Differential expression between groups using pseudobulk
Sarah Williams
Last updated: 2024-05-07
Checks: 7 0
Knit directory: spatialsnippets/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20231017) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 7b0be93. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rproj.user/
Ignored: renv/library/
Ignored: renv/staging/
Unstaged changes:
Modified: analysis/e_CompositionChange.Rmd
Modified: analysis/e_DEPseudobulk.Rmd
Modified: renv.lock
Modified: renv/activate.R
Modified: spatialsnippets.Rproj
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/e_DEPseudobulk_insitu.Rmd)
and HTML (docs/e_DEPseudobulk_insitu.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 7b0be93 | swbioinf | 2024-05-07 | wflow_publish(c("analysis/index.Rmd", "analysis/e_DEPseudobulk_insitu.Rmd", |
Overview
This requires: * Biological replicates for each group * Assigned cell types * [Optionally] Multiple fovs measured per sample
Worked example
How does gene expression change within each cell type between Ulcerative colitis or Crohn’s disease, and Healthy controls?
Load libraries and data
library(Seurat)Loading required package: SeuratObjectLoading required package: sp'SeuratObject' was built under R 4.3.0 but the current version is
4.3.2; it is recomended that you reinstall 'SeuratObject' as the ABI
for R may have changed
Attaching package: 'SeuratObject'The following object is masked from 'package:base':
intersectlibrary(speckle)Warning: replacing previous import 'S4Arrays::makeNindexFromArrayViewport' by
'DelayedArray::makeNindexFromArrayViewport' when loading 'SummarizedExperiment'library(tidyverse)── Attaching core tidyverse packages ──────────────────────── tidyverse 2.0.0 ──
✔ dplyr 1.1.4 ✔ readr 2.1.5
✔ forcats 1.0.0 ✔ stringr 1.5.1
✔ ggplot2 3.5.0 ✔ tibble 3.2.1
✔ lubridate 1.9.3 ✔ tidyr 1.3.1
✔ purrr 1.0.2 ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()
ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errorslibrary(limma)
library(DT)
Attaching package: 'DT'
The following object is masked from 'package:Seurat':
JS
The following object is masked from 'package:SeuratObject':
JSlibrary(edgeR)data_dir <- file.path("~/projects/spatialsnippets/datasets/GSE234713_IBDcosmx_GarridoTrigo2023/processed_data")
seurat_file_01_loaded <- file.path(data_dir, "GSE234713_CosMx_IBD_seurat_01_loaded.RDS")so <- readRDS(seurat_file_01_loaded)Experimental design
There are three individuals per condition (one tissue sample from each individual). With multiple fovs on each physical tissue sample.
sample_table <- select(as_tibble(so@meta.data), condition, individual_code, fov_name) %>%
unique() %>%
group_by(condition, individual_code) %>%
summarise(n_fovs= n(), item = str_c(fov_name, collapse = ", "))`summarise()` has grouped output by 'condition'. You can override using the
`.groups` argument.DT::datatable(sample_table)Count how many cells of each type in your data
Using a pseudobulk approach.
- Need at least x reads in a cell to include it
- Need at least x cells of a celltype within an fov to include that
- Can only test where we have at least 2 samples on each side of a contrast.
