levels
)
)
ft <- tryCatch(
fisher.test(mat, workspace = 2e8),
error = function(e) fisher.test(mat, simulate.p.value = TRUE, B = 1e5)
)
tibble(
TE_type = te_name,
comparison = paste(cluster2, "vs", cluster1),
odds_ratio = ft$estimate,
lower_CI = ft$conf.int[1],
upper_CI = ft$conf.int[2],
p_value = ft$p.value
)
}test_pair_TE_repName <- function(df_long, rep_name, cluster1, cluster2) {
# Subset for the specific repName
sub_df <- df_long %>%
filter(repName == rep_name) %>%
complete(
cluster = c(cluster1, cluster2),
status = c("TE", "not_TE"),
fill = list(count = 0)
)
# fixed order of statuses
status_levels <- c("TE", "not_TE")
# make sure both statuses exist
statuses <- unique(sub_df$status)
if(length(statuses) != 2) {
sub_df <- sub_df %>%
complete(cluster, status, fill = list(count = 0))
statuses <- unique(sub_df$status)
}
# counts for cluster1
c1_counts <- sub_df %>%
filter(cluster == cluster1) %>%
arrange(factor(status, levels = status_levels)) %>%
pull(count)
# counts for cluster2
c2_counts <- sub_df %>%
filter(cluster == cluster2) %>%
arrange(factor(status, levels = status_levels)) %>%
pull(count)
# 2x2 matrix for Fisher test
mat <- matrix(
c(c2_counts, c1_counts),
nrow = 2,
byrow = TRUE,
dimnames = list(
cluster = c(cluster2, cluster1),
category = status_levels
)
)
ft <- tryCatch(
fisher.test(mat, workspace = 2e8),
error = function(e) fisher.test(mat, simulate.p.value = TRUE, B = 1e5)
)
tibble(
repName = rep_name,
comparison = paste(cluster2, "vs", cluster1),
odds_ratio = ft$estimate,
lower_CI = ft$conf.int[1],
upper_CI = ft$conf.int[2],
p_value = ft$p.value
)
}SINE
Overlapping SINE family with summits to get a count
sine_hits <- findOverlaps(H3K27ac_summit_gr, SINE_gr, ignore.strand = TRUE)
SINE_overlap_df <- tibble(
summit_id = queryHits(sine_hits),
cluster = mcols(H3K27ac_summit_gr)$cluster[queryHits(sine_hits)],
TE_type = mcols(SINE_gr)$repFamily[subjectHits(sine_hits)])
SINE_counts <- SINE_overlap_df %>%
count(cluster, TE_type, name = "count") %>%
mutate(status = "TE")
total_SINE_summits <- tibble(
cluster = mcols(H3K27ac_summit_gr)$cluster
) %>% count(cluster, name = "total")
not_SINE_counts <- SINE_counts %>%
left_join(total_SINE_summits, by = "cluster") %>%
mutate(count = total - count,
status = "not_TE") %>%
dplyr::select(cluster, TE_type, status, count)
SINE_df_long <- bind_rows(SINE_counts %>%
dplyr::select(cluster, TE_type, status, count),
not_SINE_counts) %>%
filter(!is.na(cluster))
SINE_results <- comparisons %>%
mutate(results = map2(cluster2, cluster1, function(c2, c1) {
SINE_df_long %>%
distinct(TE_type) %>%
pull() %>%
map_dfr(function(te) {
test_pair_TE_generic(
SINE_df_long,
te_name = te,
cluster1 = c1,
cluster2 = c2
)
})
})
) %>%
unnest(results) %>%
mutate(FDR = p.adjust(p_value, method = "BH"))datatable(SINE_counts,
rownames = FALSE,
filter = 'top', # add filter/search boxes
options = list(
pageLength = 10,
autoWidth = TRUE,
scrollX = TRUE))total_SINE_summits# A tibble: 4 × 2
cluster total
<chr> <int>
1 Set_1 110084
2 Set_2 1385
3 Set_3 6623
4 <NA> 31979# ---- Prepare the table ----
SINE_counts_display <- SINE_results %>%
# 1. Split comparison
separate(comparison, into = c("cluster2", "cluster1"), sep = " vs ", remove = FALSE) %>%
# 2. Add log2 odds ratio
mutate(log2OR = log2(odds_ratio)) %>%
# 3. Add enrichment/depletion direction
mutate(direction = case_when(
odds_ratio > 1 ~ "enriched",
odds_ratio < 1 ~ "depleted",
TRUE ~ "neutral"
)) %>%
# 4. Flag significant
mutate(significant = FDR < 0.05) %>%
# Optional: arrange for readability
arrange(cluster2, direction, desc(log2OR))
# ---- Create interactive datatable ----
datatable(
SINE_counts_display,
rownames = FALSE,
filter = 'top', # add filter/search boxes
options = list(
pageLength = 10,
autoWidth = TRUE,
scrollX = TRUE
)
) %>%
# Conditional coloring by direction
formatStyle(
'direction',
target = 'row',
backgroundColor = styleEqual(
c('enriched', 'depleted'),
c('#FFDD99', '#99CCFF') # enriched = light orange, depleted = light blue
)
) %>%
# Bold significant rows
formatStyle(
'significant',
fontWeight = styleEqual(TRUE, 'bold')
) %>%
# Round numeric columns for readability
formatRound(columns = c('odds_ratio','log2OR','FDR'), digits = 2)ggplot(SINE_results, aes(x = (odds_ratio), y = -log10(FDR),label = TE_type)) +
geom_point(aes(color = odds_ratio > 1), size = 3) +
# geom_text_repel(data = subset(SVA_results, FDR < 0.05)) +
geom_text_repel(size=3, max.overlaps = Inf)+
geom_hline(yintercept = -log10(0.05), linetype = "dashed") +
labs(
x = "(Odds Ratio)",
y = "-log10(FDR)",
title = "SINE family enrichment at ROI summits\nH3K27ac"
) +
theme_classic() +
facet_wrap(~comparison)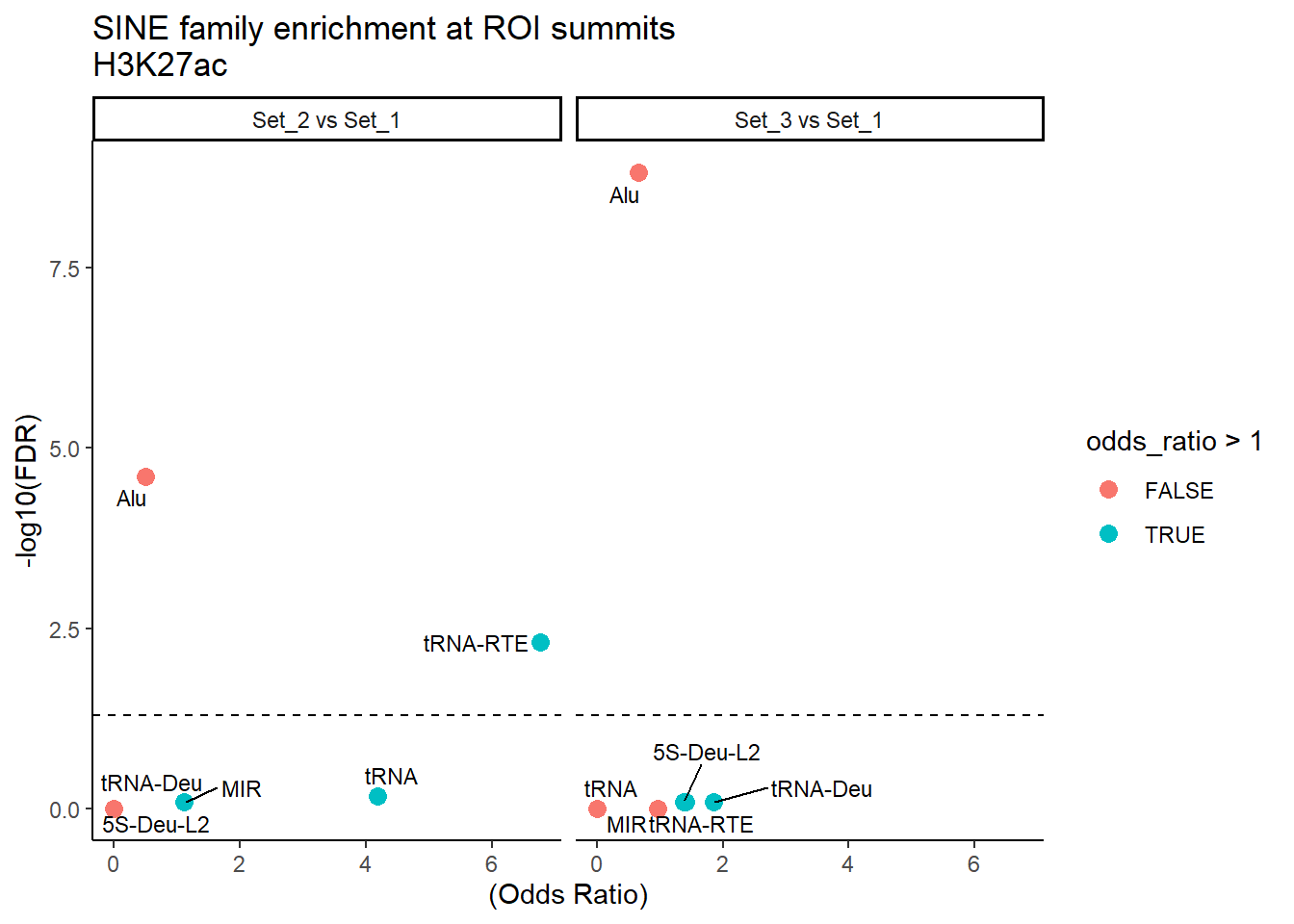
Overlapping SINE family with ROIs to get a count
count_sine_families <- function(peak_gr, sine_gr, cluster_name) {
hits <- findOverlaps(peak_gr, sine_gr)
if (length(hits) == 0) {
return(tibble(
cluster = cluster_name,
TE_type = character(),
status = character(),
count = integer()
))
}
hit_df <- tibble(
family = ifelse(
