SCZ - Brain Cerebellar Hemisphere
sheng Qian
2021-2-6
Last updated: 2022-03-14
Checks: 5 2
Knit directory: cTWAS_analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20211220) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| /project2/xinhe/shengqian/cTWAS/cTWAS_analysis/data/ | data |
| /project2/xinhe/shengqian/cTWAS/cTWAS_analysis/code/ctwas_config.R | code/ctwas_config.R |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 4c71b11. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .ipynb_checkpoints/
Ignored: data/AF/
Untracked files:
Untracked: Rplot.png
Untracked: analysis/.ipynb_checkpoints/
Untracked: analysis/SCZ_2014_EUR_Brain_Amygdala.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Anterior_cingulate_cortex_BA24.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Caudate_basal_ganglia.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Cerebellar_Hemisphere.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Cerebellum.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Cortex.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Frontal_Cortex_BA9.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Hippocampus.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Hypothalamus.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Nucleus_accumbens_basal_ganglia.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Putamen_basal_ganglia.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Spinal_cord_cervical_c-1.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Substantia_nigra.Rmd
Untracked: analysis/SCZ_2020_Brain_Cortex.Rmd
Untracked: analysis/SCZ_2020_Brain_Frontal_Cortex_BA9.Rmd
Untracked: analysis/SCZ_2020_Brain_Hypothalamus.Rmd
Untracked: analysis/SCZ_2020_Brain_Putamen_basal_ganglia.Rmd
Untracked: analysis/SCZ_Cross_Tissue_Analysis.Rmd
Untracked: code/.ipynb_checkpoints/
Untracked: code/AF_out/
Untracked: code/Autism_out/
Untracked: code/BMI_S_out/
Untracked: code/BMI_out/
Untracked: code/Glucose_out/
Untracked: code/LDL_S_out/
Untracked: code/SCZ_2014_EUR_out/
Untracked: code/SCZ_2020_out/
Untracked: code/SCZ_S_out/
Untracked: code/SCZ_out/
Untracked: code/T2D_out/
Untracked: code/ctwas_config.R
Untracked: code/mapping.R
Untracked: code/out/
Untracked: code/run_AF_analysis.sbatch
Untracked: code/run_AF_analysis.sh
Untracked: code/run_AF_ctwas_rss_LDR.R
Untracked: code/run_Autism_analysis.sbatch
Untracked: code/run_Autism_analysis.sh
Untracked: code/run_Autism_ctwas_rss_LDR.R
Untracked: code/run_BMI_analysis.sbatch
Untracked: code/run_BMI_analysis.sh
Untracked: code/run_BMI_analysis_S.sbatch
Untracked: code/run_BMI_analysis_S.sh
Untracked: code/run_BMI_ctwas_rss_LDR.R
Untracked: code/run_BMI_ctwas_rss_LDR_S.R
Untracked: code/run_Glucose_analysis.sbatch
Untracked: code/run_Glucose_analysis.sh
Untracked: code/run_Glucose_ctwas_rss_LDR.R
Untracked: code/run_LDL_analysis_S.sbatch
Untracked: code/run_LDL_analysis_S.sh
Untracked: code/run_LDL_ctwas_rss_LDR_S.R
Untracked: code/run_SCZ_2014_EUR_analysis.sbatch
Untracked: code/run_SCZ_2014_EUR_analysis.sh
Untracked: code/run_SCZ_2014_EUR_ctwas_rss_LDR.R
Untracked: code/run_SCZ_2020_analysis.sbatch
Untracked: code/run_SCZ_2020_analysis.sh
Untracked: code/run_SCZ_2020_ctwas_rss_LDR.R
Untracked: code/run_SCZ_analysis.sbatch
Untracked: code/run_SCZ_analysis.sh
Untracked: code/run_SCZ_analysis_S.sbatch
Untracked: code/run_SCZ_analysis_S.sh
Untracked: code/run_SCZ_ctwas_rss_LDR.R
Untracked: code/run_SCZ_ctwas_rss_LDR_S.R
Untracked: code/run_T2D_analysis.sbatch
Untracked: code/run_T2D_analysis.sh
Untracked: code/run_T2D_ctwas_rss_LDR.R
Untracked: code/wflow_build.R
Untracked: code/wflow_build.sbatch
Untracked: data/.ipynb_checkpoints/
Untracked: data/BMI/
Untracked: data/PGC3_SCZ_wave3_public.v2.tsv
Untracked: data/SCZ/
Untracked: data/SCZ_2014_EUR/
Untracked: data/SCZ_2020/
Untracked: data/SCZ_S/
Untracked: data/T2D/
Untracked: data/UKBB/
Untracked: data/UKBB_SNPs_Info.text
Untracked: data/gene_OMIM.txt
Untracked: data/gene_pip_0.8.txt
Untracked: data/mashr_Heart_Atrial_Appendage.db
Untracked: data/mashr_sqtl/
Untracked: data/summary_known_genes_annotations.xlsx
Untracked: data/untitled.txt
Unstaged changes:
Modified: analysis/SCZ_Brain_Amygdala.Rmd
Modified: analysis/SCZ_Brain_Anterior_cingulate_cortex_BA24.Rmd
Modified: analysis/SCZ_Brain_Caudate_basal_ganglia.Rmd
Modified: analysis/SCZ_Brain_Cerebellar_Hemisphere.Rmd
Modified: analysis/SCZ_Brain_Cerebellum.Rmd
Modified: analysis/SCZ_Brain_Cortex.Rmd
Modified: analysis/SCZ_Brain_Frontal_Cortex_BA9.Rmd
Modified: analysis/SCZ_Brain_Hippocampus.Rmd
Modified: analysis/SCZ_Brain_Hypothalamus.Rmd
Modified: analysis/SCZ_Brain_Nucleus_accumbens_basal_ganglia.Rmd
Modified: analysis/SCZ_Brain_Putamen_basal_ganglia.Rmd
Modified: analysis/SCZ_Brain_Spinal_cord_cervical_c-1.Rmd
Modified: analysis/SCZ_Brain_Substantia_nigra.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with wflow_publish() to start tracking its development.