min_reads_per_cell <- 200
ggplot(so@meta.data, aes(x=nCount_RNA)) +
geom_density() +
geom_vline(xintercept = min_reads_per_cell, lty=3) +
scale_x_log10() +
theme_bw()+
ggtitle("How many reads per cell?")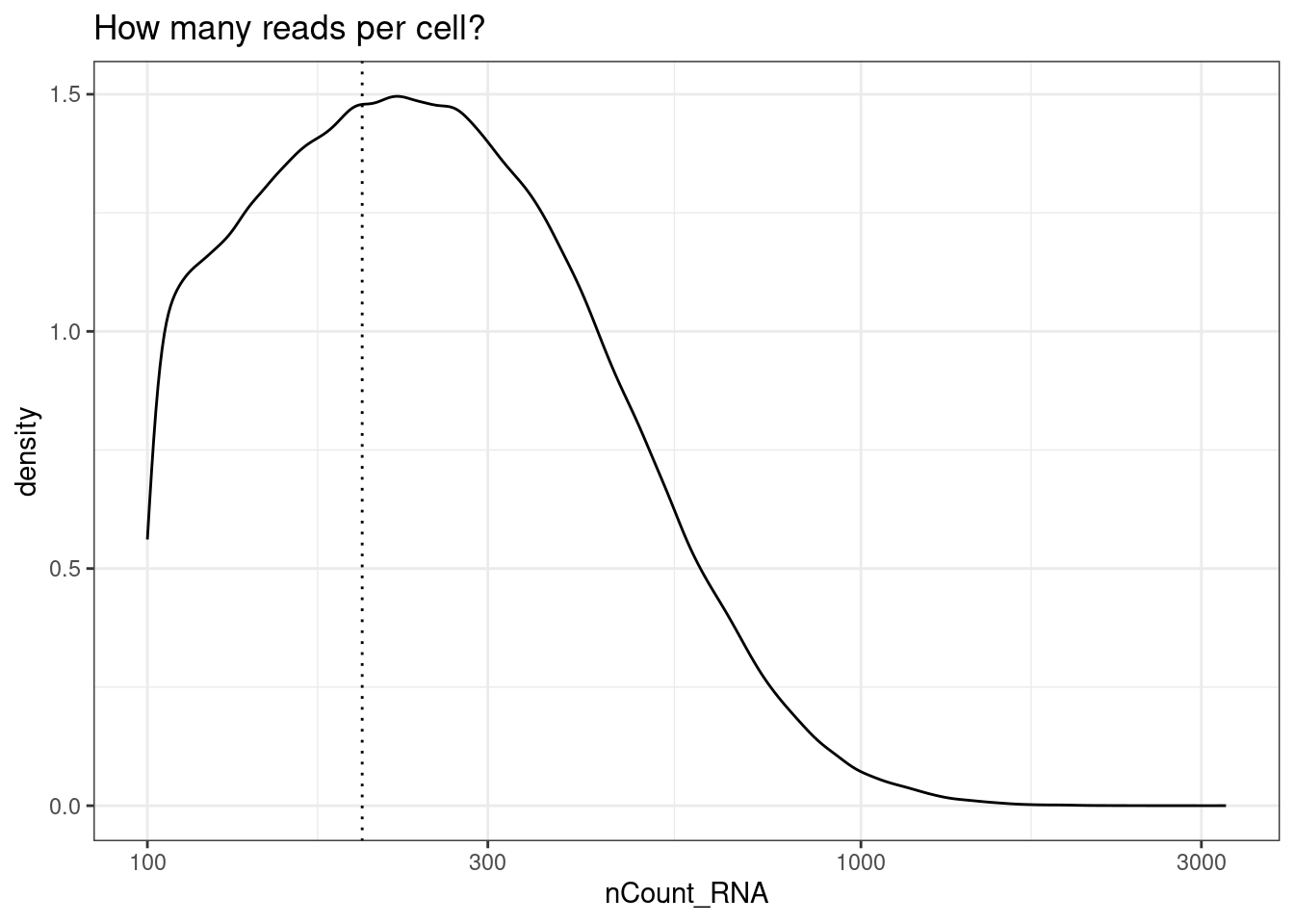
so<- so[,so$nCount_RNA >= min_reads_per_cell]We will pool each celltype within each fov (cluster_group). But there needs to be a certain number of cells for that to work.
Note there are much fewer t-cells overall, but given that we have a high number of samples, there should still be enough to include. Its typical that some of the less common cell types are difficult or impossible to reliably test.