mcols(sine_gr)$repFamily[subjectHits(hits)] == "SVA",
mcols(sine_gr)$repName[subjectHits(hits)],
mcols(sine_gr)$repFamily[subjectHits(hits)]
)
)
## TE counts per family (Alu/MIR vs SVA_A/B/C…)
te_counts <- hit_df %>%
count(family, name = "count") %>%
mutate(
cluster = cluster_name,
TE_type = family,
status = "TE"
)
## non-TE peaks (no SINE overlap)
n_total_peaks <- length(peak_gr)
n_sine_peaks <- length(unique(queryHits(hits)))
not_te <- tibble(
cluster = cluster_name,
TE_type = unique(te_counts$TE_type),
status = "not_TE",
count = n_total_peaks - n_sine_peaks
)
bind_rows(te_counts, not_te)
}count_ERV1_repNames <- function(peak_gr, TE_gr, cluster_name) {
# find overlaps
hits <- findOverlaps(peak_gr, TE_gr)
if (length(hits) == 0) {
return(tibble(
cluster = cluster_name,
repName = character(),
status = character(),
count = integer()
))
}
# subset hits to only ERV1
is_ERV1 <- mcols(TE_gr)$repFamily[subjectHits(hits)] == "ERV1"
hits <- hits[is_ERV1]
if (length(hits) == 0) {
return(tibble(
cluster = cluster_name,
repName = character(),
status = character(),
count = integer()
))
}
# make tibble of repNames
hit_df <- tibble(
repName = mcols(TE_gr)$repName[subjectHits(hits)]
)
# TE counts per repName
te_counts <- hit_df %>%
count(repName, name = "count") %>%
mutate(
cluster = cluster_name,
status = "TE"
)
# non-TE peaks
n_total_peaks <- length(peak_gr)
n_TE_peaks <- length(unique(queryHits(hits)))
not_te <- tibble(
cluster = cluster_name,
repName = unique(te_counts$repName),
status = "not_TE",
count = n_total_peaks - n_TE_peaks
)
bind_rows(te_counts, not_te)
}fullROI_long_sine <- purrr::imap_dfr(
H3K27ac_sets_gr,
~count_sine_families(.x, SINE_gr, .y)
)
SINE_results_full <- comparisons %>%
mutate(results = map2(cluster2, cluster1, function(c2, c1) {
fullROI_long_sine %>%
distinct(family) %>%
pull() %>%
map_dfr(function(te) {
test_pair_TE_generic(
fullROI_long_sine,
te_name = te,
cluster1 = c1,
cluster2 = c2
)
})
})
) %>%
unnest(results) %>%
mutate(FDR = p.adjust(p_value, method = "BH"))
ggplot(SINE_results_full, aes(x = (odds_ratio), y = -log10(FDR),label = TE_type)) +
geom_point(aes(color = odds_ratio > 1), size = 3) +
# geom_text_repel(data = subset(SVA_results, FDR < 0.05)) +
geom_text_repel(size=3, max.overlaps = Inf)+
geom_hline(yintercept = -log10(0.05), linetype = "dashed") +
geom_vline(xintercept = 1, linetype = 3) +
labs(
x = "(Odds Ratio)",
y = "-log10(FDR)",
title = "SINE family enrichment at full ROI\nH3K27ac"
) +
theme_classic() +
facet_wrap(~comparison)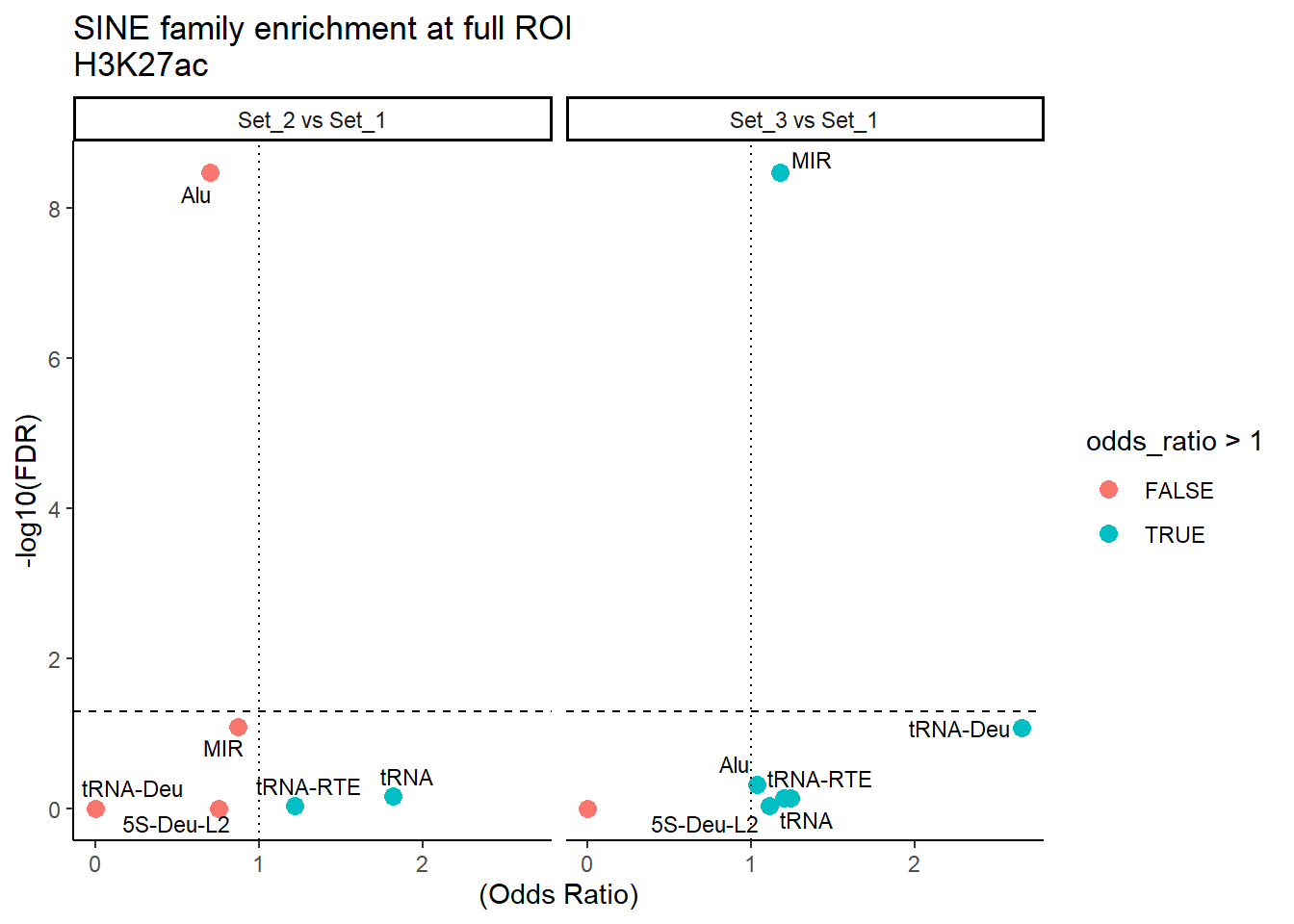
| Version | Author | Date |
|---|---|---|
| 0606abf | reneeisnowhere | 2026-01-27 |
LINE
Overlapping LINE family with summits to get a count
LINE_hits <- findOverlaps(H3K27ac_summit_gr, LINE_gr, ignore.strand = TRUE)
LINE_overlap_df <- tibble(
summit_id = queryHits(LINE_hits),
cluster = mcols(H3K27ac_summit_gr)$cluster[queryHits(LINE_hits)],
TE_type = mcols(LINE_gr)$repFamily[subjectHits(LINE_hits)])
LINE_counts <- LINE_overlap_df %>%
count(cluster, TE_type, name = "count") %>%
mutate(status = "TE")
total_LINE_summits <- tibble(
cluster = mcols(H3K27ac_summit_gr)$cluster
) %>% count(cluster, name = "total")
not_LINE_counts <- LINE_counts %>%
left_join(total_LINE_summits, by = "cluster") %>%
mutate(count = total - count,
status = "not_TE") %>%
select(cluster, TE_type, status, count)
LINE_df_long <- bind_rows(LINE_counts %>%
dplyr::select(cluster, TE_type, status, count),
not_LINE_counts) %>%
filter(!is.na(cluster))
LINE_results <- comparisons %>%
mutate(results = map2(cluster2, cluster1, function(c2, c1) {
LINE_df_long %>%
distinct(TE_type) %>%
pull() %>%
map_dfr(function(te) {
test_pair_TE_generic(
LINE_df_long,
te_name = te,
cluster1 = c1,
cluster2 = c2
)
})
})
) %>%
unnest(results) %>%
mutate(FDR = p.adjust(p_value, method = "BH"))datatable(LINE_counts,
rownames = FALSE,
filter = 'top', # add filter/search boxes
options = list(
pageLength = 10,
autoWidth = TRUE,
scrollX = TRUE))total_LINE_summits# A tibble: 4 × 2
cluster total
<chr> <int>
1 Set_1 110084
2 Set_2 1385
3 Set_3 6623
4 <NA> 31979# ---- Prepare the table ----
LINE_counts_display <- LINE_results %>%
# 1. Split comparison
separate(comparison, into = c("cluster2", "cluster1"), sep = " vs ", remove = FALSE) %>%
# 2. Add log2 odds ratio
mutate(log2OR = log2(odds_ratio)) %>%
# 3. Add enrichment/depletion direction
mutate(direction = case_when(
odds_ratio > 1 ~ "enriched",
odds_ratio < 1 ~ "depleted",
TRUE ~ "neutral"
)) %>%
# 4. Flag significant
mutate(significant = FDR < 0.05) %>%
# Optional: arrange for readability
arrange(cluster2, direction, desc(log2OR))
# ---- Create interactive datatable ----
datatable(
LINE_counts_display,
rownames = FALSE,
filter = 'top', # add filter/search boxes
options = list(
pageLength = 10,
autoWidth = TRUE,
scrollX = TRUE
)
) %>%
# Conditional coloring by direction
formatStyle(
'direction',
target = 'row',
backgroundColor = styleEqual(
c('enriched', 'depleted'),
c('#FFDD99', '#99CCFF') # enriched = light orange, depleted = light blue