Weight QC
#number of imputed weights
nrow(qclist_all)[1] 10988#number of imputed weights by chromosome
table(qclist_all$chr)
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
1044 751 636 406 524 614 548 415 425 434 669 618 208 358 360 526
17 18 19 20 21 22
701 163 854 326 130 278 #number of imputed weights without missing variants
sum(qclist_all$nmiss==0)[1] 8105#proportion of imputed weights without missing variants
mean(qclist_all$nmiss==0)[1] 0.7376Check convergence of parameters
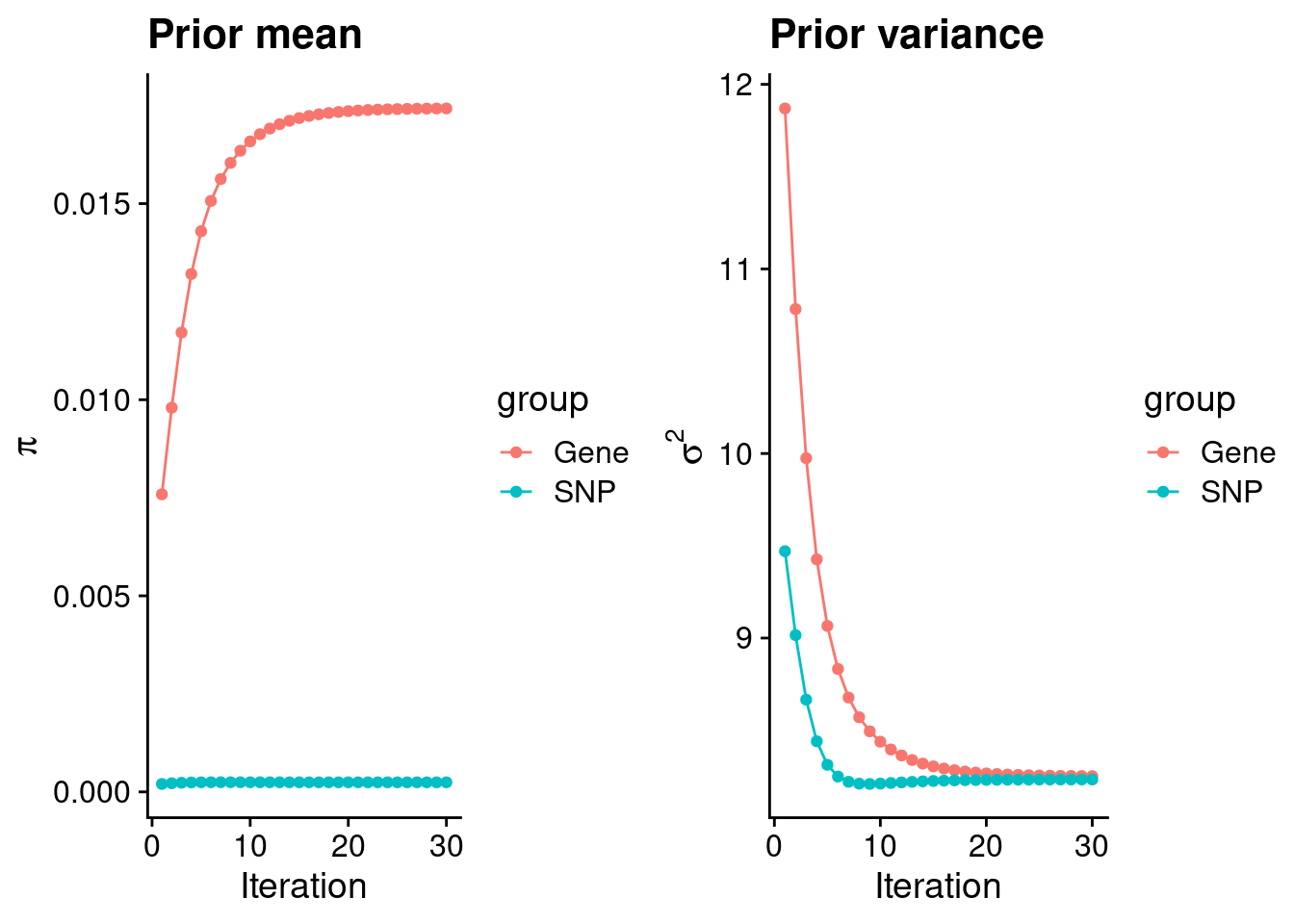
#estimated group prior
estimated_group_prior <- group_prior_rec[,ncol(group_prior_rec)]
names(estimated_group_prior) <- c("gene", "snp")
estimated_group_prior["snp"] <- estimated_group_prior["snp"]*thin #adjust parameter to account for thin argument
print(estimated_group_prior) gene snp
0.0174262 0.0002447 #estimated group prior variance
estimated_group_prior_var <- group_prior_var_rec[,ncol(group_prior_var_rec)]
names(estimated_group_prior_var) <- c("gene", "snp")
print(estimated_group_prior_var) gene snp
8.252 8.234 #report sample size
print(sample_size)[1] 77096#report group size
group_size <- c(nrow(ctwas_gene_res), n_snps)
print(group_size)[1] 10988 7352670#estimated group PVE
estimated_group_pve <- estimated_group_prior_var*estimated_group_prior*group_size/sample_size #check PVE calculation
names(estimated_group_pve) <- c("gene", "snp")
print(estimated_group_pve) gene snp
0.0205 0.1921 #compare sum(PIP*mu2/sample_size) with above PVE calculation
c(sum(ctwas_gene_res$PVE),sum(ctwas_snp_res$PVE))[1] 0.1006 1.6108Genes with highest PIPs
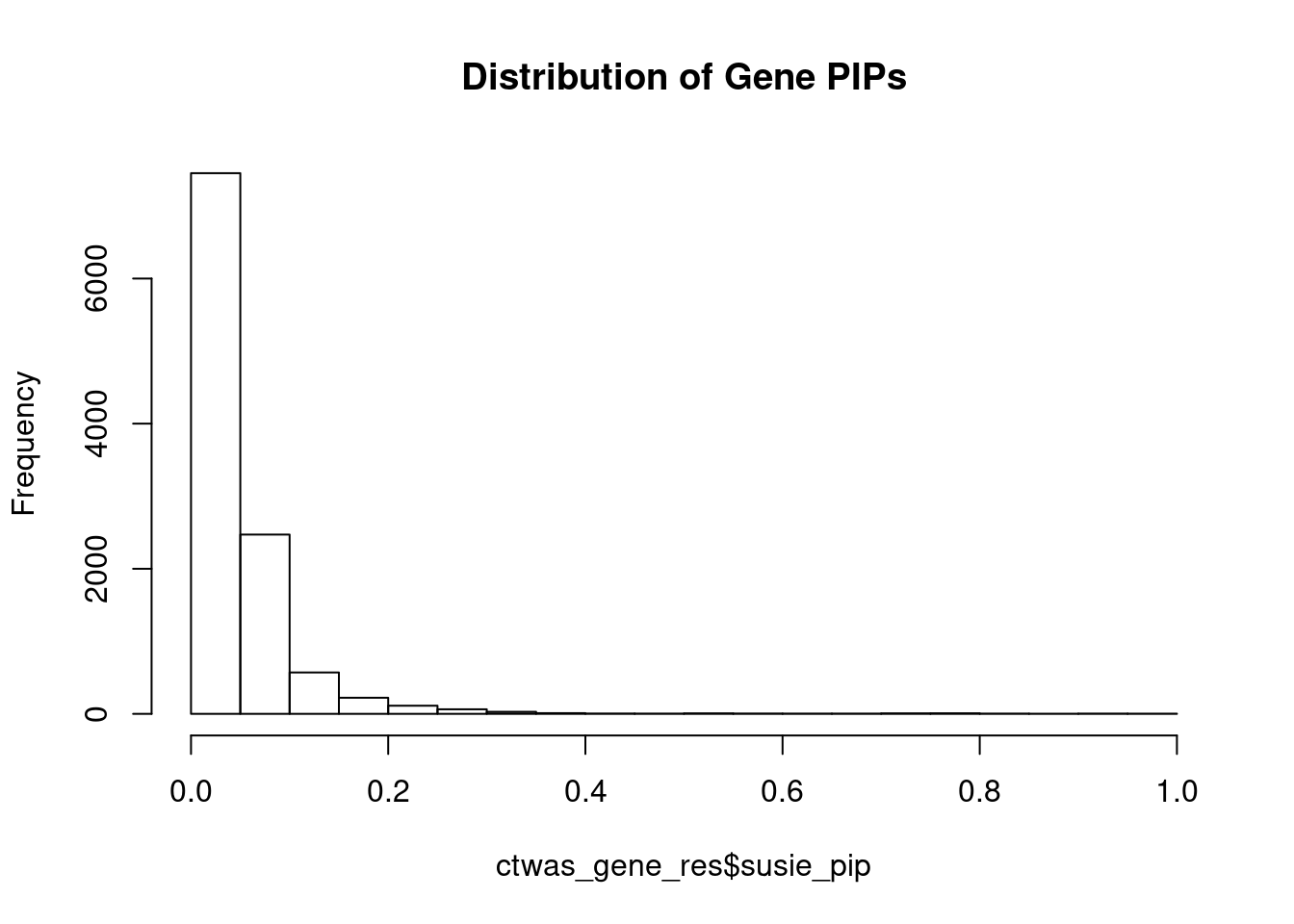
genename region_tag susie_pip mu2 PVE z num_eqtl
4143 FEZF1 7_74 0.9848 26.98 0.0003447 -5.272 1
10988 ZNF823 19_10 0.9837 28.72 0.0003664 5.472 2
6241 ARFGAP2 11_29 0.9557 24.78 0.0003072 4.839 1
2207 RUNDC3B 7_54 0.9489 24.43 0.0003006 5.373 1
12095 AC012074.2 2_15 0.9395 21.46 0.0002615 4.623 1
499 TRAPPC3 1_22 0.9258 25.13 0.0003018 5.058 1
5783 GALNT2 1_117 0.9192 23.44 0.0002795 4.843 1
11339 DISP3 1_8 0.8992 19.26 0.0002246 4.062 2