min_cells_per_fovcluster <- 20
so$fov_cluster <- paste0(so$fov_name,"_", so$celltype_subset)
celltype_summary_table <- so@meta.data %>%
group_by(condition, group, individual_code, fov_name, celltype_subset, fov_cluster) %>%
summarise(cells=n(), .groups = 'drop')
DT::datatable(celltype_summary_table)ggplot(celltype_summary_table, aes(x=cells, col=celltype_subset)) +
geom_density() +
geom_vline(xintercept=min_cells_per_fovcluster, lty=3) +
geom_rug() +
scale_x_log10() +
theme_bw() +
ggtitle("How many cells per fov-cluster?")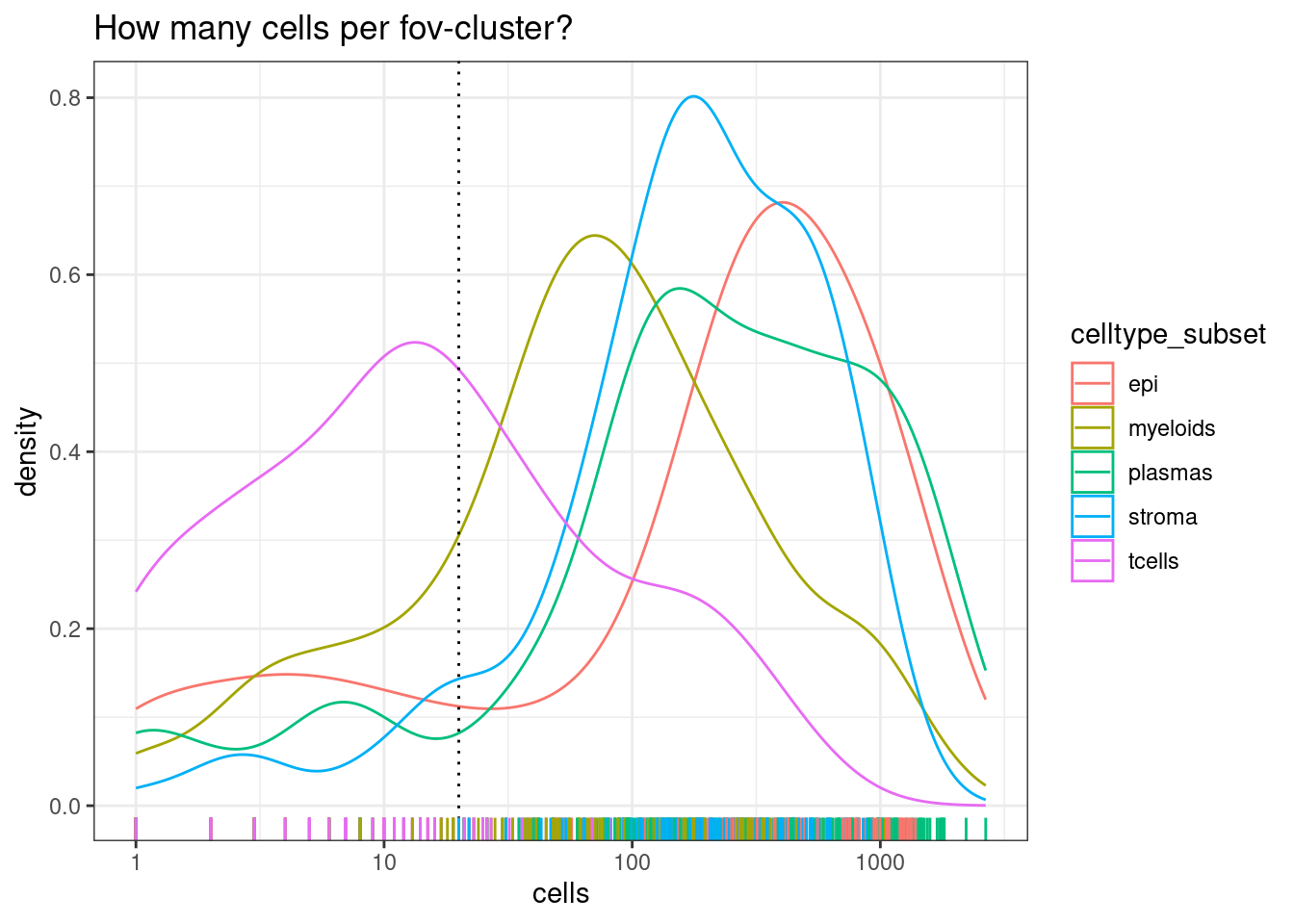
passed_fov_clusters <- celltype_summary_table$fov_cluster[celltype_summary_table$cells >= min_cells_per_fovcluster]Calculate pseudobulk
pseudobulk_counts <- PseudobulkExpression(so, assays = "RNA", layer="counts", method = 'aggregate', group.by = 'fov_cluster')Names of identity class contain underscores ('_'), replacing with dashes ('-')
This message is displayed once every 8 hours.pseudobulk_counts_matrix <- pseudobulk_counts[["RNA"]]
# CHange - back to _. Ideally we'd have neither, but - will cause problems later
colnames(pseudobulk_counts_matrix)<-gsub("-","_",colnames(pseudobulk_counts_matrix))Keep only the passed fovs
pseudobulk_counts_matrix <- pseudobulk_counts_matrix[,passed_fov_clusters]
# pull in relevant annotation in a matched order
pseudobulk_anno_table <- celltype_summary_table
match_order <- match(passed_fov_clusters, pseudobulk_anno_table$fov_cluster)
pseudobulk_anno_table <- pseudobulk_anno_table[match_order,]
stopifnot(all(colnames(pseudobulk_counts_matrix) == pseudobulk_anno_table$fov_cluster ))Calculate Differential Expression
min_samples_to_calc <- 2 # require 2 samples on on either side of contrast
de_result_list <- list()
# celltype_subset is a matrix
for (the_celltype in levels(so$celltype_subset)) {
anno_table.this <- pseudobulk_anno_table[pseudobulk_anno_table$celltype_subset == the_celltype,]
count_matrix.this <- pseudobulk_counts_matrix[,anno_table.this$fov_cluster]
print(the_celltype)
# skip clusters with nothing
if( nrow(anno_table.this) < 1 ) {next}
# Setup objects for limma
dge <- DGEList(count_matrix.this)
dge <- calcNormFactors(dge)
# Build model
group <- anno_table.this$group
individual_code <- anno_table.this$individual_code
