SCZ - Brain Hypothalamus
sheng Qian
2021-2-6
Last updated: 2022-03-14
Checks: 5 2
Knit directory: cTWAS_analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20211220) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| /project2/xinhe/shengqian/cTWAS/cTWAS_analysis/data/ | data |
| /project2/xinhe/shengqian/cTWAS/cTWAS_analysis/code/ctwas_config.R | code/ctwas_config.R |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 4c71b11. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .ipynb_checkpoints/
Ignored: data/AF/
Untracked files:
Untracked: Rplot.png
Untracked: analysis/.ipynb_checkpoints/
Untracked: analysis/SCZ_2014_EUR_Brain_Amygdala.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Anterior_cingulate_cortex_BA24.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Caudate_basal_ganglia.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Cerebellar_Hemisphere.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Cerebellum.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Cortex.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Frontal_Cortex_BA9.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Hippocampus.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Hypothalamus.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Nucleus_accumbens_basal_ganglia.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Putamen_basal_ganglia.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Spinal_cord_cervical_c-1.Rmd
Untracked: analysis/SCZ_2014_EUR_Brain_Substantia_nigra.Rmd
Untracked: analysis/SCZ_2020_Brain_Cortex.Rmd
Untracked: analysis/SCZ_2020_Brain_Frontal_Cortex_BA9.Rmd
Untracked: analysis/SCZ_2020_Brain_Hypothalamus.Rmd
Untracked: analysis/SCZ_2020_Brain_Putamen_basal_ganglia.Rmd
Untracked: analysis/SCZ_Cross_Tissue_Analysis.Rmd
Untracked: code/.ipynb_checkpoints/
Untracked: code/AF_out/
Untracked: code/Autism_out/
Untracked: code/BMI_S_out/
Untracked: code/BMI_out/
Untracked: code/Glucose_out/
Untracked: code/LDL_S_out/
Untracked: code/SCZ_2014_EUR_out/
Untracked: code/SCZ_2020_out/
Untracked: code/SCZ_S_out/
Untracked: code/SCZ_out/
Untracked: code/T2D_out/
Untracked: code/ctwas_config.R
Untracked: code/mapping.R
Untracked: code/out/
Untracked: code/run_AF_analysis.sbatch
Untracked: code/run_AF_analysis.sh
Untracked: code/run_AF_ctwas_rss_LDR.R
Untracked: code/run_Autism_analysis.sbatch
Untracked: code/run_Autism_analysis.sh
Untracked: code/run_Autism_ctwas_rss_LDR.R
Untracked: code/run_BMI_analysis.sbatch
Untracked: code/run_BMI_analysis.sh
Untracked: code/run_BMI_analysis_S.sbatch
Untracked: code/run_BMI_analysis_S.sh
Untracked: code/run_BMI_ctwas_rss_LDR.R
Untracked: code/run_BMI_ctwas_rss_LDR_S.R
Untracked: code/run_Glucose_analysis.sbatch
Untracked: code/run_Glucose_analysis.sh
Untracked: code/run_Glucose_ctwas_rss_LDR.R
Untracked: code/run_LDL_analysis_S.sbatch
Untracked: code/run_LDL_analysis_S.sh
Untracked: code/run_LDL_ctwas_rss_LDR_S.R
Untracked: code/run_SCZ_2014_EUR_analysis.sbatch
Untracked: code/run_SCZ_2014_EUR_analysis.sh
Untracked: code/run_SCZ_2014_EUR_ctwas_rss_LDR.R
Untracked: code/run_SCZ_2020_analysis.sbatch
Untracked: code/run_SCZ_2020_analysis.sh
Untracked: code/run_SCZ_2020_ctwas_rss_LDR.R
Untracked: code/run_SCZ_analysis.sbatch
Untracked: code/run_SCZ_analysis.sh
Untracked: code/run_SCZ_analysis_S.sbatch
Untracked: code/run_SCZ_analysis_S.sh
Untracked: code/run_SCZ_ctwas_rss_LDR.R
Untracked: code/run_SCZ_ctwas_rss_LDR_S.R
Untracked: code/run_T2D_analysis.sbatch
Untracked: code/run_T2D_analysis.sh
Untracked: code/run_T2D_ctwas_rss_LDR.R
Untracked: code/wflow_build.R
Untracked: code/wflow_build.sbatch
Untracked: data/.ipynb_checkpoints/
Untracked: data/BMI/
Untracked: data/PGC3_SCZ_wave3_public.v2.tsv
