An example for LDL ~ Predictdb eQTL&sQTL from Liver
XSun
2024-09-22
Last updated: 2024-09-26
Checks: 6 1
Knit directory: multigroup_ctwas_analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20231112) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version f1bc2b5. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: results/
Unstaged changes:
Modified: analysis/LDL_predictdb_esQTL.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/LDL_predictdb_esQTL.Rmd)
and HTML (docs/LDL_predictdb_esQTL.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 278395e | XSun | 2024-09-24 | update |
| html | 278395e | XSun | 2024-09-24 | update |
We present a sample cTWAS report based on real data analysis. The analyzed trait is LDL cholesterol, the prediction models are liver gene expression and splicing models trained on GTEx v8 in the PredictDB format.
Analysis settings
Input data
- GWAS Z-scores
The summary statistics for LDL are downloaded from https://gwas.mrcieu.ac.uk, using dataset ID:
ukb-d-30780_irnt. The number of SNPs it contains is
13,586,016.
The sample size is
[1] "gwas_n = 343621"- Prediction models
The prediction models used in this analysis are liver gene expression and splicing models, trained on GTEx v8 in the PredictDB format. These models can be downloaded from https://predictdb.org/post/2021/07/21/gtex-v8-models-on-eqtl-and-sqtl/
[1] "The number of eQTLs per gene = 1.5078"[1] "Total genes = 12714"[1] "The number of sQTLs per intron = 1.2151"[1] "Total introns = 29250"- Reference data
The reference data include genomic region definitions and an LD
reference. We use the genomic regions provided by the package and the LD
reference in b38, located at
/project2/mstephens/wcrouse/UKB_LDR_0.1/. Alternatively,
the LD reference can be downloaded from this link:https://uchicago.app.box.com/s/jqocacd2fulskmhoqnasrknbt59x3xkn.
Data processing and harmonization
We map the reference SNPs and LD matrices to regions following the instructions from the cTWAS tutorial.
When processing z-scores, we exclude multi-allelic and
strand-ambiguous variants by setting
drop_multiallelic = TRUE and
drop_strand_ambig = TRUE.
The process can be divided into steps below, users can expand the code snippets below to view the exact code used.
- Input and output settings
