Clustering Lareau et al (2019) bone marrow data using topic proportions with K = 13
Kaixuan Luo
Last updated: 2020-10-21
Checks: 7 0
Knit directory: scATACseq-topics/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200729) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 9058074. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Untracked files:
Untracked: analysis/clusters_pca_structure_Cusanovich2018.Rmd
Unstaged changes:
Modified: analysis/plots_Lareau2019_bonemarrow.Rmd
Modified: code/plots.R
Modified: scripts/fit_all_models_Lareau2019.sh
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/clusters_Lareau2019_bonemarrow_k13.Rmd) and HTML (docs/clusters_Lareau2019_bonemarrow_k13.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 9058074 | kevinlkx | 2020-10-21 | add interpretations for clusters |
| html | fa7f769 | kevinlkx | 2020-10-21 | Build site. |
| Rmd | 4b016ee | kevinlkx | 2020-10-21 | wflow_publish(“analysis/clusters_Lareau2019_bonemarrow_k13.Rmd”) |
| html | 697d54e | kevinlkx | 2020-10-21 | Build site. |
| Rmd | cad6c07 | kevinlkx | 2020-10-21 | clustering cells based on topic proportions from K = 13 topics |
Here we explore the structure in the Lareau et al (2019) human bone marrow scATAC-seq data inferred from the multinomial topic model with \(k = 13\).
Load packages and some functions used in this analysis.
library(Matrix)
library(dplyr)
library(ggplot2)
library(cowplot)
library(fastTopics)
source("code/plots.R")The study used dsciATAC-seq to profile bone marrow mononuclear cells (BMMCs) from two human donors before (resting, untreated controls) and after stimulation (stimulated).
resting condition
Load the data in resting condition
data.dir <- "/project2/mstephens/kevinluo/scATACseq-topics/data/Lareau_2019/bone_marrow/processed_data/"
load(file.path(data.dir, "Lareau_2019_bonemarrow_resting.RData"))
cat(sprintf("%d x %d counts matrix.\n",nrow(counts),ncol(counts)))
rm(counts)
samples$Cluster <- as.factor(samples$Cluster)
# 60495 x 146860 counts matrix.Plot PCs of the topic proportions
We first use PCA to explore the structure inferred from the multinomial topic model with \(k = 13\):
out.dir <- "/project2/mstephens/kevinluo/scATACseq-topics/output/Lareau_2019/Lareau_2019_bonemarrow_resting"
fit <- readRDS(file.path(out.dir, "/fit-Lareau2019_bonemarrow_resting-scd-ex-k=13.rds"))$fitPlot PCs of the topic proportions.
p.pca1.1 <- pca_plot(poisson2multinom(fit),pcs = 1:2,fill = "none")
p.pca1.2 <- pca_plot(poisson2multinom(fit),pcs = 3:4,fill = "none")
p.pca1.3 <- pca_plot(poisson2multinom(fit),pcs = 5:6,fill = "none")
p.pca1.4 <- pca_plot(poisson2multinom(fit),pcs = 7:8,fill = "none")
p.pca1.5 <- pca_plot(poisson2multinom(fit),pcs = 9:10,fill = "none")
p.pca1.6 <- pca_plot(poisson2multinom(fit),pcs = 11:12,fill = "none")
plot_grid(p.pca1.1,p.pca1.2,p.pca1.3,p.pca1.4,p.pca1.5,p.pca1.6)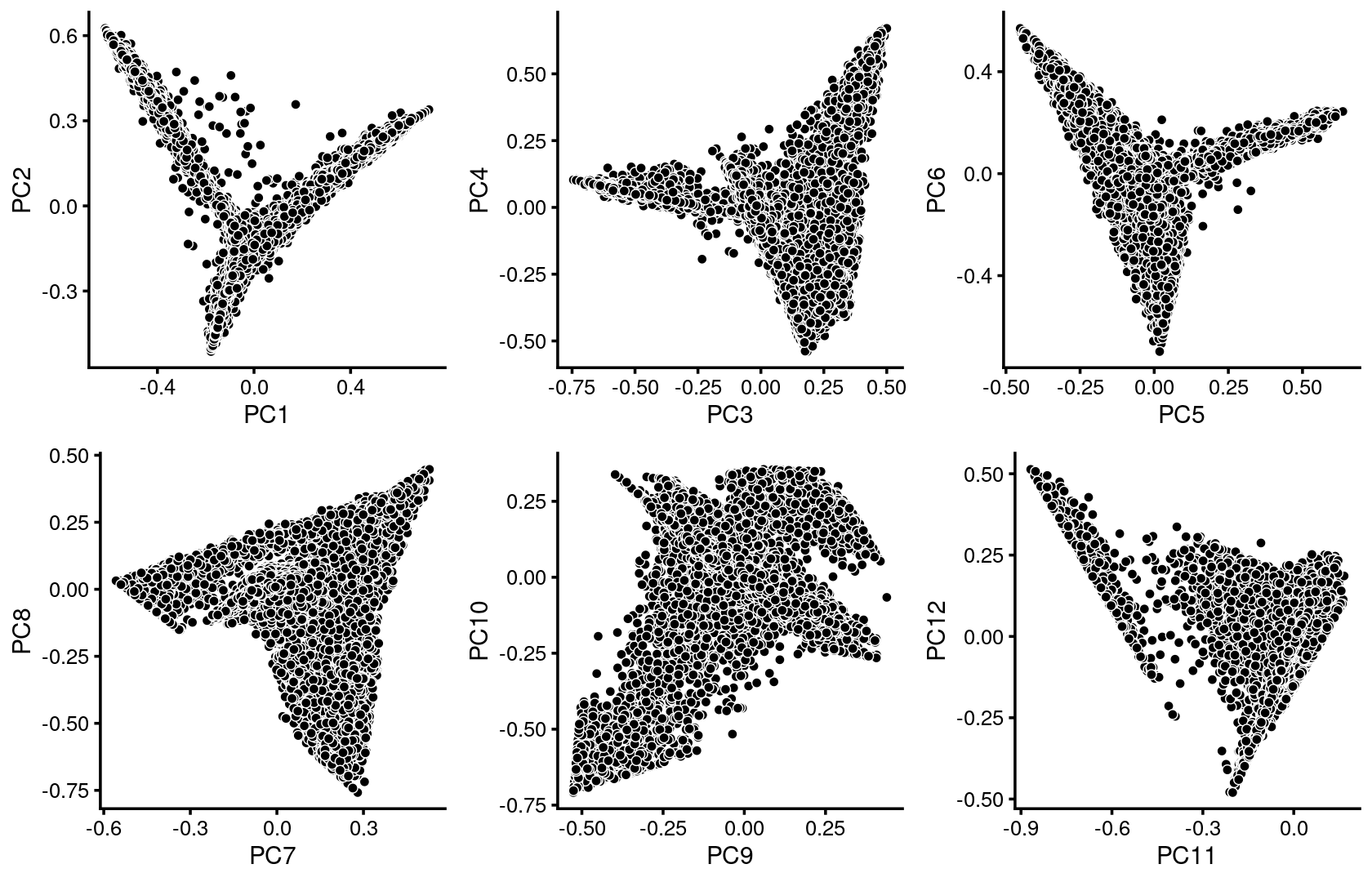
| Version | Author | Date |
|---|---|---|
| 697d54e | kevinlkx | 2020-10-21 |
Some of the structure is more evident from “hexbin” plots showing the density of the points.
breaks <- c(0,1,5,10,100,Inf)
p.pca2.1 <- pca_hexbin_plot(poisson2multinom(fit), pcs = 1:2, breaks = breaks) + guides(fill = "none")
p.pca2.2 <- pca_hexbin_plot(poisson2multinom(fit), pcs = 3:4, breaks = breaks) + guides(fill = "none")
p.pca2.3 <- pca_hexbin_plot(poisson2multinom(fit), pcs = 5:6, breaks = breaks) + guides(fill = "none")
p.pca2.4 <- pca_hexbin_plot(poisson2multinom(fit), pcs = 7:8, breaks = breaks) + guides(fill = "none")
p.pca2.5 <- pca_hexbin_plot(poisson2multinom(fit), pcs = 9:10, breaks = breaks) + guides(fill = "none")
p.pca2.6 <- pca_hexbin_plot(poisson2multinom(fit), pcs = 11:12, breaks = breaks) + guides(fill = "none")
plot_grid(p.pca2.1,p.pca2.2,p.pca2.3,p.pca2.4,p.pca2.5,p.pca2.6)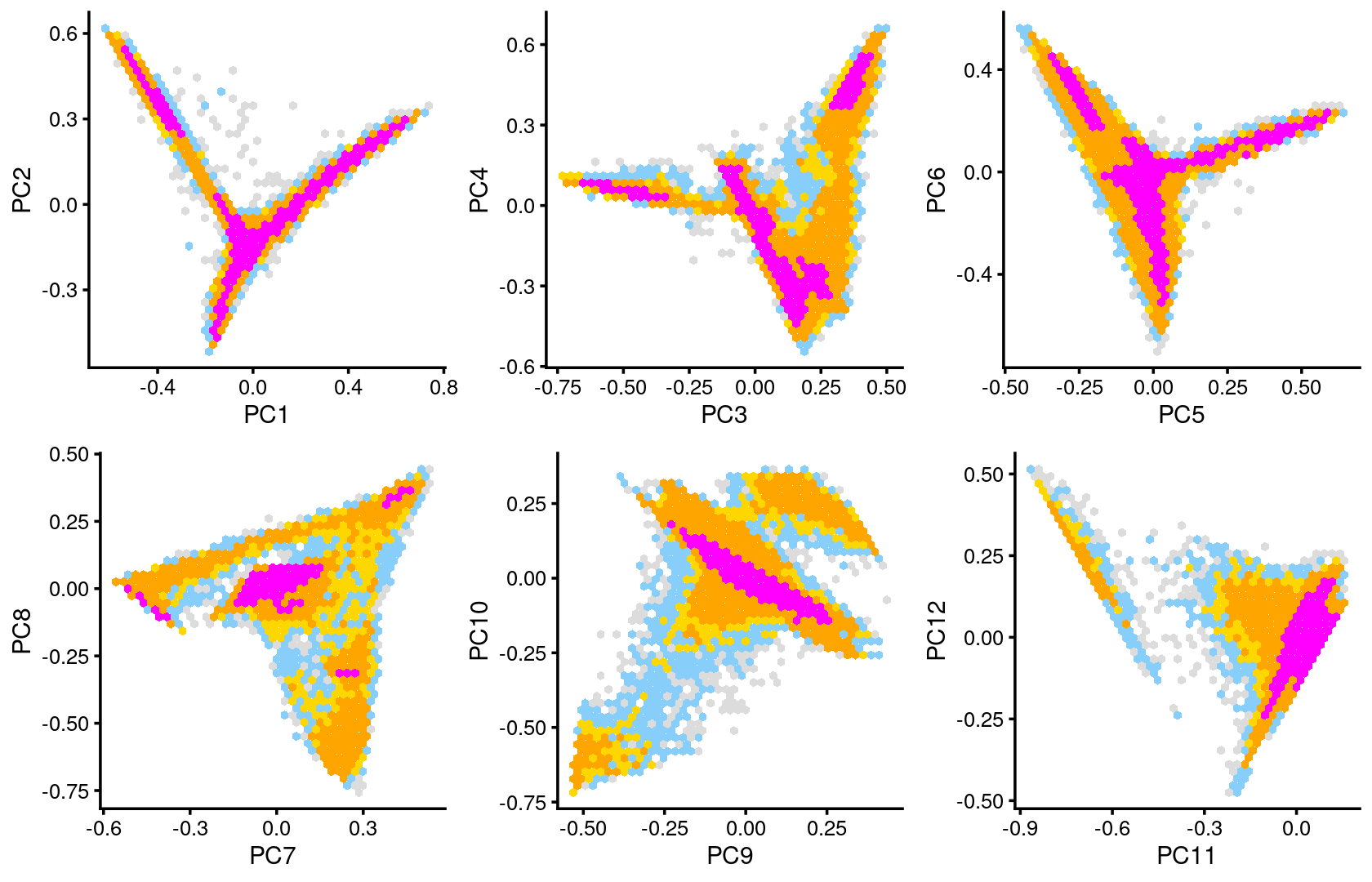
| Version | Author | Date |
|---|---|---|
| 697d54e | kevinlkx | 2020-10-21 |
Layer the cell labels onto the PC plots
Next, we layer the cell labels onto the PC plots.