Untracked: data/SCZ/
Untracked: data/SCZ_2014_EUR/
Untracked: data/SCZ_2020/
Untracked: data/SCZ_S/
Untracked: data/T2D/
Untracked: data/UKBB/
Untracked: data/UKBB_SNPs_Info.text
Untracked: data/gene_OMIM.txt
Untracked: data/gene_pip_0.8.txt
Untracked: data/mashr_Heart_Atrial_Appendage.db
Untracked: data/mashr_sqtl/
Untracked: data/summary_known_genes_annotations.xlsx
Untracked: data/untitled.txt
Unstaged changes:
Modified: analysis/SCZ_Brain_Amygdala.Rmd
Modified: analysis/SCZ_Brain_Anterior_cingulate_cortex_BA24.Rmd
Modified: analysis/SCZ_Brain_Caudate_basal_ganglia.Rmd
Modified: analysis/SCZ_Brain_Cerebellar_Hemisphere.Rmd
Modified: analysis/SCZ_Brain_Cerebellum.Rmd
Modified: analysis/SCZ_Brain_Cortex.Rmd
Modified: analysis/SCZ_Brain_Frontal_Cortex_BA9.Rmd
Modified: analysis/SCZ_Brain_Hippocampus.Rmd
Modified: analysis/SCZ_Brain_Hypothalamus.Rmd
Modified: analysis/SCZ_Brain_Nucleus_accumbens_basal_ganglia.Rmd
Modified: analysis/SCZ_Brain_Putamen_basal_ganglia.Rmd
Modified: analysis/SCZ_Brain_Spinal_cord_cervical_c-1.Rmd
Modified: analysis/SCZ_Brain_Substantia_nigra.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with wflow_publish() to start tracking its development.
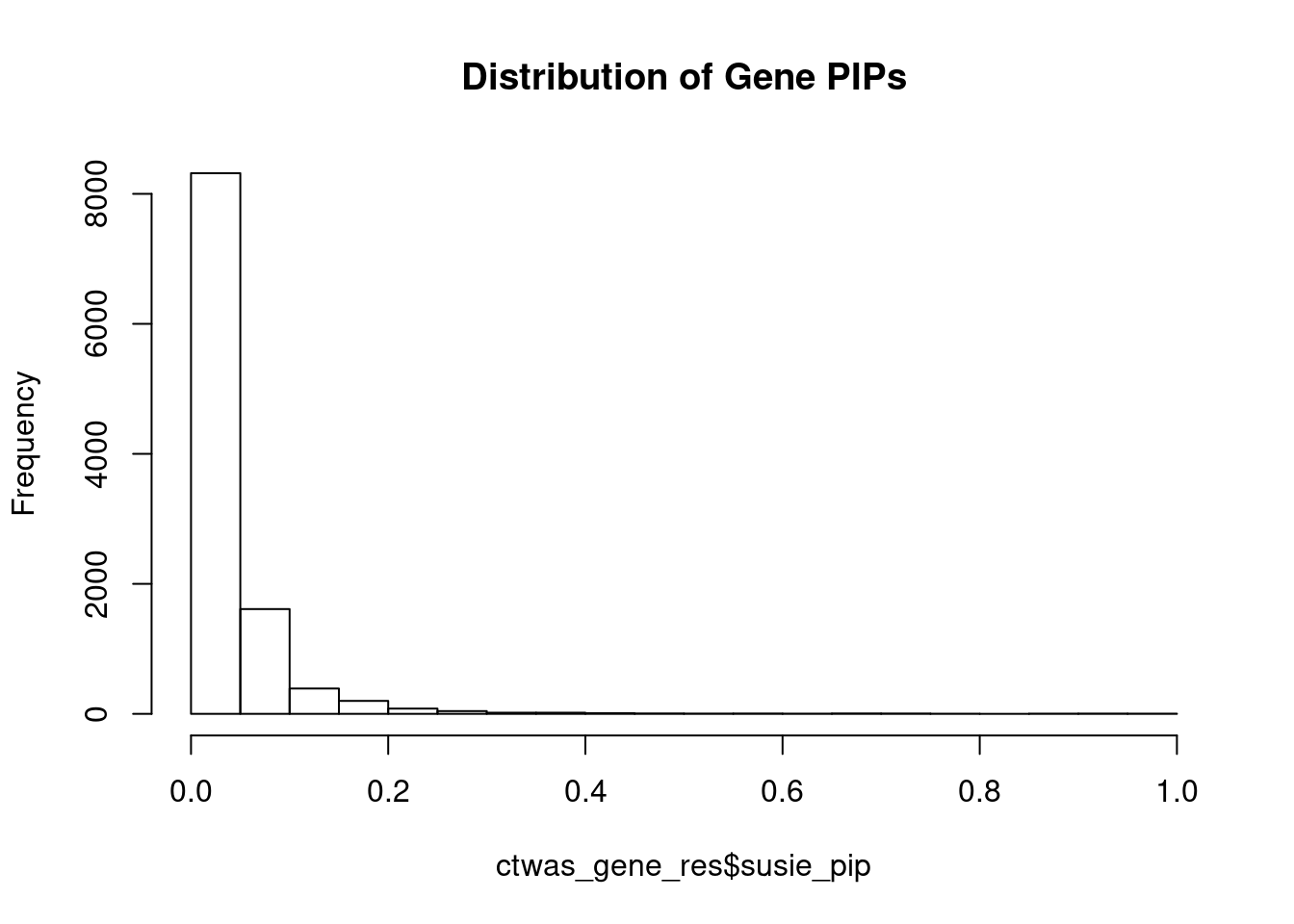
Weight QC
#number of imputed weights
nrow(qclist_all)[1] 10731#number of imputed weights by chromosome
table(qclist_all$chr)
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
1062 739 632 417 549 608 523 418 403 413 628 615 202 333 355 476
17 18 19 20 21 22
654 162 826 321 130 265 #number of imputed weights without missing variants
sum(qclist_all$nmiss==0)[1] 8242#proportion of imputed weights without missing variants
mean(qclist_all$nmiss==0)[1] 0.7681Check convergence of parameters

#estimated group prior
estimated_group_prior <- group_prior_rec[,ncol(group_prior_rec)]
names(estimated_group_prior) <- c("gene", "snp")
estimated_group_prior["snp"] <- estimated_group_prior["snp"]*thin #adjust parameter to account for thin argument
print(estimated_group_prior) gene snp
0.0150830 0.0002553 #estimated group prior variance
estimated_group_prior_var <- group_prior_var_rec[,ncol(group_prior_var_rec)]
names(estimated_group_prior_var) <- c("gene", "snp")
print(estimated_group_prior_var) gene snp
10.532 8.151 #report sample size
print(sample_size)[1] 77096#report group size
group_size <- c(nrow(ctwas_gene_res), n_snps)
print(group_size)[1] 10731 7352670#estimated group PVE
estimated_group_pve <- estimated_group_prior_var*estimated_group_prior*group_size/sample_size #check PVE calculation
names(estimated_group_pve) <- c("gene", "snp")
print(estimated_group_pve) gene snp
0.02211 0.19844 #compare sum(PIP*mu2/sample_size) with above PVE calculation
c(sum(ctwas_gene_res$PVE),sum(ctwas_snp_res$PVE))[1] 0.09444 1.71361Genes with highest PIPs
genename region_tag susie_pip mu2 PVE z num_eqtl
3993 SPECC1 17_16 0.9999 123.23 0.0015982 5.426 2
5324 FURIN 15_42 0.9884 46.00 0.0005898 -7.000 1
10843 ZNF823 19_10 0.9861 29.80 0.0003811 5.502 2
7382 THOC7 3_43 0.9389 37.10 0.0004518 -6.186 1
11997 AC012074.2 2_15 0.9381 22.10 0.0002689 4.620 2
3112 MAP7D1 1_22 0.9365 25.82 0.0003136 5.058 1
2970 SF3B1 2_117 0.9253 44.84 0.0005382 6.784 1
1089 RRN3 16_15 0.8896 24.58 0.0002836 -4.689 2
9203 DIRAS1 19_3 0.8869 22.06 0.0002538 4.504 2
10699 PCBP2 12_33 0.8826 21.89 0.0002506 4.496 1
7356 SERPINI1 3_103 0.7666 19.82 0.0001970 -4.030 1
11817 LINC00242 6_112 0.7600 20.38 0.0002009 3.871 2
1518 ZC3H7B 22_17 0.7435 43.72 0.0004217 4.922 1
2312 TBC1D12 10_60 0.7430 19.56 0.0001885 3.936 1
10218 TMEM222 1_19 0.7352 22.45 0.0002141 3.902 1
3496 TBC1D15 12_44 0.7305 22.01 0.0002086 4.436 2
1685 PPP1R16B 20_23 0.7231 34.83 0.0003267 6.009 1
3367 HELLS 10_61 0.6940 19.90 0.0001791 -3.886 1
