Montoro et al. droplet dataset analysis
Jason Willwerscheid
9/22/2019
Last updated: 2019-09-23
Checks: 6 0
Knit directory: scFLASH/
This reproducible R Markdown analysis was created with workflowr (version 1.2.0). The Report tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20181103) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: code/initialization/
Ignored: data/Ensembl2Reactome.txt
Ignored: data/droplet.rds
Ignored: data/mus_pathways.rds
Ignored: output/backfit/
Ignored: output/final_montoro/
Ignored: output/lowrank/
Ignored: output/prior_type/
Ignored: output/pseudocount/
Ignored: output/pseudocount_redux/
Ignored: output/size_factors/
Ignored: output/var_type/
Untracked files:
Untracked: analysis/NBapprox.Rmd
Untracked: analysis/trachea4.Rmd
Untracked: code/missing_data.R
Untracked: code/trachea4.R
Untracked: docs/figure/deleted.Rmd/
Unstaged changes:
Modified: code/utils.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 903d88e | Jason Willwerscheid | 2019-09-23 | wflow_publish(“analysis/final_montoro.Rmd”) |
Introduction
Here I explore a 30-factor flashier object that was fitted using the lessons learned from the previous analyses:
I scale the raw counts \(X_{ij}\) using cell-wise size factors \(\lambda_j\) estimated via Aaron Lun’s
Rpackagescran(normalizing so that the median size factor is equal to one).I divide the scaled counts by the pseudocount \(\alpha = 0.5\), add 1, and take logs to obtain \(Y_{ij} = \log \left(\frac{X_{ij}}{\alpha\lambda_j} + 1 \right)\). Note that this is equivalent to adding \(\alpha\) and taking logs (up to an additive constant), but it preserves sparsity.
It’s best to account for heteroskedasticity among both cells and genes. This can be done by estimating cell-wise variances in advance, scaling the log-transformed counts, and then fitting a
flashierobject to the matrix \(Y_{ij} / \hat{\sigma}_j\) using a gene-wise variance structure.To aid interpretability, I use semi-nonnegative priors with non-negative priors on cells.
I perform a final backfit, which accounts for by far the largest portion of the 3.4 hours needed. If fitting time were an issue, I’d consider limiting the number of backfitting iterations. Here, 180 backfitting iterations were performed, but I suspect that one could get a reasonably good fit with
maxiteras few as 10.
The code used to produce the fit can be viewed here.
source("./code/utils.R")