p.pca3.1 <- labeled_pca_plot(fit,1:2,samples$Cluster,font_size = 7,
legend_label = "Cell labels")
p.pca3.2 <- labeled_pca_plot(fit,3:4,samples$Cluster,font_size = 7,
legend_label = "Cell labels")
p.pca3.3 <- labeled_pca_plot(fit,5:6,samples$Cluster,font_size = 7,
legend_label = "Cell labels")
p.pca3.4 <- labeled_pca_plot(fit,7:8,samples$Cluster,font_size = 7,
legend_label = "Cell labels")
p.pca3.5 <- labeled_pca_plot(fit,9:10,samples$Cluster,font_size = 7,
legend_label = "Cell labels")
p.pca3.6 <- labeled_pca_plot(fit,11:12,samples$Cluster,font_size = 7,
legend_label = "Cell labels")
plot_grid(p.pca3.1,p.pca3.2,p.pca3.3,p.pca3.4,p.pca3.5,p.pca3.6)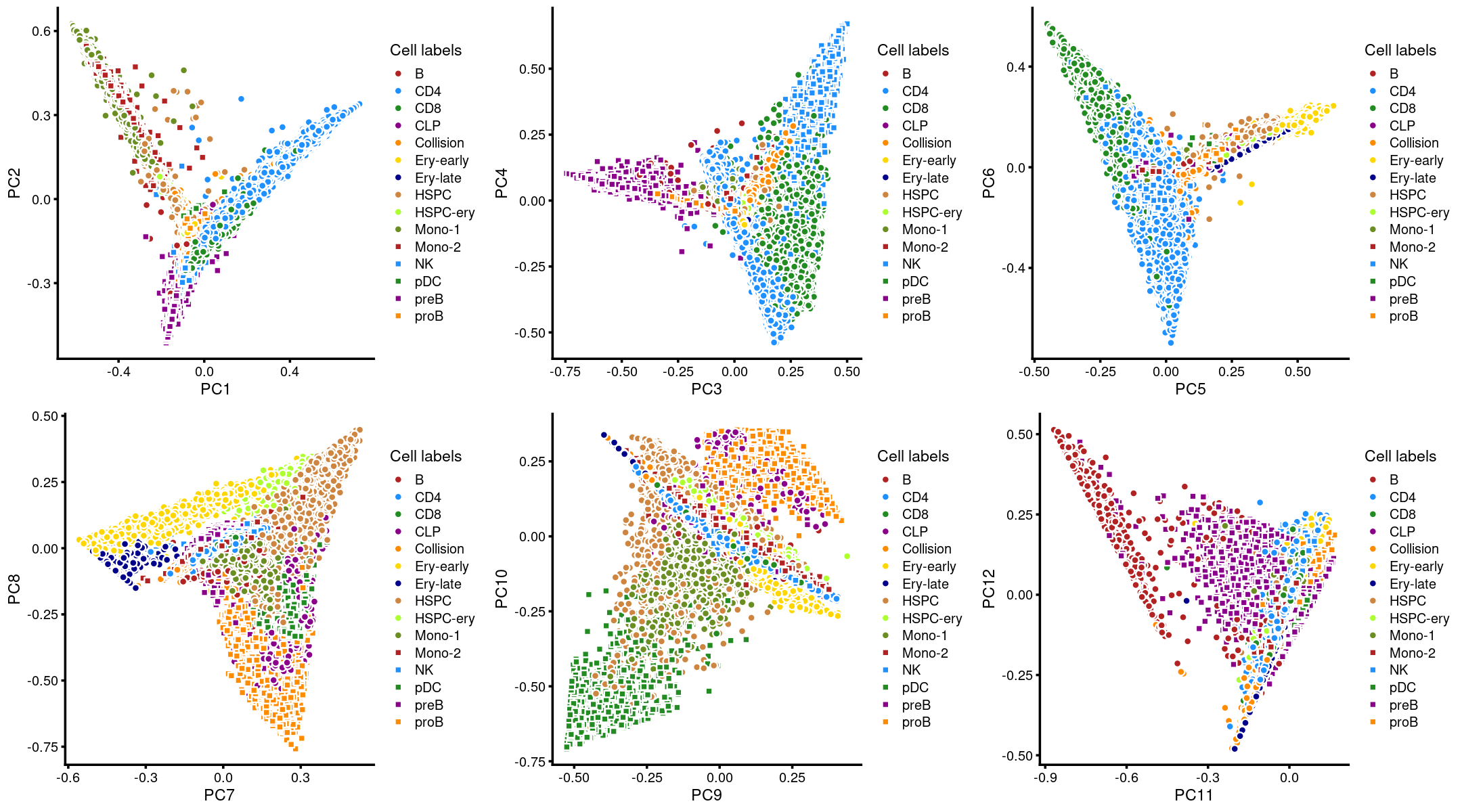
| Version | Author | Date |
|---|---|---|
| 697d54e | kevinlkx | 2020-10-21 |
Visualize by structure plot grouped by cell labels
set.seed(10)
colors_topics <- c("#a6cee3","#1f78b4","#b2df8a","#33a02c","#fb9a99","#e31a1c",
"#fdbf6f","#ff7f00","#cab2d6","#6a3d9a","#ffff99","#b15928",
"gray")
rows <- sample(nrow(fit$L),4000)
samples$Cluster <- as.factor(samples$Cluster)
p.structure.1 <- structure_plot(select(poisson2multinom(fit),loadings = rows),
grouping = samples[rows, "Cluster"],n = Inf,gap = 20,
perplexity = 50,topics = 1:13,colors = colors_topics,
num_threads = 4,verbose = FALSE)
# Perplexity automatically changed to 17 because original setting of 50 was too large for the number of samples (56)
# Perplexity automatically changed to 6 because original setting of 50 was too large for the number of samples (22)
# Perplexity automatically changed to 6 because original setting of 50 was too large for the number of samples (24)
# Perplexity automatically changed to 22 because original setting of 50 was too large for the number of samples (71)
# Perplexity automatically changed to 10 because original setting of 50 was too large for the number of samples (34)
# Perplexity automatically changed to 21 because original setting of 50 was too large for the number of samples (69)
# Perplexity automatically changed to 32 because original setting of 50 was too large for the number of samples (101)
print(p.structure.1)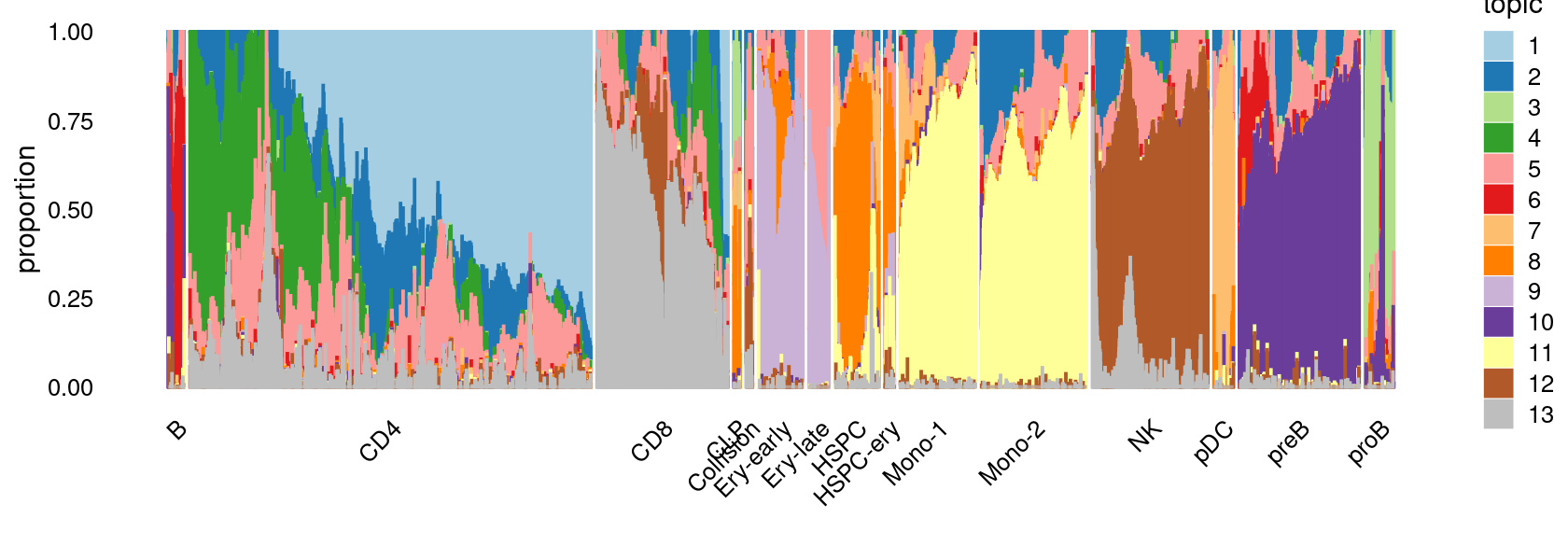
k-means clustering on topic proportions
Define clusters using k-means, and then create structure plot based on the clusters from k-means.
Define clusters using k-means with \(k = 10\):
set.seed(10)
clusters.10 <- factor(kmeans(poisson2multinom(fit)$L,centers = 10)$cluster)
print(sort(table(clusters.10),decreasing = TRUE))
# clusters.10
# 10 1 3 6 4 5 7 2 8 9
# 10753 10175 7857 6055 5803 5090 4839 3593 3339 2991Structure plot based on k-means clusters
colors_topics <- c("#a6cee3","#1f78b4","#b2df8a","#33a02c","#fb9a99","#e31a1c",
"#fdbf6f","#ff7f00","#cab2d6","#6a3d9a","#ffff99","#b15928",
"gray")
rows <- sample(nrow(fit$L),4000)
p.structure.2 <- structure_plot(select(poisson2multinom(fit),loadings = rows),
grouping = clusters.10[rows],n = Inf,gap = 20,
perplexity = 50,topics = 1:13,colors = colors_topics,
num_threads = 4,verbose = FALSE)
print(p.structure.2)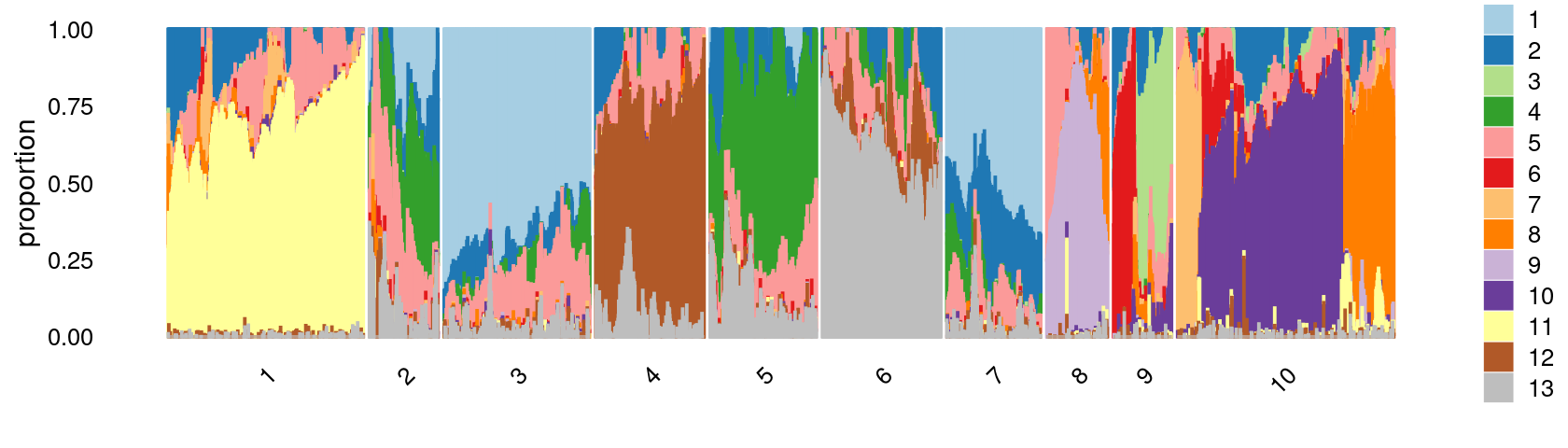
| Version | Author | Date |
|---|---|---|
| 697d54e | kevinlkx | 2020-10-21 |
Define clusters using k-means with \(k = 12\):
set.seed(10)
clusters.12 <- factor(kmeans(poisson2multinom(fit)$L,centers = 12)$cluster)
print(sort(table(clusters.12),decreasing = TRUE))
# clusters.12
# 3 1 12 5 8 10 4 7 2 6 11 9
# 12075 10014 6866 5978 5395 5045 3829 3667 3173 2147 1412 894Structure plot based on k-means clusters
colors_topics <- c("#a6cee3","#1f78b4","#b2df8a","#33a02c","#fb9a99","#e31a1c",
"#fdbf6f","#ff7f00","#cab2d6","#6a3d9a","#ffff99","#b15928",
"gray")
rows <- sample(nrow(fit$L),4000)
p.structure.3 <- structure_plot(select(poisson2multinom(fit),loadings = rows),
grouping = clusters.12[rows],n = Inf,gap = 20,
perplexity = 50,topics = 1:13,colors = colors_topics,
num_threads = 4,verbose = FALSE)
# Perplexity automatically changed to 44 because original setting of 50 was too large for the number of samples (136)
# Perplexity automatically changed to 18 because original setting of 50 was too large for the number of samples (60)
# Perplexity automatically changed to 25 because original setting of 50 was too large for the number of samples (79)
print(p.structure.3)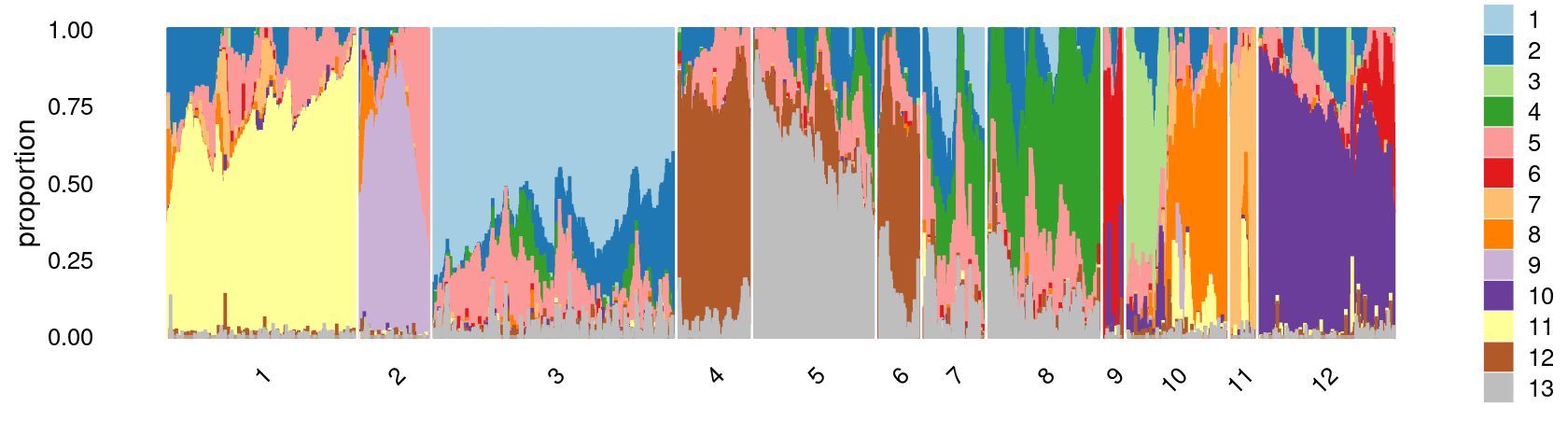
| Version | Author | Date |
|---|---|---|
| 697d54e | kevinlkx | 2020-10-21 |
Refine clusters
We then further refine the clusters based on k-means result with \(k = 12\): merge clusters 4 and 6; (maybe also merge clusters 7 and 8).