394 CTNNA1 5_82 0.6780 23.71 0.0002085 4.946 1
6111 ARFGAP2 11_29 0.6708 25.03 0.0002177 4.839 1Genes with largest effect sizes
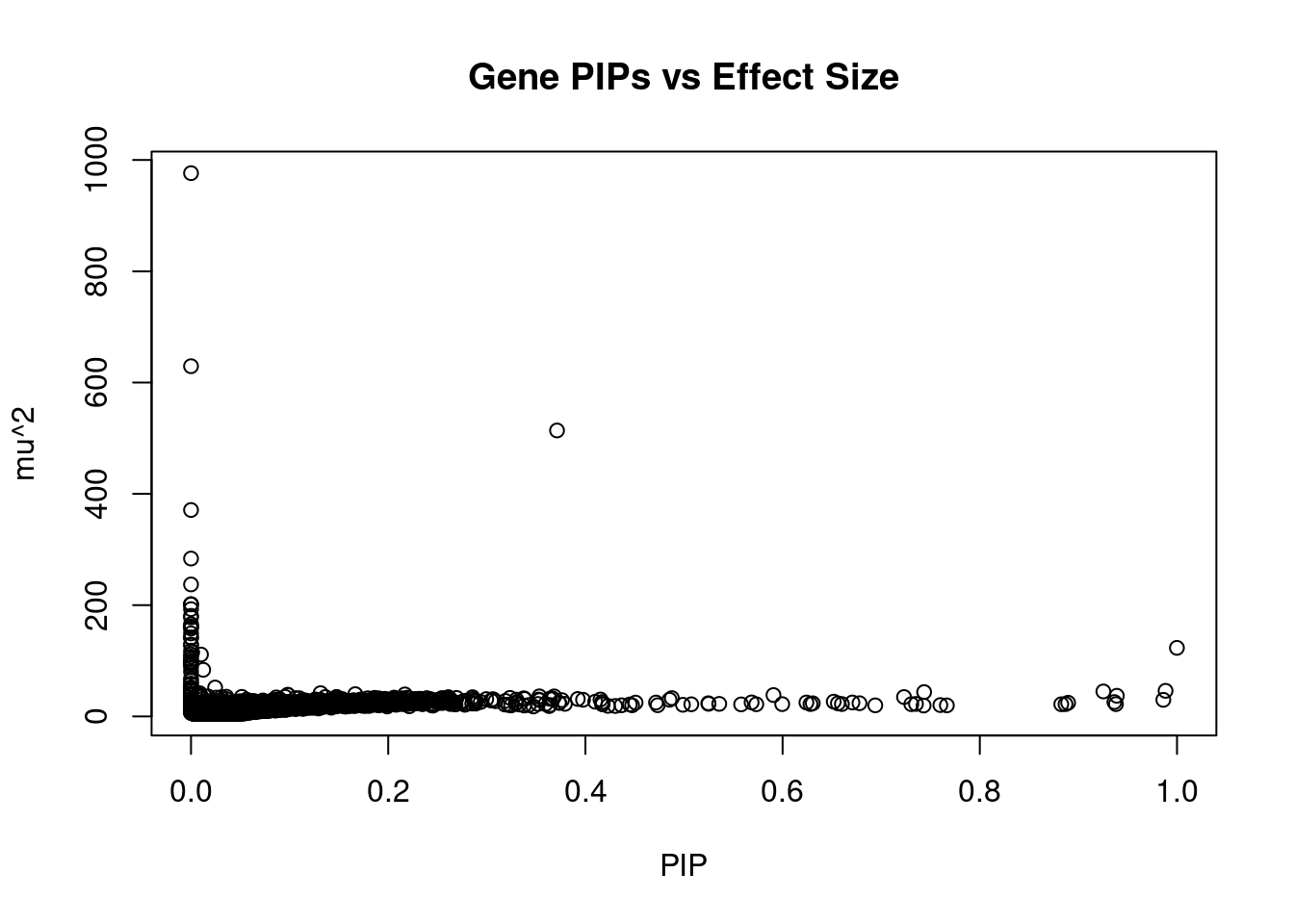
genename region_tag susie_pip mu2 PVE z num_eqtl
11948 HIST1H2BN 6_21 1.479e-06 976.3 1.873e-08 10.7729 1
10837 HIST1H2BK 6_21 0.000e+00 629.4 0.000e+00 -5.7119 1
2829 PCCB 3_84 3.712e-01 513.9 2.474e-03 -6.2860 1
2736 PRSS16 6_21 2.331e-14 371.0 1.122e-16 -8.6315 1
12073 HLA-DQA2 6_26 0.000e+00 283.7 0.000e+00 0.6583 1
13174 RP1-153G14.4 6_21 0.000e+00 237.2 0.000e+00 0.5579 2
11174 APOM 6_26 1.162e-08 201.7 3.039e-11 8.9450 1
11166 MSH5 6_26 8.766e-09 201.4 2.290e-11 8.8864 2
11169 ABHD16A 6_26 9.621e-09 201.3 2.513e-11 8.9341 1
12252 C4A 6_26 2.186e-10 192.8 5.465e-13 8.4450 1
11176 BAG6 6_26 4.441e-16 180.8 1.041e-18 5.6969 2
11172 GPANK1 6_26 4.996e-14 177.9 1.153e-16 7.9727 1
11162 HSPA1A 6_26 0.000e+00 165.7 0.000e+00 7.6575 1
9485 HLA-DQB1 6_26 0.000e+00 162.1 0.000e+00 -1.8861 1
10645 HLA-DQA1 6_26 2.220e-16 161.0 4.638e-19 -1.1464 2
11419 DDAH2 6_26 0.000e+00 157.9 0.000e+00 6.9659 2
13272 RP1-86C11.7 6_21 0.000e+00 149.2 0.000e+00 1.6735 1
9850 GRIN2A 16_10 7.518e-07 142.0 1.385e-09 -0.7161 2
10534 HLA-DRB1 6_26 0.000e+00 140.9 0.000e+00 5.1480 1
806 PPP2R3A 3_84 0.000e+00 129.0 0.000e+00 4.1188 1Genes with highest PVE
genename region_tag susie_pip mu2 PVE z num_eqtl
2829 PCCB 3_84 0.3712 513.88 0.0024743 -6.286 1
3993 SPECC1 17_16 0.9999 123.23 0.0015982 5.426 2
5324 FURIN 15_42 0.9884 46.00 0.0005898 -7.000 1
2970 SF3B1 2_117 0.9253 44.84 0.0005382 6.784 1
7382 THOC7 3_43 0.9389 37.10 0.0004518 -6.186 1
1518 ZC3H7B 22_17 0.7435 43.72 0.0004217 4.922 1
10843 ZNF823 19_10 0.9861 29.80 0.0003811 5.502 2
1685 PPP1R16B 20_23 0.7231 34.83 0.0003267 6.009 1
3112 MAP7D1 1_22 0.9365 25.82 0.0003136 5.058 1
2505 MDK 11_28 0.5907 38.37 0.0002940 -6.344 1
1089 RRN3 16_15 0.8896 24.58 0.0002836 -4.689 2
11997 AC012074.2 2_15 0.9381 22.10 0.0002689 4.620 2
9203 DIRAS1 19_3 0.8869 22.06 0.0002538 4.504 2
10699 PCBP2 12_33 0.8826 21.89 0.0002506 4.496 1
3041 ALMS1 2_48 0.6519 26.55 0.0002245 -5.177 1
6111 ARFGAP2 11_29 0.6708 25.03 0.0002177 4.839 1
10218 TMEM222 1_19 0.7352 22.45 0.0002141 3.902 1
3496 TBC1D15 12_44 0.7305 22.01 0.0002086 4.436 2
394 CTNNA1 5_82 0.6780 23.71 0.0002085 4.946 1
7454 PBRM1 3_36 0.4876 32.88 0.0002080 -5.790 1Genes with largest z scores
genename region_tag susie_pip mu2 PVE z num_eqtl
11948 HIST1H2BN 6_21 1.479e-06 976.31 1.873e-08 10.773 1
11663 LINC01623 6_22 1.009e-02 111.21 1.456e-05 -10.503 1
11174 APOM 6_26 1.162e-08 201.65 3.039e-11 8.945 1
11169 ABHD16A 6_26 9.621e-09 201.35 2.513e-11 8.934 1
11166 MSH5 6_26 8.766e-09 201.39 2.290e-11 8.886 2
10655 ZSCAN16 6_22 1.234e-02 83.76 1.341e-05 -8.813 1
2736 PRSS16 6_21 2.331e-14 370.96 1.122e-16 -8.631 1
12252 C4A 6_26 2.186e-10 192.77 5.465e-13 8.445 1
9592 HIST1H2BC 6_20 2.446e-02 51.90 1.647e-05 -7.978 1
11172 GPANK1 6_26 4.996e-14 177.89 1.153e-16 7.973 1
11142 RNF5 6_26 0.000e+00 95.74 0.000e+00 7.921 1
11162 HSPA1A 6_26 0.000e+00 165.65 0.000e+00 7.658 1
5324 FURIN 15_42 9.884e-01 46.00 5.898e-04 -7.000 1
11419 DDAH2 6_26 0.000e+00 157.89 0.000e+00 6.966 2
2970 SF3B1 2_117 9.253e-01 44.84 5.382e-04 6.784 1
11139 NOTCH4 6_26 0.000e+00 96.82 0.000e+00 6.623 3
1571 ZFYVE21 14_54 1.666e-01 39.99 8.642e-05 -6.569 1
3855 XRCC3 14_54 1.314e-01 41.74 7.114e-05 6.526 1
10694 ZSCAN26 6_22 1.708e-02 35.98 7.970e-06 6.523 1
11158 SLC44A4 6_26 0.000e+00 92.71 0.000e+00 6.361 1Comparing z scores and PIPs
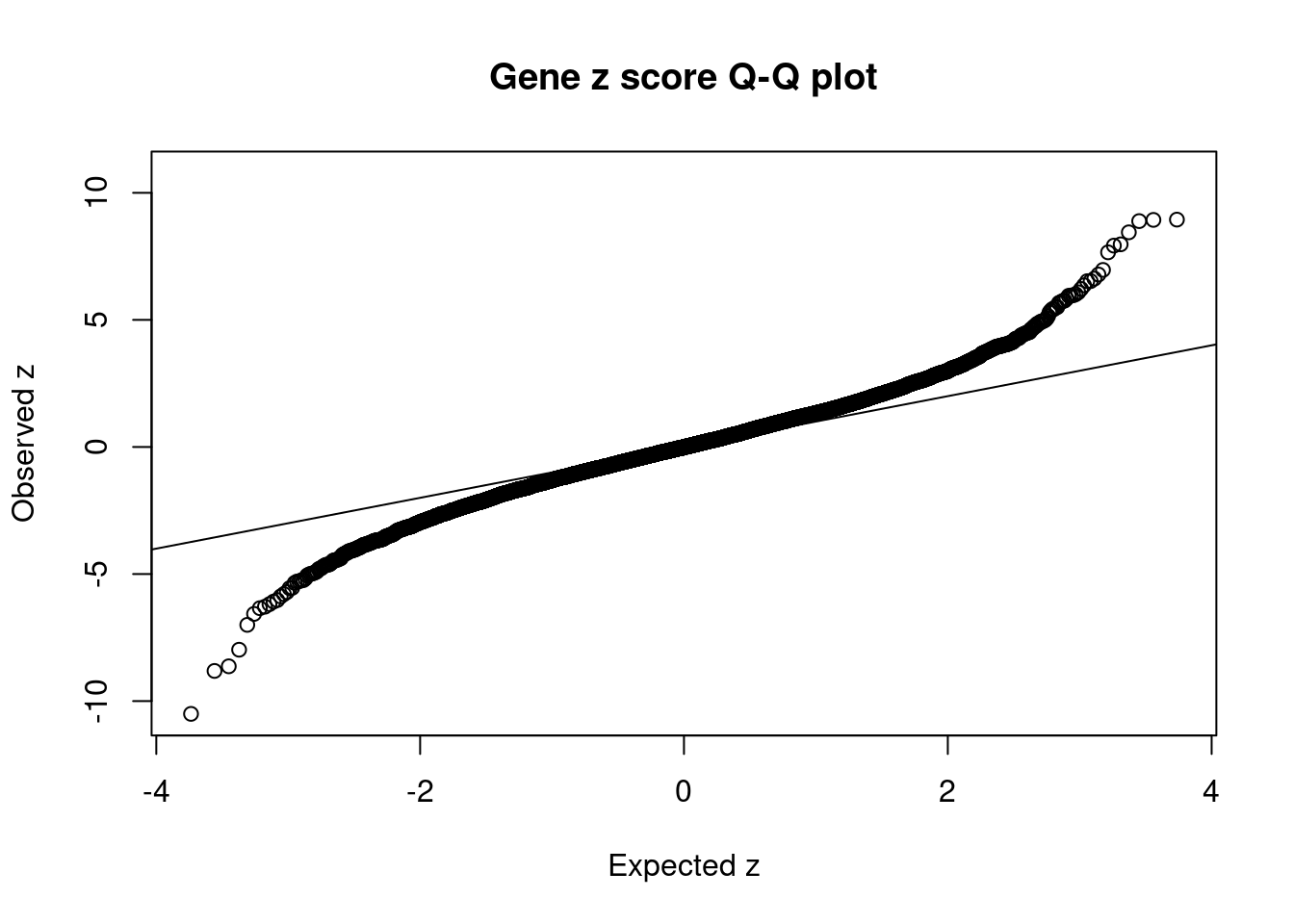
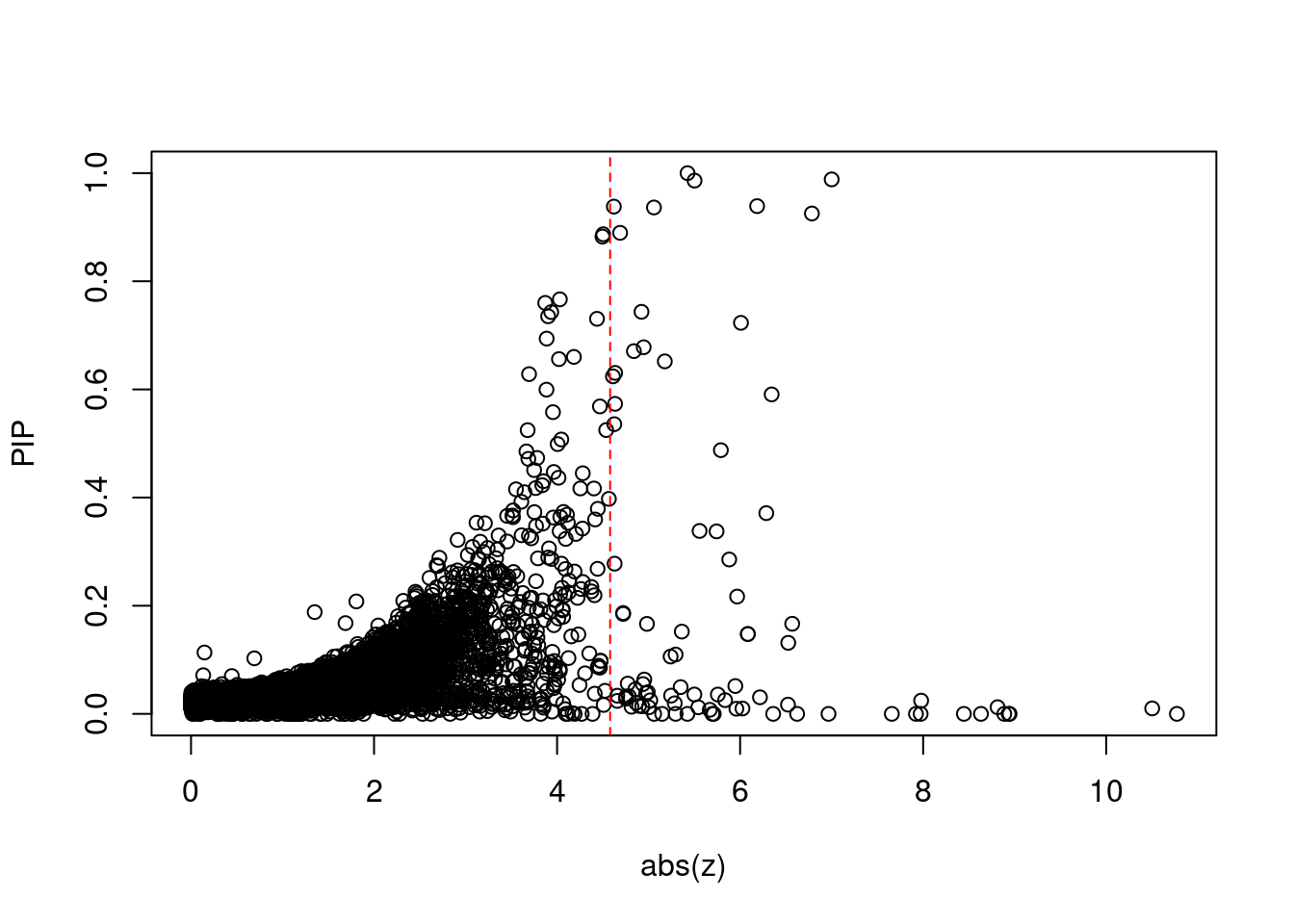
[1] 0.008014GO enrichment analysis for genes with PIP>0.5
#number of genes for gene set enrichment
length(genes)[1] 35Uploading data to Enrichr... Done.