# Do do any calculations, we need at least 2 samples per treatment.
# some clusters don't have this (e.g. c14 from mostly one biosample.)
# Also needs both sides of the contrast (e.g. c)
# if( ! ( all(table(treatment) > 2) & length(unique(treatment))==2 ) ) {next}
#<<<<<<<<<<<<<<<<<<< FIX ME for 3
###
# its fine, do design.
design <- model.matrix( ~0 + group)
vm <- voom(dge, design = design, plot = FALSE)
# Adding dupliate correlation to use individual fovs, rather than pooled per biosample
corrfit <- duplicateCorrelation(vm, design, block=individual_code)
fit <- lmFit(vm, design, correlation = corrfit$consensus, block=individual_code)
# Then fit contrasts and run ebayes
contrasts <- makeContrasts(UCvHC = groupUC - groupHC,
CDvHC = groupCD - groupHC,
levels=coef(fit))
fit <- contrasts.fit(fit, contrasts)
fit <- eBayes(fit)
for ( the_coef in colnames(contrasts) ) {
de_result.this <- topTable(fit, n = Inf, adjust.method = "BH", coef = the_coef) %>%
rownames_to_column("target") %>%
mutate(contrast=the_coef,
celltype=the_celltype) %>%
select(celltype,contrast,target,everything()) %>%
arrange(P.Value)
de_result_list[[paste0(the_celltype, the_coef, sep="_")]] <- de_result.this
}
}[1] "epi"
[1] "myeloids"
[1] "plasmas"
[1] "stroma"
[1] "tcells"
sessionInfo()R version 4.3.2 (2023-10-31)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 22.04.4 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
locale:
[1] LC_CTYPE=en_AU.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_AU.UTF-8 LC_COLLATE=en_AU.UTF-8
[5] LC_MONETARY=en_AU.UTF-8 LC_MESSAGES=en_AU.UTF-8
[7] LC_PAPER=en_AU.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_AU.UTF-8 LC_IDENTIFICATION=C
time zone: Etc/UTC
tzcode source: system (glibc)
attached base packages:
[1] stats graphics grDevices datasets utils methods base
other attached packages:
[1] edgeR_4.0.16 DT_0.33 limma_3.58.1 lubridate_1.9.3
[5] forcats_1.0.0 stringr_1.5.1 dplyr_1.1.4 purrr_1.0.2
[9] readr_2.1.5 tidyr_1.3.1 tibble_3.2.1 ggplot2_3.5.0
[13] tidyverse_2.0.0 speckle_1.2.0 Seurat_5.0.3 SeuratObject_5.0.1
[17] sp_2.1-3 workflowr_1.7.1
loaded via a namespace (and not attached):
[1] RcppAnnoy_0.0.22 splines_4.3.2
[3] later_1.3.2 bitops_1.0-7
[5] polyclip_1.10-6 fastDummies_1.7.3
[7] lifecycle_1.0.4 rprojroot_2.0.4
[9] globals_0.16.3 processx_3.8.4
[11] lattice_0.22-6 MASS_7.3-60.0.1
[13] crosstalk_1.2.1 magrittr_2.0.3