weight_files <- c("/project2/xinhe/shared_data/multigroup_ctwas/weights/expression_models/expression_Liver.db","/project2/xinhe/shared_data/multigroup_ctwas/weights/splicing_models/splicing_Liver.db")
z_snp_file <- "/project2/xinhe/shared_data/multigroup_ctwas/gwas/ctwas_inputs_zsnp/LDL-ukb-d-30780_irnt.z_snp.RDS"
genome_version <- "b38"
LD_dir <- "/project2/mstephens/wcrouse/UKB_LDR_0.1/"
region_file <- system.file("extdata/ldetect", paste0("EUR.", genome_version, ".ldetect.regions.RDS"), package = "ctwas")
region_info <- readRDS(region_file)
## output dir
outputdir <- "/project/xinhe/xsun/multi_group_ctwas/examples/results_predictdb_main/LDL-ukb-d-30780_irnt/"
dir.create(outputdir, showWarnings=F, recursive=T)
## other parameters
ncore <- 5- Preprocessing GWAS
### Preprocess LD_map & SNP_map
region_metatable <- region_info
region_metatable$LD_file <- file.path(LD_dir, paste0(LD_filestem, ".RDS"))
region_metatable$SNP_file <- file.path(LD_dir, paste0(LD_filestem, ".Rvar"))
res <- create_snp_LD_map(region_metatable)
region_info <- res$region_info
snp_map <- res$snp_map
LD_map <- res$LD_map
### Preprocess GWAS z-scores
z_snp <- readRDS(z_snp_file)
z_snp <- preprocess_z_snp(z_snp = z_snp,
snp_map = snp_map,
drop_multiallelic = TRUE,
drop_strand_ambig = TRUE)- Preprocessing weights
weights_expression1 <- preprocess_weights(weight_file = weight_files[1],
region_info = region_info,
gwas_snp_ids = z_snp$id,
snp_map = snp_map,
LD_map = LD_map,
type = "eQTL",
context = tissue,
weight_format = "PredictDB",
drop_strand_ambig = TRUE,
scale_predictdb_weights = T,
load_predictdb_LD = F, #### F for fusion converted weights
filter_protein_coding_genes = TRUE,
ncore = ncore)
weights_splicing1 <- preprocess_weights(weight_file = weight_files[2],
region_info = region_info,
gwas_snp_ids = z_snp$id,
snp_map = snp_map,
LD_map = LD_map,
type = "sQTL",
context = tissue,
weight_format = "PredictDB",
drop_strand_ambig = TRUE,
scale_predictdb_weights = T, #### F for fusion converted weights
load_predictdb_LD = F,
filter_protein_coding_genes = TRUE,
ncore = ncore)
weights <- c(weights_expression1,weights_splicing1) Running cTWAS analysis
We use the ctwas main function ctwas_sumstats() to run
the cTWAS analysis with LD. For more details on this function, refer to