3099 SF3B1 2_117 0.8960 42.31 0.0004917 6.784 1
6920 CNNM4 2_57 0.8493 39.51 0.0004353 -4.793 1
4331 TRIM28 19_39 0.8229 20.97 0.0002238 4.253 2
13214 RP11-230C9.4 6_102 0.8150 19.57 0.0002069 -3.928 3
9329 DIRAS1 19_3 0.8104 20.61 0.0002166 -4.359 1
1148 RRN3 16_15 0.7966 20.46 0.0002114 -4.310 1
7481 SERPINI1 3_103 0.7816 19.25 0.0001951 -4.030 1
13246 RP1-224A6.9 1_15 0.7795 19.35 0.0001957 -4.000 1
9372 LY6H 8_94 0.7777 21.16 0.0002135 4.236 1
3842 ABCC10 6_33 0.7726 21.94 0.0002199 -4.790 2
174 ZNF207 17_19 0.7610 20.37 0.0002011 4.200 1
4509 REEP2 5_82 0.7590 22.70 0.0002235 4.541 2Genes with largest effect sizes

genename region_tag susie_pip mu2 PVE z num_eqtl
12178 HLA-DQA2 6_26 0.000e+00 276.24 0.000e+00 0.7376 1
11986 CYP21A2 6_26 2.938e-07 215.77 8.222e-10 -7.7309 2
11296 C6orf48 6_26 2.065e-09 193.18 5.173e-12 8.9003 1
11553 CLIC1 6_26 1.193e-09 191.96 2.969e-12 8.8731 1
12355 C4A 6_26 3.523e-11 186.85 8.539e-14 8.5099 2
11554 DDAH2 6_26 0.000e+00 178.91 0.000e+00 7.6610 1
11298 HSPA1A 6_26 0.000e+00 158.24 0.000e+00 7.6575 1
9601 HLA-DQB1 6_26 0.000e+00 155.76 0.000e+00 -1.8861 1
6764 MMP16 8_63 0.000e+00 150.60 0.000e+00 -0.8409 1
2957 PCCB 3_84 0.000e+00 146.43 0.000e+00 -4.2854 1
855 PPP2R3A 3_84 0.000e+00 125.82 0.000e+00 4.1188 1
12887 CTA-254O6.1 7_54 3.569e-04 123.95 5.738e-07 -3.7199 2
11754 C4B 6_26 0.000e+00 114.25 0.000e+00 -6.9370 4
11281 RNF5 6_26 0.000e+00 108.76 0.000e+00 7.6002 2
2227 MPP6 7_21 4.725e-03 106.50 6.528e-06 -3.3024 1
11285 FKBPL 6_26 1.110e-16 104.82 1.510e-19 -4.2267 1
11078 HLA-DRB5 6_26 0.000e+00 103.53 0.000e+00 2.8311 1
10673 HLA-DRB1 6_26 0.000e+00 100.37 0.000e+00 2.4486 1
11279 NOTCH4 6_26 0.000e+00 93.85 0.000e+00 6.0856 2
11284 PRRT1 6_26 0.000e+00 91.07 0.000e+00 7.9069 1Genes with highest PVE
genename region_tag susie_pip mu2 PVE z num_eqtl
3099 SF3B1 2_117 0.8960 42.31 0.0004917 6.784 1
6920 CNNM4 2_57 0.8493 39.51 0.0004353 -4.793 1
5826 CIAO1 2_57 0.6905 43.68 0.0003912 -3.710 1
10988 ZNF823 19_10 0.9837 28.72 0.0003664 5.472 2
4143 FEZF1 7_74 0.9848 26.98 0.0003447 -5.272 1