Querying GO_Biological_Process_2021... Done.
Querying GO_Cellular_Component_2021... Done.
Querying GO_Molecular_Function_2021... Done.
Parsing results... Done.
[1] "GO_Biological_Process_2021"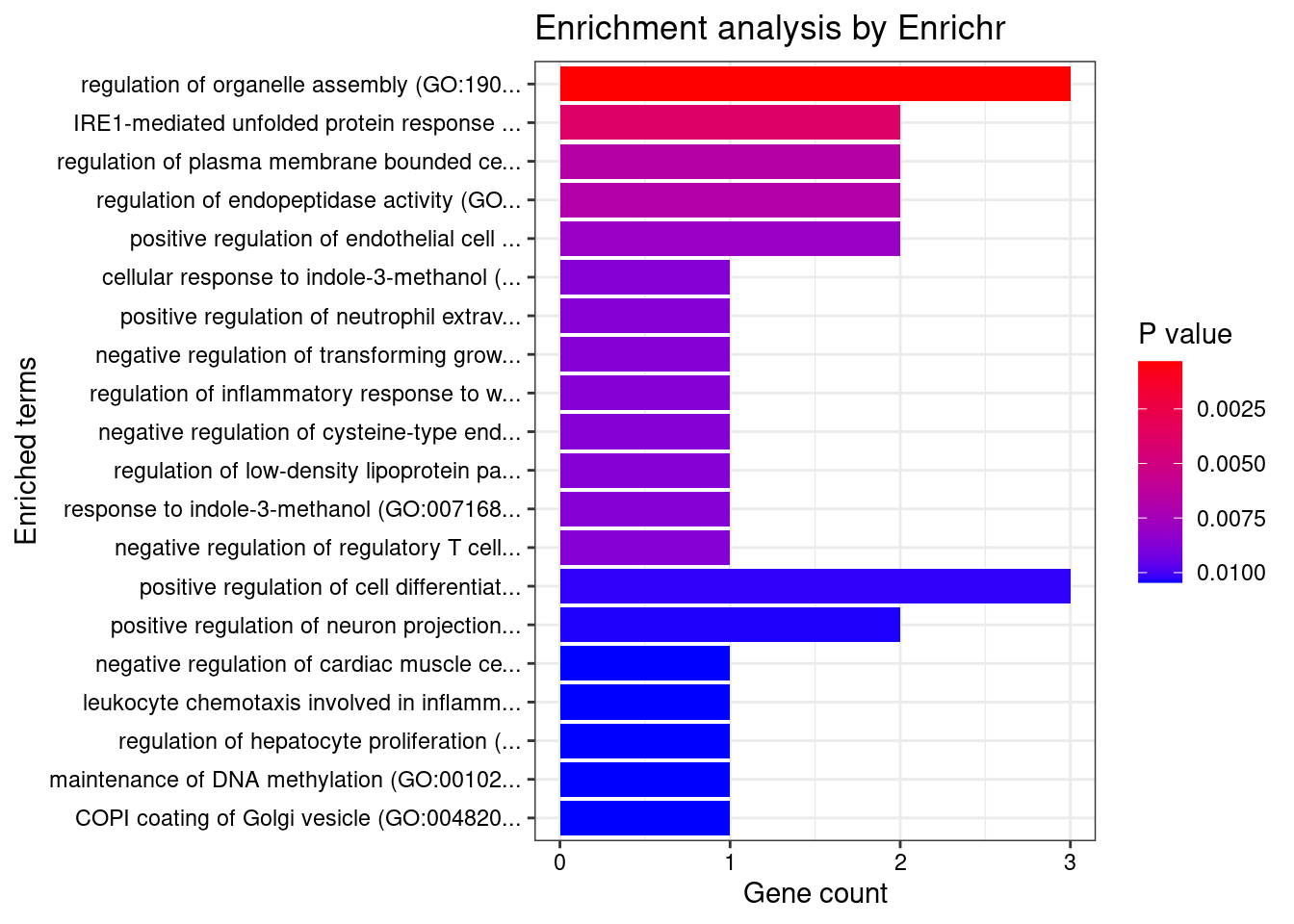
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)
[1] "GO_Cellular_Component_2021"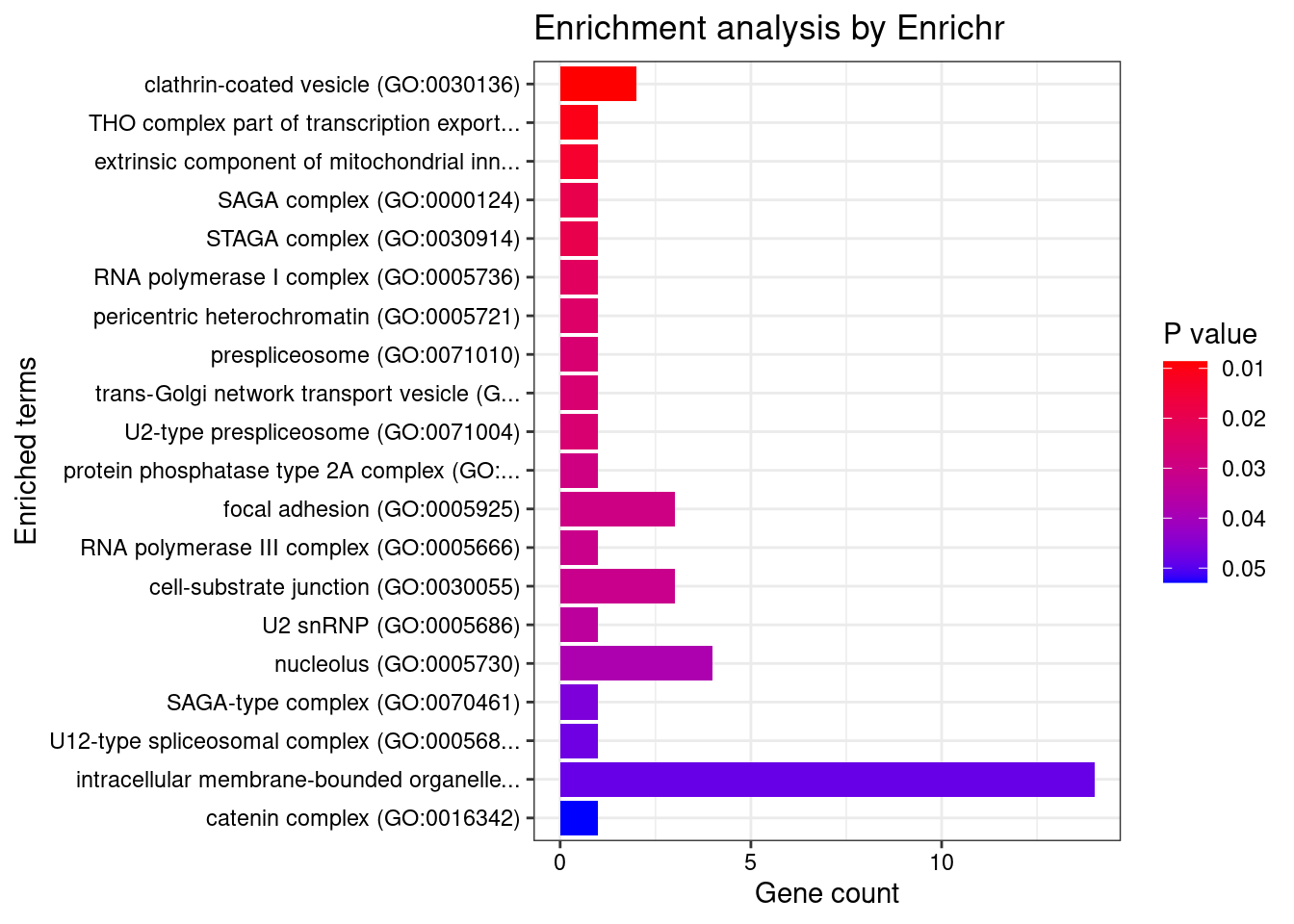
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)
[1] "GO_Molecular_Function_2021"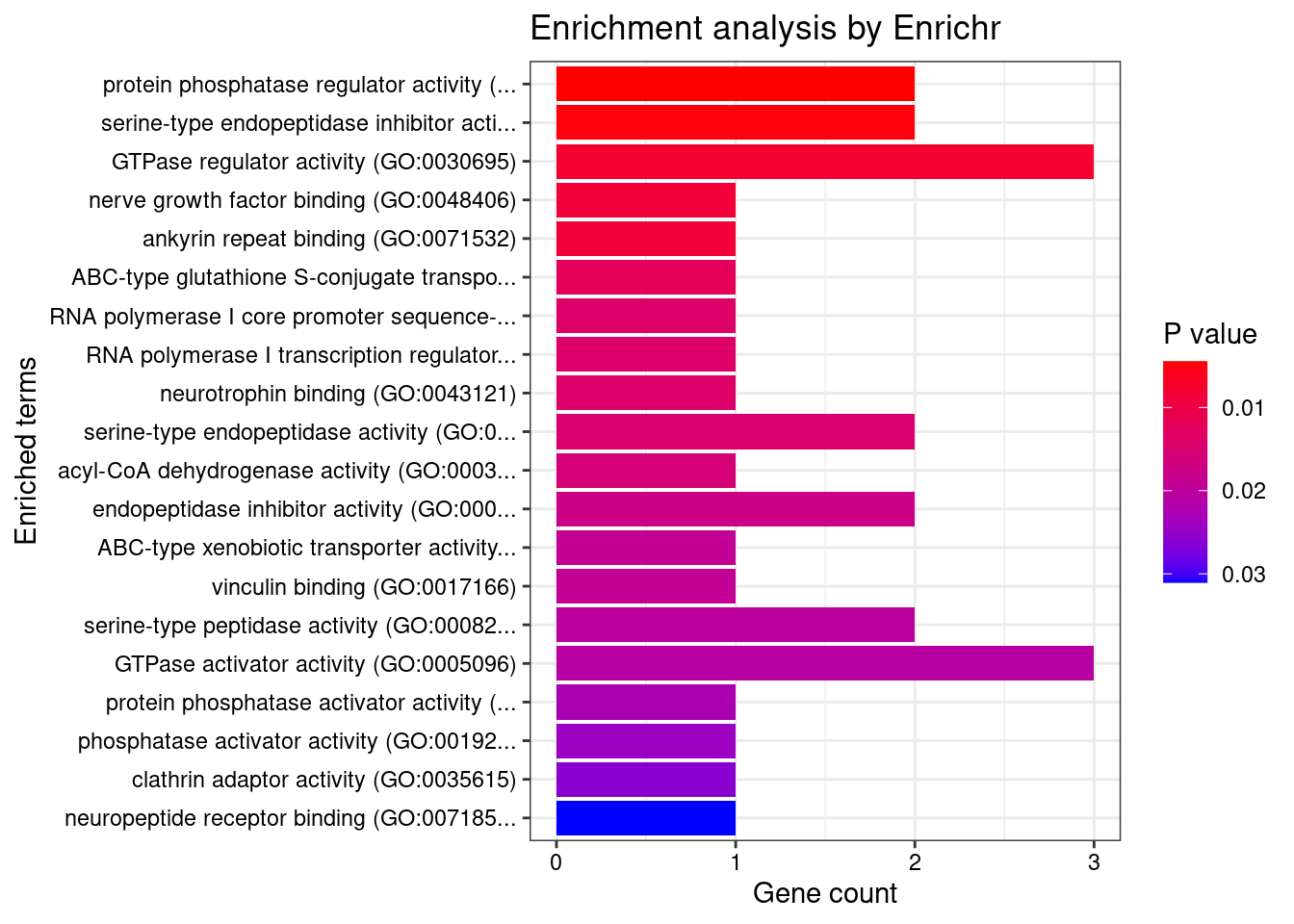
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)DisGeNET enrichment analysis for genes with PIP>0.5
Description
66 Alstrom Syndrome
117 SCHIZOPHRENIA 11
118 Familial encephalopathy with neuroserpin inclusion bodies
119 Childhood-onset truncal obesity
122 Hematopoetic Myelodysplasia
125 TREACHER COLLINS SYNDROME 2
128 Very long chain acyl-CoA dehydrogenase deficiency
129 EPILEPSY, FAMILIAL TEMPORAL LOBE, 8
130 IMMUNODEFICIENCY-CENTROMERIC INSTABILITY-FACIAL ANOMALIES SYNDROME 4
67 Metabolic myopathy
FDR Ratio BgRatio
66 0.01814 1/12 1/9703
117 0.01814 1/12 1/9703
118 0.01814 1/12 1/9703
119 0.01814 1/12 1/9703
122 0.01814 2/12 29/9703
125 0.01814 1/12 1/9703
128 0.01814 1/12 1/9703
129 0.01814 1/12 1/9703
130 0.01814 1/12 1/9703
67 0.02720 1/12 2/9703WebGestalt enrichment analysis for genes with PIP>0.5
Loading the functional categories...
Loading the ID list...
Loading the reference list...
Performing the enrichment analysis...Warning in oraEnrichment(interestGeneList, referenceGeneList, geneSet, minNum =
minNum, : No significant gene set is identified based on FDR 0.05!NULLPIP Manhattan Plot