the cTWAS tutorial: https://xinhe-lab.github.io/multigroup_ctwas/articles/running_ctwas_analysis.html#running-ctwas-main-function
All arguments are set to their default values, with the following specific settings:
group_prior_var_structure = "shared_type": Allows all groups within a molecular QTL type to share the same variance parameter.filter_L = TRUE: Estimates the number of causal signals (L) for each region.filter_nonSNP_PIP = TRUE: Prevents the computation of non-SNP PIP values.min_nonSNP_PIP = 0.5: Selects regions where the non-SNP PIP is greater than 0.5.
Users can expand the code snippets below to view the exact code used.
thin <- 0.1
maxSNP <- 20000
ctwas_res <- ctwas_sumstats(z_snp,
weights,
region_info,
LD_map,
snp_map,
thin = thin,
maxSNP = maxSNP,
group_prior_var_structure = "shared_type",
filter_L = TRUE,
filter_nonSNP_PIP = FALSE,
min_nonSNP_PIP = 0.5,
ncore = ncore,
ncore_LD = ncore,
save_cor = TRUE,
cor_dir = paste0(outputdir,"/cor_matrix"),
verbose = T)cTWAS results
ctwas_res is the object contains the outputs of
cTWAS
Parameter estimation
We extract the estimated parameters by
param <- ctwas_res$param
we make plots using the function
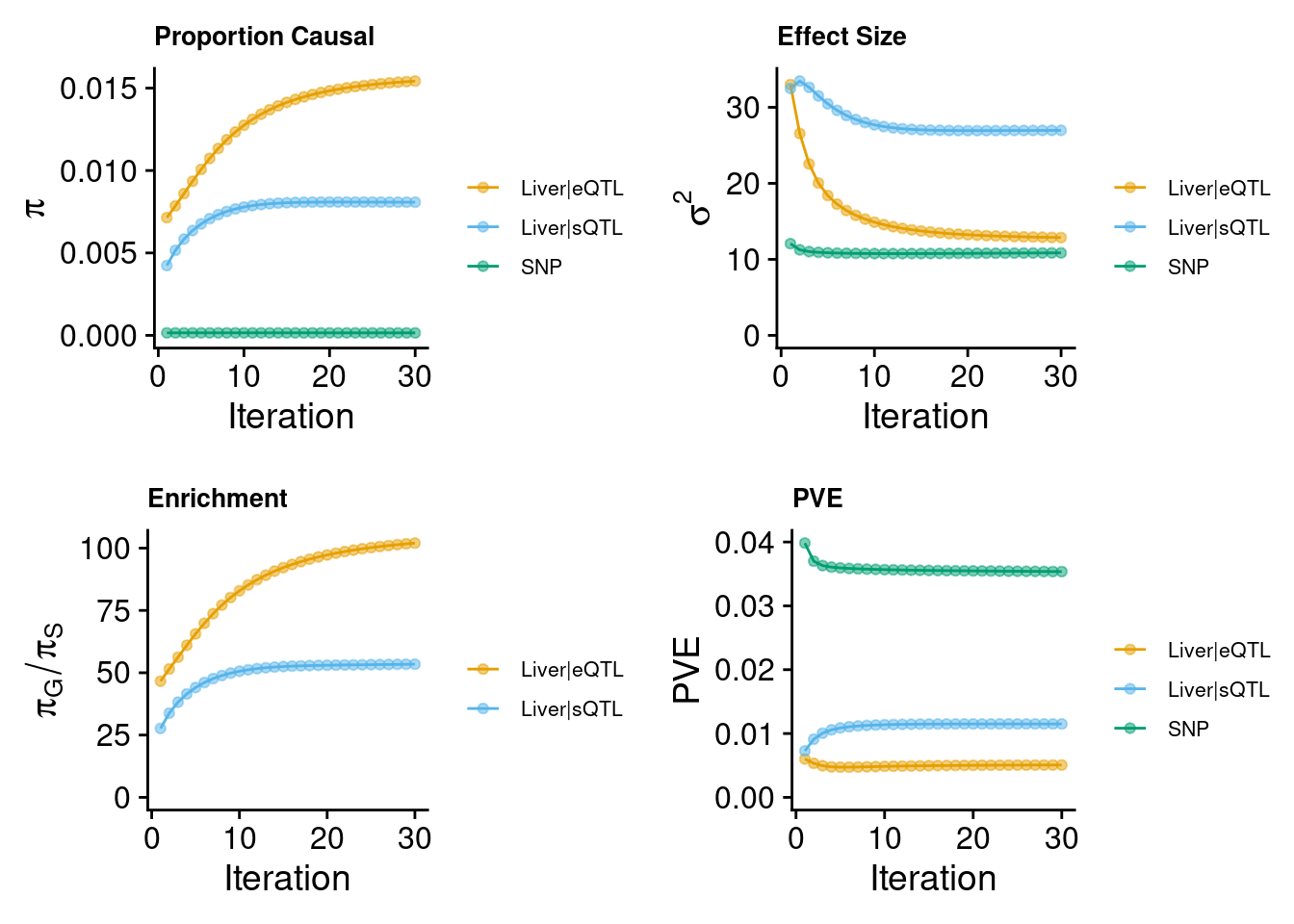
make_convergence_plots(param, gwas_n) to see how estimated
parameters converge during the execution of the program:
| Version | Author | Date |
|---|---|---|
| 278395e | XSun | 2024-09-24 |
These plots show the estimated prior inclusion probability, prior effect size variance, enrichment and proportion of variance explained (PVE) over the iterations of parameter estimation. The enrichment is defined as the ratio of the prior inclusion probability of molecular traits over the prior inclusion probability of variants. We generally expect molecular traits to have higher prior inclusion probability than variants. Enrichment values typically range from 20 - 100 for expression traits.
Then, we use summarize_param(param, gwas_n) to obtain
estimated parameters (from the last iteration) and to compute the PVE by
variants and molecular traits.
[1] "The number of genes/introns/SNPs used in the analysis is:"Liver|eQTL Liver|sQTL SNP
8775 18136 7405450 ctwas_parameters$attributable_pve contains the
proportion of heritability mediated by molecular traits and variants, we
visualize it using pie chart.