12301 HLA-DMB 6_27 0.5417 47.15 0.0003313 -7.990 1
8510 INO80E 16_24 0.6705 36.20 0.0003148 6.230 1
1612 ZC3H7B 22_19 0.5687 42.56 0.0003140 4.922 1
6241 ARFGAP2 11_29 0.9557 24.78 0.0003072 4.839 1
499 TRAPPC3 1_22 0.9258 25.13 0.0003018 5.058 1
2207 RUNDC3B 7_54 0.9489 24.43 0.0003006 5.373 1
5783 GALNT2 1_117 0.9192 23.44 0.0002795 4.843 1
12095 AC012074.2 2_15 0.9395 21.46 0.0002615 4.623 1
9199 ATG13 11_28 0.5211 34.13 0.0002307 -6.084 1
5875 FAM134A 2_129 0.7209 24.45 0.0002286 -4.940 1
11339 DISP3 1_8 0.8992 19.26 0.0002246 4.062 2
4331 TRIM28 19_39 0.8229 20.97 0.0002238 4.253 2
4509 REEP2 5_82 0.7590 22.70 0.0002235 4.541 2
3842 ABCC10 6_33 0.7726 21.94 0.0002199 -4.790 2
9329 DIRAS1 19_3 0.8104 20.61 0.0002166 -4.359 1Genes with largest z scores
genename region_tag susie_pip mu2 PVE z num_eqtl
11884 HCG11 6_20 2.774e-02 59.08 2.126e-05 8.937 1
12879 CTA-14H9.5 6_20 2.774e-02 59.08 2.126e-05 8.937 1
11296 C6orf48 6_26 2.065e-09 193.18 5.173e-12 8.900 1
11553 CLIC1 6_26 1.193e-09 191.96 2.969e-12 8.873 1
2870 PRSS16 6_21 5.521e-02 41.38 2.963e-05 -8.631 1
5086 PGBD1 6_22 1.341e-02 74.86 1.302e-05 -8.525 1
12355 C4A 6_26 3.523e-11 186.85 8.539e-14 8.510 2
12301 HLA-DMB 6_27 5.417e-01 47.15 3.313e-04 -7.990 1
6038 ABT1 6_20 4.778e-02 47.51 2.945e-05 7.913 1
11284 PRRT1 6_26 0.000e+00 91.07 0.000e+00 7.907 1
6221 CNNM2 10_66 2.427e-01 38.62 1.216e-04 -7.876 1
11986 CYP21A2 6_26 2.938e-07 215.77 8.222e-10 -7.731 2
11554 DDAH2 6_26 0.000e+00 178.91 0.000e+00 7.661 1
11298 HSPA1A 6_26 0.000e+00 158.24 0.000e+00 7.658 1
11281 RNF5 6_26 0.000e+00 108.76 0.000e+00 7.600 2
10845 ZSCAN26 6_22 1.980e-02 38.58 9.907e-06 7.054 2
11754 C4B 6_26 0.000e+00 114.25 0.000e+00 -6.937 4
10334 BTN3A2 6_20 2.185e-01 48.42 1.372e-04 6.875 1
7375 TYW5 2_118 1.251e-01 38.28 6.214e-05 -6.844 2
3099 SF3B1 2_117 8.960e-01 42.31 4.917e-04 6.784 1Comparing z scores and PIPs

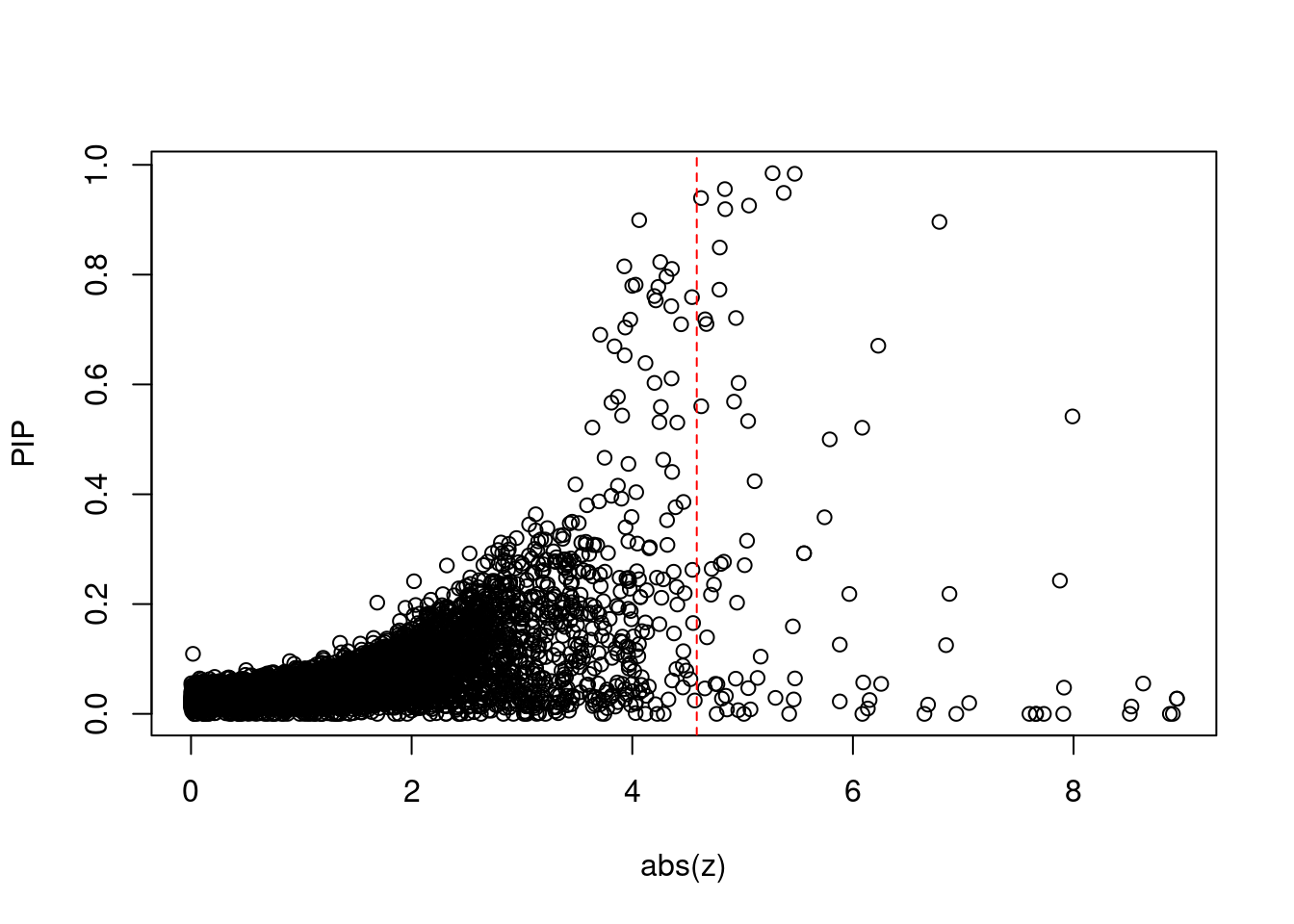
[1] 0.007372GO enrichment analysis for genes with PIP>0.5
#number of genes for gene set enrichment
length(genes)[1] 48Uploading data to Enrichr... Done.