Warning: 'timedatectl' indicates the non-existent timezone name 'n/a'Warning: Your system is mis-configured: '/etc/localtime' is not a symlinkWarning: It is strongly recommended to set envionment variable TZ to 'America/
Chicago' (or equivalent)Warning: ggrepel: 4 unlabeled data points (too many overlaps). Consider
increasing max.overlaps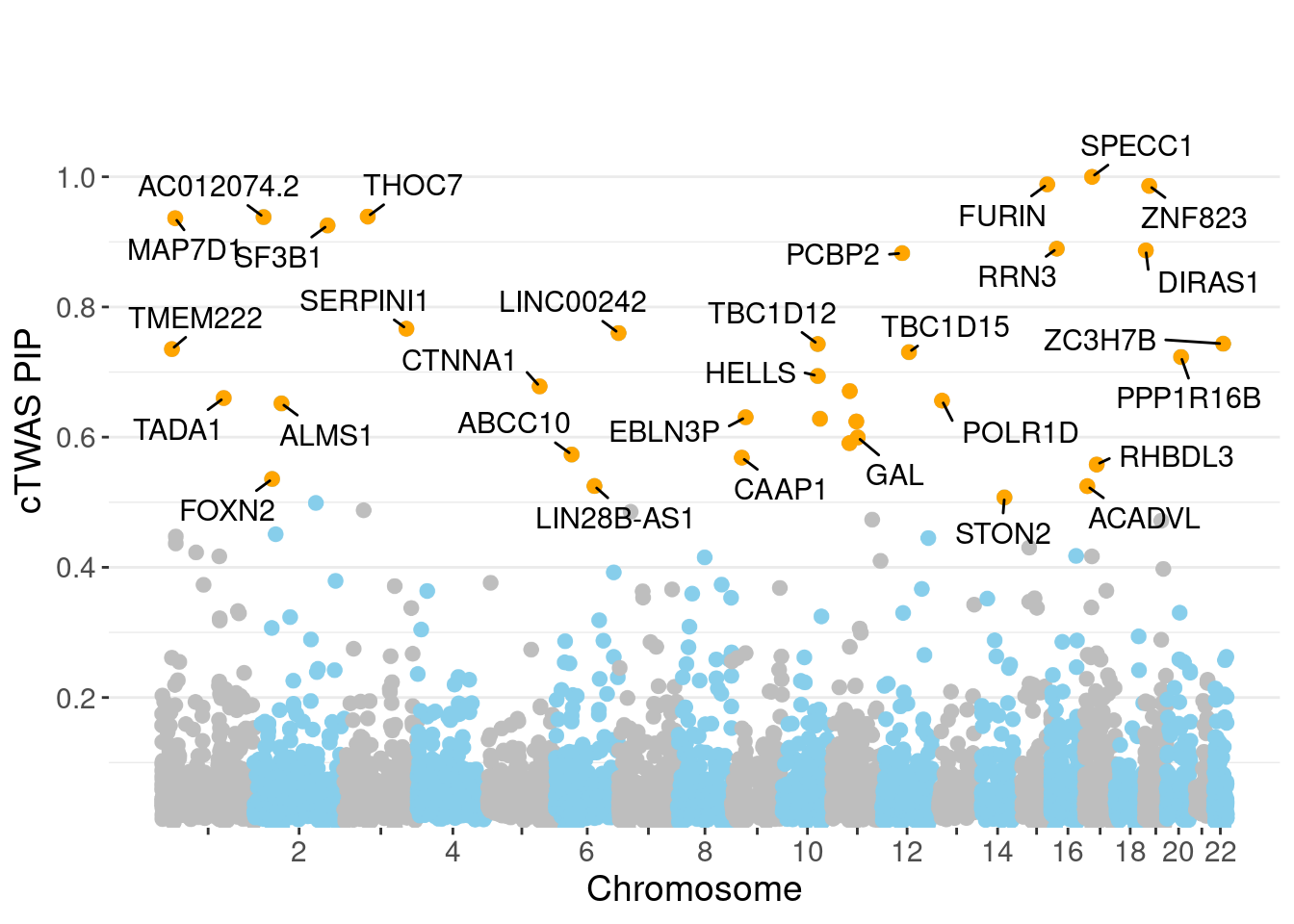
Sensitivity, specificity and precision for silver standard genes
#number of genes in known annotations
print(length(known_annotations))[1] 130#number of genes in known annotations with imputed expression
print(sum(known_annotations %in% ctwas_gene_res$genename))[1] 59#significance threshold for TWAS
print(sig_thresh)[1] 4.58#number of ctwas genes
length(ctwas_genes)[1] 10#number of TWAS genes
length(twas_genes)[1] 86#show novel genes (ctwas genes with not in TWAS genes)
ctwas_gene_res[ctwas_gene_res$genename %in% novel_genes,report_cols] genename region_tag susie_pip mu2 PVE z num_eqtl
10699 PCBP2 12_33 0.8826 21.89 0.0002506 4.496 1
9203 DIRAS1 19_3 0.8869 22.06 0.0002538 4.504 2#sensitivity / recall
print(sensitivity) ctwas TWAS
0.03077 0.06923 #specificity
print(specificity) ctwas TWAS
0.9994 0.9928 #precision / PPV
print(precision) ctwas TWAS
0.4000 0.1047 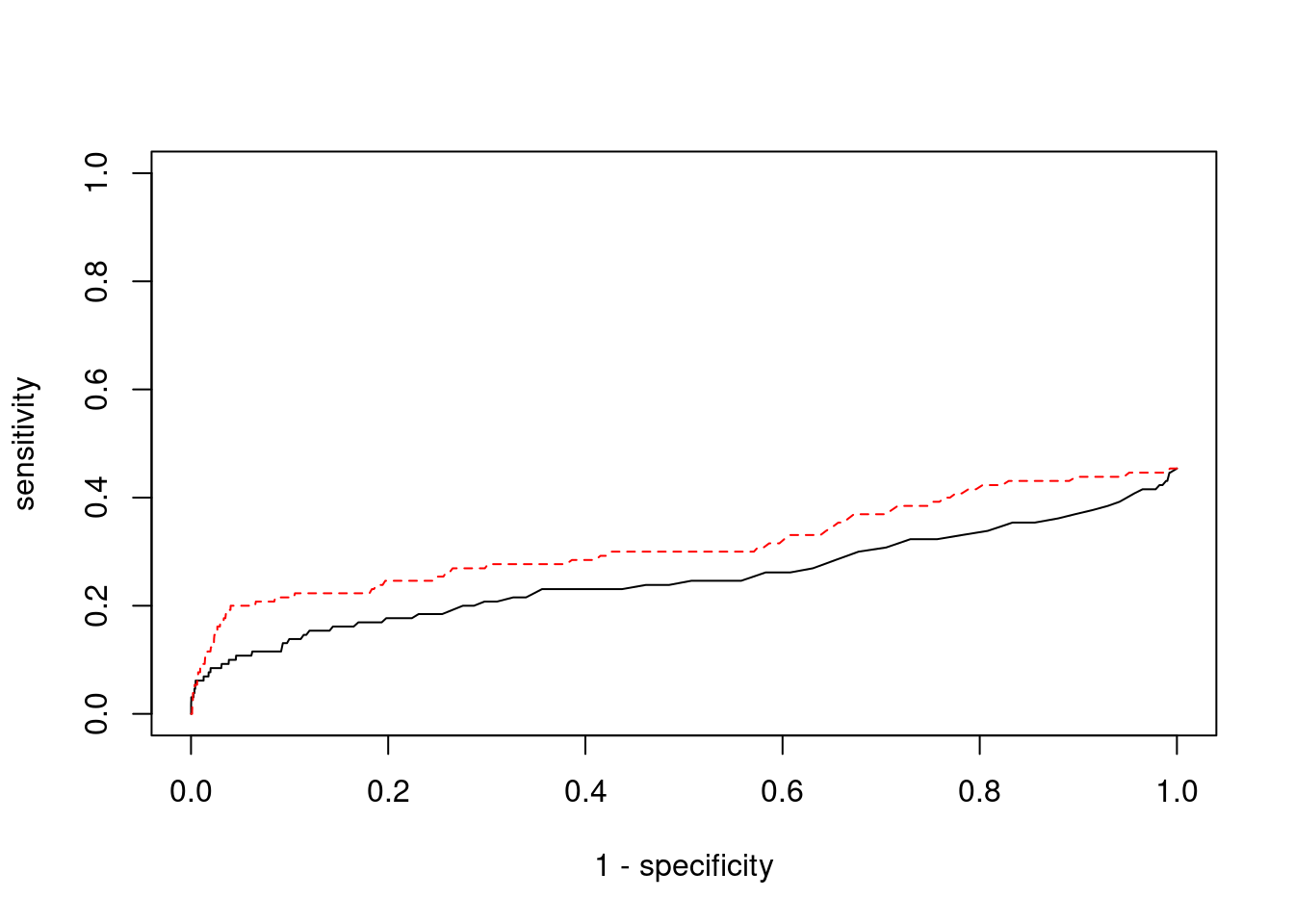
cTWAS is more precise than TWAS in distinguishing silver standard and bystander genes
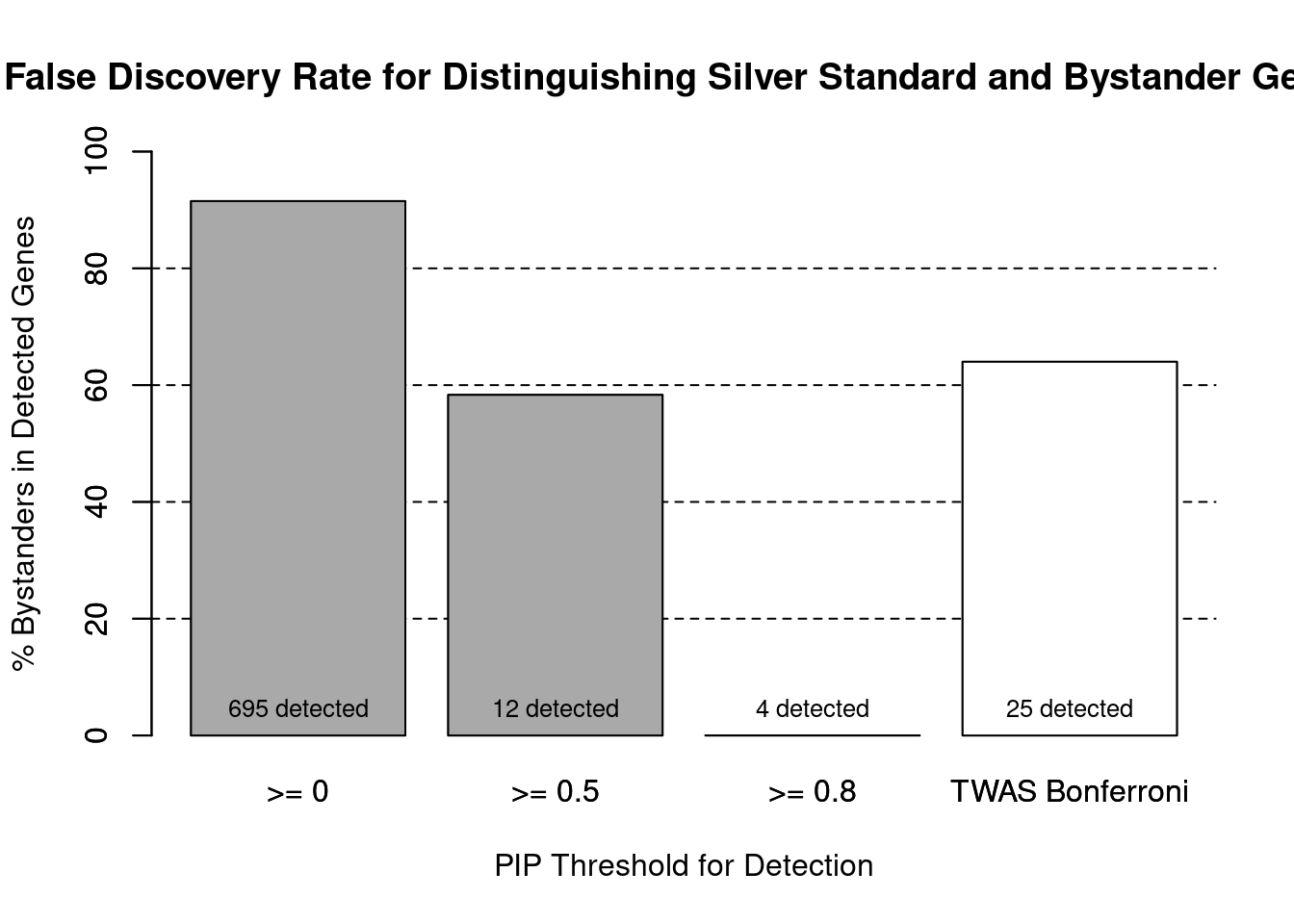
#number of genes in known annotations (with imputed expression)
print(length(known_annotations))[1] 59#number of bystander genes (with imputed expression)
print(length(unrelated_genes))[1] 635#subset results to genes in known annotations or bystanders