data <- data.frame(
category = names(ctwas_parameters$attributable_pve),
percentage = ctwas_parameters$attributable_pve
)
# Calculate percentage labels for the chart
data$percentage_label <- paste0(round(data$percentage * 100, 1), "%")
ggplot(data, aes(x = "", y = percentage, fill = category)) +
geom_bar(stat = "identity", width = 1) +
coord_polar("y", start = 0) +
theme_void() + # Remove background and axes
geom_text(aes(label = percentage_label),
position = position_stack(vjust = 0.5), size = 5) +
scale_fill_manual(values = c("#FF9999", "#66B2FF", "#99FF99")) + # Custom colors
labs(fill = "Category") +
ggtitle("Attributable PVE")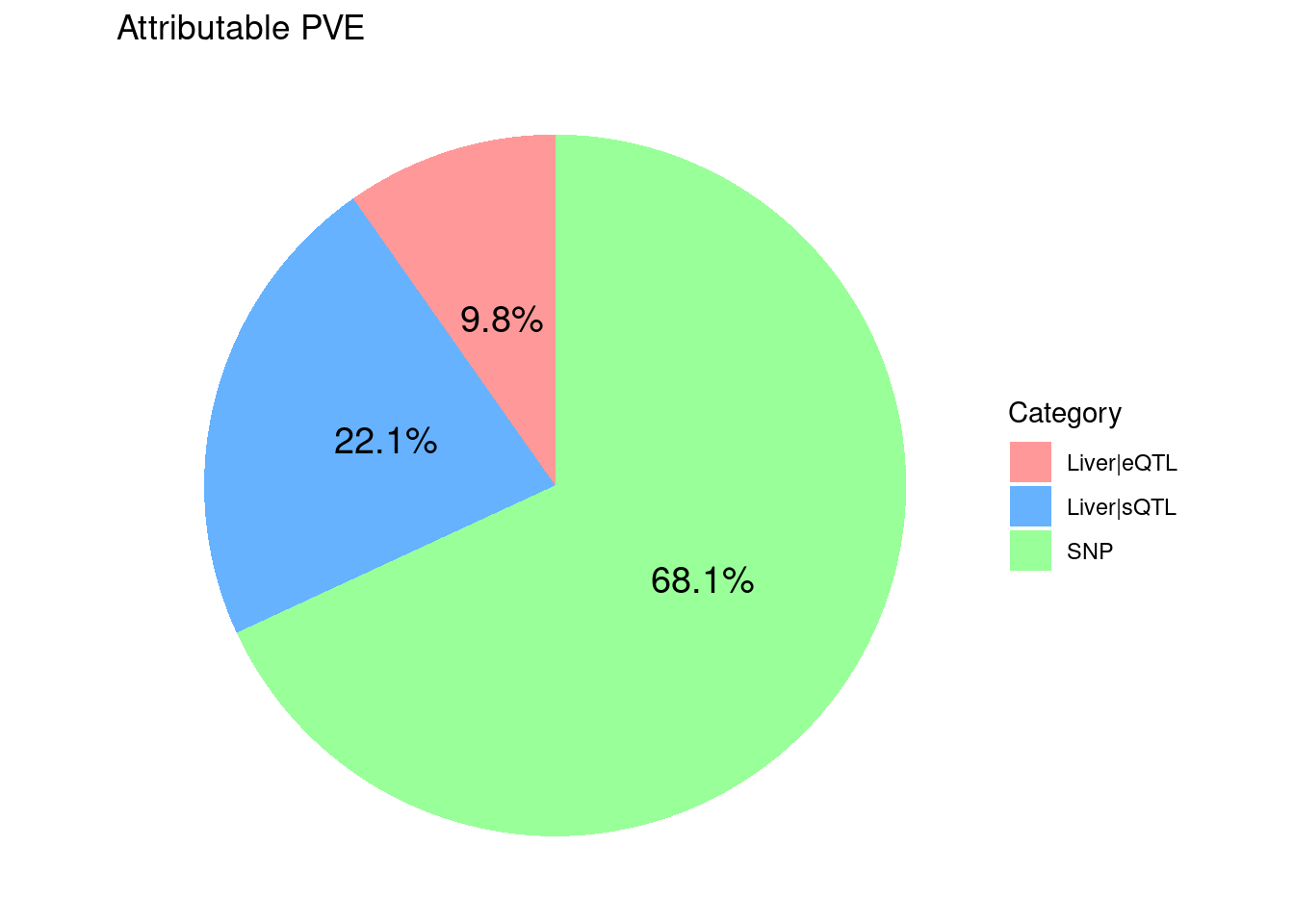
| Version | Author | Date |
|---|---|---|
| 278395e | XSun | 2024-09-24 |
Fine-mapping results
We process the fine-mapping results here.