)
) %>%
# Bold significant rows
formatStyle(
'significant',
fontWeight = styleEqual(TRUE, 'bold')
) %>%
# Round numeric columns for readability
formatRound(columns = c('odds_ratio','log2OR','FDR'), digits = 2)ggplot(LINE_results, aes(x = (odds_ratio), y = -log10(FDR),label = TE_type)) +
geom_point(aes(color = odds_ratio > 1), size = 3) +
# geom_text_repel(data = subset(SVA_results, FDR < 0.05)) +
geom_text_repel(size=3, max.overlaps = Inf)+
geom_hline(yintercept = -log10(0.05), linetype = "dashed") +
labs(
x = "(Odds Ratio)",
y = "-log10(FDR)",
title = "LINE family enrichment at ROI summits\nH3K27ac"
) +
theme_classic()+
facet_wrap(~comparison)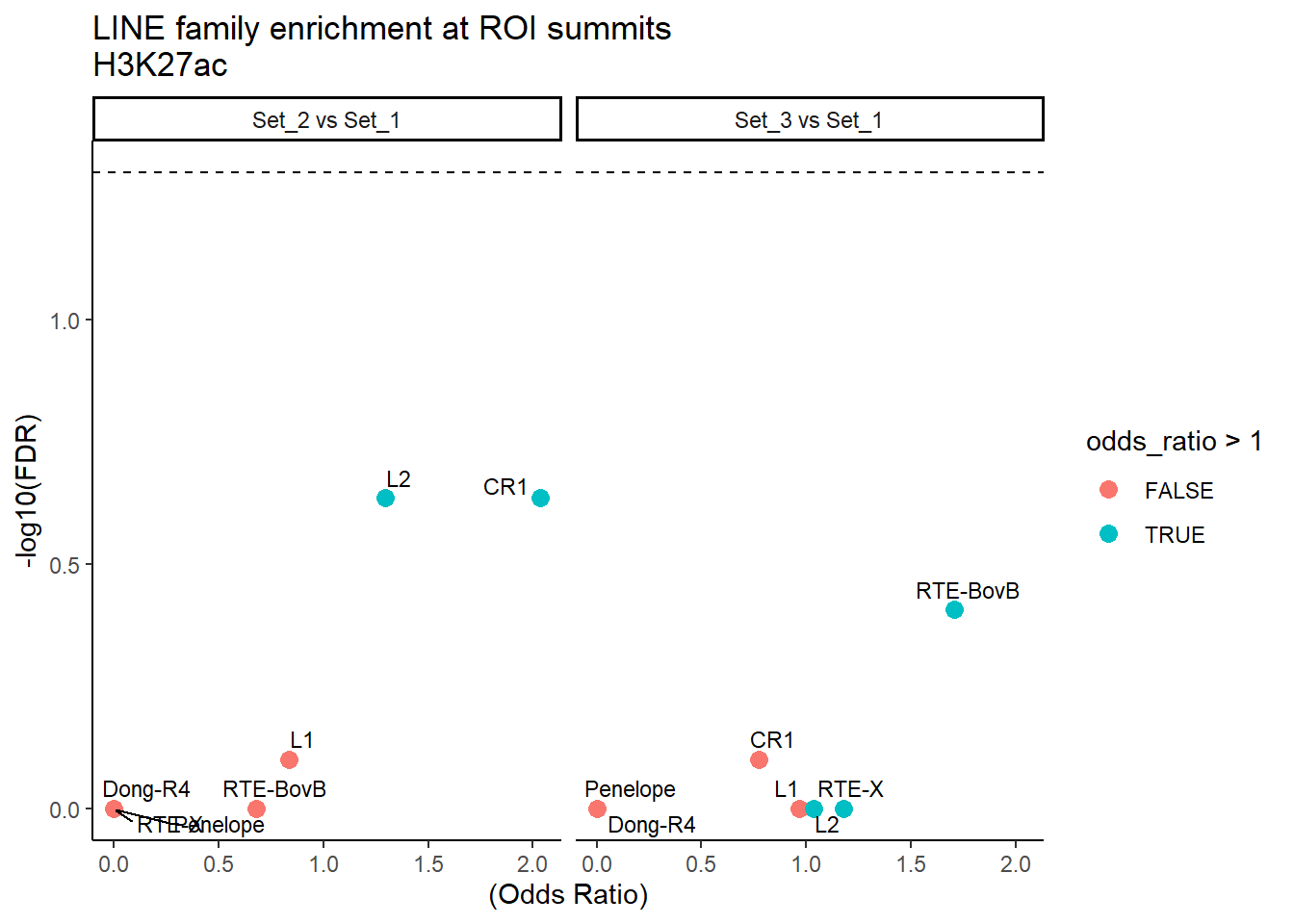
Overlapping LINE families using Full ROI
fullROI_long_LINE <- purrr::imap_dfr(
H3K27ac_sets_gr,
~count_sine_families(.x, LINE_gr, .y)
)
LINE_results_full <- comparisons %>%
mutate(results = map2(cluster2, cluster1, function(c2, c1) {
fullROI_long_LINE %>%
distinct(family) %>%
pull() %>%
map_dfr(function(te) {
test_pair_TE_generic(
fullROI_long_LINE,
te_name = te,
cluster1 = c1,
cluster2 = c2
)
})
})
) %>%
unnest(results) %>%
mutate(FDR = p.adjust(p_value, method = "BH"))
ggplot(LINE_results_full, aes(x = (odds_ratio), y = -log10(FDR),label = TE_type)) +
geom_point(aes(color = odds_ratio > 1), size = 3) +
# geom_text_repel(data = subset(SVA_results, FDR < 0.05)) +
geom_text_repel(size=3, max.overlaps = Inf)+
geom_hline(yintercept = -log10(0.05), linetype = "dashed") +
geom_vline(xintercept = 1, linetype = 3) +
labs(
x = "(Odds Ratio)",
y = "-log10(FDR)",
title = "LINE family enrichment at full ROI\nH3K27ac"
) +
theme_classic() +
facet_wrap(~comparison)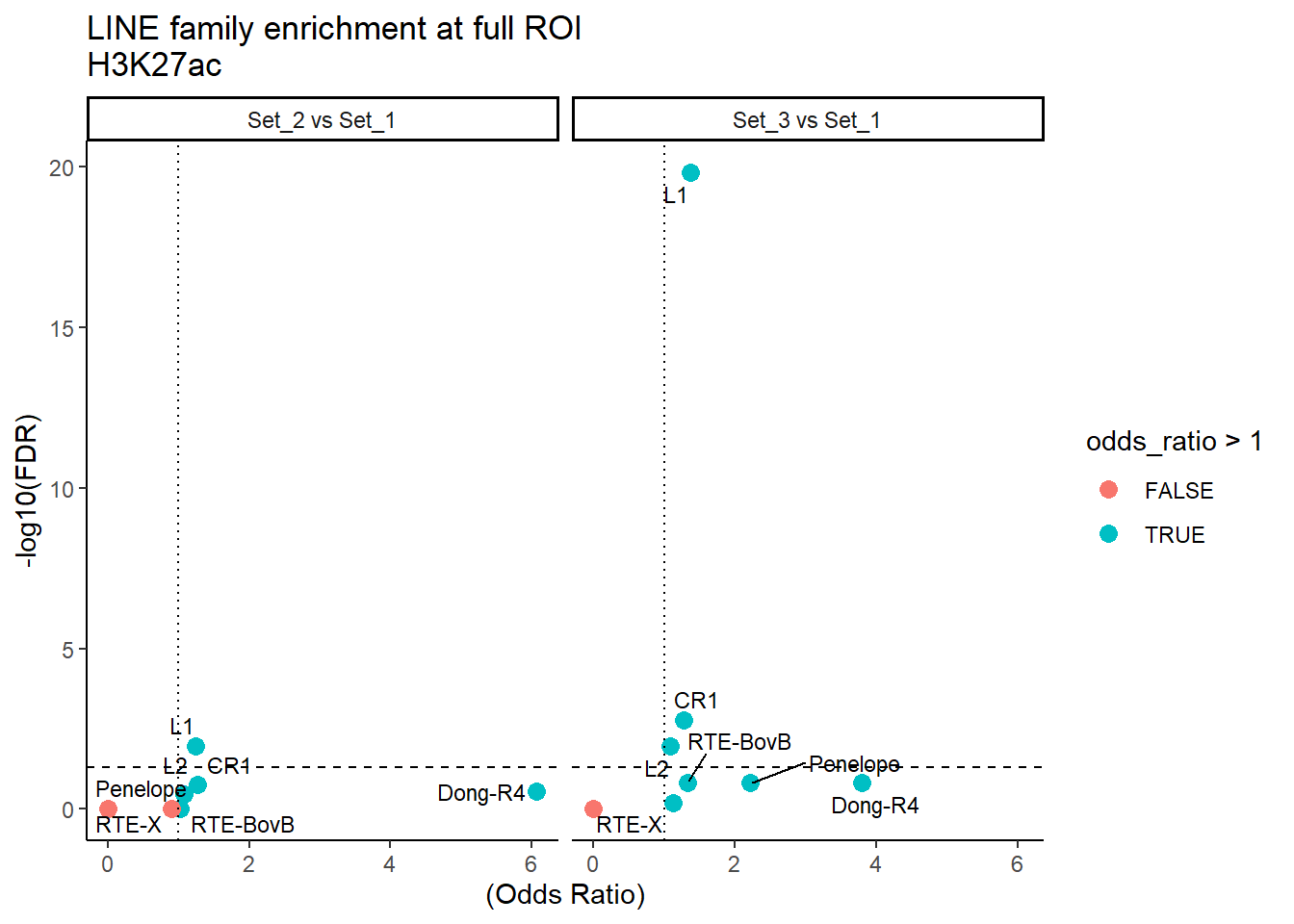
| Version | Author | Date |
|---|---|---|
| 0606abf | reneeisnowhere | 2026-01-27 |
LTR
Overlapping LTR family summits to get a count
LTR_hits <- findOverlaps(H3K27ac_summit_gr, LTR_gr, ignore.strand = TRUE)
LTR_overlap_df <- tibble(
summit_id = queryHits(LTR_hits),
cluster = mcols(H3K27ac_summit_gr)$cluster[queryHits(LTR_hits)],
TE_type = mcols(LTR_gr)$repFamily[subjectHits(LTR_hits)])
LTR_counts <- LTR_overlap_df %>%
count(cluster, TE_type, name = "count") %>%
mutate(status = "TE")
total_LTR_summits <- tibble(
cluster = mcols(H3K27ac_summit_gr)$cluster
) %>% count(cluster, name = "total")
not_LTR_counts <- LTR_counts %>%
left_join(total_LTR_summits, by = "cluster") %>%
mutate(count = total - count,
status = "not_TE") %>%
select(cluster, TE_type, status, count)
LTR_df_long <- bind_rows(LTR_counts %>%
dplyr::select(cluster, TE_type, status, count),
not_LTR_counts) %>%
filter(!is.na(cluster))
LTR_results <- comparisons %>%
mutate(results = map2(cluster2, cluster1, function(c2, c1) {
LTR_df_long %>%
distinct(TE_type) %>%
pull() %>%
map_dfr(function(te) {
test_pair_TE_generic(
LTR_df_long,
te_name = te,
cluster1 = c1,
cluster2 = c2
)
})
})
) %>%
unnest(results) %>%
mutate(FDR = p.adjust(p_value, method = "BH"))datatable(LTR_counts,
rownames = FALSE,
filter = 'top', # add filter/search boxes
options = list(
pageLength = 10,
autoWidth = TRUE,
scrollX = TRUE))total_LTR_summits# A tibble: 4 × 2
cluster total
<chr> <int>
1 Set_1 110084
2 Set_2 1385
3 Set_3 6623
4 <NA> 31979# ---- Prepare the table ----
LTR_counts_display <- LTR_results %>%
# 1. Split comparison
separate(comparison, into = c("cluster2", "cluster1"), sep = " vs ", remove = FALSE) %>%
# 2. Add log2 odds ratio
mutate(log2OR = log2(odds_ratio)) %>%
# 3. Add enrichment/depletion direction
mutate(direction = case_when(
odds_ratio > 1 ~ "enriched",
odds_ratio < 1 ~ "depleted",
TRUE ~ "neutral"
)) %>%
# 4. Flag significant
mutate(significant = FDR < 0.05) %>%
# Optional: arrange for readability
arrange(cluster2, direction, desc(log2OR))
# ---- Create interactive datatable ----
datatable(
LTR_counts_display,
rownames = FALSE,
filter = 'top', # add filter/search boxes
options = list(
pageLength = 10,
autoWidth = TRUE,
scrollX = TRUE
)
) %>%
# Conditional coloring by direction
formatStyle(
'direction',
target = 'row',
backgroundColor = styleEqual(
c('enriched', 'depleted'),
c('#FFDD99', '#99CCFF') # enriched = light orange, depleted = light blue
)
) %>%
# Bold significant rows
formatStyle(
'significant',
fontWeight = styleEqual(TRUE, 'bold')
) %>%
# Round numeric columns for readability
formatRound(columns = c('odds_ratio','log2OR','FDR'), digits = 2)ggplot(LTR_results, aes(x = (odds_ratio), y = -log10(FDR),label = TE_type)) +
geom_point(aes(color = odds_ratio > 1), size = 3) +
# geom_text_repel(data = subset(SVA_results, FDR < 0.05)) +
geom_text_repel(size=3, max.overlaps = Inf)+
geom_hline(yintercept = -log10(0.05), linetype = "dashed") +
labs(
x = "(Odds Ratio)",
y = "-log10(FDR)",
title = "LTR family enrichment at ROI summits\nH3K27ac"
) +
theme_classic()+
facet_wrap(~comparison)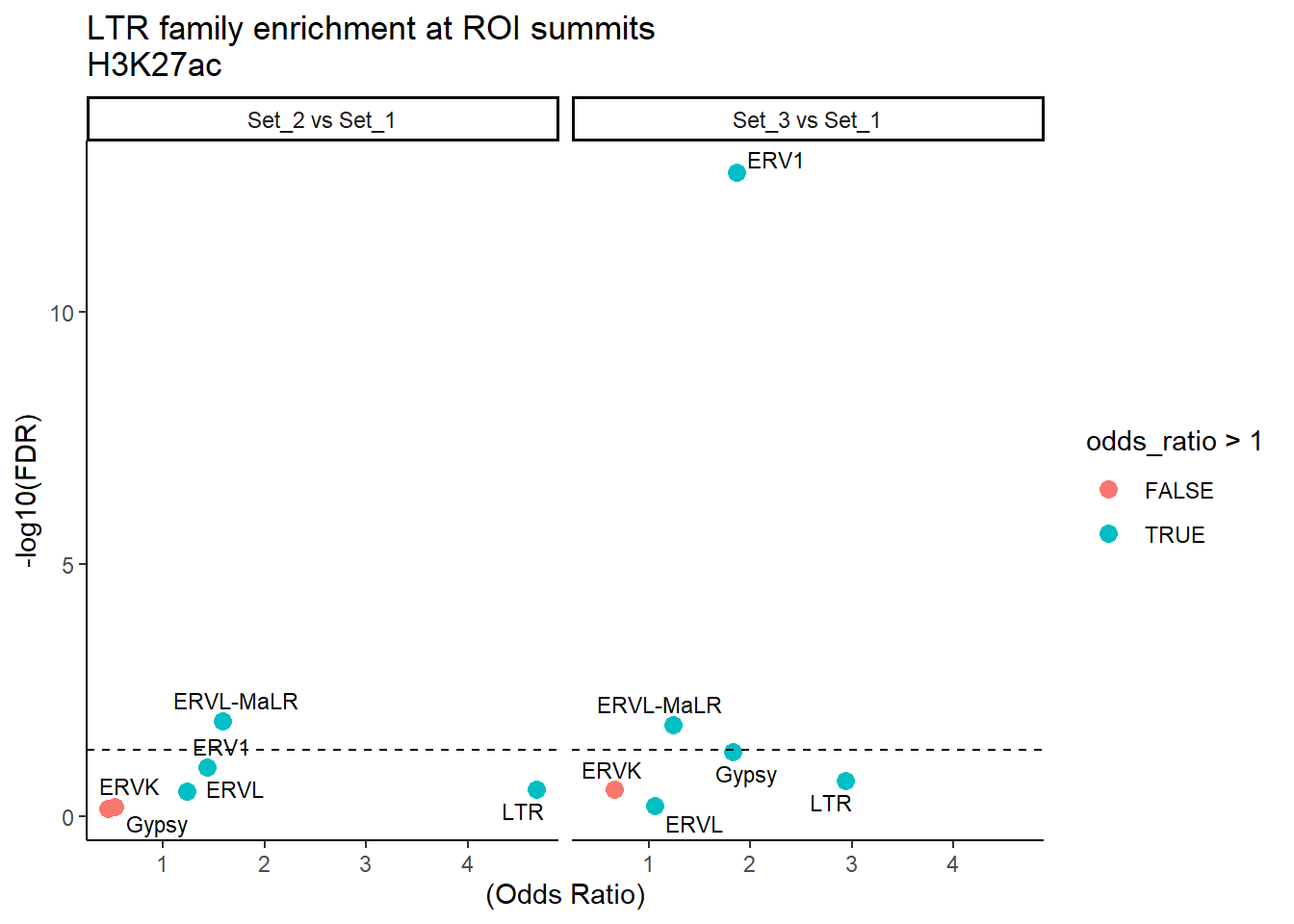
LTR_hits <- findOverlaps(H3K27ac_summit_gr, LTR_gr, ignore.strand = TRUE)
LTR_name_overlap_df <- tibble(
summit_id = queryHits(LTR_hits),