Querying GO_Biological_Process_2021... Done.
Querying GO_Cellular_Component_2021... Done.
Querying GO_Molecular_Function_2021... Done.
Parsing results... Done.
[1] "GO_Biological_Process_2021"
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)
[1] "GO_Cellular_Component_2021"
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)
[1] "GO_Molecular_Function_2021"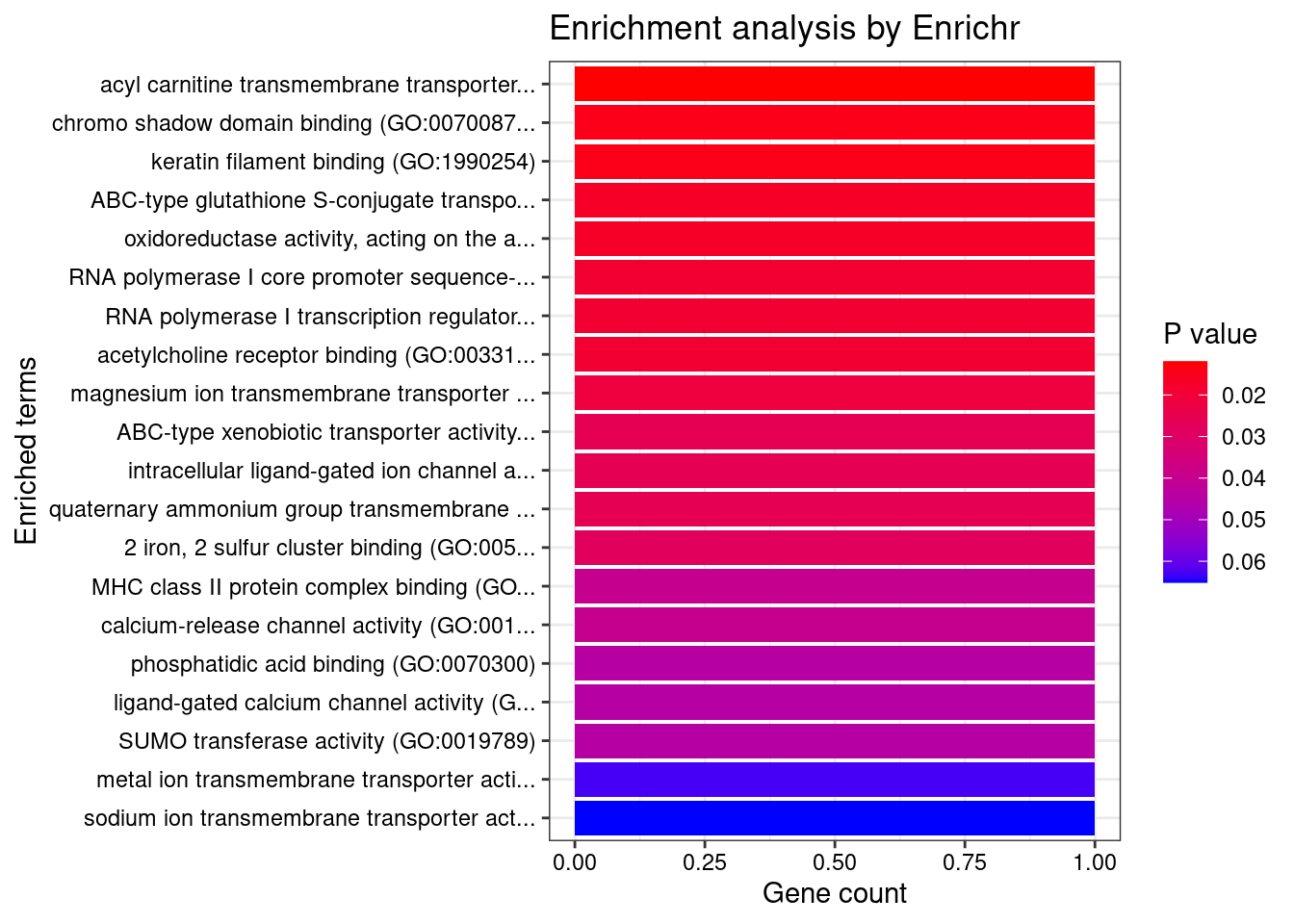
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)DisGeNET enrichment analysis for genes with PIP>0.5
Description FDR Ratio
17 Measles 0.01058 1/16
51 Amaurosis hypertrichosis 0.01058 1/16
52 Familial encephalopathy with neuroserpin inclusion bodies 0.01058 1/16
54 HEMOLYTIC UREMIC SYNDROME, ATYPICAL, SUSCEPTIBILITY TO, 2 0.01058 1/16
55 ALPHA-KETOGLUTARATE DEHYDROGENASE DEFICIENCY 0.01058 1/16
56 Cone rod dystrophy amelogenesis imperfecta 0.01058 1/16
58 JOUBERT SYNDROME 13 0.01058 1/16
61 Jalili syndrome 0.01058 1/16
67 SPASTIC PARAPLEGIA 72, AUTOSOMAL RECESSIVE 0.01058 1/16
68 SPASTIC PARAPLEGIA 72, AUTOSOMAL DOMINANT 0.01058 1/16
BgRatio
17 1/9703
51 1/9703
52 1/9703
54 1/9703
55 1/9703
56 1/9703
58 1/9703
61 1/9703
67 1/9703
68 1/9703WebGestalt enrichment analysis for genes with PIP>0.5
Loading the functional categories...
Loading the ID list...
Loading the reference list...