We first add gene annotations to cTWAS results
load("/project2/xinhe/shared_data/multigroup_ctwas/weights/E_S_A_mapping_updated.RData")
colnames(E_S_A_mapping)[1] <- "molecular_id"
finemap_res <- ctwas_res$finemap_res
finemap_res$molecular_id <- get_molecular_ids(finemap_res)
snp_map <- readRDS(paste0(results_dir,trait,".snp_map.RDS"))
finemap_res <- anno_finemap_res(finemap_res,
snp_map = snp_map,
mapping_table = E_S_A_mapping,
add_gene_annot = TRUE,
map_by = "molecular_id",
drop_unmapped = TRUE,
add_position = TRUE,
use_gene_pos = "mid")2024-09-26 17:46:05 INFO::Annotating fine-mapping result ...
2024-09-26 17:46:06 INFO::Map molecular traits to genes
2024-09-26 17:46:08 INFO::Split PIPs for molecular traits mapped to multiple genes
2024-09-26 17:46:13 INFO::Add gene positions
2024-09-26 17:46:14 INFO::Add SNP positionsfinemap_res_show <- finemap_res[finemap_res$cs_index!=0 &finemap_res$type !="SNP",]
DT::datatable(finemap_res_show,caption = htmltools::tags$caption( style = 'caption-side: topleft; text-align = left; color:black;','The annotated fine-mapping results, ones within credible sets are shown'),options = list(pageLength = 5) )For all genes analyzed, we compare the z-scores and fine-mapping PIPs
ggplot(data = finemap_res[finemap_res$type!="SNP",], aes(x = abs(z), y = susie_pip)) +
geom_point() +
labs(x = "abs(z-scores)", y = "PIPs") +
theme_minimal()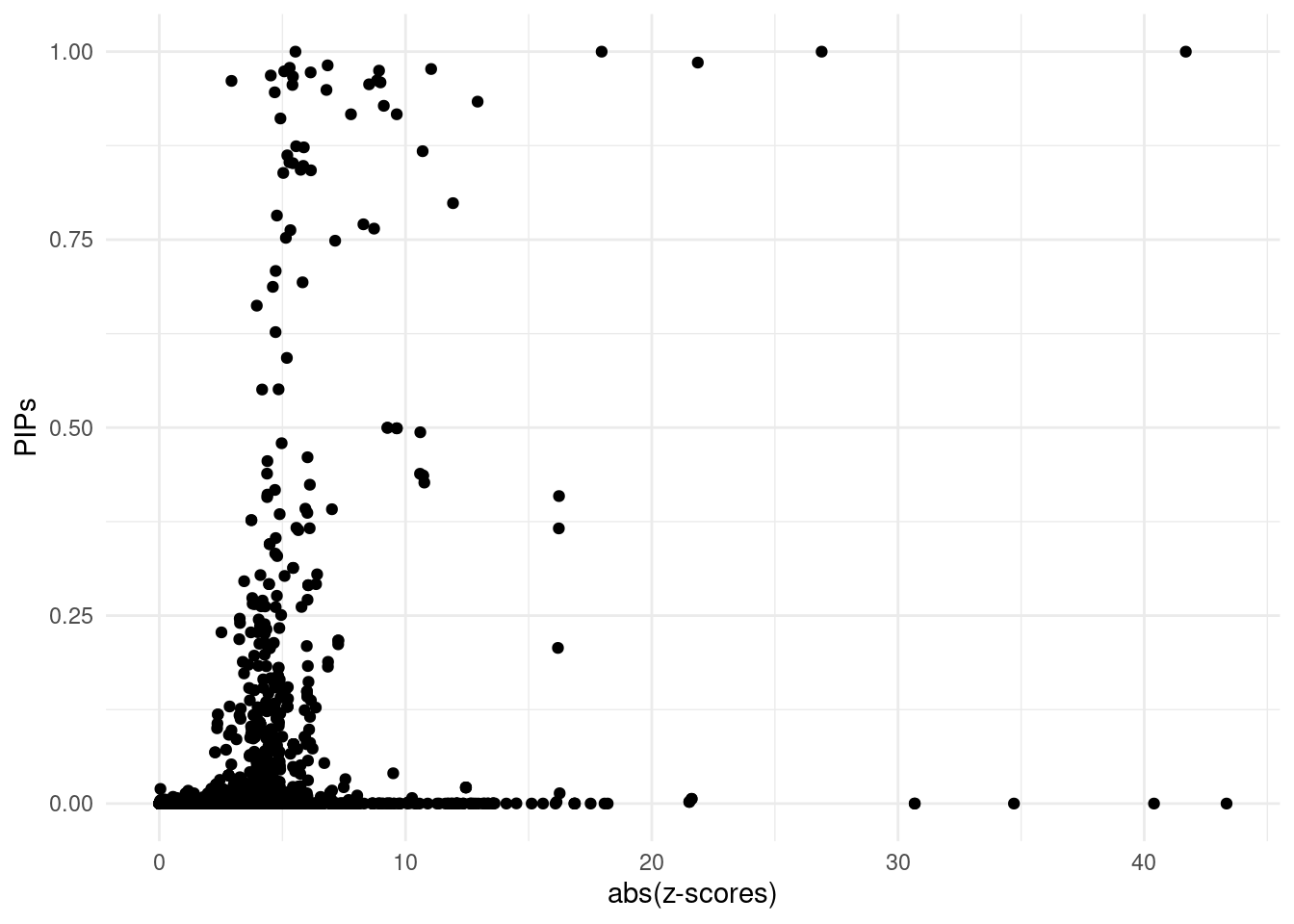
Next, we compute gene PIPs across different types of molecular traits
library(dplyr)
Attaching package: 'dplyr'The following objects are masked from 'package:ensembldb':
filter, selectThe following object is masked from 'package:AnnotationDbi':
selectThe following object is masked from 'package:Biobase':
combineThe following objects are masked from 'package:GenomicRanges':
intersect, setdiff, unionThe following object is masked from 'package:GenomeInfoDb':
intersectThe following objects are masked from 'package:IRanges':