droplet <- readRDS("./data/droplet.rds")
droplet <- preprocess.droplet(droplet)
res <- readRDS("./output/final_montoro/final_fit.rds")Overview
I first plot the factors in the order they were added.
plot.factors(res, droplet$cell.type, 1:res$fl$n.factors, nonnegative = TRUE)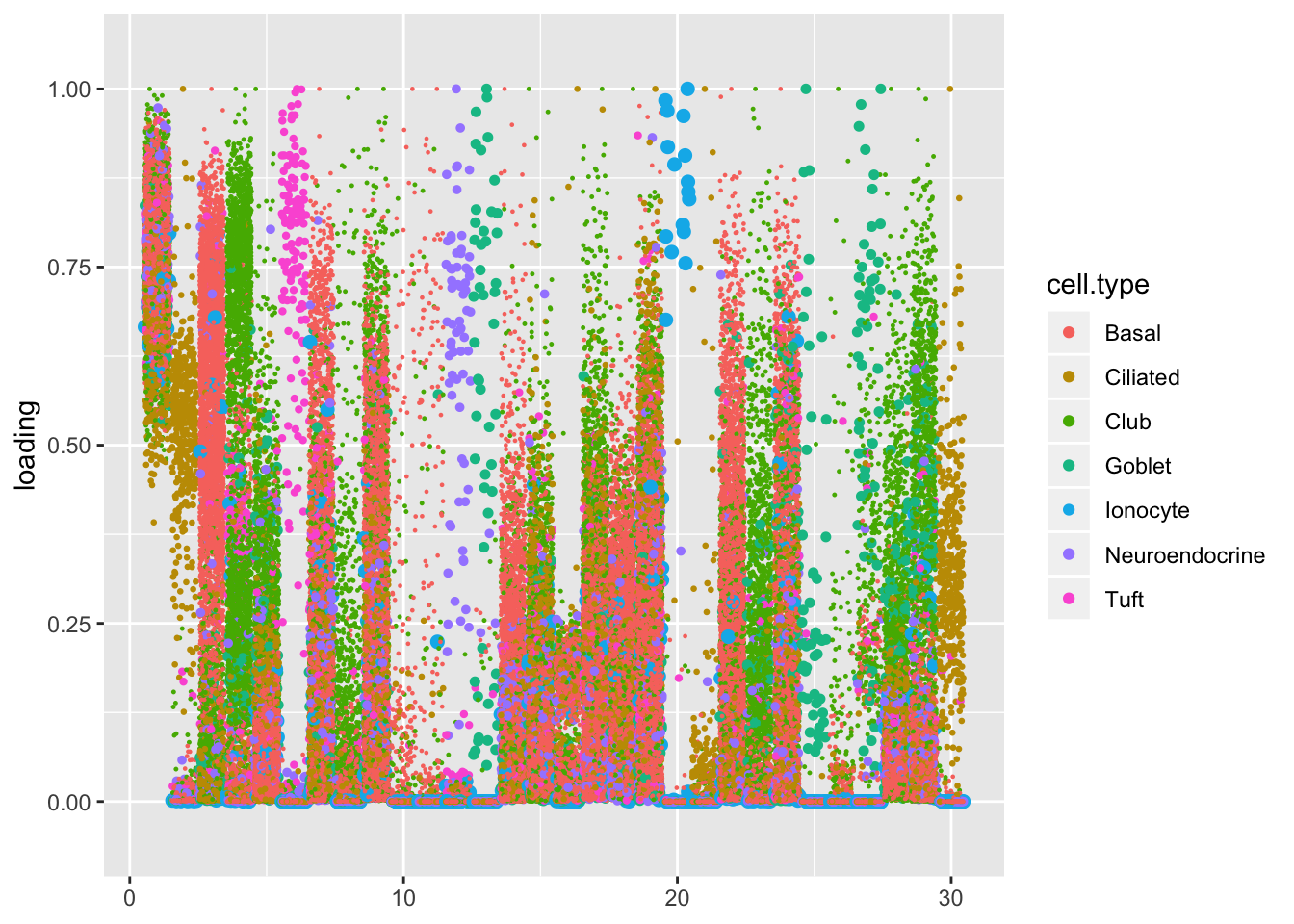
A heatmap of the correlation matrix \(F^T F\) (where \(F\) is the matrix of \(\ell_2\)-normalized cell loadings) can usefully orient a discussion of the results.
heat <- heatmap(crossprod(res$fl$loadings.pm[[2]]))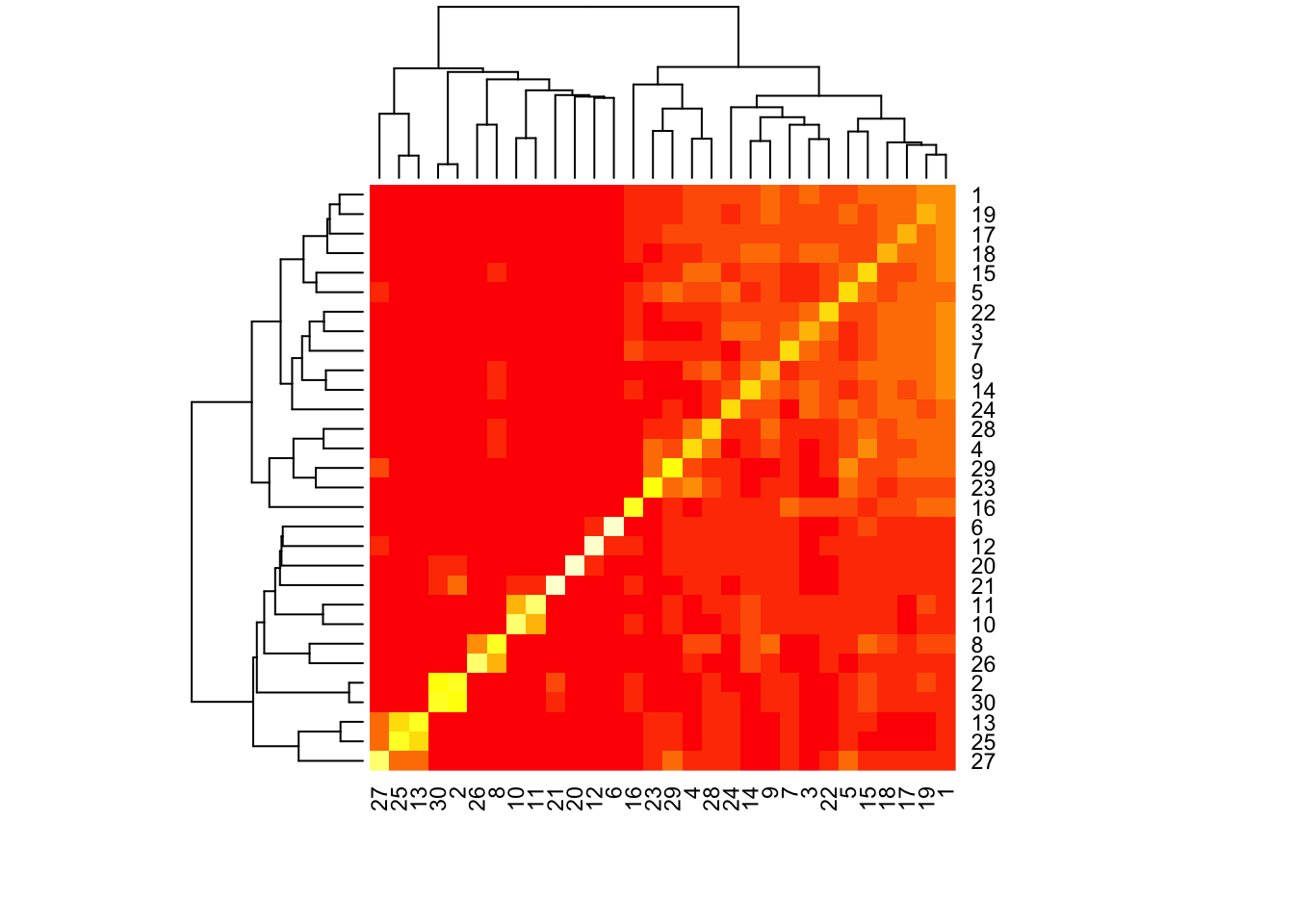
I re-order the factors to match the order given in the heatmap. The first branch of the dendrogram separates a large mass of club and basal cells from more specialized cell types. From left to right, there are easily identifiable clusters of goblet cells, ciliated cells, hillock cells, a small subset of basal cells, more ciliated cells, ionocytes, neuroendocrine cells, and tuft cells. Among the mass of club and basal cells, one can distinguish a lone factor that’s primarily loaded on genes that code for ribosomal proteins (Factor 16), four factors loaded on club cells, six factors loaded on basal cells, and six factors that are a wintry mix of cell types.