clusters.merged <- clusters.12
clusters.merged[clusters.12 %in% c(4,6)] <- 4
# clusters.merged[clusters.12 %in% c(7,8)] <- 7
clusters.merged <- factor(clusters.merged, labels = paste0("c", c(1:length(unique(clusters.merged)))))
print(sort(table(clusters.merged),decreasing = TRUE))
samples$cluster_kmeans <- clusters.merged
# clusters.merged
# c3 c1 c11 c5 c4 c7 c9 c6 c2 c10 c8
# 12075 10014 6866 5978 5976 5395 5045 3667 3173 1412 894colors_topics <- c("#a6cee3","#1f78b4","#b2df8a","#33a02c","#fb9a99","#e31a1c",
"#fdbf6f","#ff7f00","#cab2d6","#6a3d9a","#ffff99","#b15928",
"gray")
rows <- sample(nrow(fit$L),4000)
p.structure.refined <- structure_plot(select(poisson2multinom(fit),loadings = rows),
grouping = clusters.merged[rows],n = Inf,gap = 20,
perplexity = 50,topics = 1:13,colors = colors_topics,
num_threads = 4,verbose = FALSE)
# Perplexity automatically changed to 18 because original setting of 50 was too large for the number of samples (59)
# Perplexity automatically changed to 26 because original setting of 50 was too large for the number of samples (84)
print(p.structure.refined)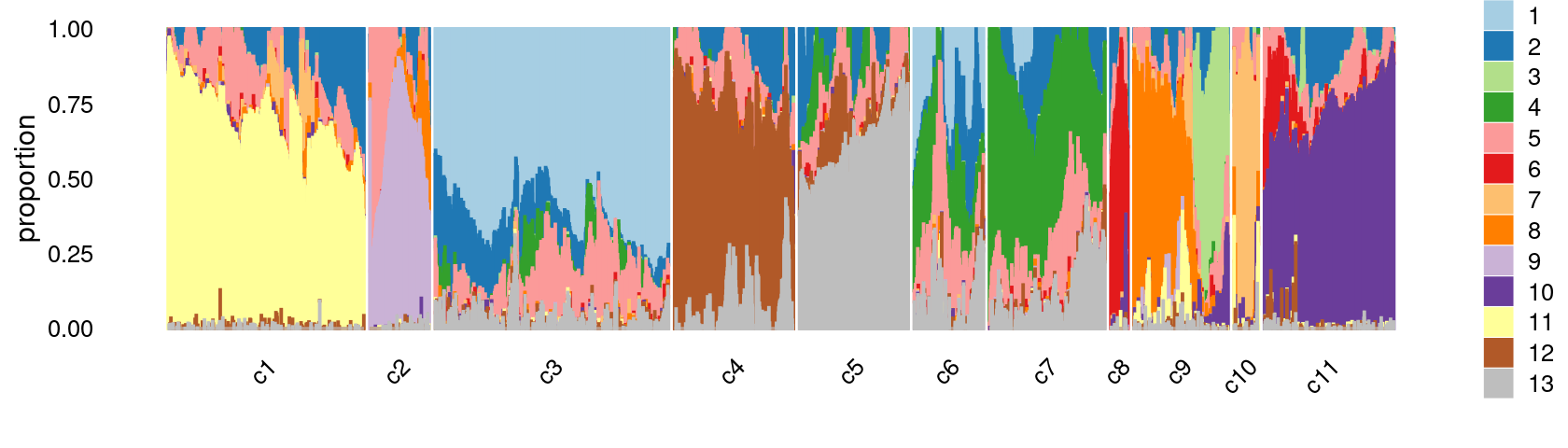
| Version | Author | Date |
|---|---|---|
| 697d54e | kevinlkx | 2020-10-21 |
The clusters defined by k-means on topic proportions reasonably identify the clusters shown in the PCA hexbin plots (below).
p.pca.4.1 <- labeled_pca_plot(fit,1:2,samples$cluster_kmeans,font_size = 7,
legend_label = "cluster_kmeans")
p.pca.4.2 <- labeled_pca_plot(fit,3:4,samples$cluster_kmeans,font_size = 7,
legend_label = "cluster_kmeans")
p.pca.4.3 <- labeled_pca_plot(fit,5:6,samples$cluster_kmeans,font_size = 7,
legend_label = "cluster_kmeans")
p.pca.4.4 <- labeled_pca_plot(fit,7:8,samples$cluster_kmeans,font_size = 7,
legend_label = "cluster_kmeans")
p.pca.4.5 <- labeled_pca_plot(fit,9:10,samples$cluster_kmeans,font_size = 7,
legend_label = "cluster_kmeans")
p.pca.4.6 <- labeled_pca_plot(fit,11:12,samples$cluster_kmeans,font_size = 7,
legend_label = "cluster_kmeans")
plot_grid(p.pca.4.1,p.pca.4.2,p.pca.4.3,p.pca.4.4,p.pca.4.5,p.pca.4.6)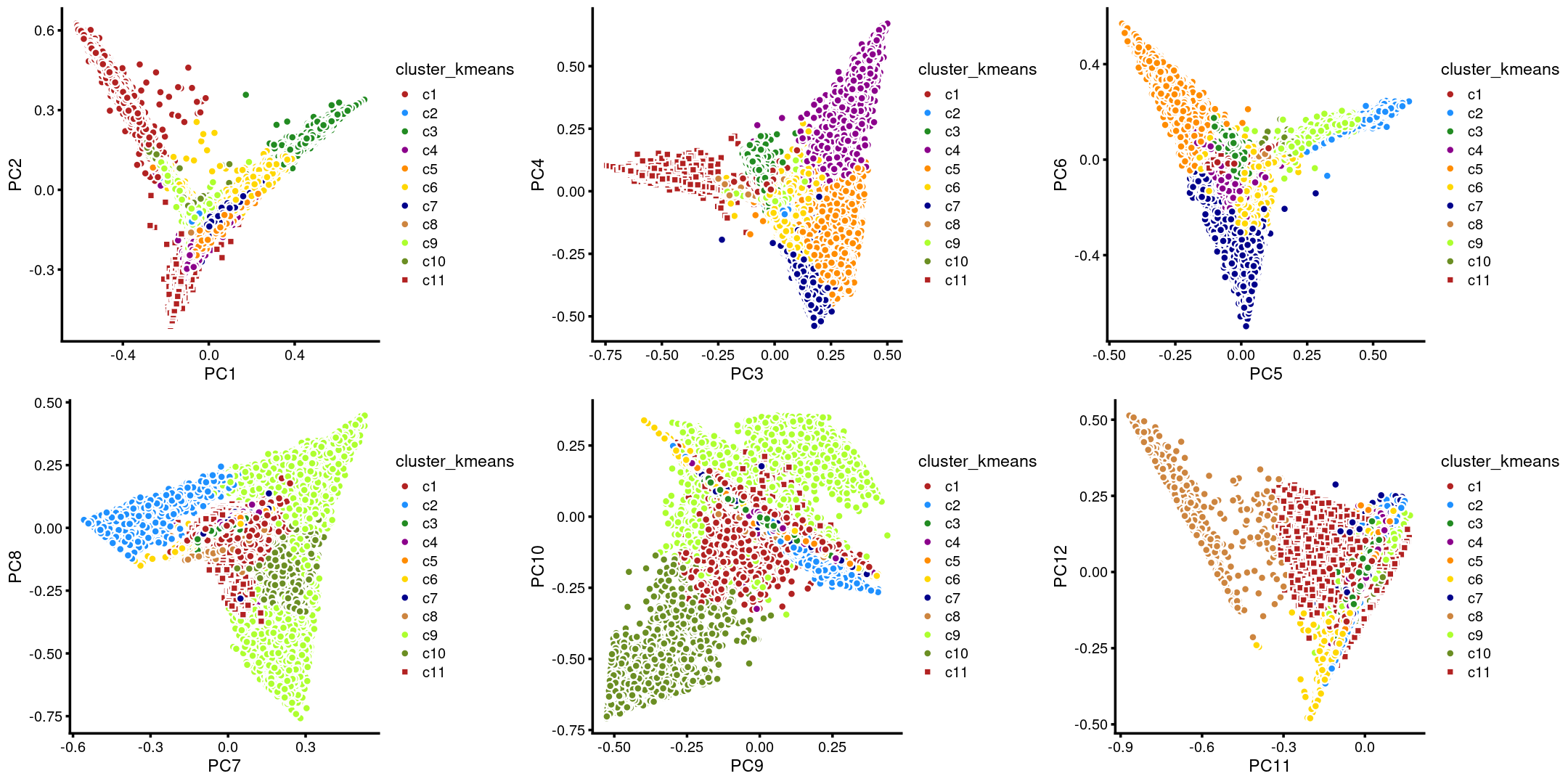
| Version | Author | Date |
|---|---|---|
| 697d54e | kevinlkx | 2020-10-21 |
plot_grid(p.pca2.1,p.pca2.2,p.pca2.3,p.pca2.4,p.pca2.5,p.pca2.6)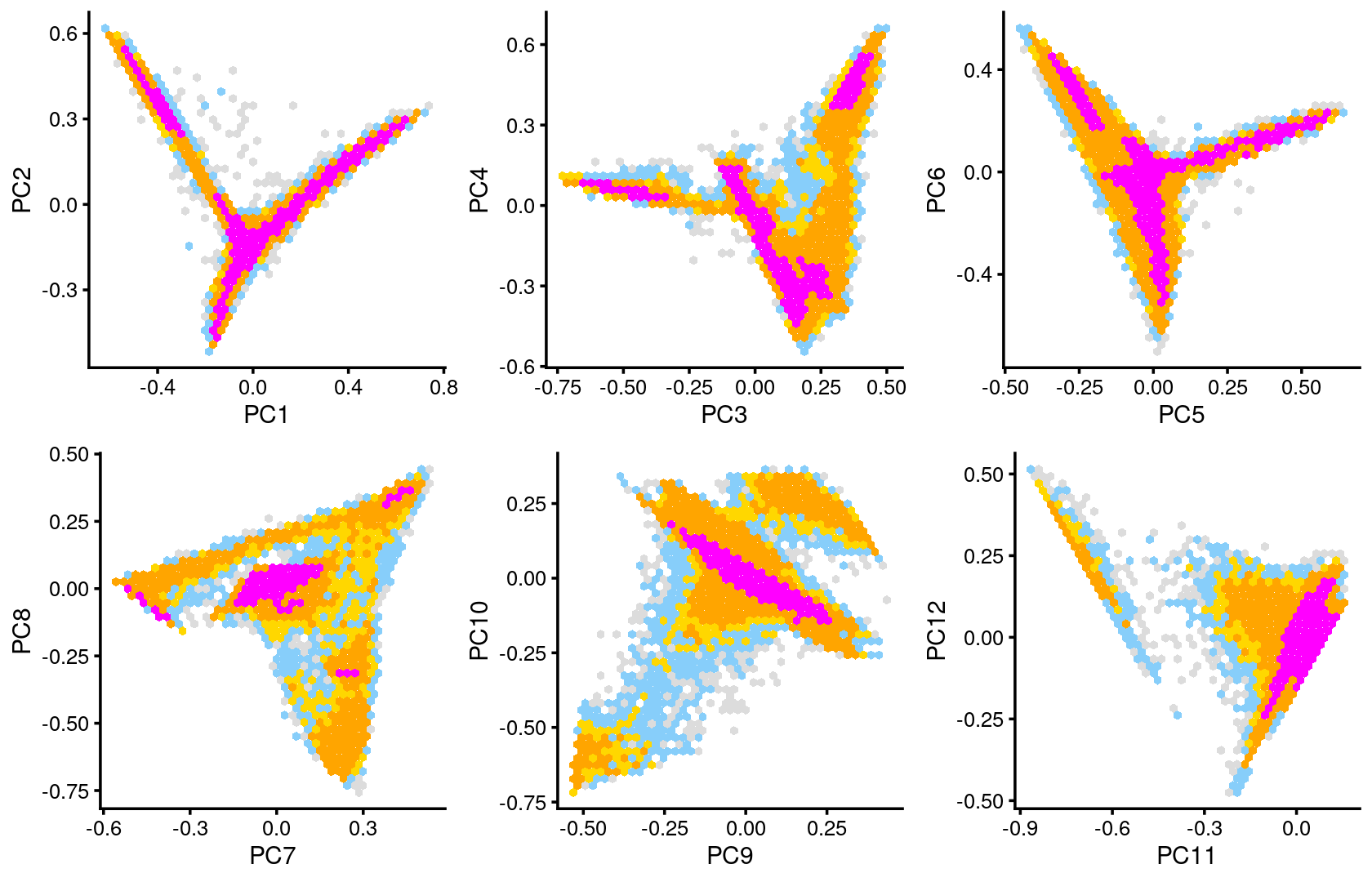
| Version | Author | Date |
|---|---|---|
| 697d54e | kevinlkx | 2020-10-21 |
We then label the cells in each cluster with the known cell labels.