collapse, desc, intersect, setdiff, slice, unionThe following objects are masked from 'package:S4Vectors':
first, intersect, rename, setdiff, setequal, unionThe following objects are masked from 'package:BiocGenerics':
combine, intersect, setdiff, unionThe following objects are masked from 'package:stats':
filter, lagThe following objects are masked from 'package:base':
intersect, setdiff, setequal, unioncombined_pip_by_type <- combine_gene_pips(finemap_res,
group_by = "gene_name",
by = "type",
method = "combine_cs",
filter_cs = TRUE)2024-09-26 17:46:28 INFO::Limit gene results to credible setscombined_pip_by_type$sQTL_pip_partition <- sapply(combined_pip_by_type$gene_name, function(gene) {
# Find rows in finemap_res_show matching the gene_name
matching_rows <- finemap_res_show %>%
filter(gene_name == gene, type == "sQTL") # Match gene_name and filter by type == "sQTL"
# If no matching rows, return NA
if (nrow(matching_rows) == 0) {
return(NA)
}
# Create the desired string format: molecular_id-round(susie_pip, digits = 4)
paste(matching_rows$molecular_id, "-PIP=", round(matching_rows$susie_pip, digits = 4), sep = "", collapse = ", ")
})
DT::datatable(combined_pip_by_type,caption = htmltools::tags$caption( style = 'caption-side: topleft; text-align = left; color:black;','Gene PIPs, only genes within credible sets are shown'),options = list(pageLength = 5) )Visualization
We make locuz plot for the region(“16_71020125_72901251”) containing
the gene HPR. we limit PIP results to credible sets in the PIP track by
setting filter_cs = TRUE.
- The top one shows -log10(p-value) of the association of variants (from LDL GWAS) and molecular traits (from the package computed z-scores) with the phenotype
- The next track shows the PIPs of variants and molecular traits. By
default, we limit PIP results to credible sets in the PIP track
(
filter_cs = TRUE) - The next track shows the QTLs of the focal gene.