plot.factors(res, droplet$cell.type, heat$rowInd, nonnegative = TRUE)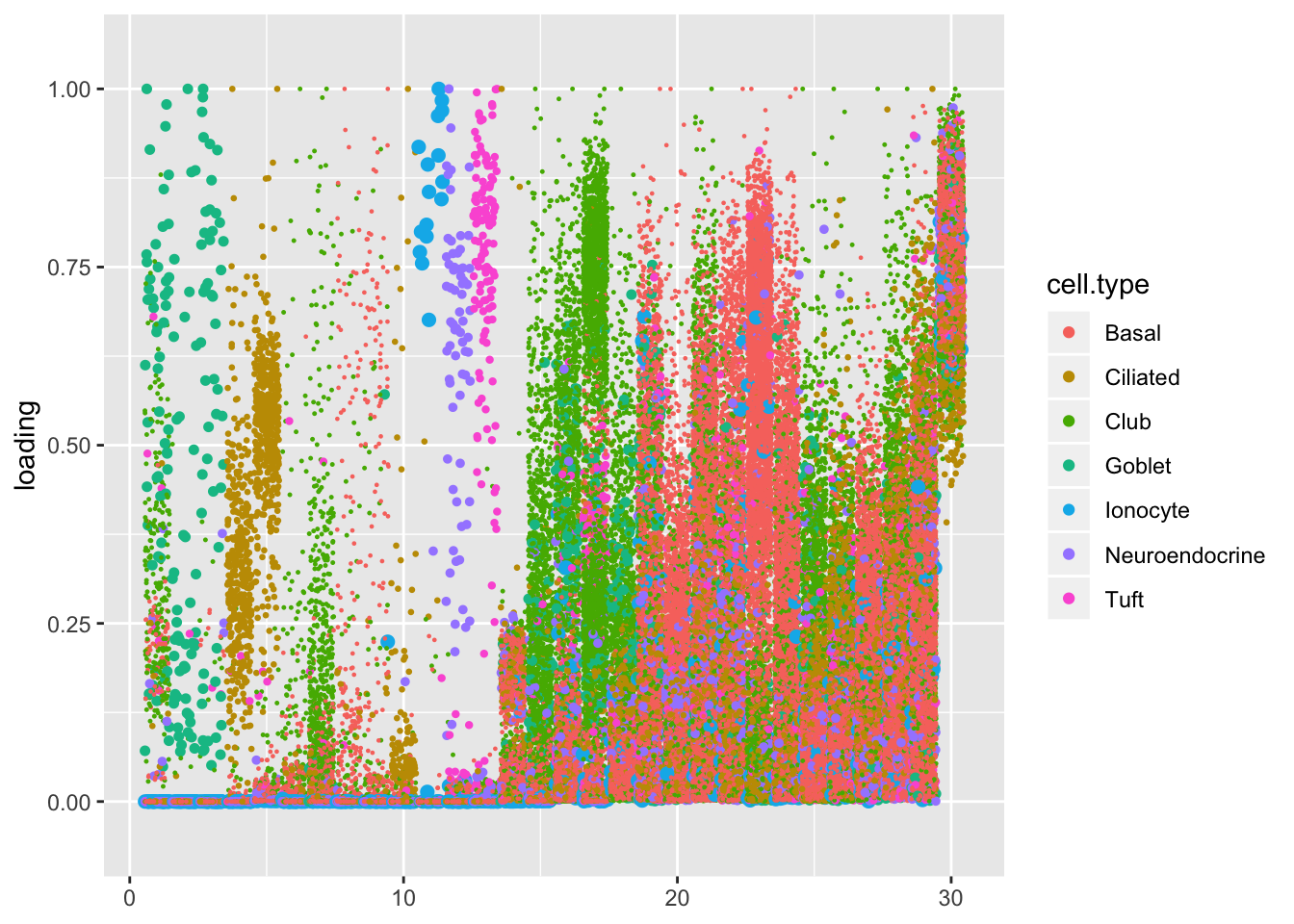
Goblet factors
Factors 13 and 25 nicely distinguish among the “goblet-1” and “goblet-2” subpopulations identified by Montoro et al. (Genes that mark goblet-1 cells are indicated in blue, while goblet-2 marker genes are in red.) I’m not sure what factor 27 is doing. It’s possible that it is indexing cellular functions that are distributed among goblet cells and other cell types (primarily club), but it’s also possible that it is an artefact caused by incomplete convergence.