Cell labels:
samples$Cluster <- as.factor(samples$Cluster)
cat(length(levels(samples$Cluster)), "cell labels. \n")
table(samples$Cluster)
# 15 cell labels.
#
# B CD4 CD8 CLP Collision Ery-early Ery-late HSPC
# 1001 20378 6647 411 271 2176 1174 2536
# HSPC-ery Mono-1 Mono-2 NK pDC preB proB
# 479 3913 6012 5741 1218 6658 1880Plot the distribution of cell labels by clusters.
Stacked barplot for the counts of cell labels by clusters:
library(plyr);library(dplyr)
# ------------------------------------------------------------------------------
# You have loaded plyr after dplyr - this is likely to cause problems.
# If you need functions from both plyr and dplyr, please load plyr first, then dplyr:
# library(plyr); library(dplyr)
# ------------------------------------------------------------------------------
#
# Attaching package: 'plyr'
# The following objects are masked from 'package:dplyr':
#
# arrange, count, desc, failwith, id, mutate, rename, summarise,
# summarize
library(RColorBrewer)
freq_cluster_tissue <- count(samples, vars=c("cluster_kmeans","Cluster"))
n_colors <- length(levels(samples$Cluster))
colors_labels <- colorRampPalette(brewer.pal(9, "Set1"))(n_colors)
# stacked barplot for the counts of cell labels by clusters
p.barplot.1 <- ggplot(freq_cluster_tissue, aes(fill=Cluster, y=freq, x=cluster_kmeans)) +
geom_bar(position="stack", stat="identity") +
theme_classic() + xlab("Cluster") + ylab("Number of cells") +
scale_fill_manual(values = colors_labels) +
guides(fill=guide_legend(ncol=2)) +
theme(
legend.title = element_text(size = 10),
legend.text = element_text(size = 8)
)
print(p.barplot.1)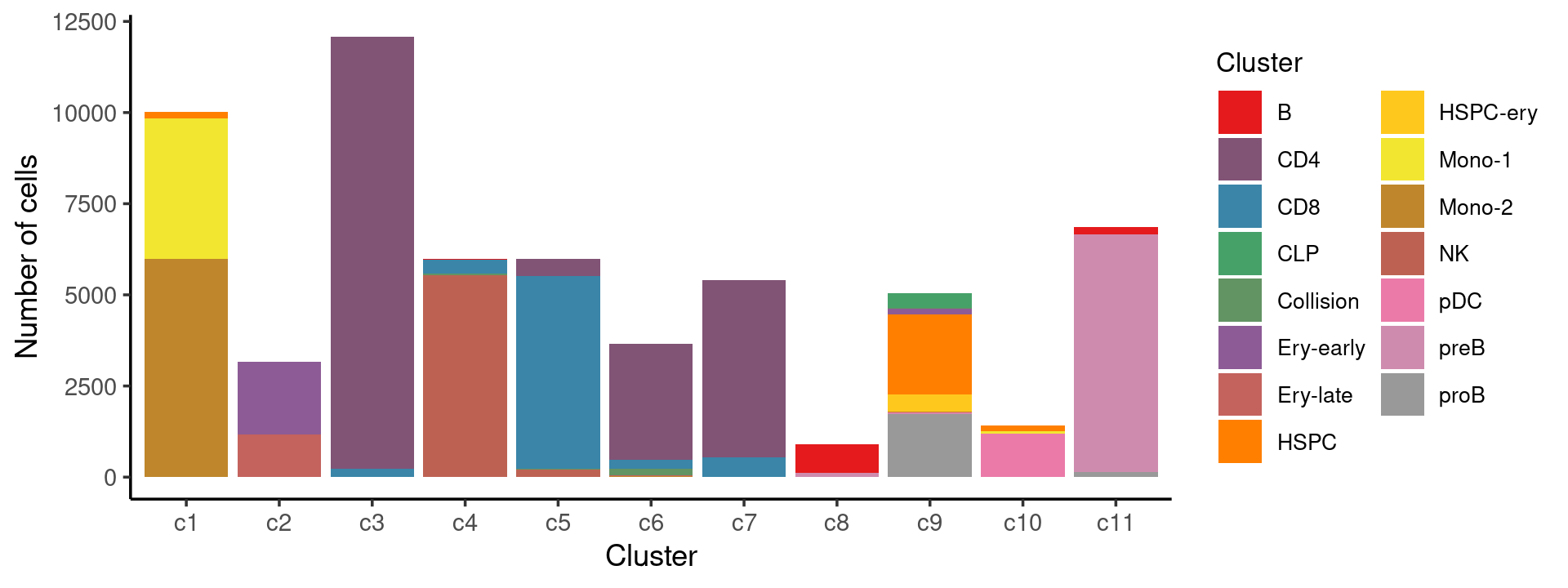
| Version | Author | Date |
|---|---|---|
| 697d54e | kevinlkx | 2020-10-21 |
Percent stacked barplot for the counts of cell labels by clusters:
freq_cluster_tissue <- count(samples, vars=c("cluster_kmeans","Cluster"))
n_colors <- length(levels(samples$Cluster))
colors_labels <- colorRampPalette(brewer.pal(9, "Set1"))(n_colors)
p.barplot.2 <- ggplot(freq_cluster_tissue, aes(fill=Cluster, y=freq, x=cluster_kmeans)) +
geom_bar(position="fill", stat="identity") +
theme_classic() + xlab("Cluster") + ylab("Proportion of cells") +
scale_fill_manual(values = colors_labels) +
guides(fill=guide_legend(ncol=2)) +
theme(
legend.title = element_text(size = 10),
legend.text = element_text(size = 8)
)
print(p.barplot.2)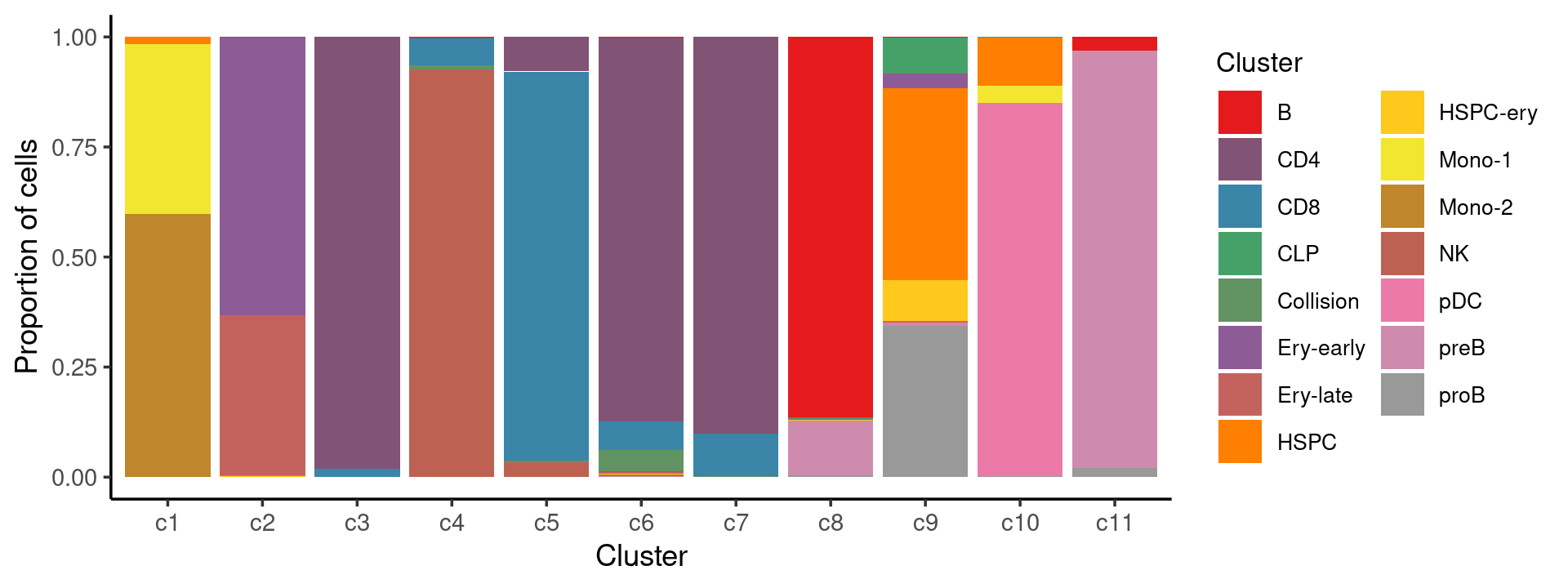
| Version | Author | Date |
|---|---|---|
| 697d54e | kevinlkx | 2020-10-21 |
We can see a few clusters are dominated by one major cell type: * cells in cluster c3 are primarily CD4 cells; * cells in cluster c4 are primarily NK cells; * cells in cluster c5 are primarily CD8 cells; * the majority of cells in cluster c6 and c7 are CD4 cells; * the majority of cells in cluster c8 are CD4 cells (and B cells); * the majority of cells in cluster c10 are pDC cells (and HSPC cells); * the majority of cells in cluster c11 are preB cells.