ctwas_gene_res_subset <- ctwas_gene_res[ctwas_gene_res$genename %in% c(known_annotations, unrelated_genes),]
#assign ctwas and TWAS genes
ctwas_genes <- ctwas_gene_res_subset$genename[ctwas_gene_res_subset$susie_pip>0.8]
twas_genes <- ctwas_gene_res_subset$genename[abs(ctwas_gene_res_subset$z)>sig_thresh]
#significance threshold for TWAS
print(sig_thresh)[1] 4.58#number of ctwas genes (in known annotations or bystanders)
length(ctwas_genes)[1] 4#number of TWAS genes (in known annotations or bystanders)
length(twas_genes)[1] 25#sensitivity / recall
sensitivity ctwas TWAS
0.0678 0.1525 #specificity / (1 - False Positive Rate)
specificity ctwas TWAS
1.0000 0.9748 #precision / PPV / (1 - False Discovery Rate)
precisionctwas TWAS
1.00 0.36 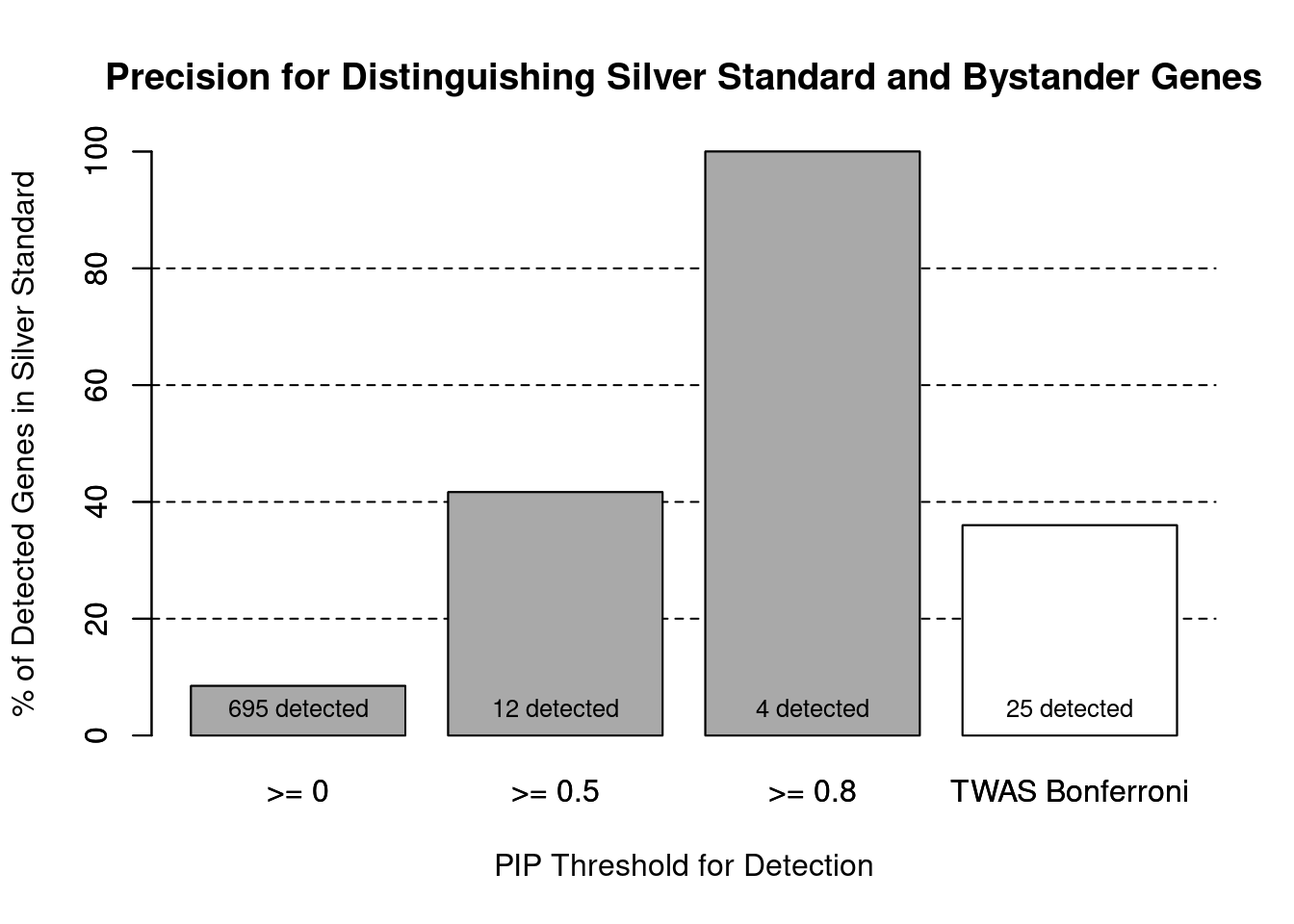
pip_range <- (0:1000)/1000
sensitivity <- rep(NA, length(pip_range))
specificity <- rep(NA, length(pip_range))
for (index in 1:length(pip_range)){
pip <- pip_range[index]
ctwas_genes <- ctwas_gene_res_subset$genename[ctwas_gene_res_subset$susie_pip>=pip]
sensitivity[index] <- sum(ctwas_genes %in% known_annotations)/length(known_annotations)
specificity[index] <- sum(!(unrelated_genes %in% ctwas_genes))/length(unrelated_genes)
}
plot(1-specificity, sensitivity, type="l", xlim=c(0,1), ylim=c(0,1), main="", xlab="1 - Specificity", ylab="Sensitivity")
title(expression("ROC Curve for cTWAS (black) and TWAS (" * phantom("red") * ")"))
title(expression(phantom("ROC Curve for cTWAS (black) and TWAS (") * "red" * phantom(")")), col.main="red")
sig_thresh_range <- seq(from=0, to=max(abs(ctwas_gene_res_subset$z)), length.out=length(pip_range))
for (index in 1:length(sig_thresh_range)){
sig_thresh_plot <- sig_thresh_range[index]