cluster = mcols(H3K27ac_summit_gr)$cluster[queryHits(LTR_hits)],
TE_type = mcols(LTR_gr)$repFamily[subjectHits(LTR_hits)],
repName= mcols(LTR_gr)$repName[subjectHits(LTR_hits)])
LTR_ERV1_counts <-
LTR_name_overlap_df %>%
dplyr::filter(TE_type=="ERV1") %>%
count(cluster, repName, name = "count") %>%
mutate(status = "TE")
total_LTR_summits <- tibble(
cluster = mcols(H3K27ac_summit_gr)$cluster
) %>% count(cluster, name = "total")
not_LTR_ERV1_counts <- LTR_ERV1_counts %>%
left_join(total_LTR_summits, by = "cluster") %>%
mutate(count = total - count,
status = "not_TE") %>%
select(cluster, repName, status, count)
LTR_ERV1_df_long <- bind_rows(LTR_ERV1_counts %>%
dplyr::select(cluster, repName, status, count),
not_LTR_ERV1_counts) %>%
filter(!is.na(cluster))
ERV1_LTR_results <- comparisons %>%
mutate(results = map2(cluster2, cluster1, function(c2, c1) {
LTR_ERV1_df_long %>%
distinct(repName) %>%
pull() %>%
map_dfr(function(rep) {
test_pair_TE_repName(
LTR_ERV1_df_long,
rep_name = rep,
cluster1 = c1,
cluster2 = c2
)
})
})
) %>%
unnest(results) %>%
mutate(FDR = p.adjust(p_value, method = "BH"))ggplot(ERV1_LTR_results, aes(x = (odds_ratio), y = -log10(FDR),label = repName)) +
geom_point(aes(color = odds_ratio > 1), size = 3) +
# geom_text_repel(data = subset(SVA_results, FDR < 0.05)) +
geom_text_repel(data = subset(ERV1_LTR_results, FDR < 0.05), # only significant points
size = 3,
max.overlaps = 100
) +
geom_hline(yintercept = -log10(0.05), linetype = "dashed") +
labs(
x = "(Odds Ratio)",
y = "-log10(FDR)",
title = "LTR Name (type) enrichment at ROI summits\nH3K27ac"
) +
theme_classic()+
facet_wrap(~comparison)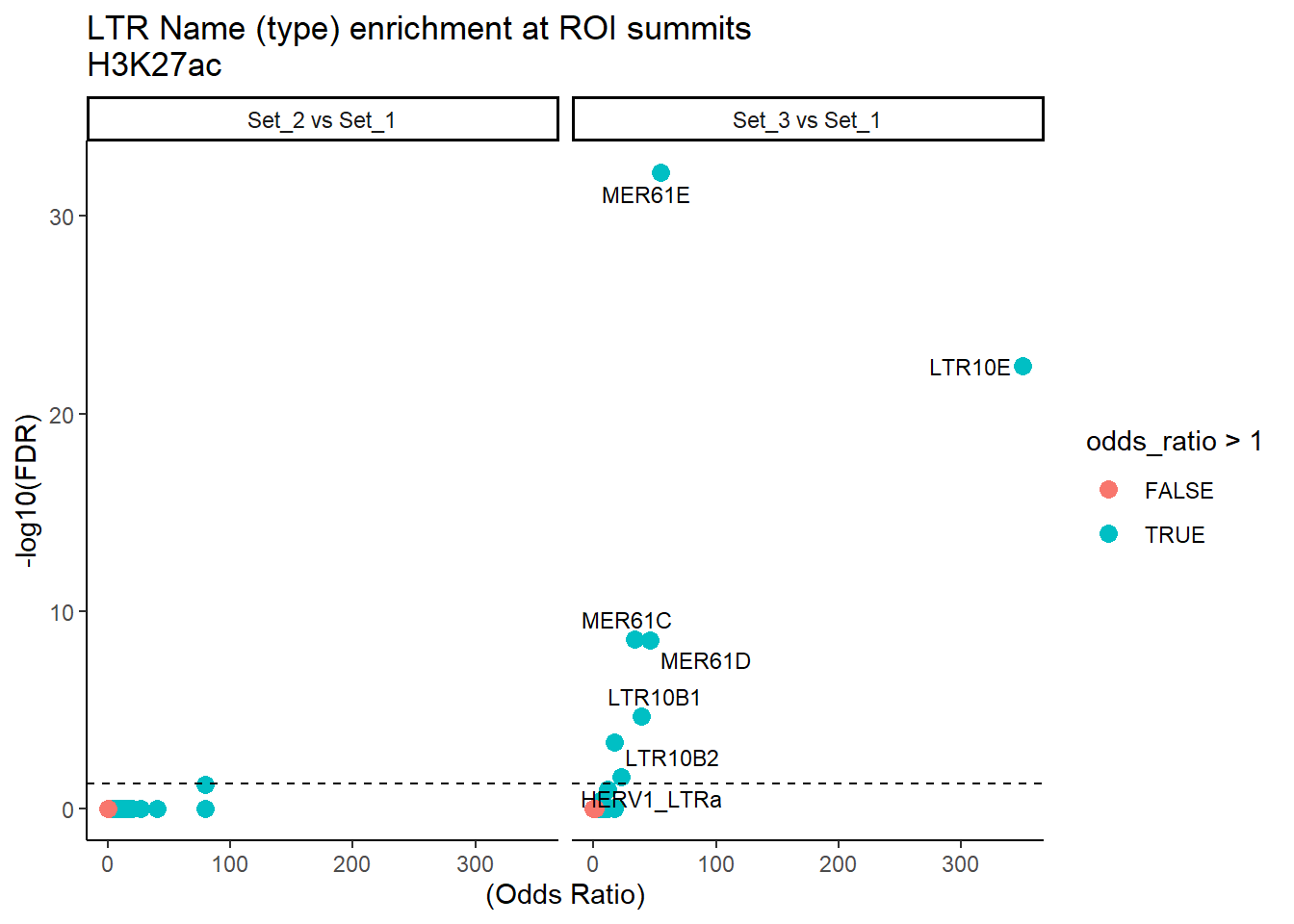
Overlapping LTR families using Full ROI
fullROI_long_LTR <- purrr::imap_dfr(
H3K27ac_sets_gr,
~count_sine_families(.x, LTR_gr, .y)
)
LTR_results_full <- comparisons %>%
mutate(results = map2(cluster2, cluster1, function(c2, c1) {
fullROI_long_LTR %>%
distinct(family) %>%
pull() %>%
map_dfr(function(te) {
test_pair_TE_generic(
fullROI_long_LTR,
te_name = te,
cluster1 = c1,
cluster2 = c2
)
})
})
) %>%
unnest(results) %>%
mutate(FDR = p.adjust(p_value, method = "BH"))
ggplot(LTR_results_full, aes(x = (odds_ratio), y = -log10(FDR),label = TE_type)) +
geom_point(aes(color = odds_ratio > 1), size = 3) +
# geom_text_repel(data = subset(SVA_results, FDR < 0.05)) +
geom_text_repel(size=3, max.overlaps = Inf)+
geom_hline(yintercept = -log10(0.05), linetype = "dashed") +
geom_vline(xintercept = 1, linetype = 3) +
labs(
x = "(Odds Ratio)",
y = "-log10(FDR)",
title = "LTR family enrichment at full ROI\nH3K27ac"
) +
theme_classic() +
facet_wrap(~comparison)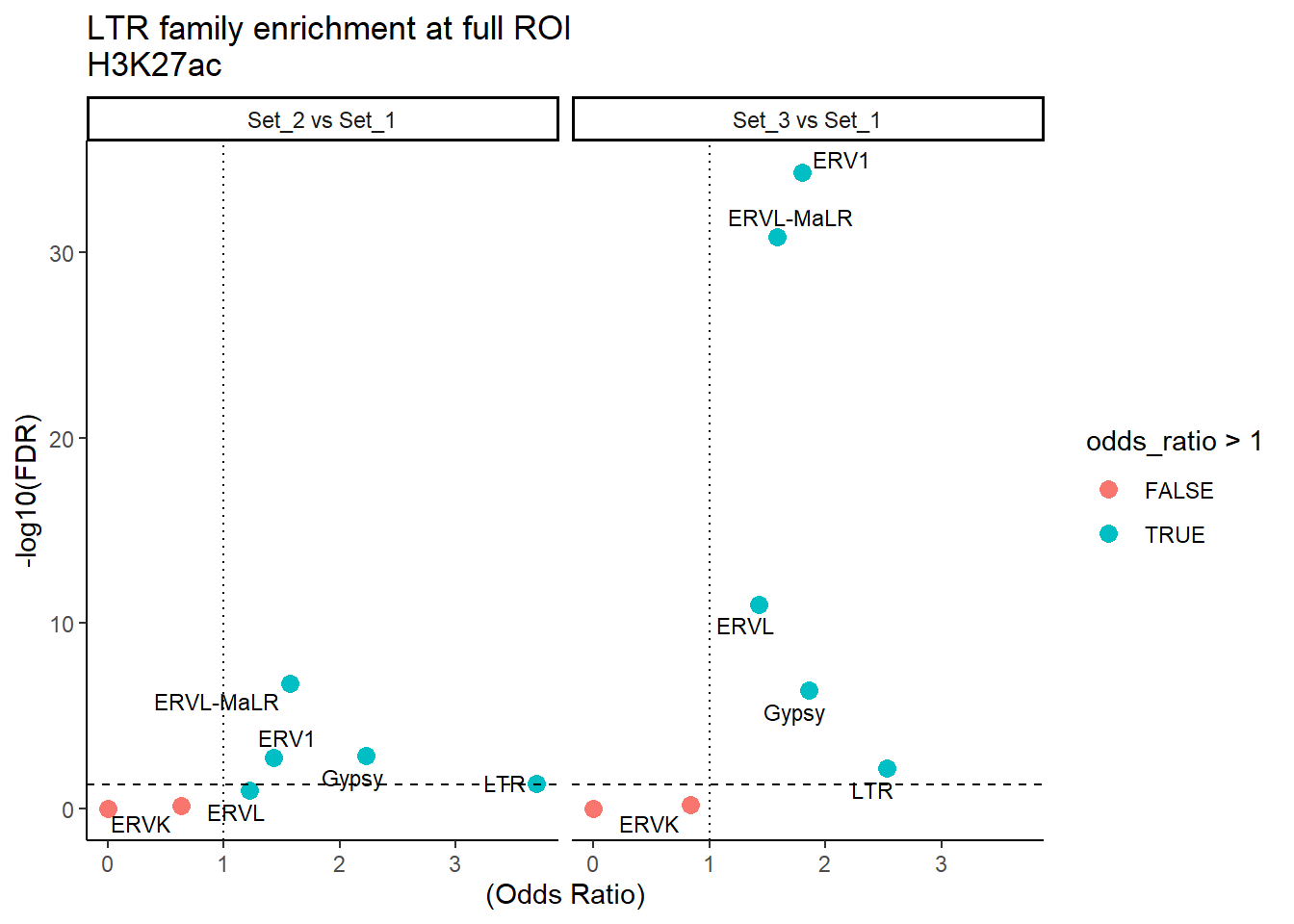
| Version | Author | Date |
|---|---|---|
| 0606abf | reneeisnowhere | 2026-01-27 |
fullROI_long_LTR_ERV1 <- purrr::imap_dfr(
H3K27ac_sets_gr,
~count_ERV1_repNames(.x, LTR_gr, .y)
)
LTRERV1_results_full <- comparisons %>%
mutate(results = map2(cluster2, cluster1, function(c2, c1) {
fullROI_long_LTR_ERV1 %>%
distinct(repName) %>%
pull() %>%
map_dfr(function(rep) {
test_pair_TE_repName(
LTR_ERV1_df_long,
rep_name = rep,
cluster1 = c1,
cluster2 = c2
)
})
})
) %>%
unnest(results) %>%
mutate(FDR = p.adjust(p_value, method = "BH"))
ggplot(LTRERV1_results_full, aes(x = (odds_ratio), y = -log10(FDR),label = repName)) +
geom_point(aes(color = odds_ratio > 1), size = 3) +
geom_text_repel(data = subset(LTRERV1_results_full, FDR < 0.05), # only significant points
size = 3,
max.overlaps = Inf
) +
geom_hline(yintercept = -log10(0.05), linetype = "dashed") +
geom_vline(xintercept = 1, linetype = 3) +
labs(
x = "(Odds Ratio)",
y = "-log10(FDR)",
title = "LTR:ERV1 type enrichment at full ROI\nH3K27ac"
) +
theme_classic() +
facet_wrap(~comparison)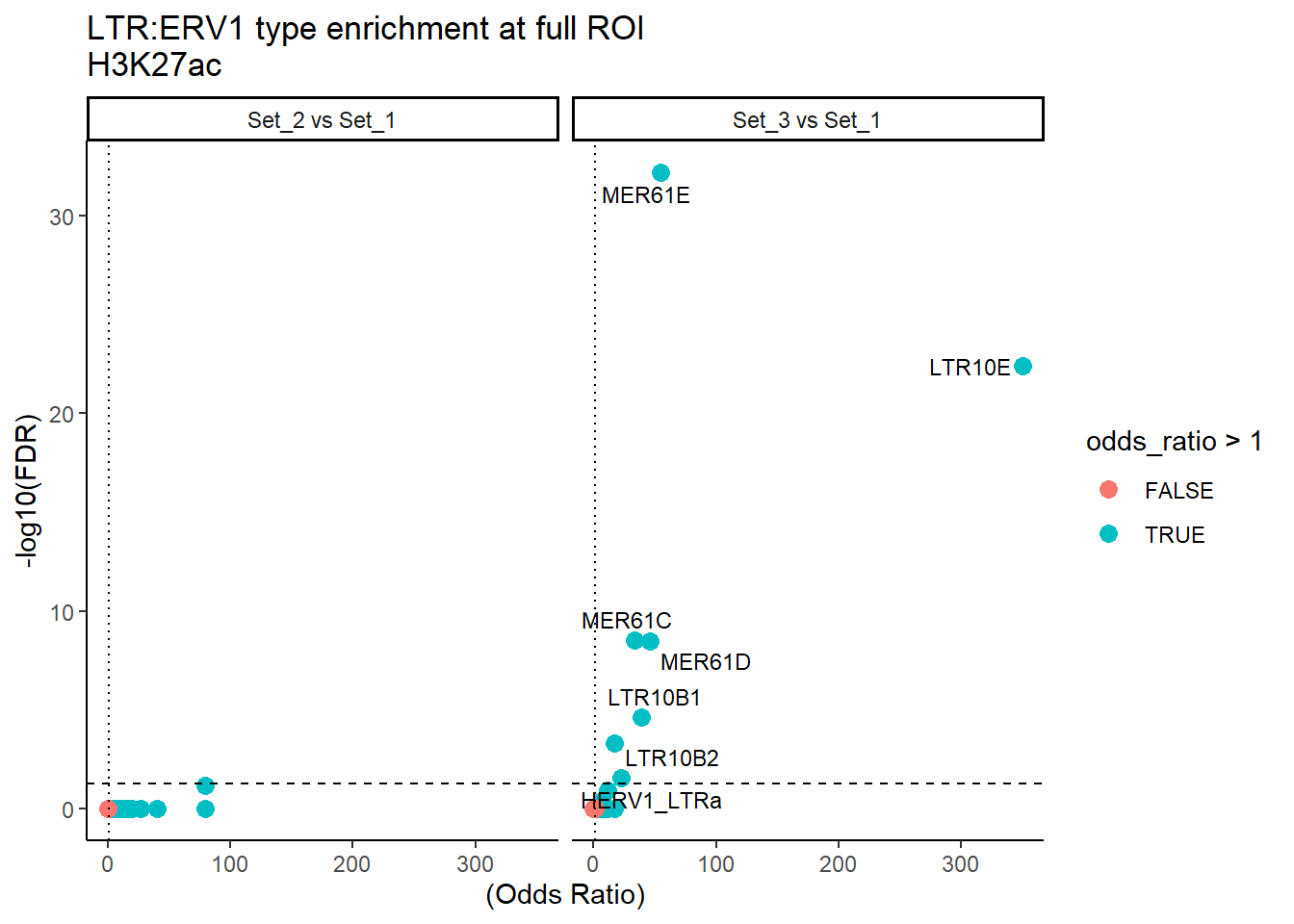 ### SVA
### SVA
Overlapping SVA family with summits to get a count
SVA_hits <- findOverlaps(H3K27ac_summit_gr, SVA_gr, ignore.strand = TRUE)
SVA_overlap_df <- tibble(
summit_id = queryHits(SVA_hits),
cluster = mcols(H3K27ac_summit_gr)$cluster[queryHits(SVA_hits)],
TE_type = mcols(SVA_gr)$repName[subjectHits(SVA_hits)])
SVA_counts <- SVA_overlap_df %>%
count(cluster, TE_type, name = "count") %>%
mutate(status = "TE")
total_SVA_summits <- tibble(
cluster = mcols(H3K27ac_summit_gr)$cluster
) %>% count(cluster, name = "total")
not_SVA_counts <- SVA_counts %>%
left_join(total_SVA_summits, by = "cluster") %>%
mutate(count = total - count,
status = "not_TE") %>%
select(cluster, TE_type, status, count)
SVA_df_long <- bind_rows(SVA_counts %>%
dplyr::select(cluster, TE_type, status, count),
not_SVA_counts) %>%
filter(!is.na(cluster))
SVA_results <- comparisons %>%
mutate(results = map2(cluster2, cluster1, function(c2, c1) {
SVA_df_long %>%
distinct(TE_type) %>%
pull() %>%
map_dfr(function(te) {
test_pair_TE_generic(
SVA_df_long,
te_name = te,
cluster1 = c1,
cluster2 = c2
)
})
})
) %>%
unnest(results) %>%
mutate(FDR = p.adjust(p_value, method = "BH"))datatable(SVA_counts,
rownames = FALSE,
filter = 'top', # add filter/search boxes