Some clusters are combinations of related cell types: * cells in cluster c1 are a combination of Mono-1 and Mono-2 cells; * cells in cluster c2 are a combination of Ery-early and Ery-late cells;
Cluster c9 is more heterogeneous with HSPC, HSPC-ery, proB, and CLP cells.
freq_cluster_cells <- with(samples,table(Cluster,cluster_kmeans))
print(freq_cluster_cells)
# cluster_kmeans
# Cluster c1 c2 c3 c4 c5 c6 c7 c8 c9 c10 c11
# B 0 0 0 9 0 8 0 773 3 0 208
# CD4 0 2 11836 14 469 3192 4862 0 1 0 2
# CD8 0 0 234 363 5289 240 520 1 0 0 0
# CLP 0 0 0 0 0 0 0 0 410 1 0
# Collision 2 1 4 54 18 181 7 2 2 0 0
# Ery-early 0 2002 0 0 0 2 1 1 170 0 0
# Ery-late 0 1157 0 0 0 16 0 1 0 0 0
# HSPC 169 2 0 2 3 2 1 0 2202 155 0
# HSPC-ery 1 7 0 0 0 5 0 1 465 0 0
# Mono-1 3851 0 0 0 0 0 1 1 5 55 0
# Mono-2 5990 0 0 3 0 8 0 0 11 0 0
# NK 1 2 0 5531 196 8 1 0 2 0 0
# pDC 0 0 1 0 0 0 0 0 18 1199 0
# preB 0 0 0 0 3 5 2 110 19 0 6519
# proB 0 0 0 0 0 0 0 4 1737 2 137stimulated condition
Load the data in stimulated condition
data.dir <- "/project2/mstephens/kevinluo/scATACseq-topics/data/Lareau_2019/bone_marrow/processed_data/"
load(file.path(data.dir, "Lareau_2019_bonemarrow_stimulated.RData"))
cat(sprintf("%d x %d counts matrix.\n",nrow(counts),ncol(counts)))
rm(counts)
samples$Cluster <- as.factor(samples$Cluster)
# 75968 x 146860 counts matrix.Plot PCs of the topic proportions
We first use PCA to explore the structure inferred from the multinomial topic model with \(k = 13\):
out.dir <- "/project2/mstephens/kevinluo/scATACseq-topics/output/Lareau_2019/Lareau_2019_bonemarrow_stimulated"
fit <- readRDS(file.path(out.dir, "/fit-Lareau2019_bonemarrow_stimulated-scd-ex-k=13.rds"))$fitPlot PCs of the topic proportions.
p.pca1.1 <- pca_plot(poisson2multinom(fit),pcs = 1:2,fill = "none")
p.pca1.2 <- pca_plot(poisson2multinom(fit),pcs = 3:4,fill = "none")
p.pca1.3 <- pca_plot(poisson2multinom(fit),pcs = 5:6,fill = "none")
p.pca1.4 <- pca_plot(poisson2multinom(fit),pcs = 7:8,fill = "none")
p.pca1.5 <- pca_plot(poisson2multinom(fit),pcs = 9:10,fill = "none")
p.pca1.6 <- pca_plot(poisson2multinom(fit),pcs = 11:12,fill = "none")
plot_grid(p.pca1.1,p.pca1.2,p.pca1.3,p.pca1.4,p.pca1.5,p.pca1.6)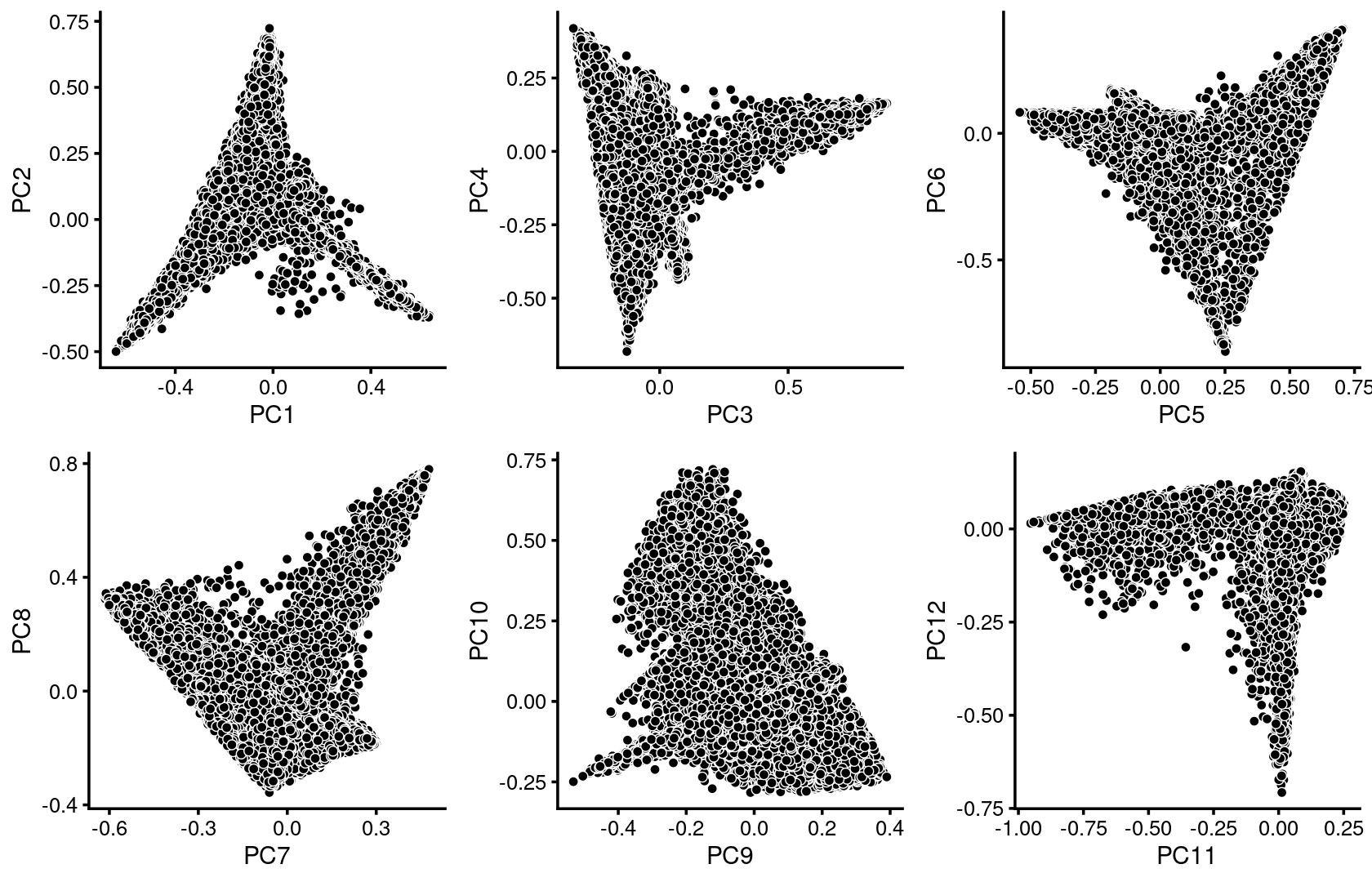
| Version | Author | Date |
|---|---|---|
| 697d54e | kevinlkx | 2020-10-21 |
Some of the structure is more evident from “hexbin” plots showing the density of the points.
breaks <- c(0,1,5,10,100,Inf)
p.pca2.1 <- pca_hexbin_plot(poisson2multinom(fit), pcs = 1:2, breaks = breaks) + guides(fill = "none")
p.pca2.2 <- pca_hexbin_plot(poisson2multinom(fit), pcs = 3:4, breaks = breaks) + guides(fill = "none")
p.pca2.3 <- pca_hexbin_plot(poisson2multinom(fit), pcs = 5:6, breaks = breaks) + guides(fill = "none")
p.pca2.4 <- pca_hexbin_plot(poisson2multinom(fit), pcs = 7:8, breaks = breaks) + guides(fill = "none")
p.pca2.5 <- pca_hexbin_plot(poisson2multinom(fit), pcs = 9:10, breaks = breaks) + guides(fill = "none")
p.pca2.6 <- pca_hexbin_plot(poisson2multinom(fit), pcs = 11:12, breaks = breaks) + guides(fill = "none")
plot_grid(p.pca2.1,p.pca2.2,p.pca2.3,p.pca2.4,p.pca2.5,p.pca2.6)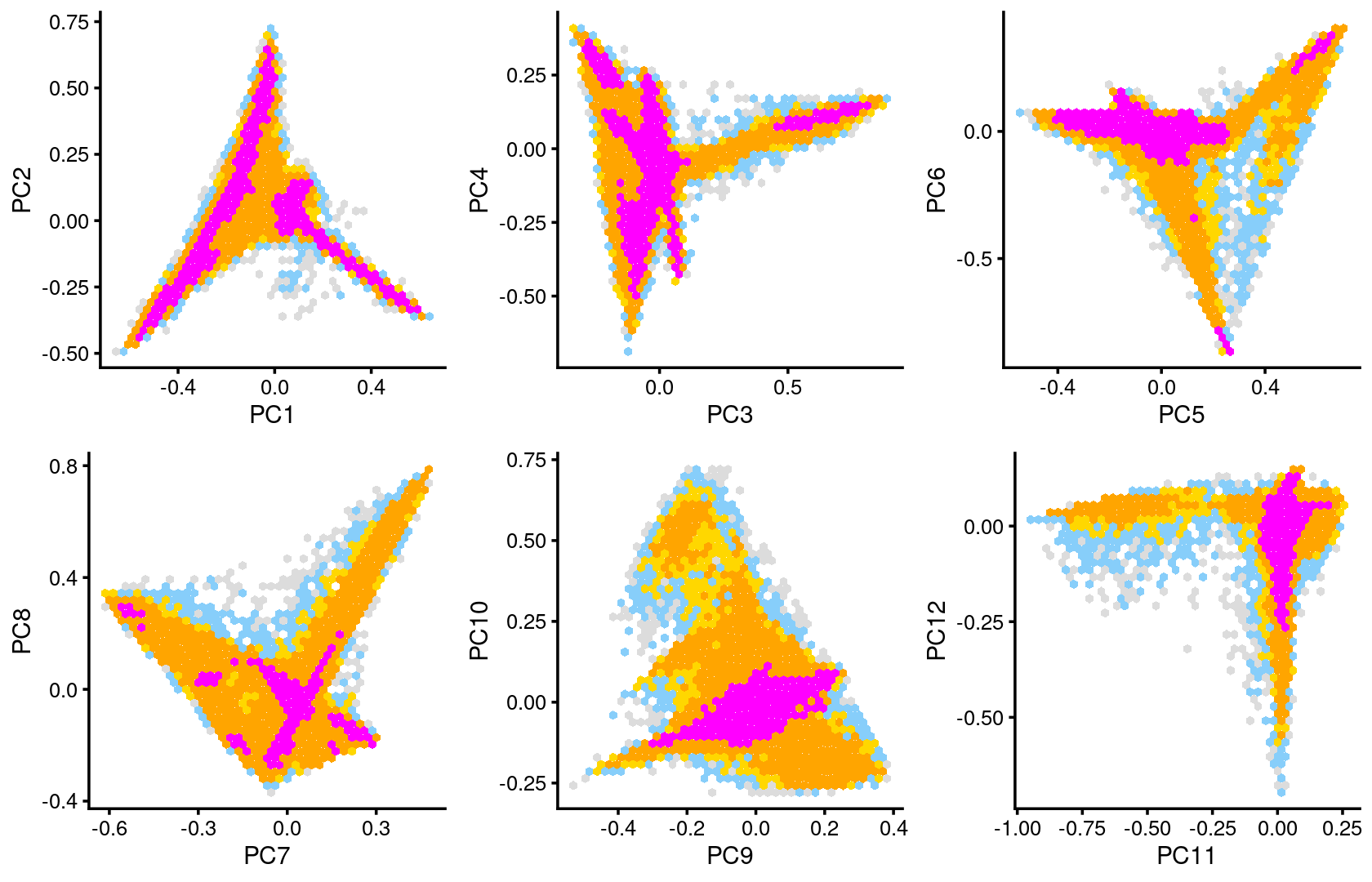
| Version | Author | Date |
|---|---|---|
| 697d54e | kevinlkx | 2020-10-21 |
Layer the cell labels onto the PC plots
Next, we layer the cell labels onto the PC plots.