twas_genes <- ctwas_gene_res_subset$genename[abs(ctwas_gene_res_subset$z)>=sig_thresh_plot]
sensitivity[index] <- sum(twas_genes %in% known_annotations)/length(known_annotations)
specificity[index] <- sum(!(unrelated_genes %in% twas_genes))/length(unrelated_genes)
}
lines(1-specificity, sensitivity, xlim=c(0,1), ylim=c(0,1), col="red", lty=1)
abline(a=0,b=1,lty=3)
#add previously computed points from the analysis
ctwas_genes <- ctwas_gene_res_subset$genename[ctwas_gene_res_subset$susie_pip>0.8]
twas_genes <- ctwas_gene_res_subset$genename[abs(ctwas_gene_res_subset$z)>sig_thresh]
points(1-specificity_plot["ctwas"], sensitivity_plot["ctwas"], pch=21, bg="black")
points(1-specificity_plot["TWAS"], sensitivity_plot["TWAS"], pch=21, bg="red")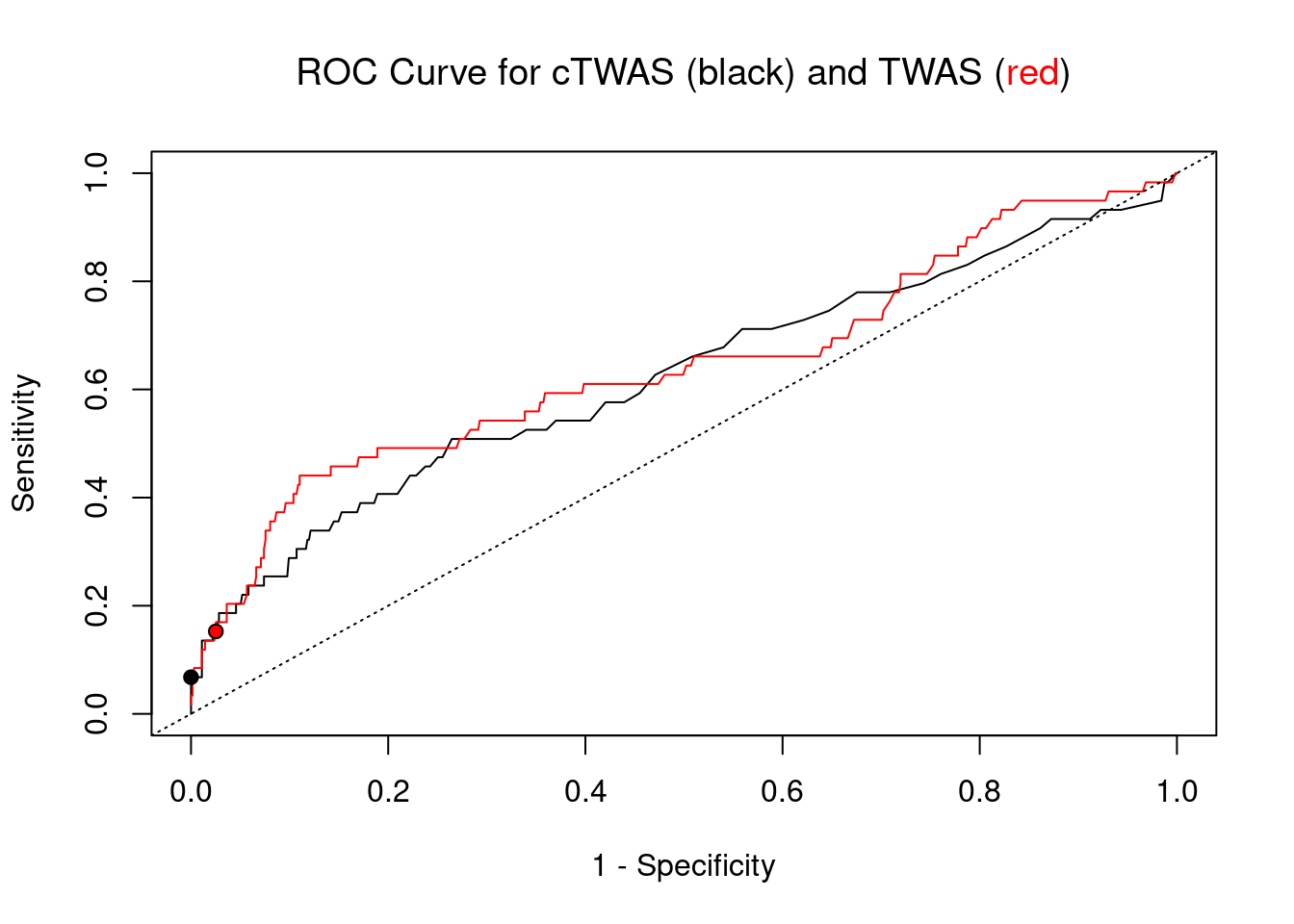
Undetected silver standard genes have low TWAS z-scores or stronger signal from nearby variants
#table of outcomes for silver standard genes
-sort(-table(silver_standard_case))silver_standard_case
Not Imputed Insignificant z-score Nearby SNP(s)
71 50 5
Detected (PIP > 0.8)
4 #show inconclusive genes
silver_standard_case[silver_standard_case=="Inconclusive"]named character(0)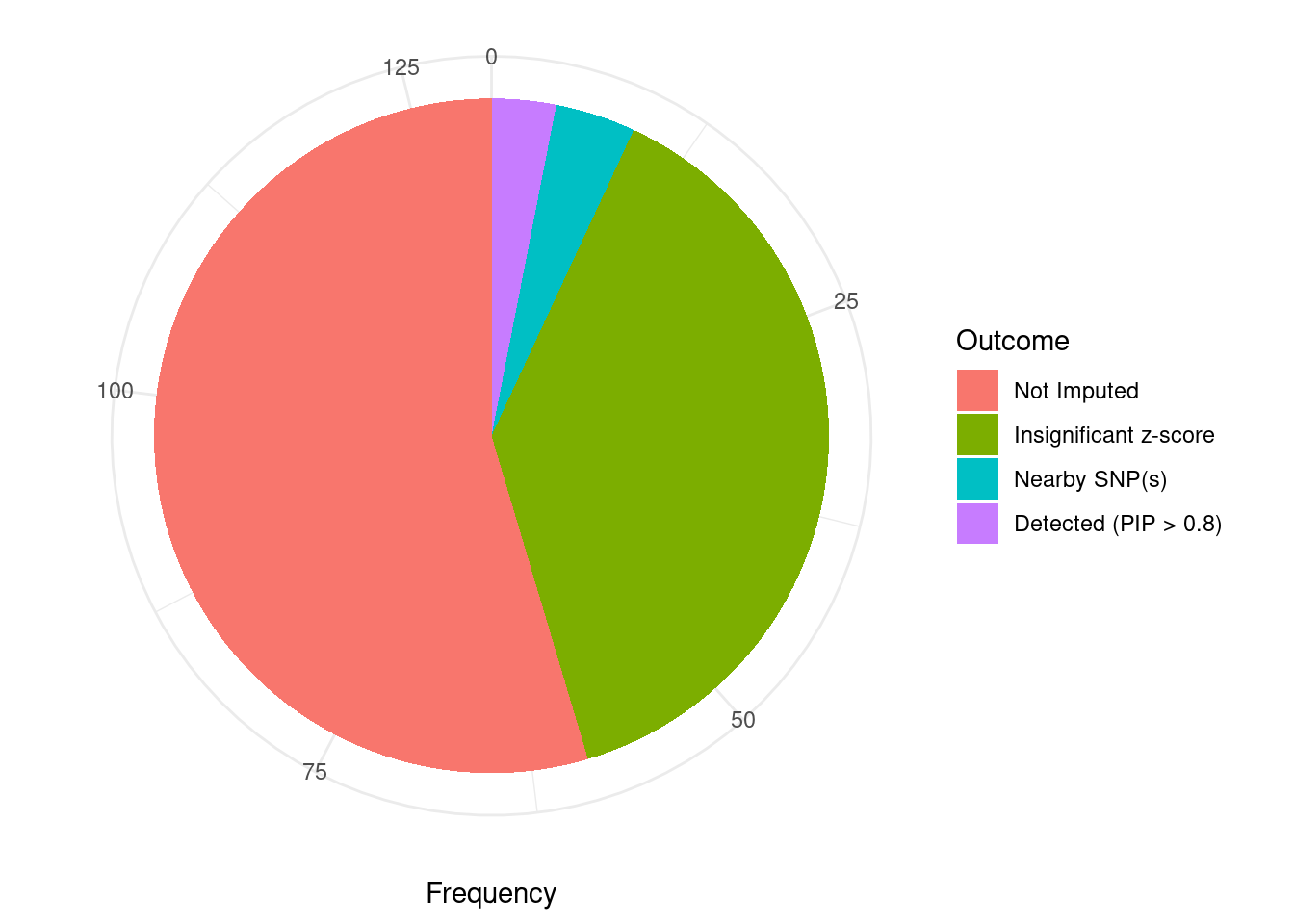
sessionInfo()R version 3.6.1 (2019-07-05)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] GenomicRanges_1.36.1 GenomeInfoDb_1.20.0 IRanges_2.18.1
[4] S4Vectors_0.22.1 BiocGenerics_0.30.0 biomaRt_2.40.1
[7] readxl_1.3.1 forcats_0.5.1 stringr_1.4.0
[10] dplyr_1.0.7 purrr_0.3.4 readr_2.1.1
[13] tidyr_1.1.4 tidyverse_1.3.1 tibble_3.1.6
[16] WebGestaltR_0.4.4 disgenet2r_0.99.2 enrichR_3.0