options = list(
pageLength = 10,
autoWidth = TRUE,
scrollX = TRUE))total_SVA_summits# A tibble: 4 × 2
cluster total
<chr> <int>
1 Set_1 110084
2 Set_2 1385
3 Set_3 6623
4 <NA> 31979# ---- Prepare the table ----
SVA_counts_display <- SVA_results %>%
# 1. Split comparison
separate(comparison, into = c("cluster2", "cluster1"), sep = " vs ", remove = FALSE) %>%
# 2. Add log2 odds ratio
mutate(log2OR = log2(odds_ratio)) %>%
# 3. Add enrichment/depletion direction
mutate(direction = case_when(
odds_ratio > 1 ~ "enriched",
odds_ratio < 1 ~ "depleted",
TRUE ~ "neutral"
)) %>%
# 4. Flag significant
mutate(significant = FDR < 0.05) %>%
# Optional: arrange for readability
arrange(cluster2, direction, desc(log2OR))
# ---- Create interactive datatable ----
datatable(
SVA_counts_display,
rownames = FALSE,
filter = 'top', # add filter/search boxes
options = list(
pageLength = 10,
autoWidth = TRUE,
scrollX = TRUE
)
) %>%
# Conditional coloring by direction
formatStyle(
'direction',
target = 'row',
backgroundColor = styleEqual(
c('enriched', 'depleted'),
c('#FFDD99', '#99CCFF') # enriched = light orange, depleted = light blue
)
) %>%
# Bold significant rows
formatStyle(
'significant',
fontWeight = styleEqual(TRUE, 'bold')
) %>%
# Round numeric columns for readability
formatRound(columns = c('odds_ratio','log2OR','FDR'), digits = 2)ggplot(SVA_results, aes(x = (odds_ratio), y = -log10(FDR),label = TE_type)) +
geom_point(aes(color = odds_ratio > 1), size = 3) +
# geom_text_repel(data = subset(SVA_results, FDR < 0.05)) +
geom_text_repel(size=3, max.overlaps = Inf)+
geom_hline(yintercept = -log10(0.05), linetype = "dashed") +
labs(
x = "(Odds Ratio)",
y = "-log10(FDR)",
title = "SVA family enrichment at ROI summits\nH3K27ac"
) +
theme_classic()+
facet_wrap(~comparison)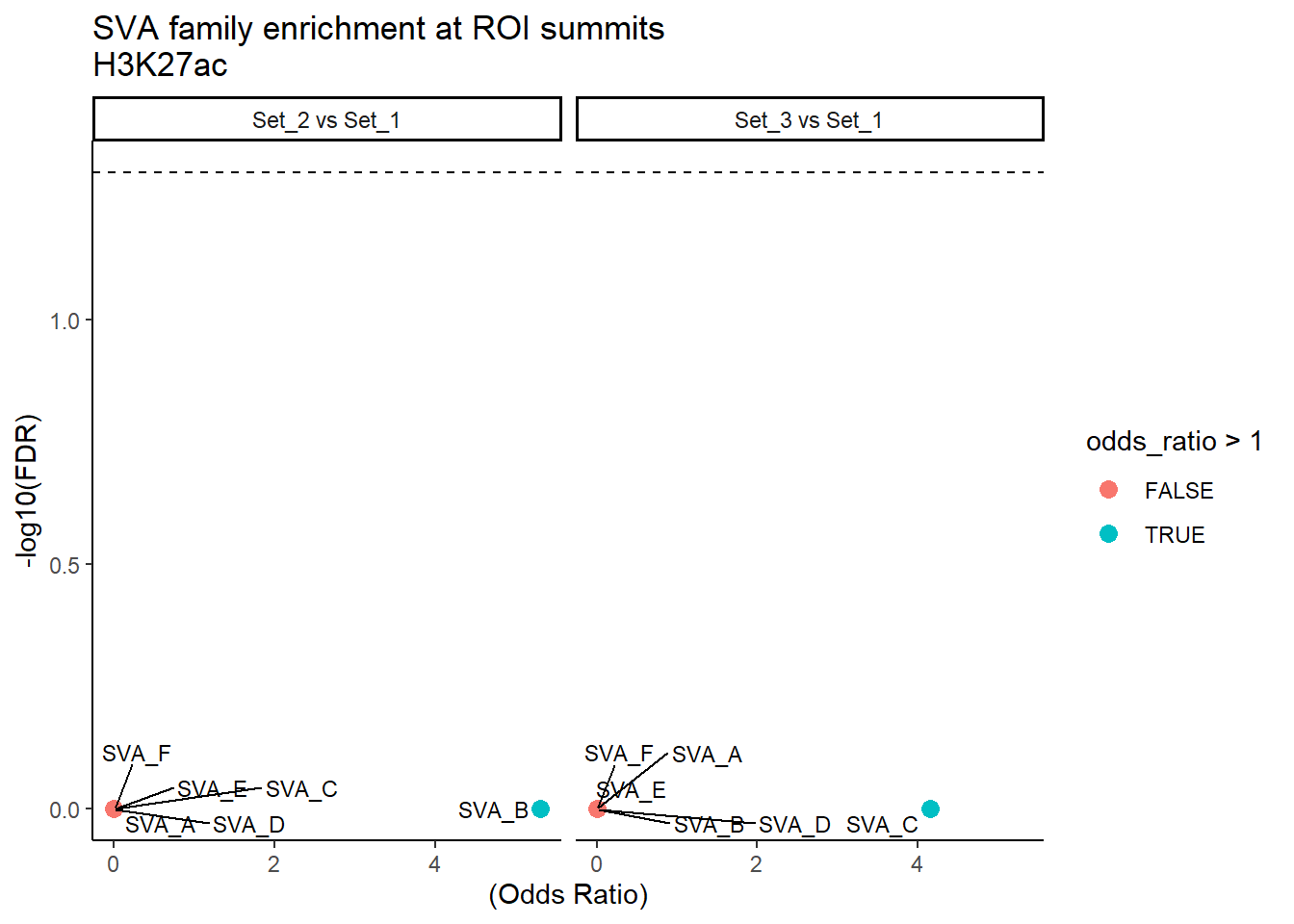
Overlapping SVA families using Full ROI
fullROI_long_SVA <- purrr::imap_dfr(
H3K27ac_sets_gr,
~count_sine_families(.x, SVA_gr, .y)
)
SVA_results_full <- comparisons %>%
mutate(results = map2(cluster2, cluster1, function(c2, c1) {
fullROI_long_SVA %>%
distinct(family) %>%
pull() %>%
map_dfr(function(te) {
test_pair_TE_generic(
fullROI_long_SVA,
te_name = te,
cluster1 = c1,
cluster2 = c2
)
})
})
) %>%
unnest(results) %>%
mutate(FDR = p.adjust(p_value, method = "BH"))
ggplot(SVA_results_full, aes(x = (odds_ratio), y = -log10(FDR),label = TE_type)) +
geom_point(aes(color = odds_ratio > 1), size = 3) +
# geom_text_repel(data = subset(SVA_results, FDR < 0.05)) +
geom_text_repel(size=3, max.overlaps = Inf)+
geom_hline(yintercept = -log10(0.05), linetype = "dashed") +
geom_vline(xintercept = 1, linetype = 3) +
labs(
x = "(Odds Ratio)",
y = "-log10(FDR)",
title = "SVA family enrichment at full ROI\nH3K27ac"
) +
theme_classic() +
facet_wrap(~comparison)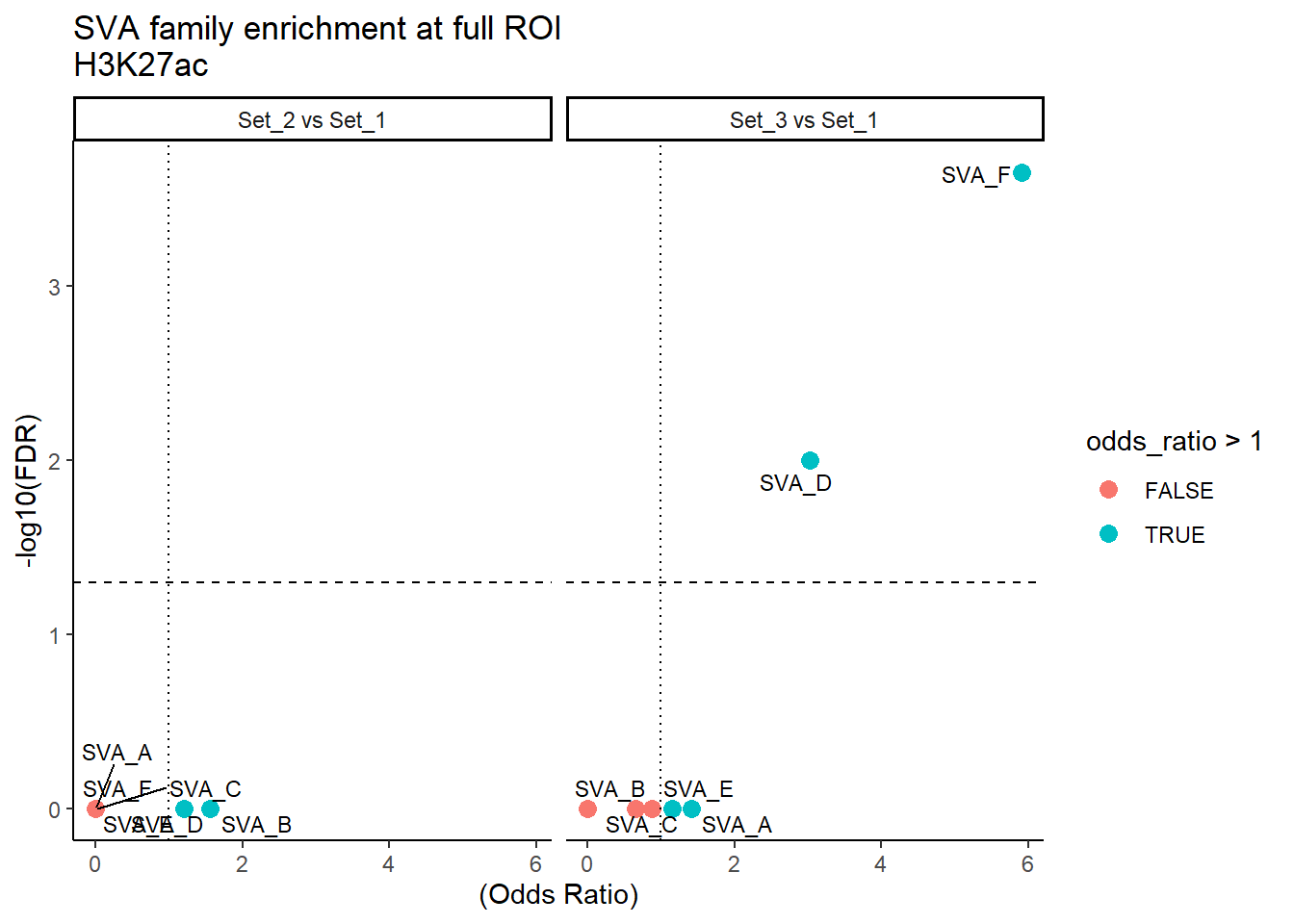
| Version | Author | Date |
|---|---|---|
| 0606abf | reneeisnowhere | 2026-01-27 |
DNA
Overlapping DNA family with summits to get a count
DNA_hits <- findOverlaps(H3K27ac_summit_gr, DNA_gr, ignore.strand = TRUE)
DNA_overlap_df <- tibble(
summit_id = queryHits(DNA_hits),
cluster = mcols(H3K27ac_summit_gr)$cluster[queryHits(DNA_hits)],
TE_type = mcols(DNA_gr)$repFamily[subjectHits(DNA_hits)])
DNA_counts <- DNA_overlap_df %>%
count(cluster, TE_type, name = "count") %>%
mutate(status = "TE")
total_DNA_summits <- tibble(
cluster = mcols(H3K27ac_summit_gr)$cluster
) %>% count(cluster, name = "total")
not_DNA_counts <- DNA_counts %>%
left_join(total_DNA_summits, by = "cluster") %>%
mutate(count = total - count,
status = "not_TE") %>%
select(cluster, TE_type, status, count)
DNA_df_long <- bind_rows(DNA_counts %>%
dplyr::select(cluster, TE_type, status, count),
not_DNA_counts) %>%
filter(!is.na(cluster))
DNA_results <- comparisons %>%
mutate(results = map2(cluster2, cluster1, function(c2, c1) {
DNA_df_long %>%
distinct(TE_type) %>%
pull() %>%
map_dfr(function(te) {
test_pair_TE_generic(
DNA_df_long,
te_name = te,
cluster1 = c1,
cluster2 = c2
)
})
})
) %>%
unnest(results) %>%
mutate(FDR = p.adjust(p_value, method = "BH"))datatable(DNA_counts,
rownames = FALSE,
filter = 'top', # add filter/search boxes
options = list(
pageLength = 10,
autoWidth = TRUE,
scrollX = TRUE))total_DNA_summits# A tibble: 4 × 2
cluster total
<chr> <int>
1 Set_1 110084
2 Set_2 1385
3 Set_3 6623
4 <NA> 31979# ---- Prepare the table ----
DNA_counts_display <- DNA_results %>%
# 1. Split comparison
separate(comparison, into = c("cluster2", "cluster1"), sep = " vs ", remove = FALSE) %>%
# 2. Add log2 odds ratio
mutate(log2OR = log2(odds_ratio)) %>%
# 3. Add enrichment/depletion direction
mutate(direction = case_when(
odds_ratio > 1 ~ "enriched",
odds_ratio < 1 ~ "depleted",
TRUE ~ "neutral"
)) %>%
# 4. Flag significant
mutate(significant = FDR < 0.05) %>%
# Optional: arrange for readability
arrange(cluster2, direction, desc(log2OR))
# ---- Create interactive datatable ----
datatable(
DNA_counts_display,
rownames = FALSE,
filter = 'top', # add filter/search boxes
options = list(
pageLength = 10,