top.n <- 50
label.top.n <- 20
plot.one.factor(res$fl, 13, droplet$all.goblet.genes,
title = "Factor 13 (goblet)", gene.colors = droplet$all.goblet.colors,
top.n = top.n, label.top.n = label.top.n)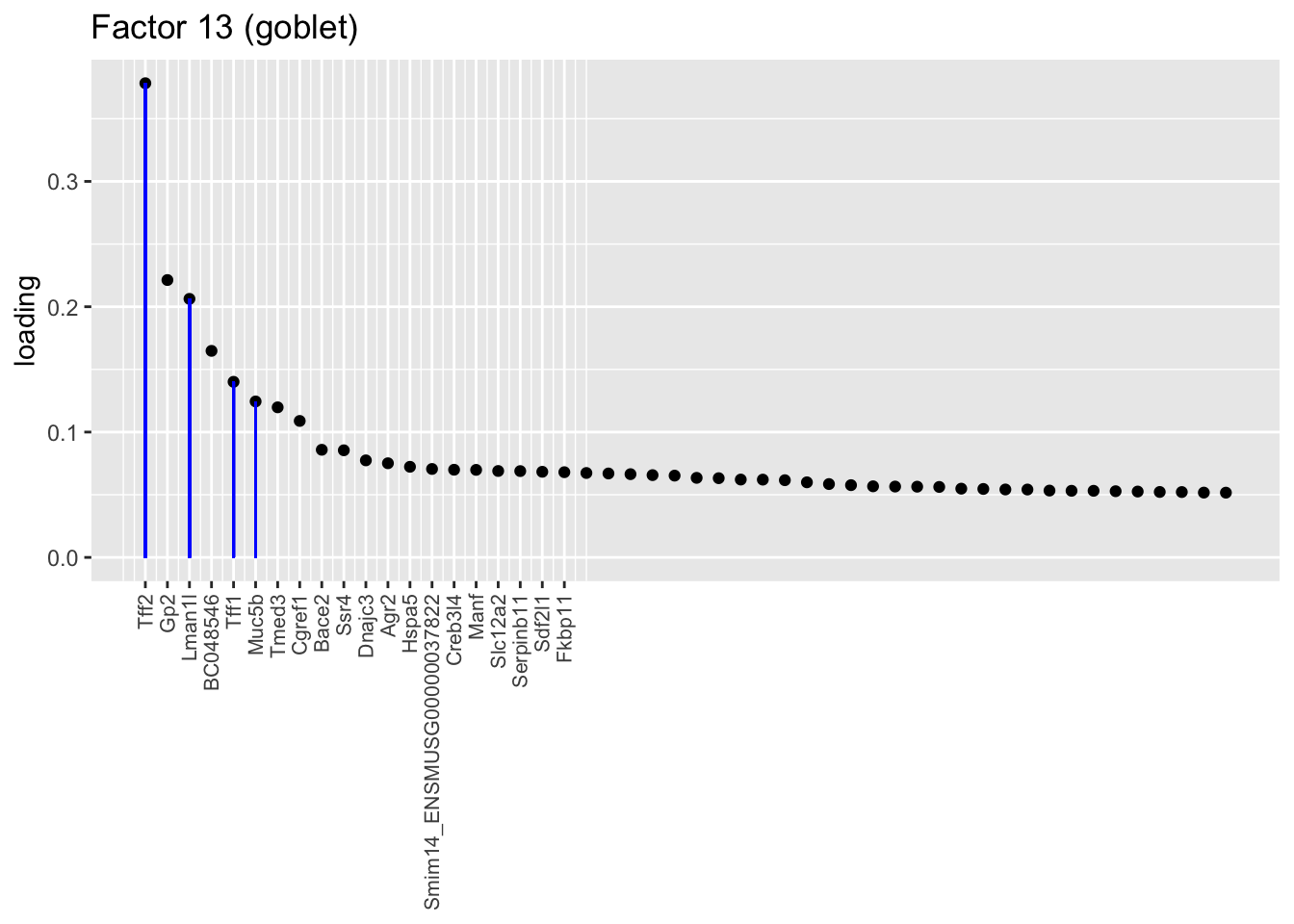