[19] cowplot_1.0.0 ggplot2_3.3.5 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] ggbeeswarm_0.6.0 colorspace_2.0-2 rjson_0.2.20
[4] ellipsis_0.3.2 rprojroot_2.0.2 XVector_0.24.0
[7] fs_1.5.2 rstudioapi_0.13 farver_2.1.0
[10] ggrepel_0.9.1 bit64_4.0.5 AnnotationDbi_1.46.0
[13] fansi_1.0.2 lubridate_1.8.0 xml2_1.3.3
[16] codetools_0.2-16 doParallel_1.0.17 cachem_1.0.6
[19] knitr_1.36 jsonlite_1.7.2 apcluster_1.4.8
[22] Cairo_1.5-12.2 broom_0.7.10 dbplyr_2.1.1
[25] compiler_3.6.1 httr_1.4.2 backports_1.4.1
[28] assertthat_0.2.1 Matrix_1.2-18 fastmap_1.1.0
[31] cli_3.1.0 later_0.8.0 prettyunits_1.1.1
[34] htmltools_0.5.2 tools_3.6.1 igraph_1.2.10
[37] GenomeInfoDbData_1.2.1 gtable_0.3.0 glue_1.6.2
[40] reshape2_1.4.4 doRNG_1.8.2 Rcpp_1.0.8
[43] Biobase_2.44.0 cellranger_1.1.0 jquerylib_0.1.4
[46] vctrs_0.3.8 svglite_1.2.2 iterators_1.0.14
[49] xfun_0.29 ps_1.6.0 rvest_1.0.2
[52] lifecycle_1.0.1 rngtools_1.5.2 XML_3.99-0.3
[55] zlibbioc_1.30.0 getPass_0.2-2 scales_1.1.1
[58] vroom_1.5.7 hms_1.1.1 promises_1.0.1
[61] yaml_2.2.1 curl_4.3.2 memoise_2.0.1
[64] ggrastr_1.0.1 gdtools_0.1.9 stringi_1.7.6
[67] RSQLite_2.2.8 highr_0.9 foreach_1.5.2
[70] rlang_1.0.1 pkgconfig_2.0.3 bitops_1.0-7
[73] evaluate_0.14 lattice_0.20-38 labeling_0.4.2
[76] bit_4.0.4 processx_3.5.2 tidyselect_1.1.1
[79] plyr_1.8.6 magrittr_2.0.2 R6_2.5.1
[82] generics_0.1.1 DBI_1.1.2 pillar_1.6.4
[85] haven_2.4.3 whisker_0.3-2 withr_2.4.3
[88] RCurl_1.98-1.5 modelr_0.1.8 crayon_1.5.0
[91] utf8_1.2.2 tzdb_0.2.0 rmarkdown_2.11
[94] progress_1.2.2 grid_3.6.1 data.table_1.14.2
[97] blob_1.2.2 callr_3.7.0 git2r_0.26.1
[100] reprex_2.0.1 digest_0.6.29 httpuv_1.5.1
[103] munsell_0.5.0 beeswarm_0.2.3 vipor_0.4.5