autoWidth = TRUE,
scrollX = TRUE
)
) %>%
# Conditional coloring by direction
formatStyle(
'direction',
target = 'row',
backgroundColor = styleEqual(
c('enriched', 'depleted'),
c('#FFDD99', '#99CCFF') # enriched = light orange, depleted = light blue
)
) %>%
# Bold significant rows
formatStyle(
'significant',
fontWeight = styleEqual(TRUE, 'bold')
) %>%
# Round numeric columns for readability
formatRound(columns = c('odds_ratio','log2OR','FDR'), digits = 2)ggplot(DNA_results, aes(x = (odds_ratio), y = -log10(FDR),label = TE_type)) +
geom_point(aes(color = odds_ratio > 1), size = 3) +
# geom_text_repel(data = subset(SVA_results, FDR < 0.05)) +
geom_text_repel(size=3, max.overlaps = Inf)+
geom_hline(yintercept = -log10(0.05), linetype = "dashed") +
labs(
x = "(Odds Ratio)",
y = "-log10(FDR)",
title = "DNA family enrichment at ROI summits\nH3K27ac"
) +
theme_classic()+
facet_wrap(~comparison)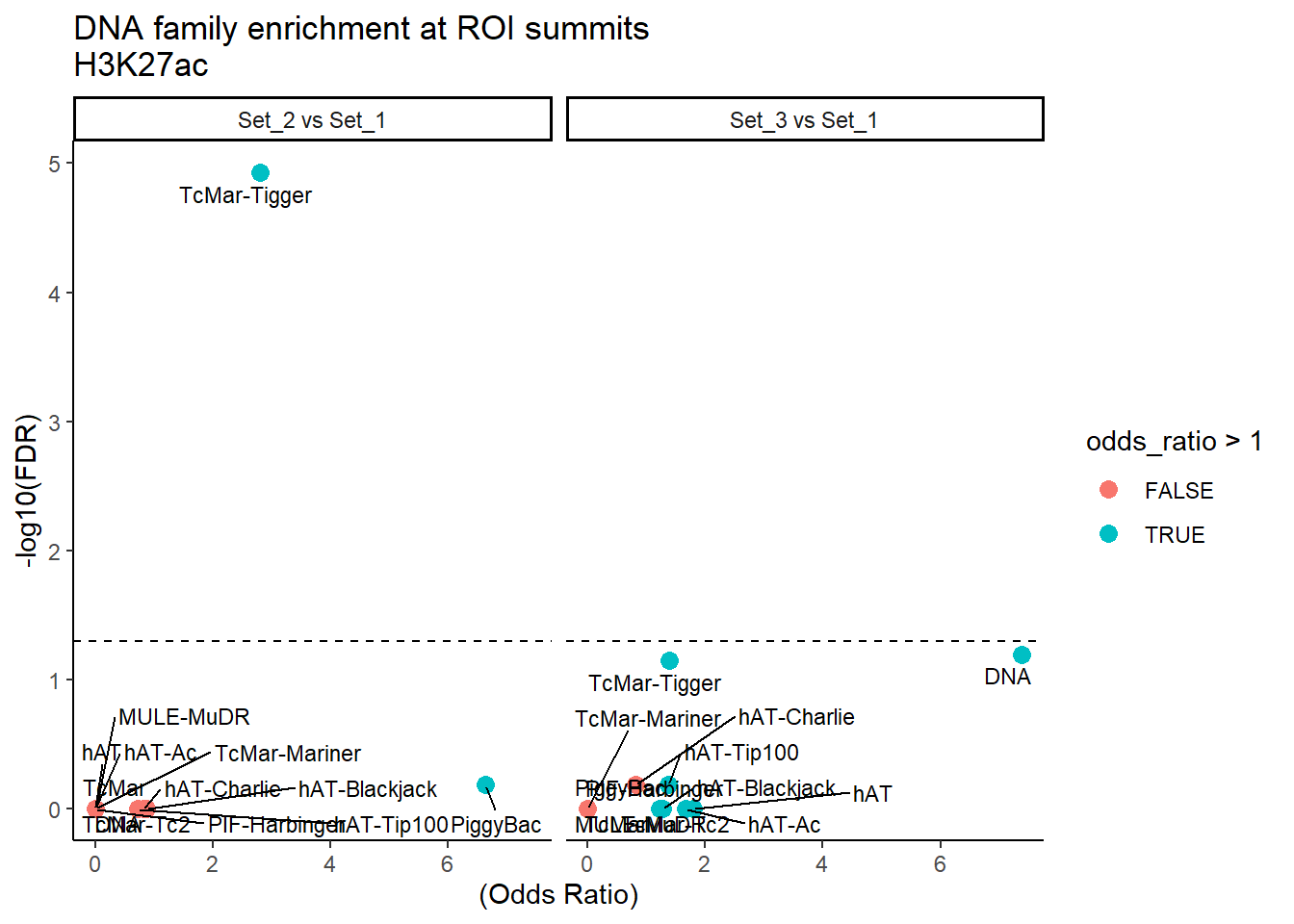
peakAnnoList_H3K27ac <- readRDS("data/motif_lists/H3K27ac_annotated_peaks.RDS")
out_dir <- "data/Bed_exports/H3K27ac_sets"
set_list <- names(peakAnnoList_H3K27ac)
for (group_name in set_list) {
cs <- peakAnnoList_H3K27ac[[group_name]]
gr <- cs@anno
# Set BED name column
mcols(gr)$name <- mcols(gr)$Peakid
# Optional: if you want a score column
if(!"score" %in% colnames(mcols(gr))) {
mcols(gr)$score <- 0
}
# Export to BED
bed_file <- file.path(out_dir, paste0(group_name, "_H3K27ac.bed"))
# Export
export(gr, bed_file, format = "BED")
cat("Exported:", bed_file, "\n")
}Overlapping DNA families using Full ROI
fullROI_long_DNA <- purrr::imap_dfr(
H3K27ac_sets_gr,
~count_sine_families(.x, DNA_gr, .y)
)
DNA_results_full <- comparisons %>%
mutate(results = map2(cluster2, cluster1, function(c2, c1) {
fullROI_long_DNA %>%
distinct(family) %>%
pull() %>%
map_dfr(function(te) {
test_pair_TE_generic(
fullROI_long_DNA,
te_name = te,
cluster1 = c1,
cluster2 = c2
)
})
})
) %>%
unnest(results) %>%
mutate(FDR = p.adjust(p_value, method = "BH"))
ggplot(DNA_results_full, aes(x = (odds_ratio), y = -log10(FDR),label = TE_type)) +
geom_point(aes(color = odds_ratio > 1), size = 3) +
# geom_text_repel(data = subset(SVA_results, FDR < 0.05)) +
geom_text_repel(size=3, max.overlaps = Inf)+
geom_hline(yintercept = -log10(0.05), linetype = "dashed") +
geom_vline(xintercept = 1, linetype = 3) +
labs(
x = "(Odds Ratio)",
y = "-log10(FDR)",
title = "DNA family enrichment at full ROI\nH3K27ac"
) +
theme_classic() +
facet_wrap(~comparison)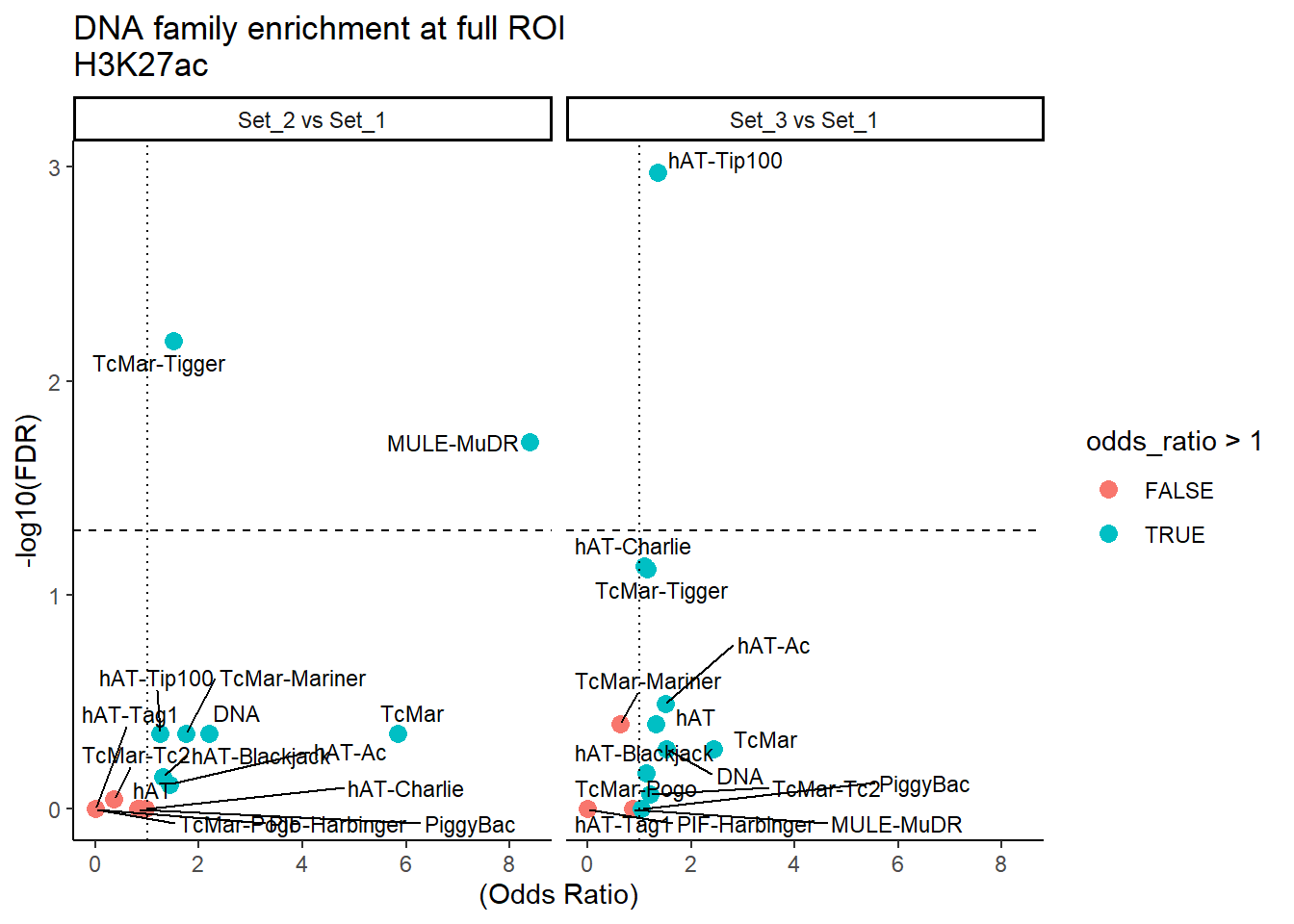
| Version | Author | Date |
|---|---|---|
| 0606abf | reneeisnowhere | 2026-01-27 |
Proportions of SETs and SETS of TEs
H3K27ac_set_case_lfc <- readRDS("data/RDS_files/H3K27ac_set_case_lfc.RDS")
annotated_tables <- readRDS("data/TE_annotation/annotated_tables_1bp_ol.RDS")
anno_H3K27ac <- annotated_tables$annotated_H3K27ac
H3K27ac_lookup# A tibble: 118,092 × 2
Peakid cluster
<chr> <chr>
1 chr1:778197-779527 Set_1
2 chr1:819712-820327 Set_1
3 chr1:821438-822526 Set_1
4 chr1:825652-826234 Set_1
5 chr1:826639-827783 Set_1
6 chr1:831254-832817 Set_1
7 chr1:910490-911487 Set_1
8 chr1:913389-914255 Set_1
9 chr1:918697-919102 Set_1
10 chr1:920182-924328 Set_1
# ℹ 118,082 more rowsH3K27ac_set_case_lookup <- bind_rows(H3K27ac_set_case_lfc, .id = "set_case") %>%
dplyr::select(Peakid,cluster:set_case)
anno_H3K27ac %>%
left_join(H3K27ac_set_case_lookup) %>%
group_by(cluster) %>%
count() %>%
ungroup() %>%
mutate(percent = n / sum(n)) %>%
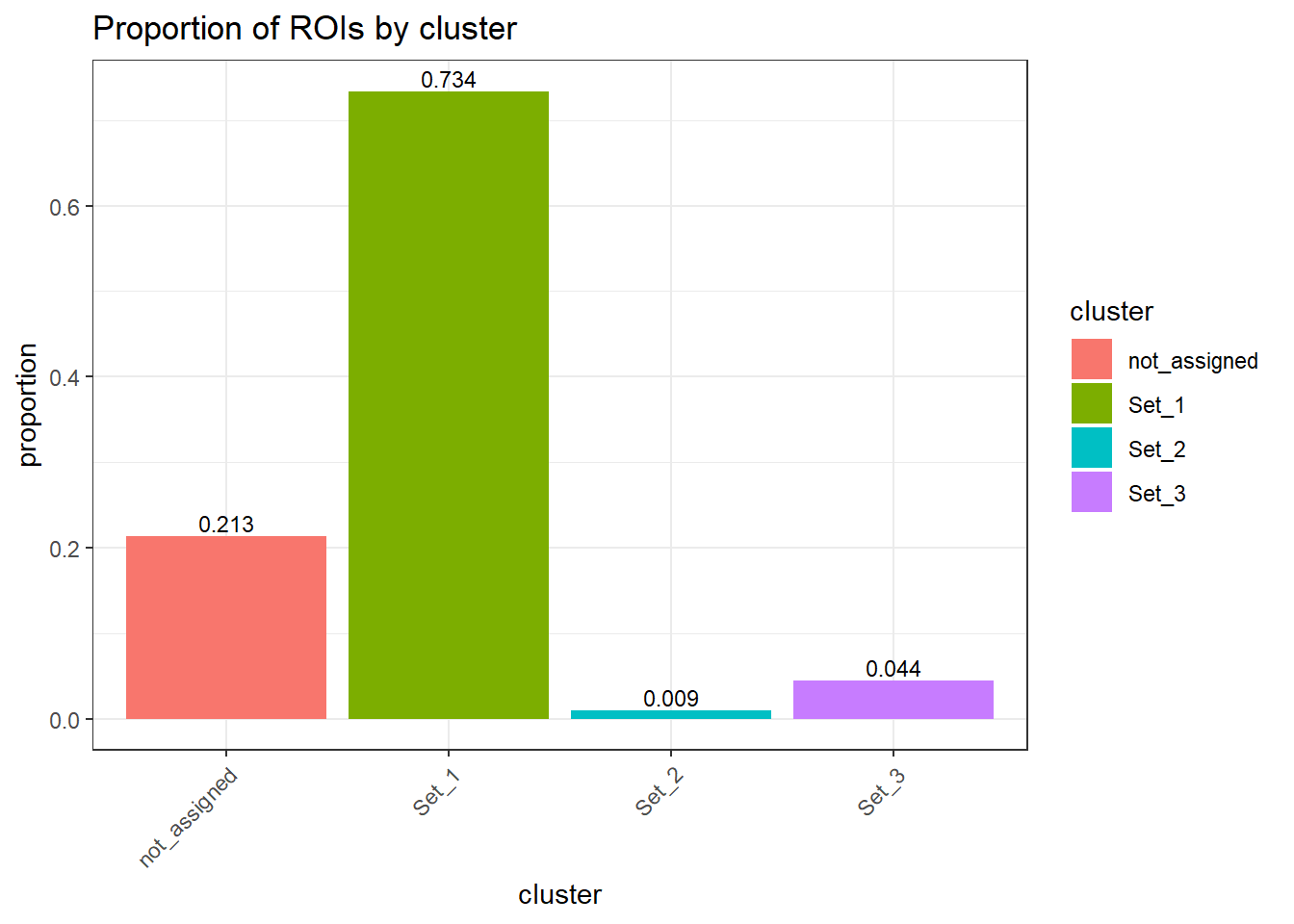
ggplot(.,aes(x=cluster, y=percent, fill=cluster)) +
geom_col() +
geom_text(
aes(label = sprintf("%.3f", percent)),
vjust = -0.3,
size = 3
) +
ylab("proportion")+
ggtitle("Proportion of ROIs by cluster")+
theme_bw() +
theme(
axis.text.x = element_text(angle = 45, hjust = 1),
strip.text = element_text(face = "bold")
)
| Version | Author | Date |
|---|---|---|
| 26f72a7 | reneeisnowhere | 2026-02-05 |
# LTR_counts %>%
# group_by(cluster) %>%
# summarize(n_LTR = sum(count),
# .groups = "drop") %>%
# mutate(percent = n_LTR/ sum(n_LTR))
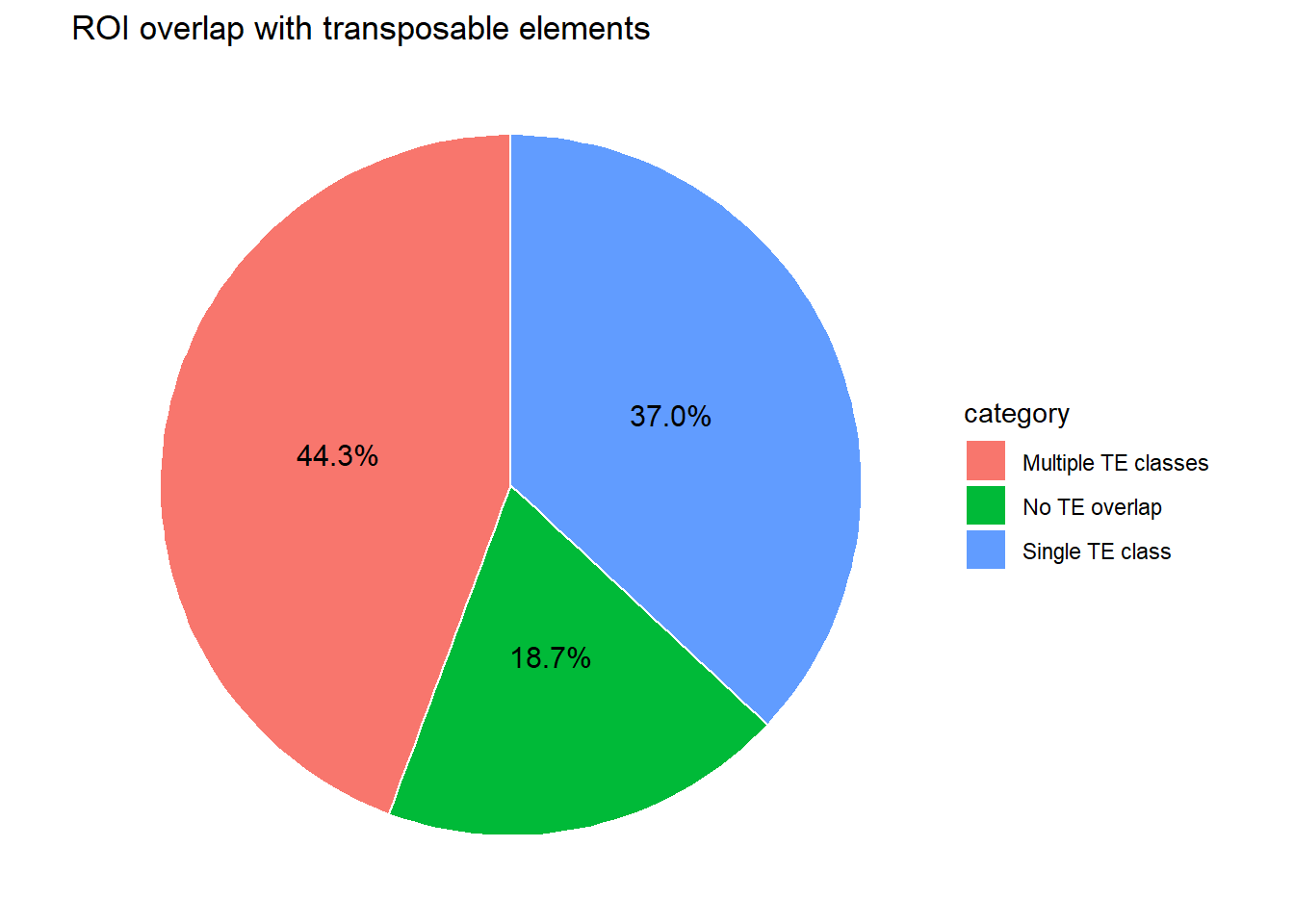
H3K27ac_te_summary <- anno_H3K27ac %>%
left_join(H3K27ac_set_case_lookup) %>%
mutate(repClass = na_if(repClass, ""), # treat empty as NA
n_TE_class = case_when(
is.na(repClass) ~ 0L,
TRUE ~ lengths(strsplit(repClass, ";")))) %>%
mutate( category = case_when(n_TE_class == 0 ~ "No TE overlap",
n_TE_class == 1 ~ "Single TE class",
n_TE_class > 1 ~ "Multiple TE classes"))
pie_df <- H3K27ac_te_summary %>%
count(category) %>%
mutate(
percent = n / sum(n) * 100
)
ggplot(pie_df, aes(x = "", y = percent, fill = category)) +
geom_col(width = 1, color = "white") +
coord_polar(theta = "y") +
theme_void() +
geom_text(
aes(label = sprintf("%.1f%%", percent)),
position = position_stack(vjust = 0.5),
size = 4
) +
labs(
title = "ROI overlap with transposable elements"
)
| Version | Author | Date |
|---|---|---|
| 26f72a7 | reneeisnowhere | 2026-02-05 |
of ROIs that overlap TEs, what is the distrubution across TE families?