p.pca3.1 <- labeled_pca_plot(fit,1:2,samples$Cluster,font_size = 7,
legend_label = "Cell labels")
p.pca3.2 <- labeled_pca_plot(fit,3:4,samples$Cluster,font_size = 7,
legend_label = "Cell labels")
p.pca3.3 <- labeled_pca_plot(fit,5:6,samples$Cluster,font_size = 7,
legend_label = "Cell labels")
p.pca3.4 <- labeled_pca_plot(fit,7:8,samples$Cluster,font_size = 7,
legend_label = "Cell labels")
p.pca3.5 <- labeled_pca_plot(fit,9:10,samples$Cluster,font_size = 7,
legend_label = "Cell labels")
p.pca3.6 <- labeled_pca_plot(fit,11:12,samples$Cluster,font_size = 7,
legend_label = "Cell labels")
plot_grid(p.pca3.1,p.pca3.2,p.pca3.3,p.pca3.4,p.pca3.5,p.pca3.6)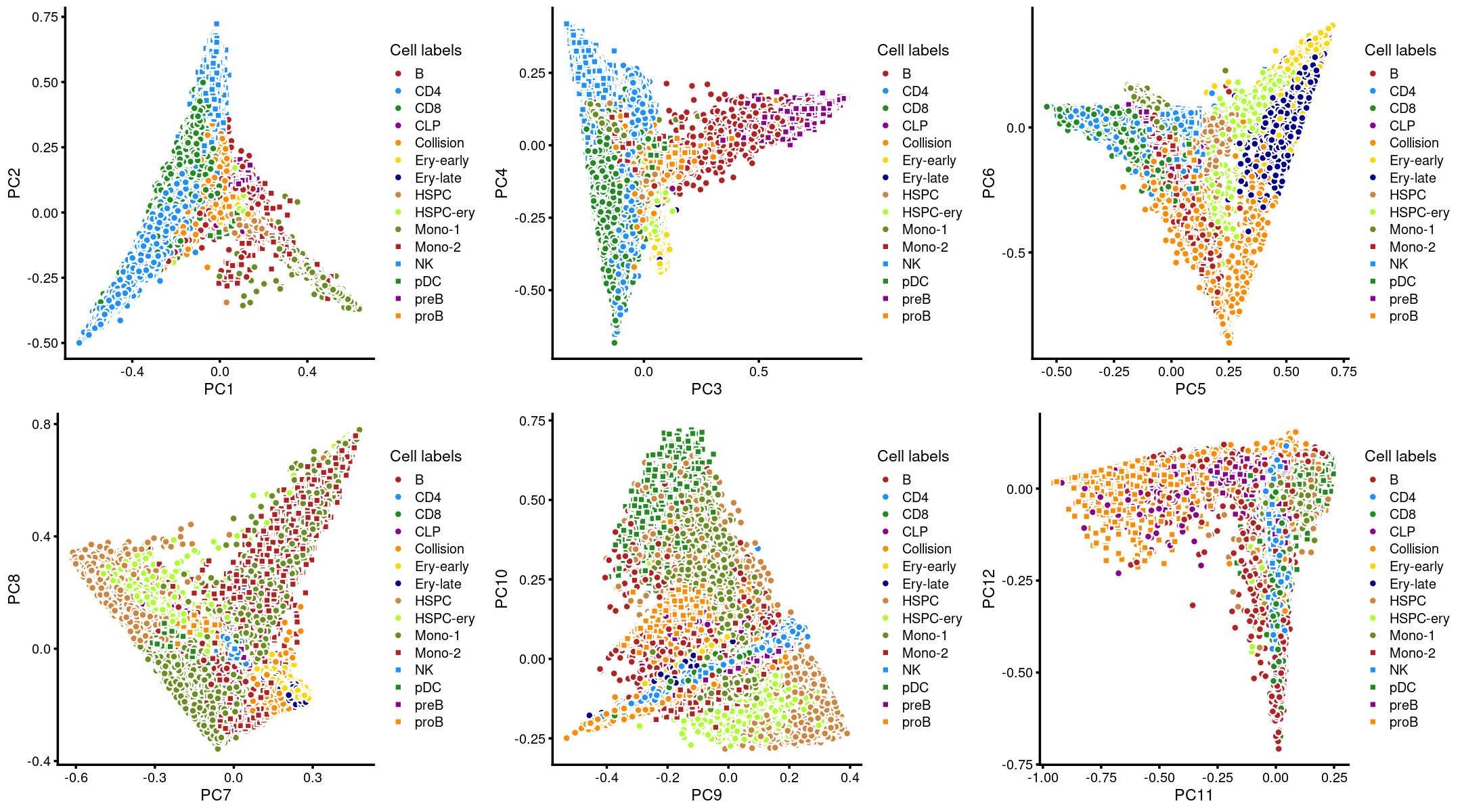
| Version | Author | Date |
|---|---|---|
| 697d54e | kevinlkx | 2020-10-21 |
Visualize by structure plot grouped by cell labels
set.seed(10)
colors_topics <- c("#a6cee3","#1f78b4","#b2df8a","#33a02c","#fb9a99","#e31a1c",
"#fdbf6f","#ff7f00","#cab2d6","#6a3d9a","#ffff99","#b15928",
"gray")
rows <- sample(nrow(fit$L),4000)
samples$Cluster <- as.factor(samples$Cluster)
p.structure.1 <- structure_plot(select(poisson2multinom(fit),loadings = rows),
grouping = samples[rows, "Cluster"],n = Inf,gap = 20,
perplexity = 50,topics = 1:13,colors = colors_topics,
num_threads = 4,verbose = FALSE)
# Perplexity automatically changed to 2 because original setting of 50 was too large for the number of samples (12)
# Perplexity automatically changed to 46 because original setting of 50 was too large for the number of samples (143)
# Perplexity automatically changed to 16 because original setting of 50 was too large for the number of samples (54)
# Perplexity automatically changed to 33 because original setting of 50 was too large for the number of samples (103)
# Perplexity automatically changed to 15 because original setting of 50 was too large for the number of samples (50)
# Perplexity automatically changed to 19 because original setting of 50 was too large for the number of samples (63)
print(p.structure.1)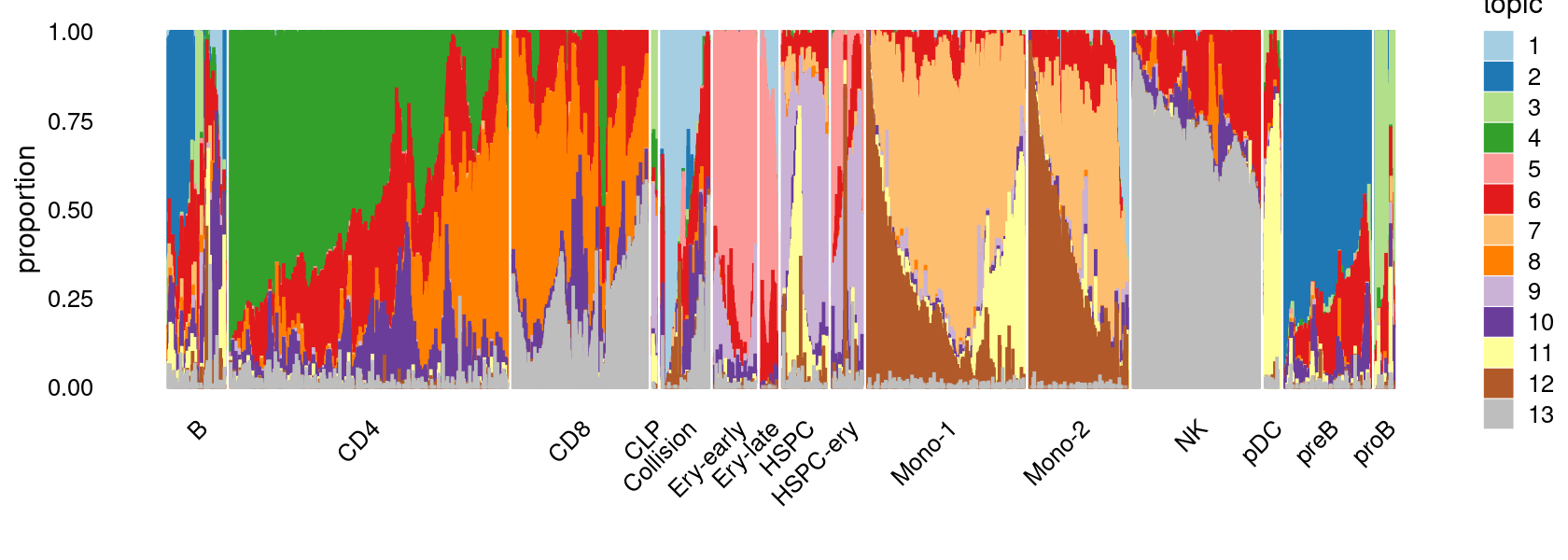
k-means clustering on topic proportions
Define clusters using k-means, and then create structure plot based on the clusters from k-means.
Define clusters using k-means with \(k = 10\):
set.seed(10)
clusters.10 <- factor(kmeans(poisson2multinom(fit)$L,centers = 10)$cluster)
print(sort(table(clusters.10),decreasing = TRUE))
# clusters.10
# 3 8 2 10 4 7 5 6 1 9
# 15209 11641 8174 8161 7711 6990 5376 4713 4556 3437Structure plot based on k-means clusters
colors_topics <- c("#a6cee3","#1f78b4","#b2df8a","#33a02c","#fb9a99","#e31a1c",
"#fdbf6f","#ff7f00","#cab2d6","#6a3d9a","#ffff99","#b15928",
"gray")
rows <- sample(nrow(fit$L),4000)
p.structure.2 <- structure_plot(select(poisson2multinom(fit),loadings = rows),
grouping = clusters.10[rows],n = Inf,gap = 20,
perplexity = 50,topics = 1:13,colors = colors_topics,
num_threads = 4,verbose = FALSE)
print(p.structure.2)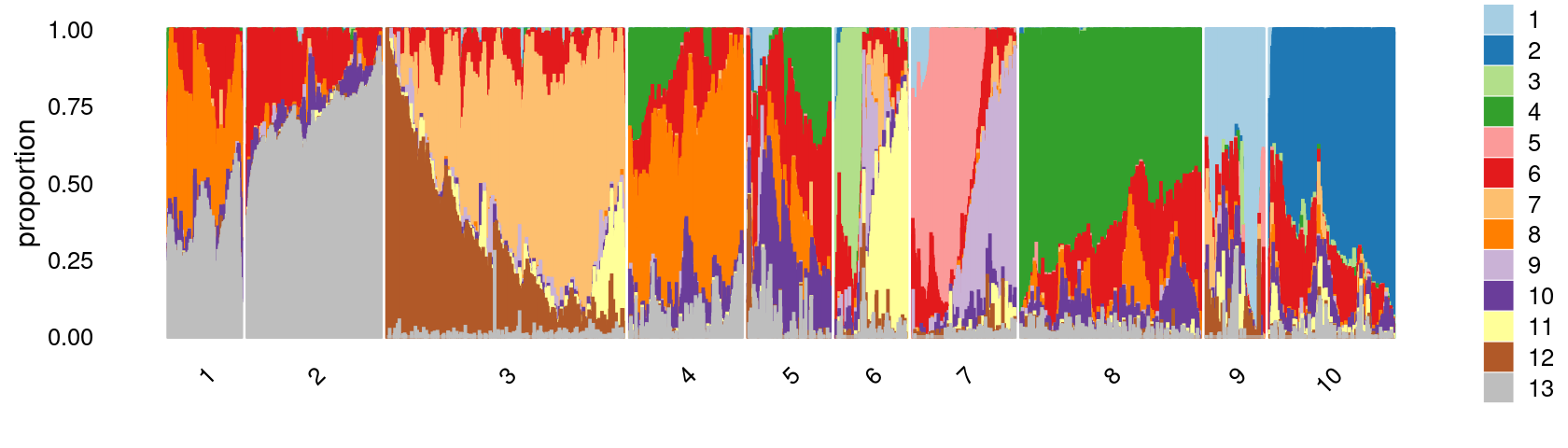
| Version | Author | Date |
|---|---|---|
| 697d54e | kevinlkx | 2020-10-21 |
Define clusters using k-means with \(k = 12\):
set.seed(10)
clusters.12 <- factor(kmeans(poisson2multinom(fit)$L,centers = 12)$cluster)
print(sort(table(clusters.12),decreasing = TRUE))
# clusters.12
# 3 8 2 10 4 5 12 1 11 6 7 9
# 15412 8326 8133 8036 7826 6055 5953 4474 4044 3584 2577 1548Structure plot based on k-means clusters
colors_topics <- c("#a6cee3","#1f78b4","#b2df8a","#33a02c","#fb9a99","#e31a1c",
"#fdbf6f","#ff7f00","#cab2d6","#6a3d9a","#ffff99","#b15928",
"gray")
rows <- sample(nrow(fit$L),4000)
p.structure.3 <- structure_plot(select(poisson2multinom(fit),loadings = rows),
grouping = clusters.12[rows],n = Inf,gap = 20,
perplexity = 50,topics = 1:13,colors = colors_topics,
num_threads = 4,verbose = FALSE)
# Perplexity automatically changed to 47 because original setting of 50 was too large for the number of samples (145)
# Perplexity automatically changed to 25 because original setting of 50 was too large for the number of samples (79)
print(p.structure.3)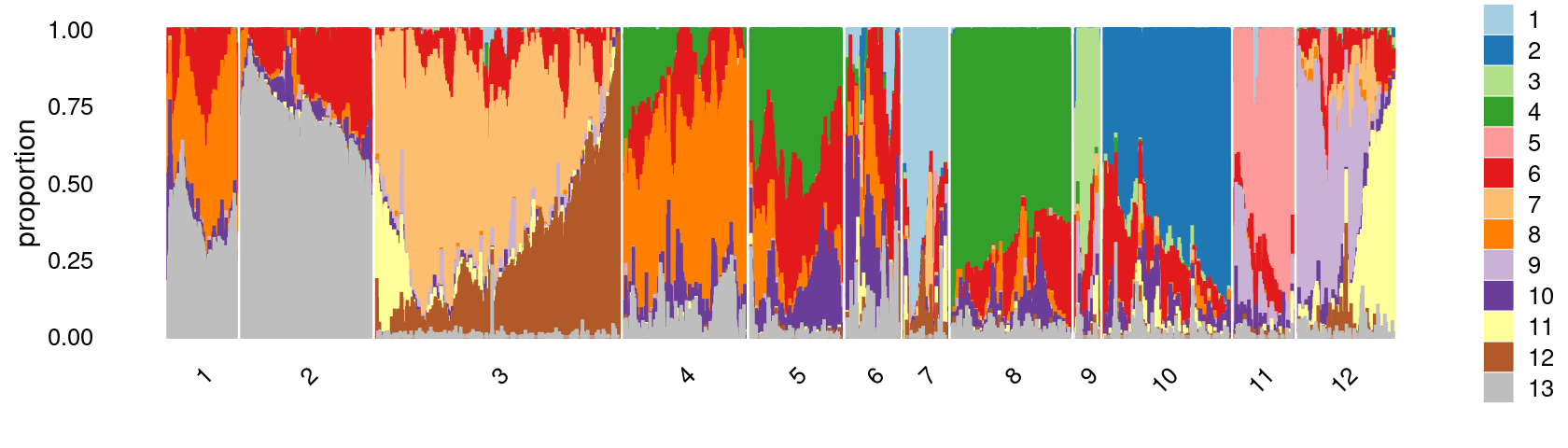
| Version | Author | Date |
|---|---|---|
| 697d54e | kevinlkx | 2020-10-21 |
Refine clusters
We then further refine the clusters based on k-means result with \(k = 12\): merge clusters 5 and 8.