[15] plotly_4.10.4 sass_0.4.9
[17] rmarkdown_2.26 jquerylib_0.1.4
[19] yaml_2.3.8 httpuv_1.6.15
[21] sctransform_0.4.1 spam_2.10-0
[23] spatstat.sparse_3.0-3 reticulate_1.35.0
[25] cowplot_1.1.3 pbapply_1.7-2
[27] RColorBrewer_1.1-3 abind_1.4-5
[29] zlibbioc_1.48.2 Rtsne_0.17
[31] GenomicRanges_1.54.1 BiocGenerics_0.48.1
[33] RCurl_1.98-1.14 git2r_0.33.0
[35] GenomeInfoDbData_1.2.11 IRanges_2.36.0
[37] S4Vectors_0.40.2 ggrepel_0.9.5
[39] irlba_2.3.5.1 listenv_0.9.1
[41] spatstat.utils_3.0-4 goftest_1.2-3
[43] RSpectra_0.16-1 spatstat.random_3.2-3
[45] fitdistrplus_1.1-11 parallelly_1.37.1
[47] leiden_0.4.3.1 codetools_0.2-20
[49] DelayedArray_0.28.0 tidyselect_1.2.1
[51] farver_2.1.1 matrixStats_1.2.0
[53] stats4_4.3.2 spatstat.explore_3.2-7
[55] jsonlite_1.8.8 progressr_0.14.0
[57] ggridges_0.5.6 survival_3.5-8
[59] tools_4.3.2 ica_1.0-3
[61] Rcpp_1.0.12 glue_1.7.0
[63] gridExtra_2.3 SparseArray_1.2.4
[65] xfun_0.43 MatrixGenerics_1.14.0
[67] GenomeInfoDb_1.38.8 withr_3.0.0
[69] BiocManager_1.30.22 fastmap_1.1.1
[71] fansi_1.0.6 callr_3.7.6
[73] digest_0.6.35 timechange_0.3.0
[75] R6_2.5.1 mime_0.12
[77] colorspace_2.1-0 scattermore_1.2
[79] tensor_1.5 spatstat.data_3.0-4
[81] utf8_1.2.4 generics_0.1.3
[83] renv_1.0.5 data.table_1.15.4
[85] httr_1.4.7 htmlwidgets_1.6.4
[87] S4Arrays_1.2.1 whisker_0.4.1
[89] uwot_0.1.16 pkgconfig_2.0.3
[91] gtable_0.3.4 lmtest_0.9-40
[93] SingleCellExperiment_1.24.0 XVector_0.42.0
[95] htmltools_0.5.8 dotCall64_1.1-1
[97] scales_1.3.0 Biobase_2.62.0
[99] png_0.1-8 knitr_1.45
[101] rstudioapi_0.16.0 tzdb_0.4.0
[103] reshape2_1.4.4 nlme_3.1-164
[105] cachem_1.0.8 zoo_1.8-12
[107] KernSmooth_2.23-22 parallel_4.3.2
[109] miniUI_0.1.1.1 pillar_1.9.0
[111] grid_4.3.2 vctrs_0.6.5
[113] RANN_2.6.1 promises_1.2.1
[115] xtable_1.8-4 cluster_2.1.6
[117] evaluate_0.23 cli_3.6.2
[119] locfit_1.5-9.9 compiler_4.3.2
[121] rlang_1.1.3 crayon_1.5.2
[123] future.apply_1.11.2 labeling_0.4.3
[125] ps_1.7.6 getPass_0.2-4
[127] plyr_1.8.9 fs_1.6.3
[129] stringi_1.8.3 viridisLite_0.4.2
[131] deldir_2.0-4 munsell_0.5.1
[133] lazyeval_0.2.2 spatstat.geom_3.2-9
[135] Matrix_1.6-5 RcppHNSW_0.6.0
[137] hms_1.1.3 patchwork_1.2.0
[139] future_1.33.2 statmod_1.5.0
[141] shiny_1.8.1.1 highr_0.10
[143] SummarizedExperiment_1.32.0 ROCR_1.0-11
[145] igraph_2.0.3 bslib_0.7.0