Performing the enrichment analysis...Warning in oraEnrichment(interestGeneList, referenceGeneList, geneSet, minNum =
minNum, : No significant gene set is identified based on FDR 0.05!NULLPIP Manhattan Plot
Warning: 'timedatectl' indicates the non-existent timezone name 'n/a'Warning: Your system is mis-configured: '/etc/localtime' is not a symlinkWarning: It is strongly recommended to set envionment variable TZ to 'America/
Chicago' (or equivalent)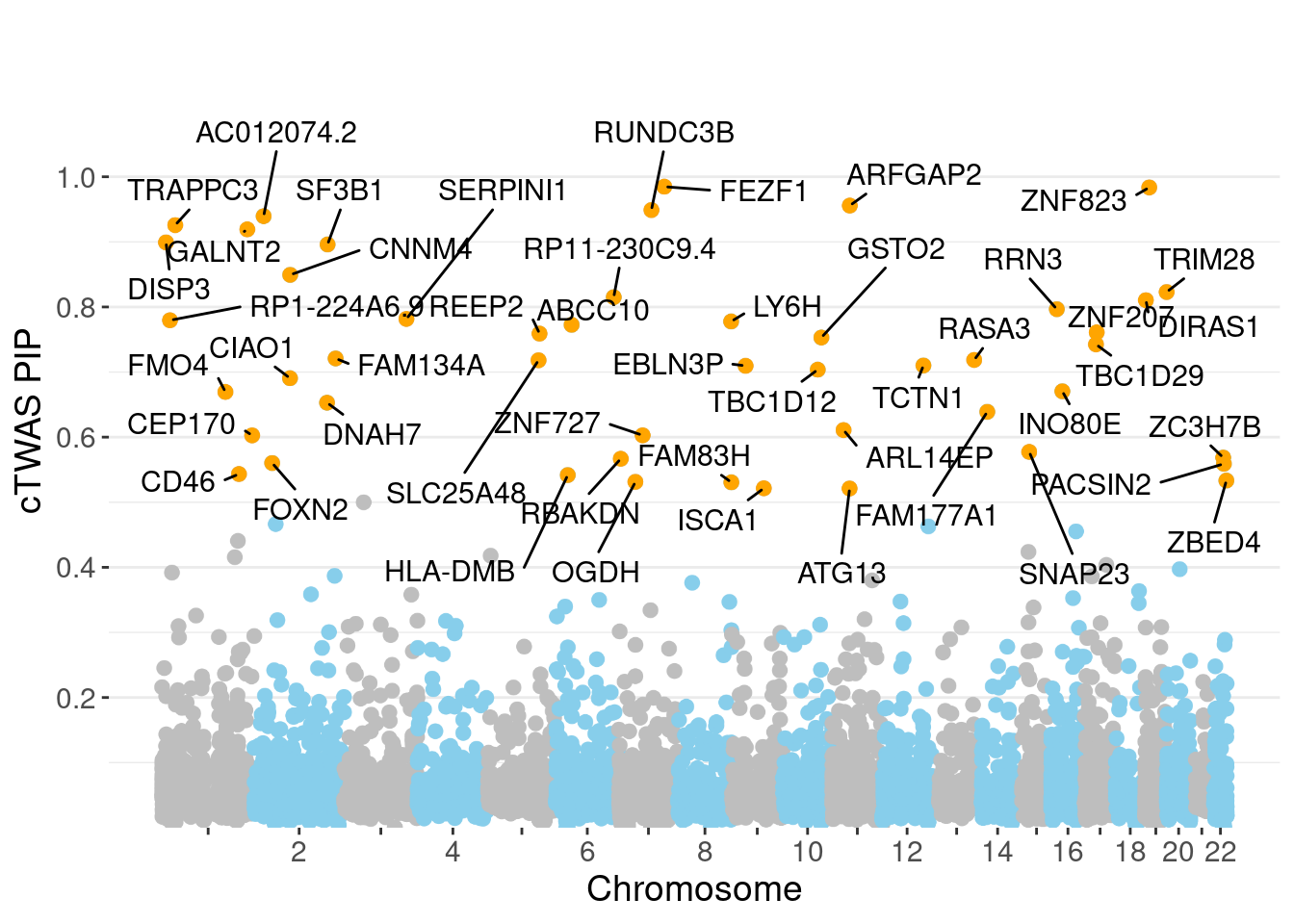
Sensitivity, specificity and precision for silver standard genes
#number of genes in known annotations
print(length(known_annotations))[1] 130#number of genes in known annotations with imputed expression
print(sum(known_annotations %in% ctwas_gene_res$genename))[1] 60#significance threshold for TWAS
print(sig_thresh)[1] 4.585#number of ctwas genes
length(ctwas_genes)[1] 13#number of TWAS genes
length(twas_genes)[1] 81#show novel genes (ctwas genes with not in TWAS genes)
ctwas_gene_res[ctwas_gene_res$genename %in% novel_genes,report_cols] genename region_tag susie_pip mu2 PVE z num_eqtl
11339 DISP3 1_8 0.8992 19.26 0.0002246 4.062 2
13214 RP11-230C9.4 6_102 0.8150 19.57 0.0002069 -3.928 3
9329 DIRAS1 19_3 0.8104 20.61 0.0002166 -4.359 1
4331 TRIM28 19_39 0.8229 20.97 0.0002238 4.253 2#sensitivity / recall
print(sensitivity) ctwas TWAS
0.01538 0.06154 #specificity
print(specificity) ctwas TWAS
0.9990 0.9933 #precision / PPV
print(precision) ctwas TWAS
0.15385 0.09877 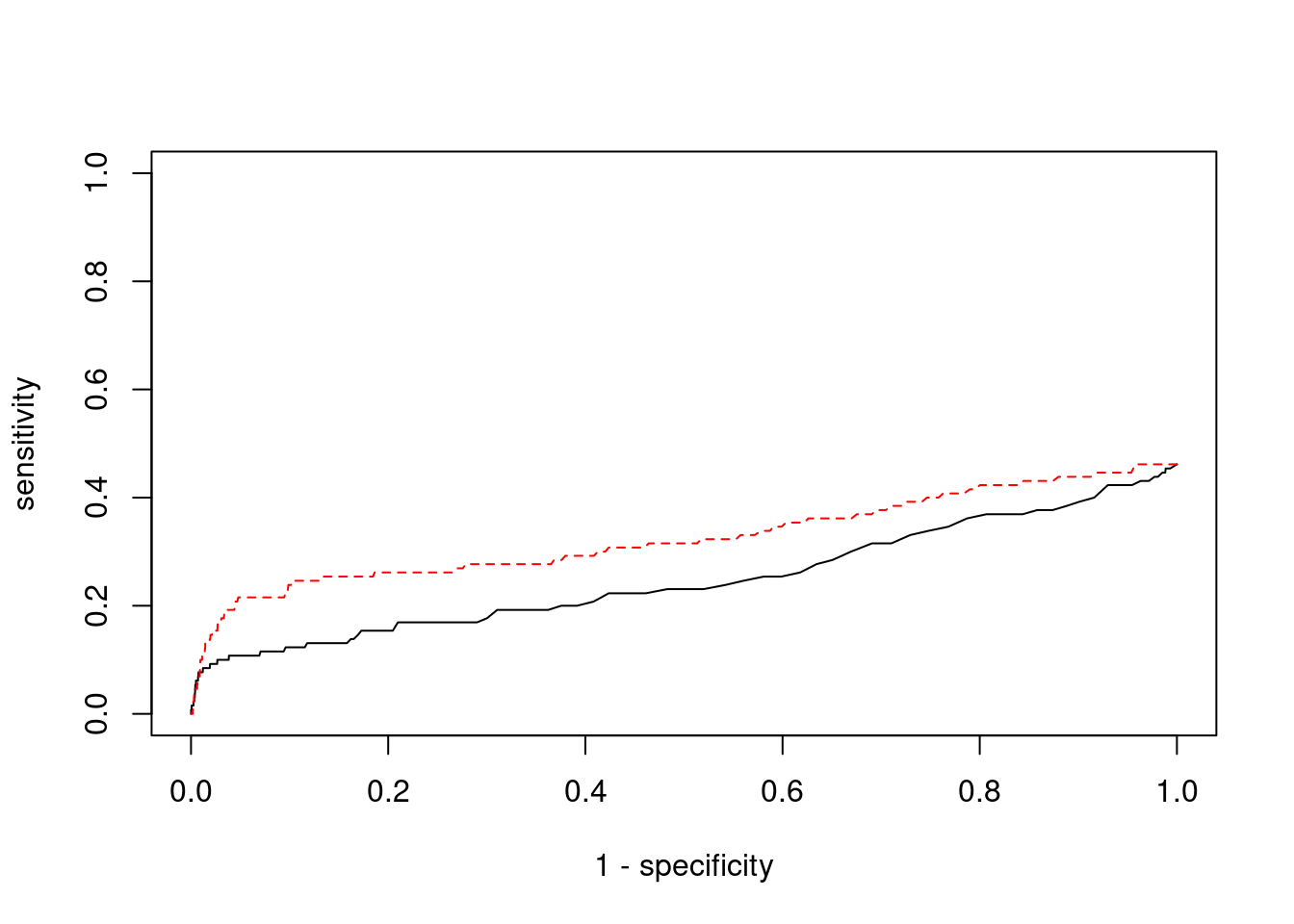
cTWAS is more precise than TWAS in distinguishing silver standard and bystander genes
#number of genes in known annotations (with imputed expression)
print(length(known_annotations))[1] 60#number of bystander genes (with imputed expression)
print(length(unrelated_genes))[1] 710#subset results to genes in known annotations or bystanders
ctwas_gene_res_subset <- ctwas_gene_res[ctwas_gene_res$genename %in% c(known_annotations, unrelated_genes),]
#assign ctwas and TWAS genes
ctwas_genes <- ctwas_gene_res_subset$genename[ctwas_gene_res_subset$susie_pip>0.8]
twas_genes <- ctwas_gene_res_subset$genename[abs(ctwas_gene_res_subset$z)>sig_thresh]
#significance threshold for TWAS
print(sig_thresh)[1] 4.585#number of ctwas genes (in known annotations or bystanders)