clusters.merged <- clusters.12
clusters.merged[clusters.12 %in% c(5,8)] <- 5
clusters.merged <- factor(clusters.merged, labels = paste0("c", c(1:length(unique(clusters.merged)))))
print(sort(table(clusters.merged),decreasing = TRUE))
samples$cluster_kmeans <- clusters.merged
# clusters.merged
# c3 c5 c2 c9 c4 c11 c1 c10 c6 c7 c8
# 15412 14381 8133 8036 7826 5953 4474 4044 3584 2577 1548colors_topics <- c("#a6cee3","#1f78b4","#b2df8a","#33a02c","#fb9a99","#e31a1c",
"#fdbf6f","#ff7f00","#cab2d6","#6a3d9a","#ffff99","#b15928",
"gray")
rows <- sample(nrow(fit$L),4000)
p.structure.refined <- structure_plot(select(poisson2multinom(fit),loadings = rows),
grouping = clusters.merged[rows],n = Inf,gap = 20,
perplexity = 50,topics = 1:13,colors = colors_topics,
num_threads = 4,verbose = FALSE)
# Perplexity automatically changed to 42 because original setting of 50 was too large for the number of samples (130)
# Perplexity automatically changed to 26 because original setting of 50 was too large for the number of samples (82)
print(p.structure.refined)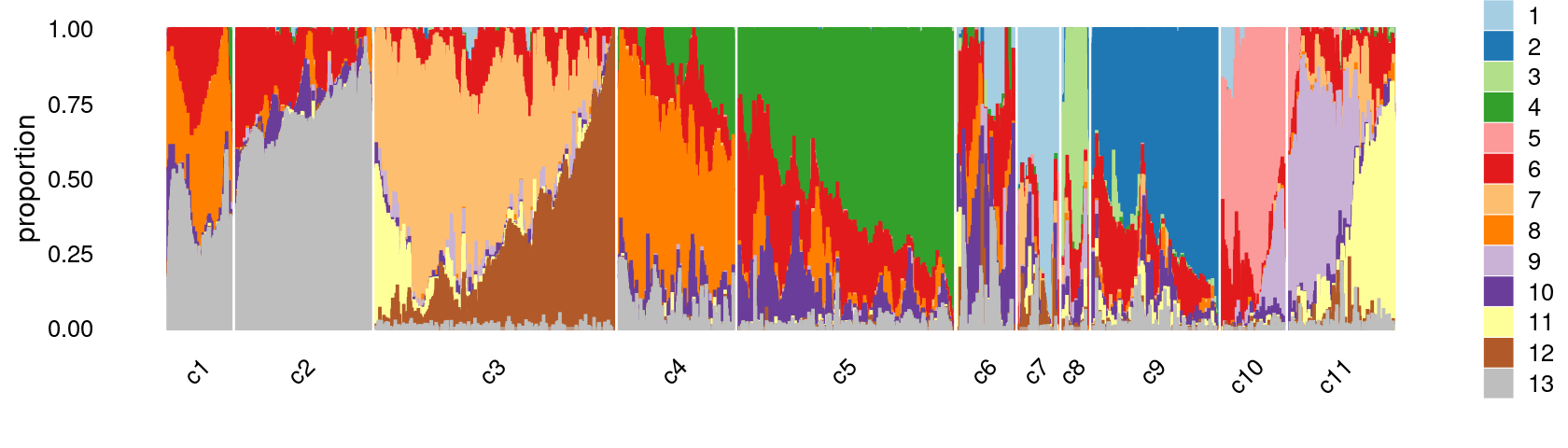
| Version | Author | Date |
|---|---|---|
| 697d54e | kevinlkx | 2020-10-21 |
The clusters defined by k-means on topic proportions reasonably identify the clusters shown in the PCA hexbin plots (below).
p.pca.4.1 <- labeled_pca_plot(fit,1:2,samples$cluster_kmeans,font_size = 7,
legend_label = "cluster_kmeans")
p.pca.4.2 <- labeled_pca_plot(fit,3:4,samples$cluster_kmeans,font_size = 7,
legend_label = "cluster_kmeans")
p.pca.4.3 <- labeled_pca_plot(fit,5:6,samples$cluster_kmeans,font_size = 7,
legend_label = "cluster_kmeans")
p.pca.4.4 <- labeled_pca_plot(fit,7:8,samples$cluster_kmeans,font_size = 7,
legend_label = "cluster_kmeans")
p.pca.4.5 <- labeled_pca_plot(fit,9:10,samples$cluster_kmeans,font_size = 7,
legend_label = "cluster_kmeans")
p.pca.4.6 <- labeled_pca_plot(fit,11:12,samples$cluster_kmeans,font_size = 7,
legend_label = "cluster_kmeans")
plot_grid(p.pca.4.1,p.pca.4.2,p.pca.4.3,p.pca.4.4,p.pca.4.5,p.pca.4.6)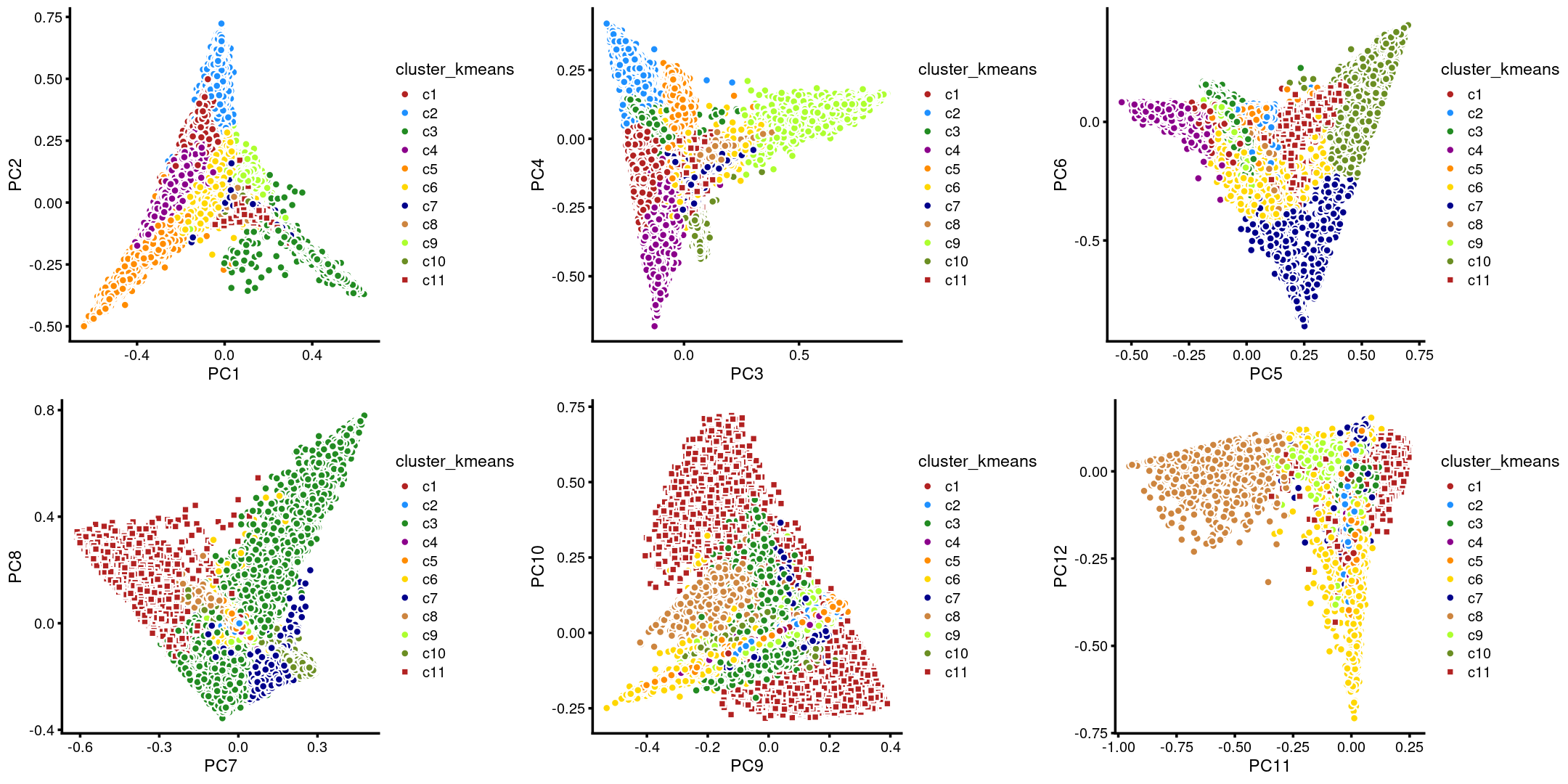
| Version | Author | Date |
|---|---|---|
| 697d54e | kevinlkx | 2020-10-21 |
plot_grid(p.pca2.1,p.pca2.2,p.pca2.3,p.pca2.4,p.pca2.5,p.pca2.6)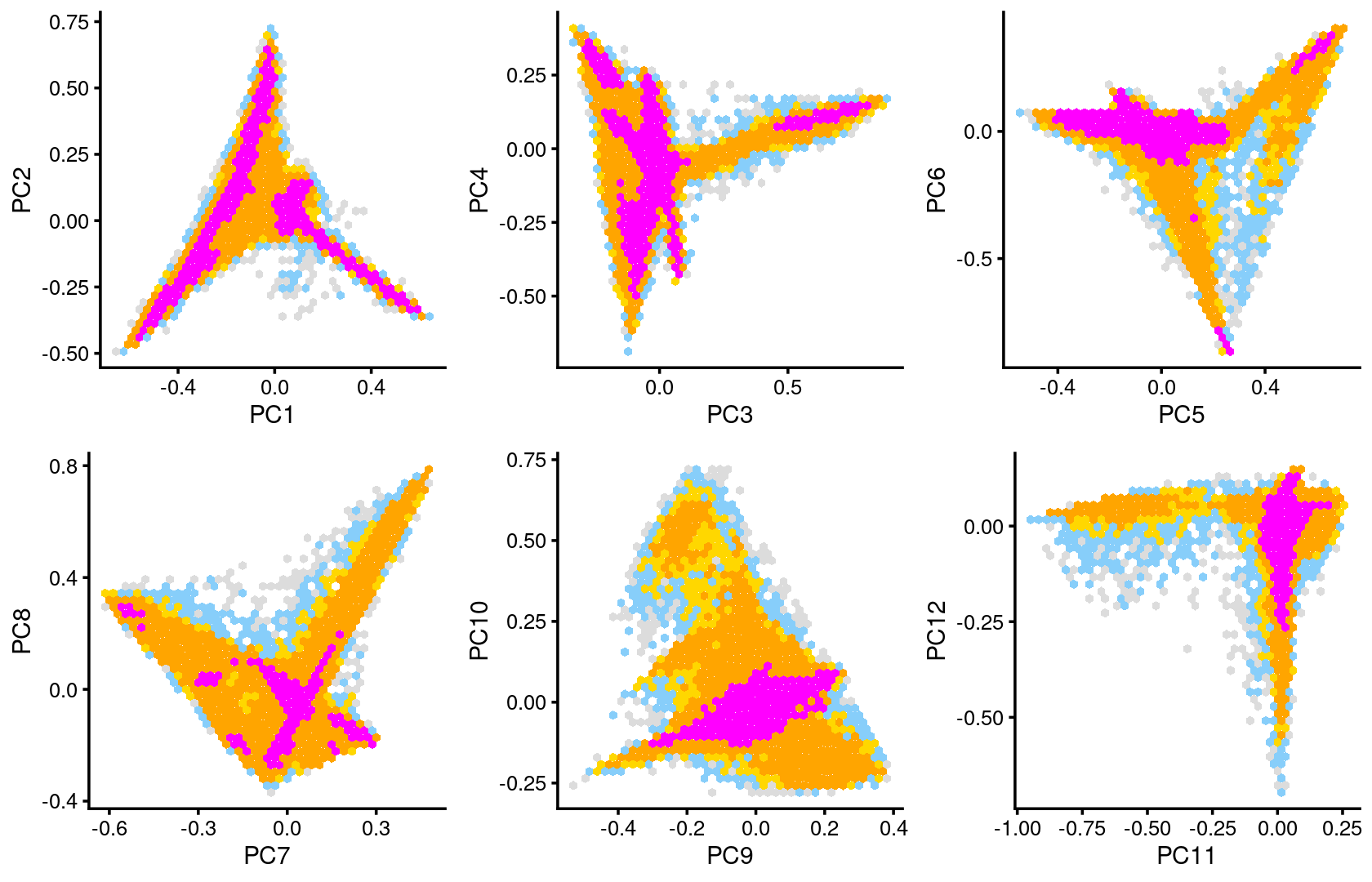
| Version | Author | Date |
|---|---|---|
| 697d54e | kevinlkx | 2020-10-21 |
We then label the cells in each cluster with the known cell labels.