plot.one.factor(res$fl, 25,droplet$all.goblet.genes,
title = "Factor 25 (goblet)", gene.colors = droplet$all.goblet.colors,
top.n = top.n, label.top.n = label.top.n)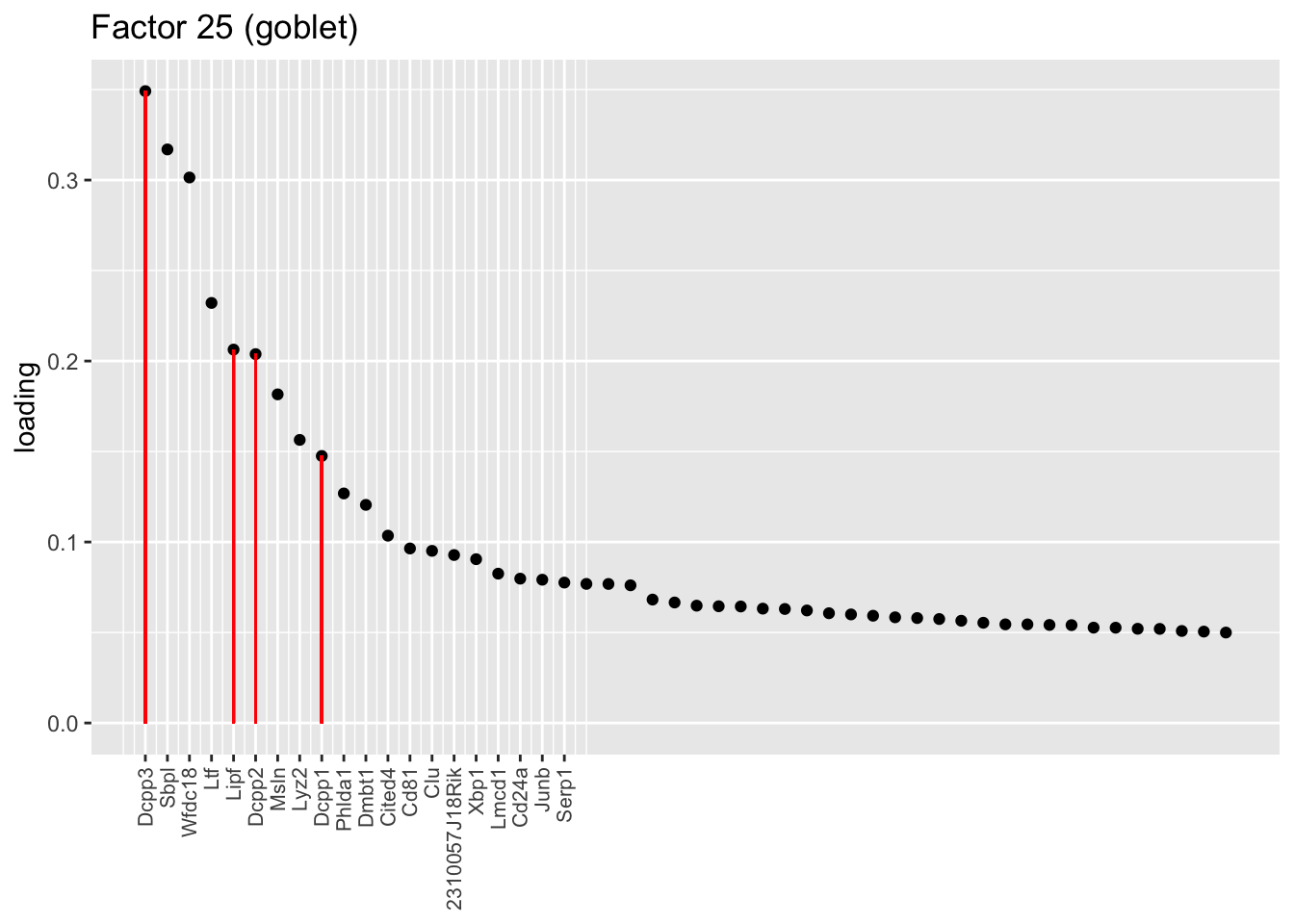
plot.one.factor(res$fl, 27, droplet$all.goblet.genes,