- The bottom is the gene track.
sessionInfo()R version 4.2.0 (2022-04-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.3.13-el7-x86_64/lib/libopenblas_haswellp-r0.3.13.so
locale:
[1] C
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] dplyr_1.1.4 ggplot2_3.5.1
[3] EnsDb.Hsapiens.v86_2.99.0 ensembldb_2.20.2
[5] AnnotationFilter_1.20.0 GenomicFeatures_1.48.3
[7] AnnotationDbi_1.58.0 Biobase_2.56.0
[9] GenomicRanges_1.48.0 GenomeInfoDb_1.39.9
[11] IRanges_2.30.0 S4Vectors_0.34.0
[13] BiocGenerics_0.42.0 ctwas_0.4.14
loaded via a namespace (and not attached):
[1] colorspace_2.0-3 rjson_0.2.21
[3] ellipsis_0.3.2 rprojroot_2.0.3
[5] XVector_0.36.0 locuszoomr_0.2.1
[7] fs_1.5.2 rstudioapi_0.13
[9] farver_2.1.0 DT_0.22
[11] ggrepel_0.9.1 bit64_4.0.5
[13] fansi_1.0.3 xml2_1.3.3
[15] codetools_0.2-18 logging_0.10-108
[17] cachem_1.0.6 knitr_1.39
[19] jsonlite_1.8.0 workflowr_1.7.0
[21] Rsamtools_2.12.0 dbplyr_2.1.1
[23] png_0.1-7 readr_2.1.2
[25] compiler_4.2.0 httr_1.4.3
[27] assertthat_0.2.1 Matrix_1.5-3
[29] fastmap_1.1.0 lazyeval_0.2.2
[31] cli_3.6.1 later_1.3.0
[33] htmltools_0.5.2 prettyunits_1.1.1
[35] tools_4.2.0 gtable_0.3.0
[37] glue_1.6.2 GenomeInfoDbData_1.2.8
[39] rappdirs_0.3.3 Rcpp_1.0.12
[41] jquerylib_0.1.4 vctrs_0.6.5
[43] Biostrings_2.64.0 rtracklayer_1.56.0
[45] crosstalk_1.2.0 xfun_0.41
[47] stringr_1.5.1 lifecycle_1.0.4
[49] irlba_2.3.5 restfulr_0.0.14
[51] XML_3.99-0.14 zlibbioc_1.42.0
[53] zoo_1.8-10 scales_1.3.0
[55] gggrid_0.2-0 hms_1.1.1
[57] promises_1.2.0.1 MatrixGenerics_1.8.0
[59] ProtGenerics_1.28.0 parallel_4.2.0
[61] SummarizedExperiment_1.26.1 LDlinkR_1.2.3
[63] yaml_2.3.5 curl_4.3.2
[65] memoise_2.0.1 sass_0.4.1
[67] biomaRt_2.54.1 stringi_1.7.6
[69] RSQLite_2.3.1 highr_0.9
[71] BiocIO_1.6.0 filelock_1.0.2
[73] BiocParallel_1.30.3 rlang_1.1.2
[75] pkgconfig_2.0.3 matrixStats_0.62.0
[77] bitops_1.0-7 evaluate_0.15
[79] lattice_0.20-45 purrr_1.0.2
[81] labeling_0.4.2 GenomicAlignments_1.32.0
[83] htmlwidgets_1.5.4 cowplot_1.1.1
[85] bit_4.0.4 tidyselect_1.2.0
[87] magrittr_2.0.3 R6_2.5.1
[89] generics_0.1.2 DelayedArray_0.22.0
[91] DBI_1.2.2 withr_2.5.0
[93] pgenlibr_0.3.3 pillar_1.9.0
[95] whisker_0.4 KEGGREST_1.36.3
[97] RCurl_1.98-1.7 mixsqp_0.3-43
[99] tibble_3.2.1 crayon_1.5.1
[101] utf8_1.2.2 BiocFileCache_2.4.0
[103] plotly_4.10.0 tzdb_0.4.0
[105] rmarkdown_2.25 progress_1.2.2
[107] grid_4.2.0 data.table_1.14.2
[109] blob_1.2.3 git2r_0.30.1
[111] digest_0.6.29 tidyr_1.3.0
[113] httpuv_1.6.5 munsell_0.5.0
[115] viridisLite_0.4.0 bslib_0.3.1