Cell labels:
samples$Cluster <- as.factor(samples$Cluster)
cat(length(levels(samples$Cluster)), "cell labels. \n")
table(samples$Cluster)
# 15 cell labels.
#
# B CD4 CD8 CLP Collision Ery-early Ery-late HSPC
# 3731 18042 9304 216 3235 2453 1095 3033
# HSPC-ery Mono-1 Mono-2 NK pDC preB proB
# 1774 10020 6687 8386 936 5954 1102Plot the distribution of cell labels by clusters.
Stacked barplot for the counts of cell labels by clusters:
library(plyr);library(dplyr)
library(RColorBrewer)
freq_cluster_tissue <- count(samples, vars=c("cluster_kmeans","Cluster"))
n_colors <- length(levels(samples$Cluster))
colors_labels <- colorRampPalette(brewer.pal(9, "Set1"))(n_colors)
# stacked barplot for the counts of cell labels by clusters
p.barplot.1 <- ggplot(freq_cluster_tissue, aes(fill=Cluster, y=freq, x=cluster_kmeans)) +
geom_bar(position="stack", stat="identity") +
theme_classic() + xlab("Cluster") + ylab("Number of cells") +
scale_fill_manual(values = colors_labels) +
guides(fill=guide_legend(ncol=2)) +
theme(
legend.title = element_text(size = 10),
legend.text = element_text(size = 8)
)
print(p.barplot.1)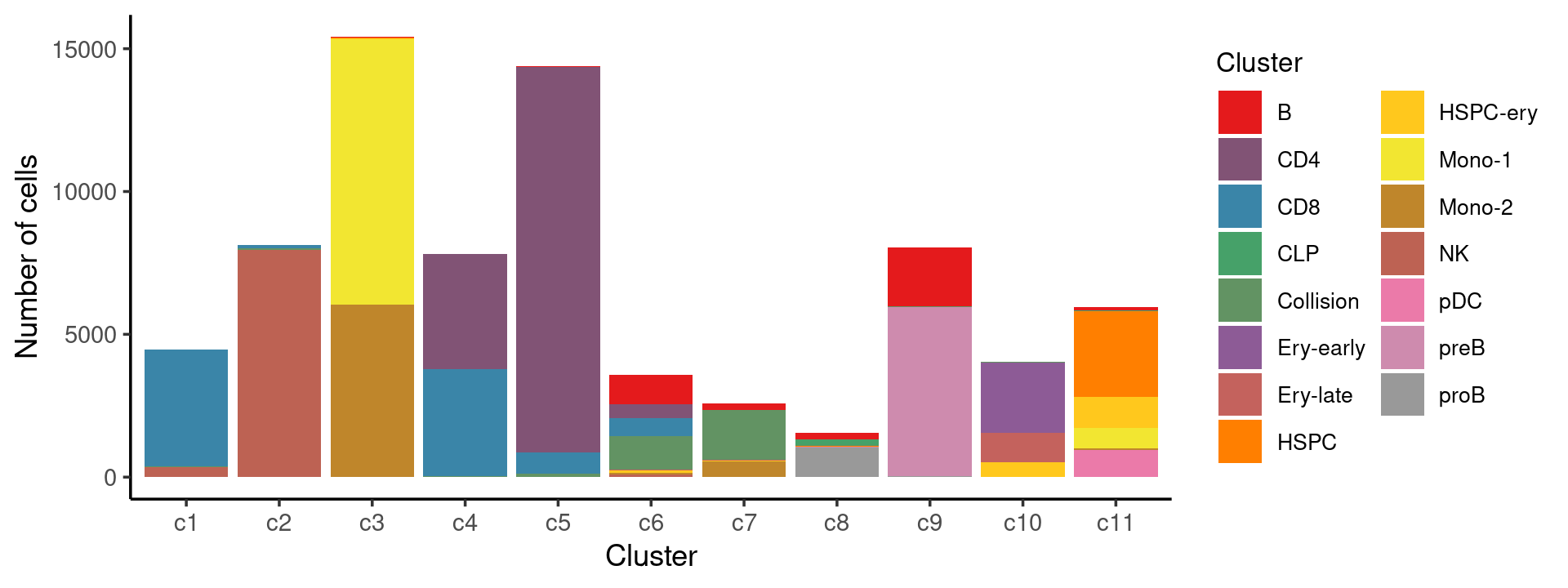
| Version | Author | Date |
|---|---|---|
| 697d54e | kevinlkx | 2020-10-21 |
Percent stacked barplot for the counts of cell labels by clusters:
freq_cluster_tissue <- count(samples, vars=c("cluster_kmeans","Cluster"))
n_colors <- length(levels(samples$Cluster))
colors_labels <- colorRampPalette(brewer.pal(9, "Set1"))(n_colors)
p.barplot.2 <- ggplot(freq_cluster_tissue, aes(fill=Cluster, y=freq, x=cluster_kmeans)) +
geom_bar(position="fill", stat="identity") +
theme_classic() + xlab("Cluster") + ylab("Proportion of cells") +
scale_fill_manual(values = colors_labels) +
guides(fill=guide_legend(ncol=2)) +
theme(
legend.title = element_text(size = 10),
legend.text = element_text(size = 8)
)
print(p.barplot.2)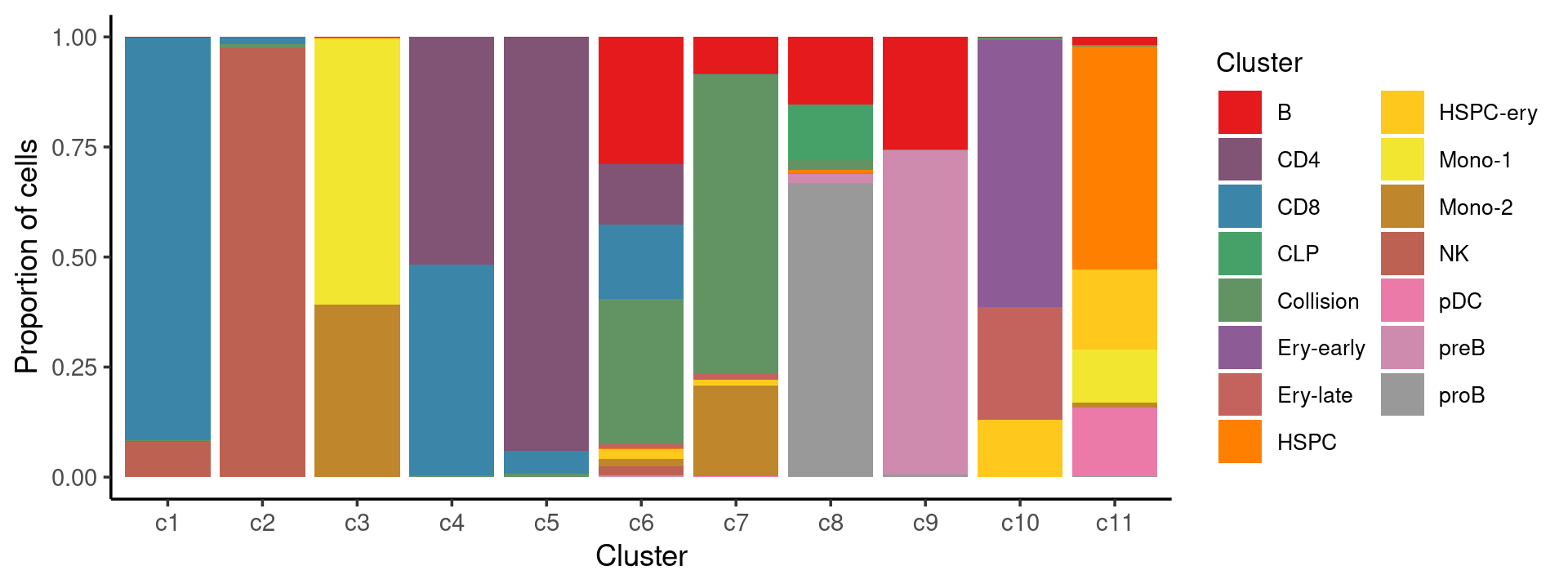
| Version | Author | Date |
|---|---|---|
| 697d54e | kevinlkx | 2020-10-21 |
We can see a few clusters are dominated by one major cell type: * cells in cluster c1 are primarily CD8 cells; * cells in cluster c2 are primarily NK cells; * cells in cluster c5 are primarily CD4 cells.
Some clusters are combinations of related cell types: * cells in cluster c3 are a combination of Mono-1 and Mono-2 cells; * cells in cluster c4 are a combination of CD4 and CD8 cells; * cells in cluster c9 are a combination of B and preB cells; * cells in cluster c10 are a combination of Ery-early, Ery-late and HSPC-ery cells;
Clusters c6, c7, c8, c11 are more heterogeneous with multiple cell types.
freq_cluster_cells <- with(samples,table(Cluster,cluster_kmeans))
print(freq_cluster_cells)
# cluster_kmeans
# Cluster c1 c2 c3 c4 c5 c6 c7 c8 c9 c10 c11
# B 5 4 33 3 28 1033 216 238 2060 3 108
# CD4 4 0 0 4046 13495 494 1 0 0 0 2
# CD8 4086 127 1 3739 742 608 1 0 0 0 0
# CLP 0 0 0 0 0 0 0 195 0 0 21
# Collision 16 55 0 36 111 1182 1757 35 11 29 3
# Ery-early 1 0 0 0 0 0 0 0 0 2452 0
# Ery-late 0 0 0 0 0 31 34 0 0 1030 0
# HSPC 0 1 5 0 0 7 0 11 0 0 3009
# HSPC-ery 0 1 53 0 3 81 26 1 0 529 1080
# Mono-1 0 0 9287 0 0 1 6 0 0 0 726
# Mono-2 1 1 6032 0 2 57 529 1 0 0 64
# NK 361 7944 0 2 0 77 1 0 0 1 0
# pDC 0 0 0 0 0 8 4 0 0 0 924
# preB 0 0 0 0 0 5 0 32 5917 0 0
# proB 0 0 1 0 0 0 2 1035 48 0 16
sessionInfo()
# R version 3.5.1 (2018-07-02)
# Platform: x86_64-pc-linux-gnu (64-bit)
# Running under: Scientific Linux 7.4 (Nitrogen)
#
# Matrix products: default
# BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
#
# locale:
# [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
# [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
# [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
# [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
# [9] LC_ADDRESS=C LC_TELEPHONE=C
# [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#
# attached base packages:
# [1] stats graphics grDevices utils datasets methods base
#
# other attached packages:
# [1] RColorBrewer_1.1-2 plyr_1.8.6 fastTopics_0.3-180 cowplot_1.0.0
# [5] ggplot2_3.3.0 dplyr_0.8.5 Matrix_1.2-15 workflowr_1.6.2
#
# loaded via a namespace (and not attached):
# [1] ggrepel_0.8.2 Rcpp_1.0.4.6 lattice_0.20-38 tidyr_0.8.3
# [5] prettyunits_1.1.1 assertthat_0.2.1 rprojroot_1.3-2 digest_0.6.25
# [9] R6_2.4.1 backports_1.1.7 MatrixModels_0.4-1 evaluate_0.14
# [13] coda_0.19-2 httr_1.4.1 pillar_1.4.4 rlang_0.4.6
# [17] progress_1.2.2 lazyeval_0.2.2 data.table_1.12.8 irlba_2.3.3
# [21] SparseM_1.77 whisker_0.4 hexbin_1.28.1 rmarkdown_2.1
# [25] labeling_0.3 Rtsne_0.15 stringr_1.4.0 htmlwidgets_1.5.1
# [29] munsell_0.5.0 compiler_3.5.1 httpuv_1.5.3.1 xfun_0.14
# [33] pkgconfig_2.0.3 mcmc_0.9-7 htmltools_0.4.0 tidyselect_0.2.5
# [37] tibble_3.0.1 quadprog_1.5-5 viridisLite_0.3.0 crayon_1.3.4
# [41] withr_2.1.2 later_1.0.0 MASS_7.3-51.6 grid_3.5.1
# [45] jsonlite_1.6 gtable_0.3.0 lifecycle_0.2.0 git2r_0.27.1
# [49] magrittr_1.5 scales_1.1.1 RcppParallel_4.4.3 stringi_1.4.6
# [53] farver_2.0.3 fs_1.3.1 promises_1.1.0 ellipsis_0.3.1
# [57] vctrs_0.3.0 tools_3.5.1 glue_1.4.1 purrr_0.3.4
# [61] hms_0.4.2 yaml_2.2.0 colorspace_1.4-1 plotly_4.8.0
# [65] knitr_1.28 quantreg_5.36 MCMCpack_1.4-4