title = "Factor 27 (goblet)", gene.colors = droplet$all.goblet.colors,
top.n = top.n, label.top.n = label.top.n)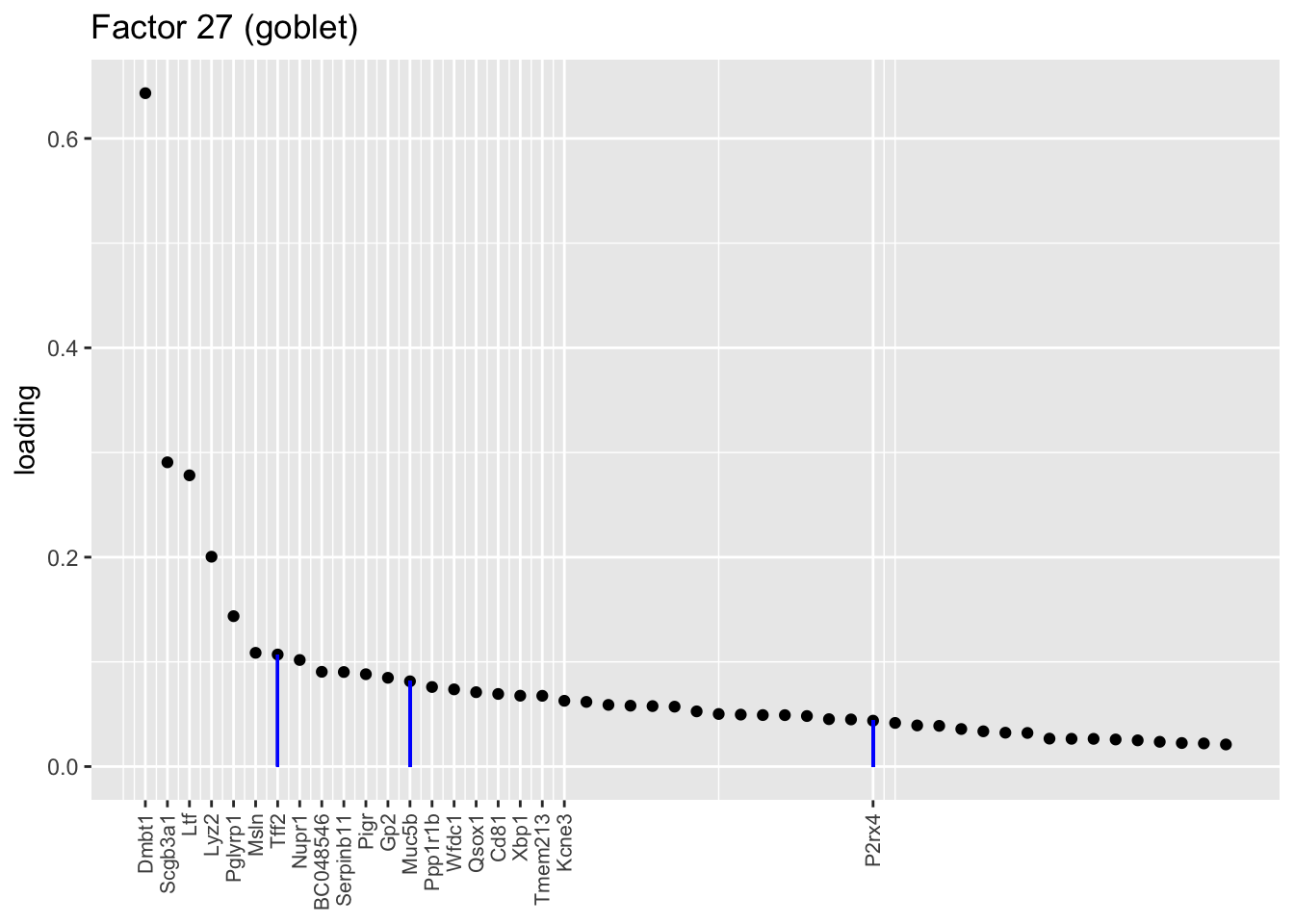
Ciliated factors
I don’t know enough to go into detail, but the two ciliated factors are clearly indexing two very different gene sets.