length(ctwas_genes)[1] 4#number of TWAS genes (in known annotations or bystanders)
length(twas_genes)[1] 24#sensitivity / recall
sensitivity ctwas TWAS
0.03333 0.13333 #specificity / (1 - False Positive Rate)
specificity ctwas TWAS
0.9972 0.9775 #precision / PPV / (1 - False Discovery Rate)
precision ctwas TWAS
0.5000 0.3333 

pip_range <- (0:1000)/1000
sensitivity <- rep(NA, length(pip_range))
specificity <- rep(NA, length(pip_range))
for (index in 1:length(pip_range)){
pip <- pip_range[index]
ctwas_genes <- ctwas_gene_res_subset$genename[ctwas_gene_res_subset$susie_pip>=pip]
sensitivity[index] <- sum(ctwas_genes %in% known_annotations)/length(known_annotations)
specificity[index] <- sum(!(unrelated_genes %in% ctwas_genes))/length(unrelated_genes)
}
plot(1-specificity, sensitivity, type="l", xlim=c(0,1), ylim=c(0,1), main="", xlab="1 - Specificity", ylab="Sensitivity")
title(expression("ROC Curve for cTWAS (black) and TWAS (" * phantom("red") * ")"))
title(expression(phantom("ROC Curve for cTWAS (black) and TWAS (") * "red" * phantom(")")), col.main="red")
sig_thresh_range <- seq(from=0, to=max(abs(ctwas_gene_res_subset$z)), length.out=length(pip_range))
for (index in 1:length(sig_thresh_range)){
sig_thresh_plot <- sig_thresh_range[index]
twas_genes <- ctwas_gene_res_subset$genename[abs(ctwas_gene_res_subset$z)>=sig_thresh_plot]
sensitivity[index] <- sum(twas_genes %in% known_annotations)/length(known_annotations)
specificity[index] <- sum(!(unrelated_genes %in% twas_genes))/length(unrelated_genes)
}
lines(1-specificity, sensitivity, xlim=c(0,1), ylim=c(0,1), col="red", lty=1)
abline(a=0,b=1,lty=3)
#add previously computed points from the analysis
ctwas_genes <- ctwas_gene_res_subset$genename[ctwas_gene_res_subset$susie_pip>0.8]
twas_genes <- ctwas_gene_res_subset$genename[abs(ctwas_gene_res_subset$z)>sig_thresh]
points(1-specificity_plot["ctwas"], sensitivity_plot["ctwas"], pch=21, bg="black")
points(1-specificity_plot["TWAS"], sensitivity_plot["TWAS"], pch=21, bg="red")
Undetected silver standard genes have low TWAS z-scores or stronger signal from nearby variants
#table of outcomes for silver standard genes
-sort(-table(silver_standard_case))silver_standard_case
Not Imputed Insignificant z-score Nearby SNP(s)