H3K27ac_te_familiy_long <- H3K27ac_te_summary %>%
filter(!is.na(repClass), repClass != "") %>% # TE-positive only
separate_rows(repClass, sep = ";") %>%
distinct(Peakid, repClass,cluster)
fam_pie_df <- H3K27ac_te_familiy_long %>%
count(repClass, name = "n") %>%
mutate(percent = n / sum(n)*100) %>%
mutate(
repClass = if_else(percent < 2, "Other", repClass)
) %>%
group_by(repClass) %>%
summarise(n = sum(n), .groups = "drop") %>%
mutate(percent = n / sum(n) * 100)
ggplot(fam_pie_df, aes(x = "", y = percent, fill = repClass)) +
geom_col(width = 1, color = "white") +
coord_polar(theta = "y") +
theme_void() +
geom_text(
aes(label = sprintf("%s\n%.1f%%", repClass, percent)),
position = position_stack(vjust = 0.5),
size = 3
) +
labs(
title = "TE Class composition among TE-overlapping ROIs",
fill = "TE Class")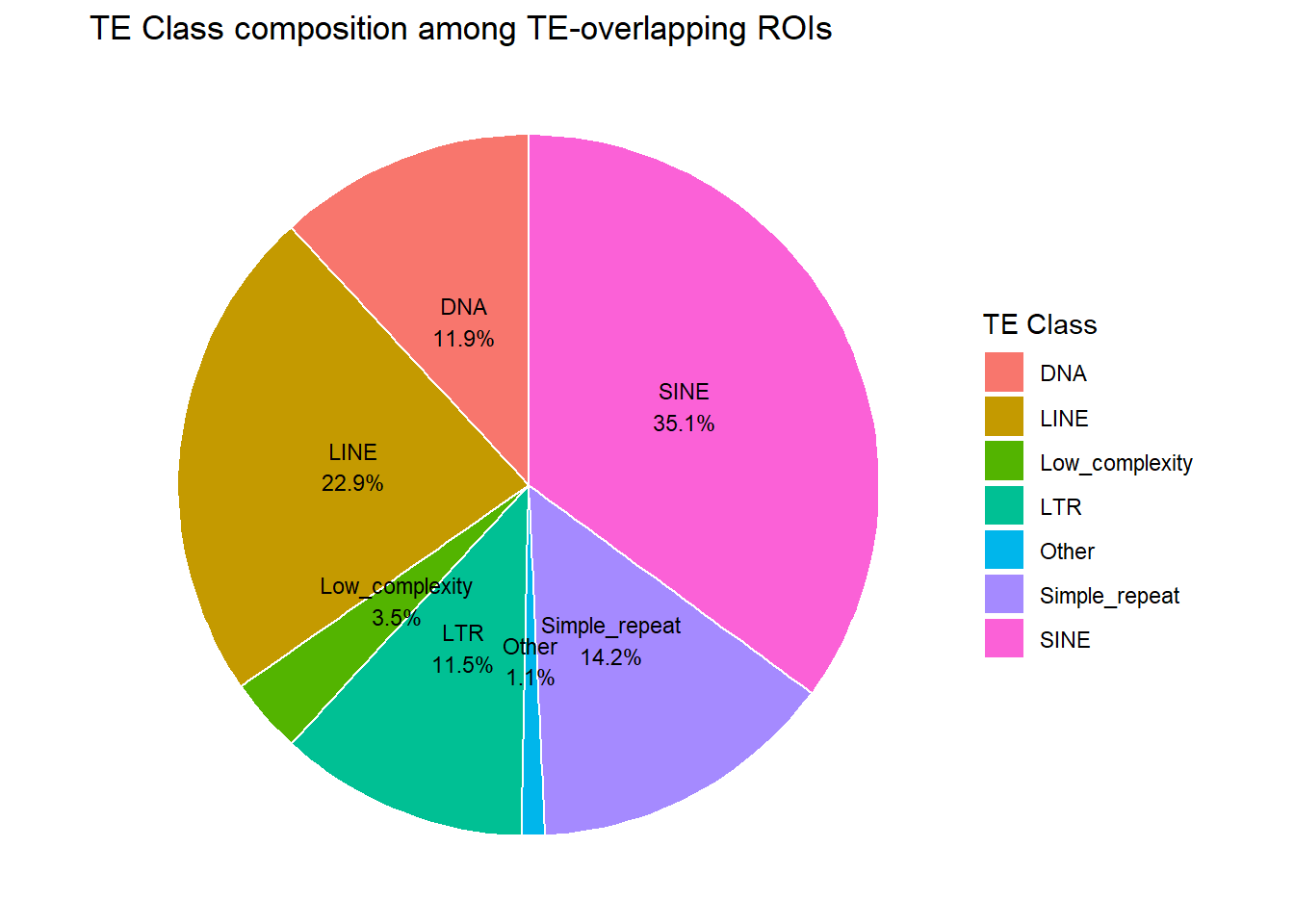
| Version | Author | Date |
|---|---|---|
| 26f72a7 | reneeisnowhere | 2026-02-05 |
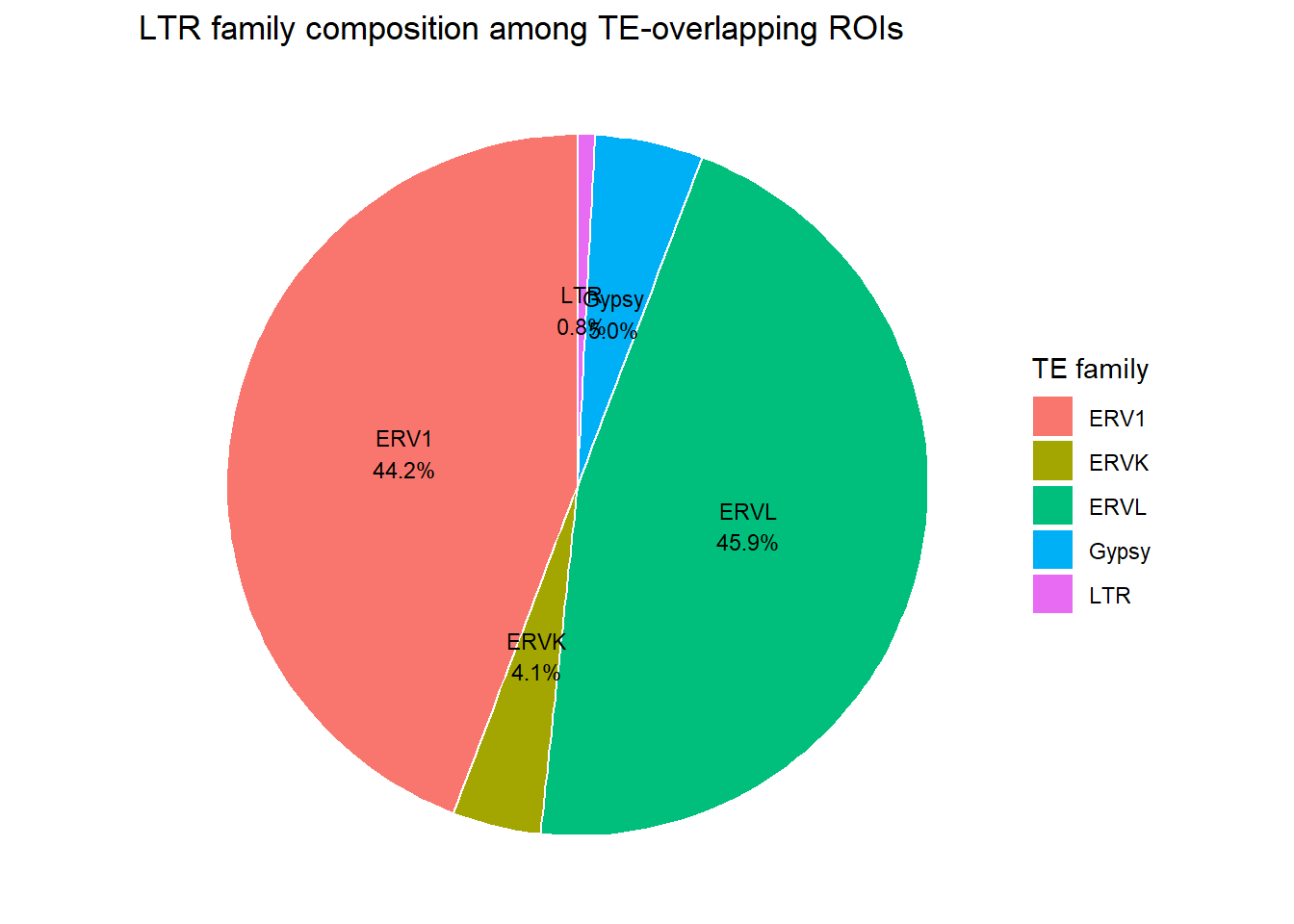
# unique(rpt_split_gr_list$LTR$repFamily)H3K27ac_long_repFamily <- H3K27ac_te_summary %>%
filter(!is.na(repFamily), repFamily != "") %>% # TE-positive only
separate_rows(repFamily, sep = ";") %>%
distinct(Peakid, repFamily)
H3K27ac_LTR_pie_df <- H3K27ac_long_repFamily %>%
dplyr::filter(repFamily %in% c("ERV1", "ERVK","ERVL", "EVRL-MaLR","Gypsy","LTR")) %>%
count(repFamily, name = "n") %>%
mutate(percent= n/ sum(n) * 100)
ggplot(H3K27ac_LTR_pie_df, aes(x = "", y = percent, fill = repFamily)) +
geom_col(width = 1, color = "white") +
coord_polar(theta = "y") +
theme_void() +
geom_text(
aes(label = sprintf("%s\n%.1f%%", repFamily, percent)),
position = position_stack(vjust = 0.5),
size = 3
) +
labs(
title = "LTR family composition among TE-overlapping ROIs",
fill = "TE family"
)
| Version | Author | Date |
|---|---|---|
| 26f72a7 | reneeisnowhere | 2026-02-05 |
H3K27ac_te_family_clust <- H3K27ac_te_familiy_long %>%
dplyr::filter(cluster != "not_assigned") %>%
distinct(Peakid, cluster, repClass) %>%
count(cluster, repClass, name = "n") %>%
group_by(cluster) %>%
mutate(percent = n / sum(n)*100) %>%
mutate(
repClass = if_else(percent < 2, "Other", repClass)
) %>%
group_by(cluster,repClass) %>%
summarise(n = sum(n), .groups = "drop") %>%
group_by(cluster) %>%
mutate(percent = n / sum(n) * 100)
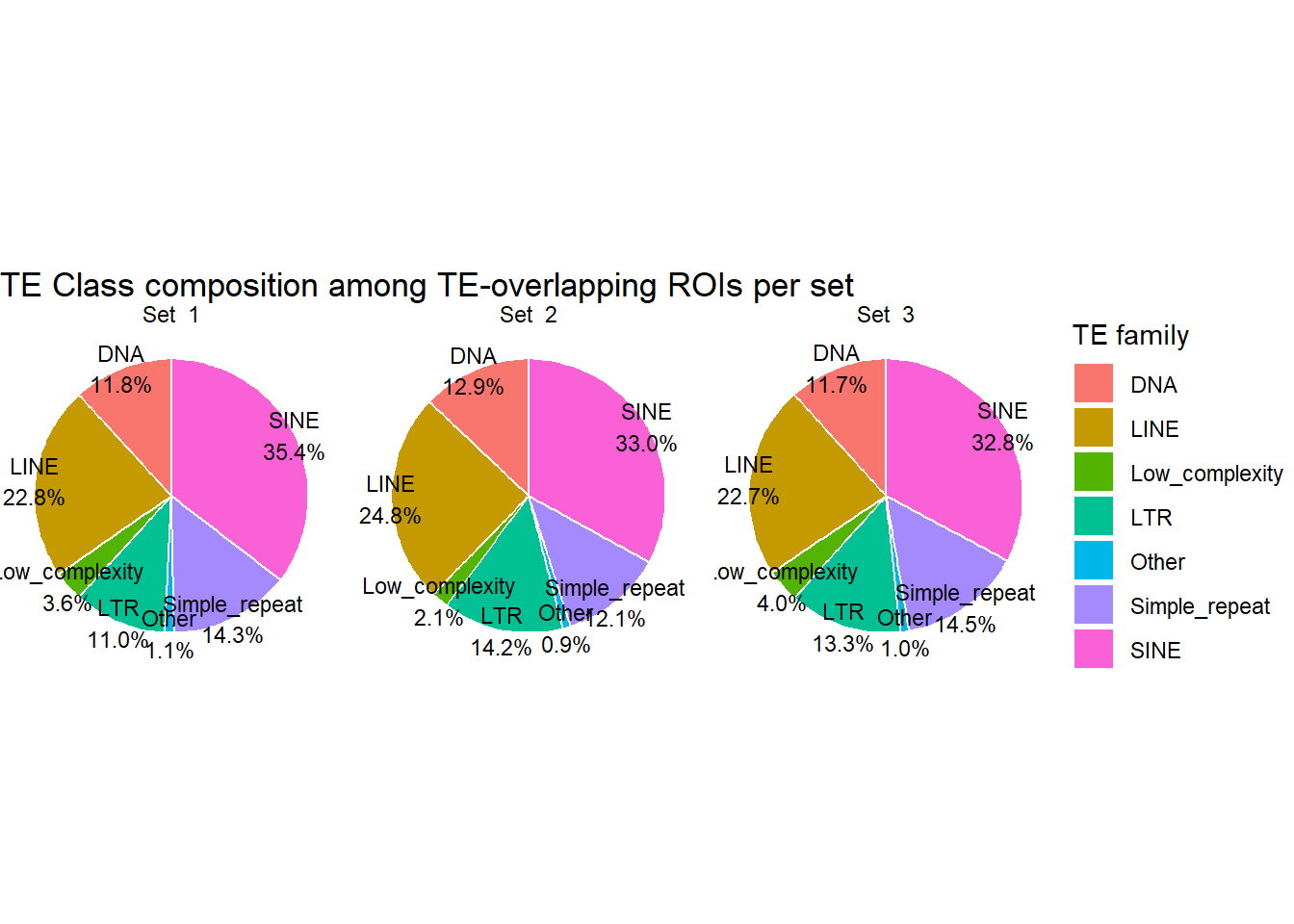
ggplot(H3K27ac_te_family_clust, aes(x = "", y=percent, fill=repClass))+
geom_col(width =1, color="white")+
coord_polar(theta = "y")+
geom_text(
aes(x= 1.5,label = sprintf("%s\n%.1f%%", repClass, percent)),
position = position_stack(vjust = 0.5),
size = 3
) +
facet_wrap(~cluster)+
theme_void()+
labs(
title = "TE Class composition among TE-overlapping ROIs per set",
fill = "TE family"
)
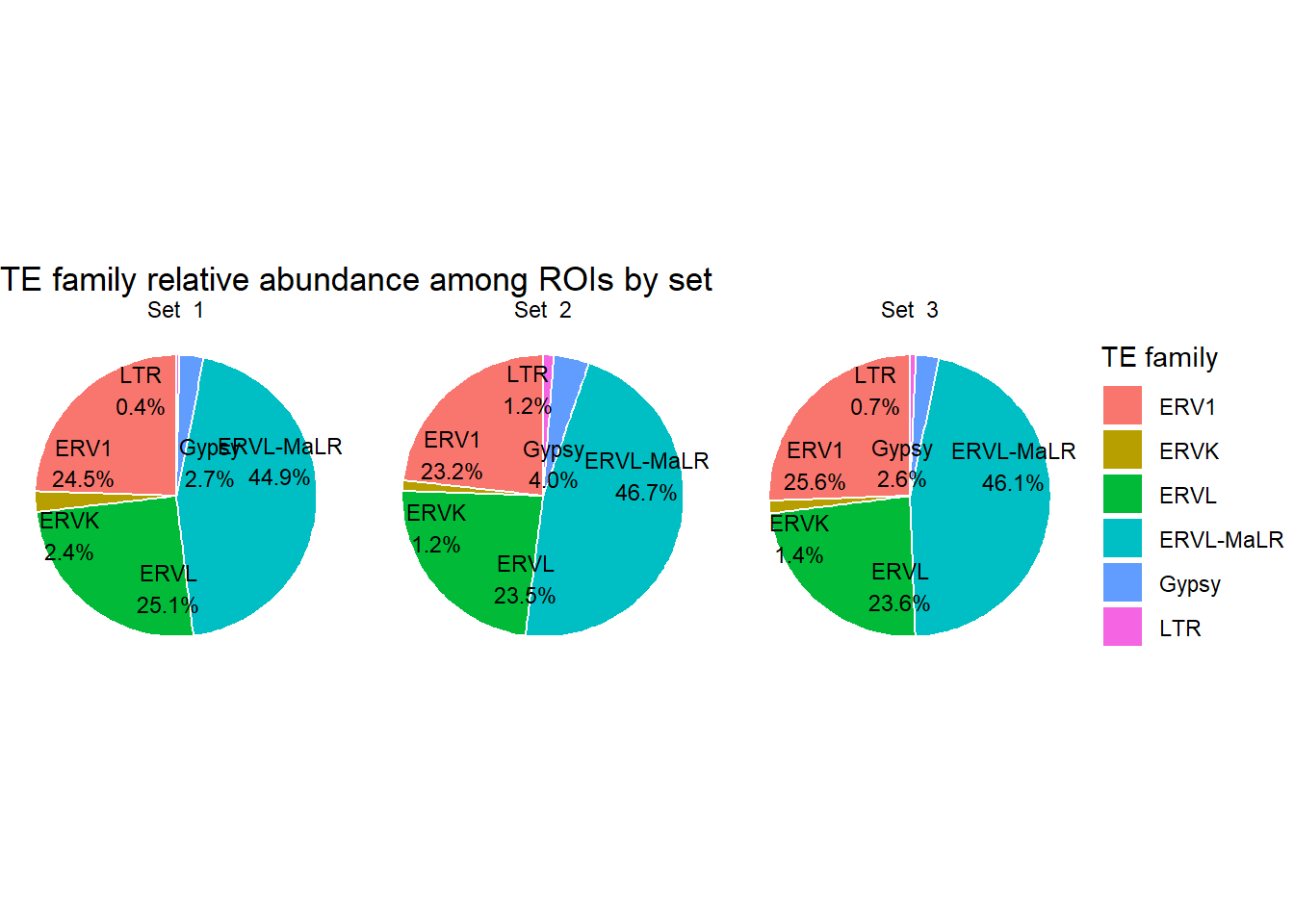
H3K27ac_long_repFamily <-
H3K27ac_te_summary %>%
dplyr::filter(cluster != "not_assigned") %>%
filter(!is.na(repFamily), repFamily != "") %>% # TE-positive only
separate_rows(repFamily, sep = ";") %>%
distinct(cluster,Peakid, repFamily) %>%
dplyr::filter(repFamily %in% c("ERV1", "ERVK","ERVL", "ERVL-MaLR","Gypsy","LTR")) %>%
count(cluster,repFamily, name = "n") %>%
group_by(cluster) %>%
mutate(percent = n / sum(n)*100) %>%
# mutate(
# repFamily = if_else(percent < 2, "Other", repFamily)
# ) %>%
group_by(cluster,repFamily) %>%
summarise(n = sum(n), .groups = "drop") %>%
group_by(cluster) %>%
mutate(percent = n / sum(n) * 100)
ggplot(H3K27ac_long_repFamily, aes(x = "", y=percent, fill=repFamily))+
geom_col(width =1, color="white")+
coord_polar(theta = "y")+
geom_text_repel(
aes(label = sprintf("%s\n%.1f%%", repFamily, percent)),
position = position_stack(vjust = 0.5),
size = 3,
show.legend = FALSE
) +
facet_wrap(~cluster)+
theme_void()+
labs(
title = "TE family relative abundance among ROIs by set",
fill = "TE family"
)
LTR_bar_df <-H3K27ac_te_summary %>%
filter(!is.na(repFamily), repFamily != "") %>% # TE-positive only
separate_rows(repFamily, sep = ";") %>%
distinct(cluster,Peakid, repFamily) %>%
dplyr::filter(repFamily %in% c("ERV1", "ERVK","ERVL", "ERVL-MaLR","Gypsy","LTR")) %>%
group_by(repFamily, cluster) %>%