70 52 6
Detected (PIP > 0.8)
2 #show inconclusive genes
silver_standard_case[silver_standard_case=="Inconclusive"]named character(0)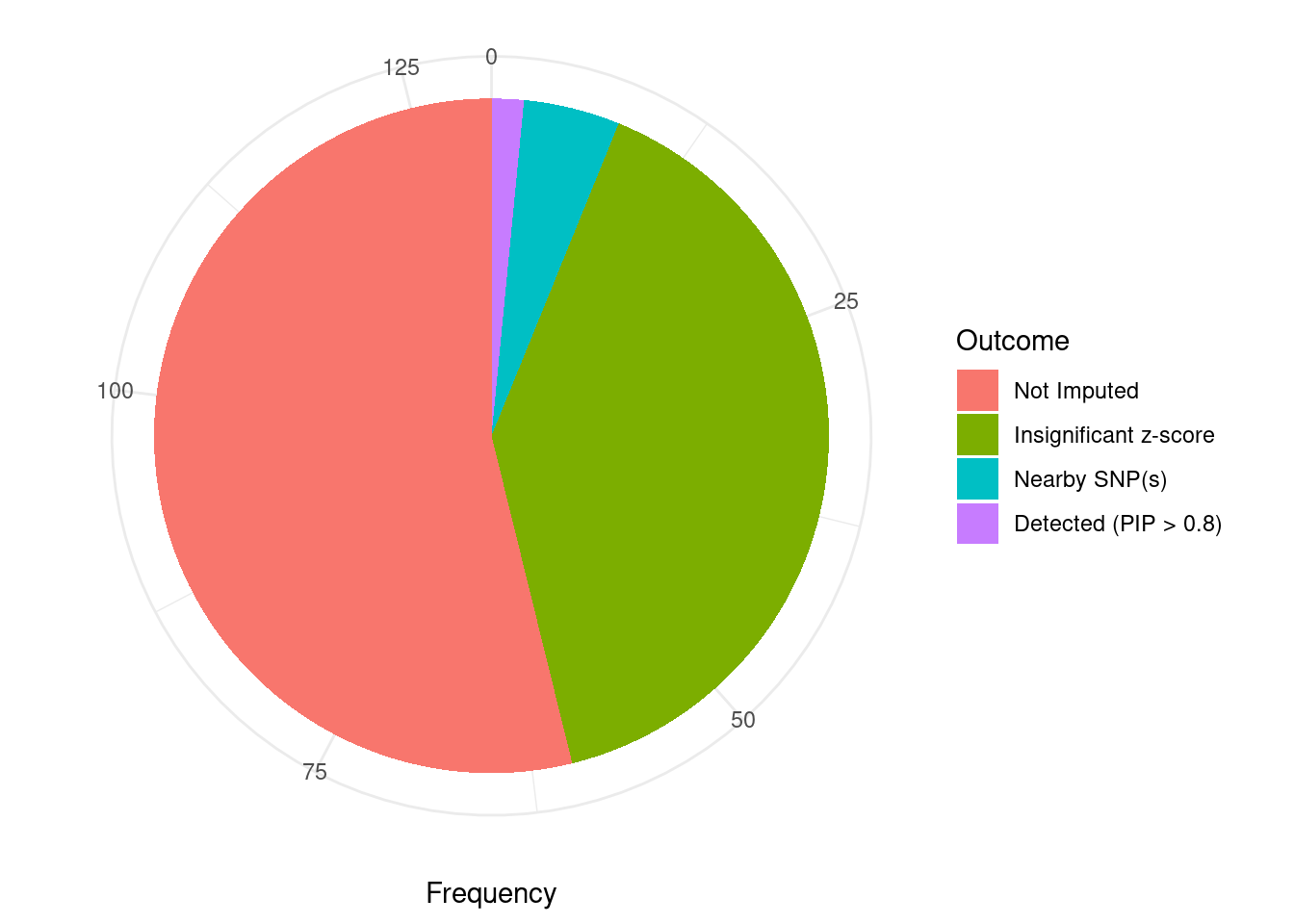
sessionInfo()R version 3.6.1 (2019-07-05)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] GenomicRanges_1.36.1 GenomeInfoDb_1.20.0 IRanges_2.18.1
[4] S4Vectors_0.22.1 BiocGenerics_0.30.0 biomaRt_2.40.1
[7] readxl_1.3.1 forcats_0.5.1 stringr_1.4.0
[10] dplyr_1.0.7 purrr_0.3.4 readr_2.1.1
[13] tidyr_1.1.4 tidyverse_1.3.1 tibble_3.1.6
[16] WebGestaltR_0.4.4 disgenet2r_0.99.2 enrichR_3.0
[19] cowplot_1.0.0 ggplot2_3.3.5 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] ggbeeswarm_0.6.0 colorspace_2.0-2 rjson_0.2.20
[4] ellipsis_0.3.2 rprojroot_2.0.2 XVector_0.24.0
[7] fs_1.5.2 rstudioapi_0.13 farver_2.1.0
[10] ggrepel_0.9.1 bit64_4.0.5 AnnotationDbi_1.46.0
[13] fansi_1.0.2 lubridate_1.8.0 xml2_1.3.3
[16] codetools_0.2-16 doParallel_1.0.17 cachem_1.0.6
[19] knitr_1.36 jsonlite_1.7.2 apcluster_1.4.8
[22] Cairo_1.5-12.2 broom_0.7.10 dbplyr_2.1.1
[25] compiler_3.6.1 httr_1.4.2 backports_1.4.1
[28] assertthat_0.2.1 Matrix_1.2-18 fastmap_1.1.0
[31] cli_3.1.0 later_0.8.0 prettyunits_1.1.1
[34] htmltools_0.5.2 tools_3.6.1 igraph_1.2.10
[37] GenomeInfoDbData_1.2.1 gtable_0.3.0 glue_1.6.2
[40] reshape2_1.4.4 doRNG_1.8.2 Rcpp_1.0.8
[43] Biobase_2.44.0 cellranger_1.1.0 jquerylib_0.1.4
[46] vctrs_0.3.8 svglite_1.2.2 iterators_1.0.14
[49] xfun_0.29 ps_1.6.0 rvest_1.0.2
[52] lifecycle_1.0.1 rngtools_1.5.2 XML_3.99-0.3
[55] zlibbioc_1.30.0 getPass_0.2-2 scales_1.1.1
[58] vroom_1.5.7 hms_1.1.1 promises_1.0.1
[61] yaml_2.2.1 curl_4.3.2 memoise_2.0.1
[64] ggrastr_1.0.1 gdtools_0.1.9 stringi_1.7.6
[67] RSQLite_2.2.8 highr_0.9 foreach_1.5.2
[70] rlang_1.0.1 pkgconfig_2.0.3 bitops_1.0-7
[73] evaluate_0.14 lattice_0.20-38 labeling_0.4.2
[76] bit_4.0.4 processx_3.5.2 tidyselect_1.1.1
[79] plyr_1.8.6 magrittr_2.0.2 R6_2.5.1
[82] generics_0.1.1 DBI_1.1.2 pillar_1.6.4
[85] haven_2.4.3 whisker_0.3-2 withr_2.4.3
[88] RCurl_1.98-1.5 modelr_0.1.8 crayon_1.5.0
[91] utf8_1.2.2 tzdb_0.2.0 rmarkdown_2.11
[94] progress_1.2.2 grid_3.6.1 data.table_1.14.2
[97] blob_1.2.2 callr_3.7.0 git2r_0.26.1
[100] reprex_2.0.1 digest_0.6.29 httpuv_1.5.1
[103] munsell_0.5.0 beeswarm_0.2.3 vipor_0.4.5