summarise(n = dplyr::n(), .groups = "drop") %>%
# optionally convert NA to a string
mutate(cluster = if_else(is.na(cluster), "NA", cluster))
ggplot(LTR_bar_df, aes(x = repFamily, y = n, fill = cluster)) +
geom_col(position = "fill") +
theme_classic() +
labs(
x = "LTR Family",
y = "Proportion of ROIs",
fill = "Cluster",
title = "Proportion of ROI clusters for each LTR family"
) +
theme(axis.text.x = element_text(angle = 45, hjust = 1))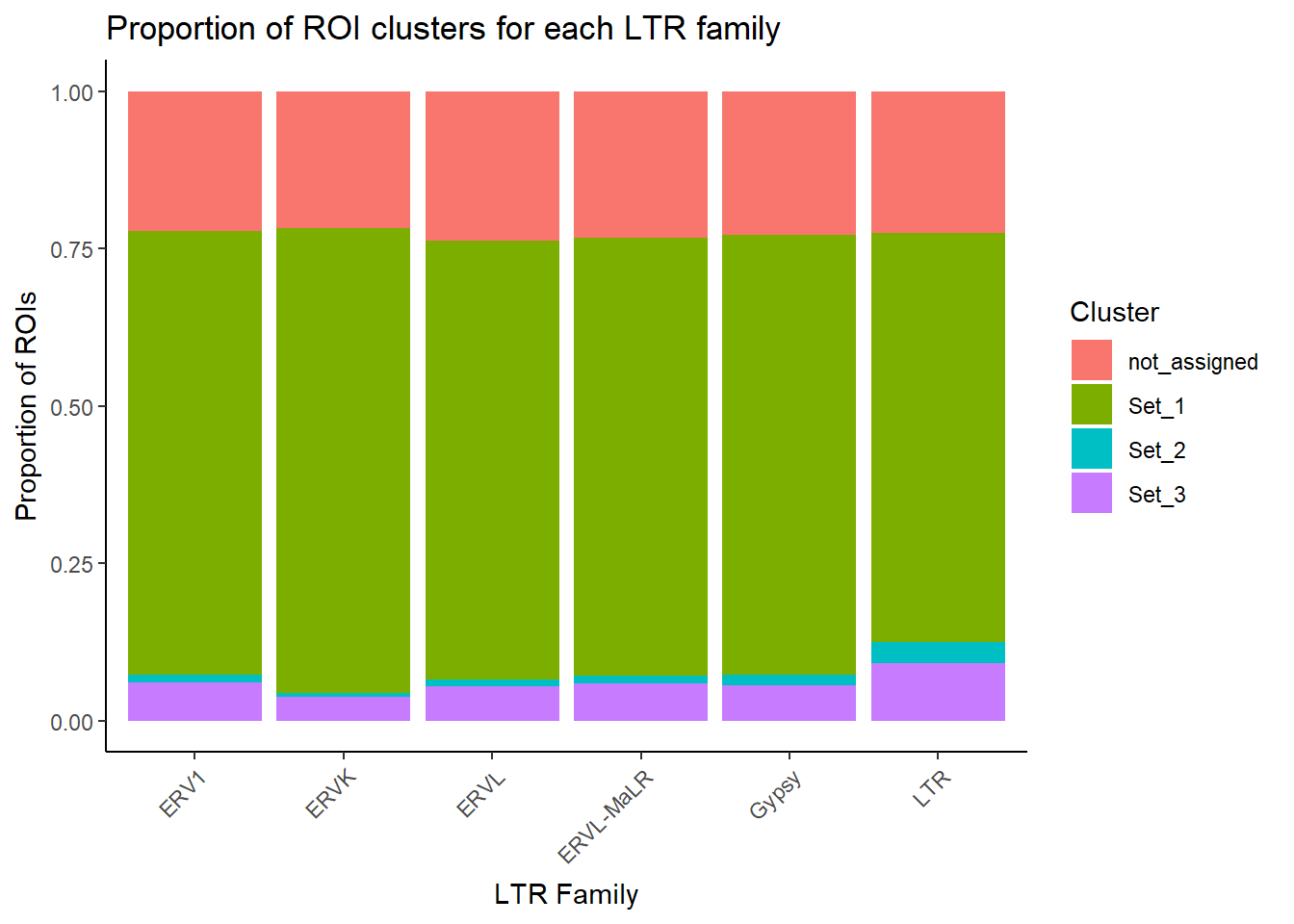
sessionInfo()R version 4.4.2 (2024-10-31 ucrt)
Platform: x86_64-w64-mingw32/x64
Running under: Windows 11 x64 (build 26200)
Matrix products: default
locale:
[1] LC_COLLATE=English_United States.utf8
[2] LC_CTYPE=English_United States.utf8
[3] LC_MONETARY=English_United States.utf8
[4] LC_NUMERIC=C
[5] LC_TIME=English_United States.utf8
time zone: America/Chicago
tzcode source: internal
attached base packages:
[1] grid stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] ChIPseeker_1.42.1 DT_0.33 ggrepel_0.9.6
[4] rtracklayer_1.66.0 genomation_1.38.0 plyranges_1.26.0
[7] GenomicRanges_1.58.0 GenomeInfoDb_1.42.3 IRanges_2.40.1
[10] S4Vectors_0.44.0 BiocGenerics_0.52.0 lubridate_1.9.4
[13] forcats_1.0.0 stringr_1.5.1 dplyr_1.1.4
[16] purrr_1.1.0 readr_2.1.5 tidyr_1.3.1
[19] tibble_3.3.0 ggplot2_3.5.2 tidyverse_2.0.0
[22] workflowr_1.7.1
loaded via a namespace (and not attached):
[1] RColorBrewer_1.1-3
[2] rstudioapi_0.17.1
[3] jsonlite_2.0.0
[4] magrittr_2.0.3
[5] ggtangle_0.0.7
[6] GenomicFeatures_1.58.0
[7] farver_2.1.2
[8] rmarkdown_2.29
[9] fs_1.6.6
[10] BiocIO_1.16.0
[11] zlibbioc_1.52.0
[12] vctrs_0.6.5
[13] memoise_2.0.1
[14] Rsamtools_2.22.0
[15] RCurl_1.98-1.17
[16] ggtree_3.14.0
[17] htmltools_0.5.8.1
[18] S4Arrays_1.6.0
[19] TxDb.Hsapiens.UCSC.hg19.knownGene_3.2.2
[20] plotrix_3.8-4
[21] curl_7.0.0
[22] SparseArray_1.6.2
[23] gridGraphics_0.5-1
[24] sass_0.4.10
[25] KernSmooth_2.23-26
[26] bslib_0.9.0
[27] htmlwidgets_1.6.4
[28] plyr_1.8.9
[29] impute_1.80.0
[30] cachem_1.1.0
[31] GenomicAlignments_1.42.0
[32] igraph_2.1.4
[33] whisker_0.4.1
[34] lifecycle_1.0.4
[35] pkgconfig_2.0.3
[36] Matrix_1.7-3
[37] R6_2.6.1
[38] fastmap_1.2.0
[39] GenomeInfoDbData_1.2.13
[40] MatrixGenerics_1.18.1
[41] enrichplot_1.26.6
[42] digest_0.6.37
[43] aplot_0.2.8
[44] colorspace_2.1-1
[45] patchwork_1.3.2
[46] AnnotationDbi_1.68.0
[47] ps_1.9.1
[48] rprojroot_2.1.1
[49] crosstalk_1.2.2
[50] RSQLite_2.4.3
[51] labeling_0.4.3
[52] timechange_0.3.0
[53] httr_1.4.7
[54] abind_1.4-8
[55] compiler_4.4.2
[56] bit64_4.6.0-1
[57] withr_3.0.2
[58] BiocParallel_1.40.2
[59] DBI_1.2.3
[60] gplots_3.2.0
[61] R.utils_2.13.0
[62] rappdirs_0.3.3
[63] DelayedArray_0.32.0
[64] rjson_0.2.23
[65] caTools_1.18.3
[66] gtools_3.9.5
[67] tools_4.4.2
[68] ape_5.8-1
[69] httpuv_1.6.16
[70] R.oo_1.27.1
[71] glue_1.8.0
[72] restfulr_0.0.16
[73] callr_3.7.6
[74] nlme_3.1-168
[75] GOSemSim_2.32.0
[76] promises_1.3.3
[77] getPass_0.2-4
[78] gridBase_0.4-7
[79] reshape2_1.4.4
[80] fgsea_1.32.4
[81] generics_0.1.4
[82] gtable_0.3.6
[83] BSgenome_1.74.0
[84] tzdb_0.5.0
[85] R.methodsS3_1.8.2
[86] seqPattern_1.38.0
[87] data.table_1.17.8
[88] hms_1.1.3
[89] utf8_1.2.6
[90] XVector_0.46.0
[91] pillar_1.11.0
[92] yulab.utils_0.2.1
[93] vroom_1.6.5
[94] later_1.4.2
[95] splines_4.4.2
[96] treeio_1.30.0
[97] lattice_0.22-7
[98] bit_4.6.0
[99] tidyselect_1.2.1
[100] GO.db_3.20.0
[101] Biostrings_2.74.1
[102] knitr_1.50
[103] git2r_0.36.2
[104] SummarizedExperiment_1.36.0
[105] xfun_0.52
[106] Biobase_2.66.0
[107] matrixStats_1.5.0
[108] stringi_1.8.7
[109] UCSC.utils_1.2.0
[110] lazyeval_0.2.2
[111] boot_1.3-32
[112] ggfun_0.2.0
[113] yaml_2.3.10
[114] evaluate_1.0.5
[115] codetools_0.2-20
[116] qvalue_2.38.0
[117] ggplotify_0.1.2
[118] cli_3.6.5
[119] processx_3.8.6
[120] jquerylib_0.1.4
[121] dichromat_2.0-0.1
[122] Rcpp_1.1.0
[123] png_0.1-8
[124] XML_3.99-0.18
[125] parallel_4.4.2
[126] blob_1.2.4
[127] DOSE_4.0.1
[128] bitops_1.0-9
[129] tidytree_0.4.6
[130] scales_1.4.0
[131] crayon_1.5.3
[132] rlang_1.1.6
[133] fastmatch_1.1-6
[134] cowplot_1.2.0
[135] KEGGREST_1.46.0