plot.one.factor(res$fl, 2, NULL,
title = "Factor 2 (ciliated)",
top.n = top.n, label.top.n = label.top.n)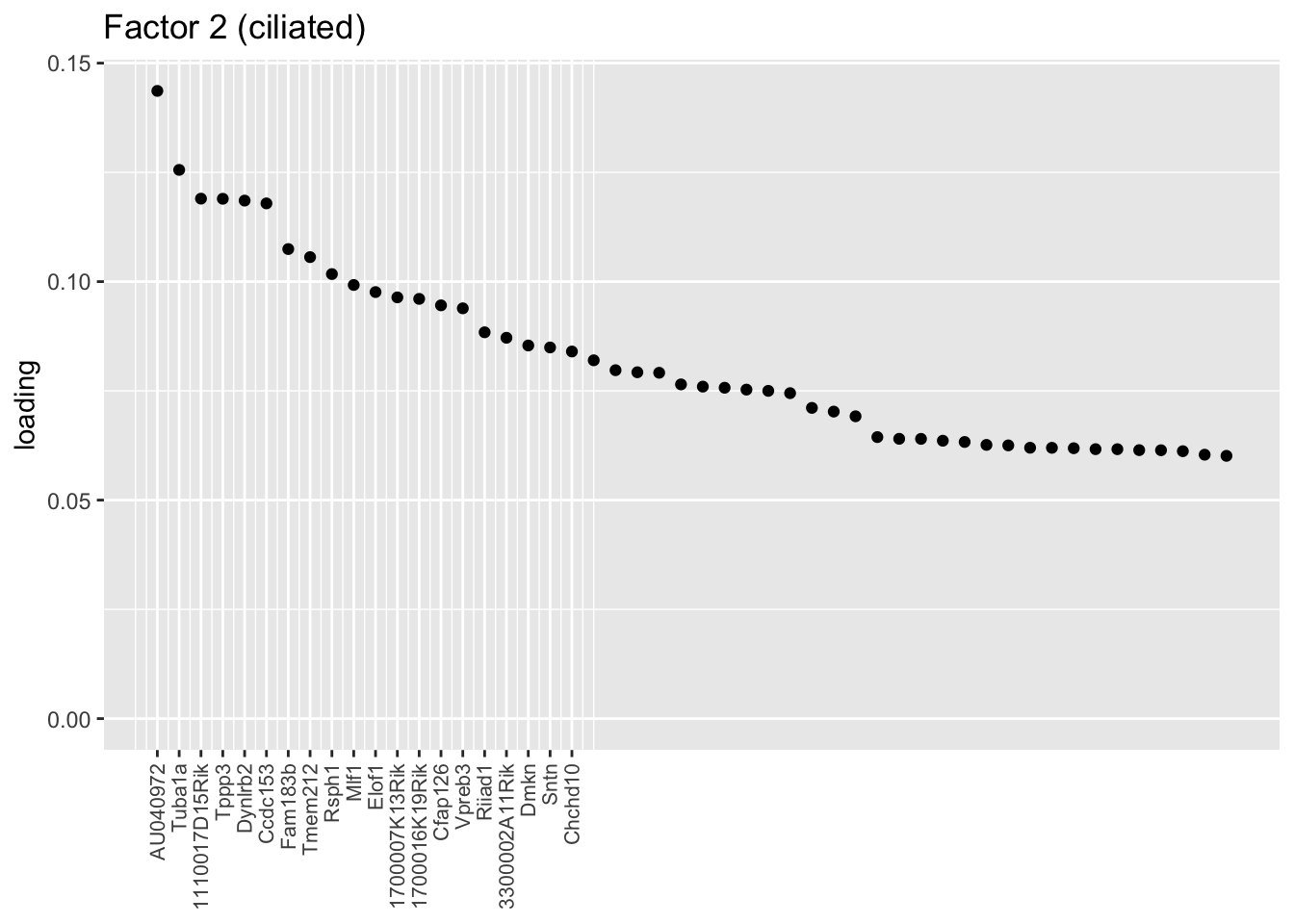
plot.one.factor(res$fl, 30, NULL,
title = "Factor 30 (ciliated)",
top.n = top.n, label.top.n = label.top.n)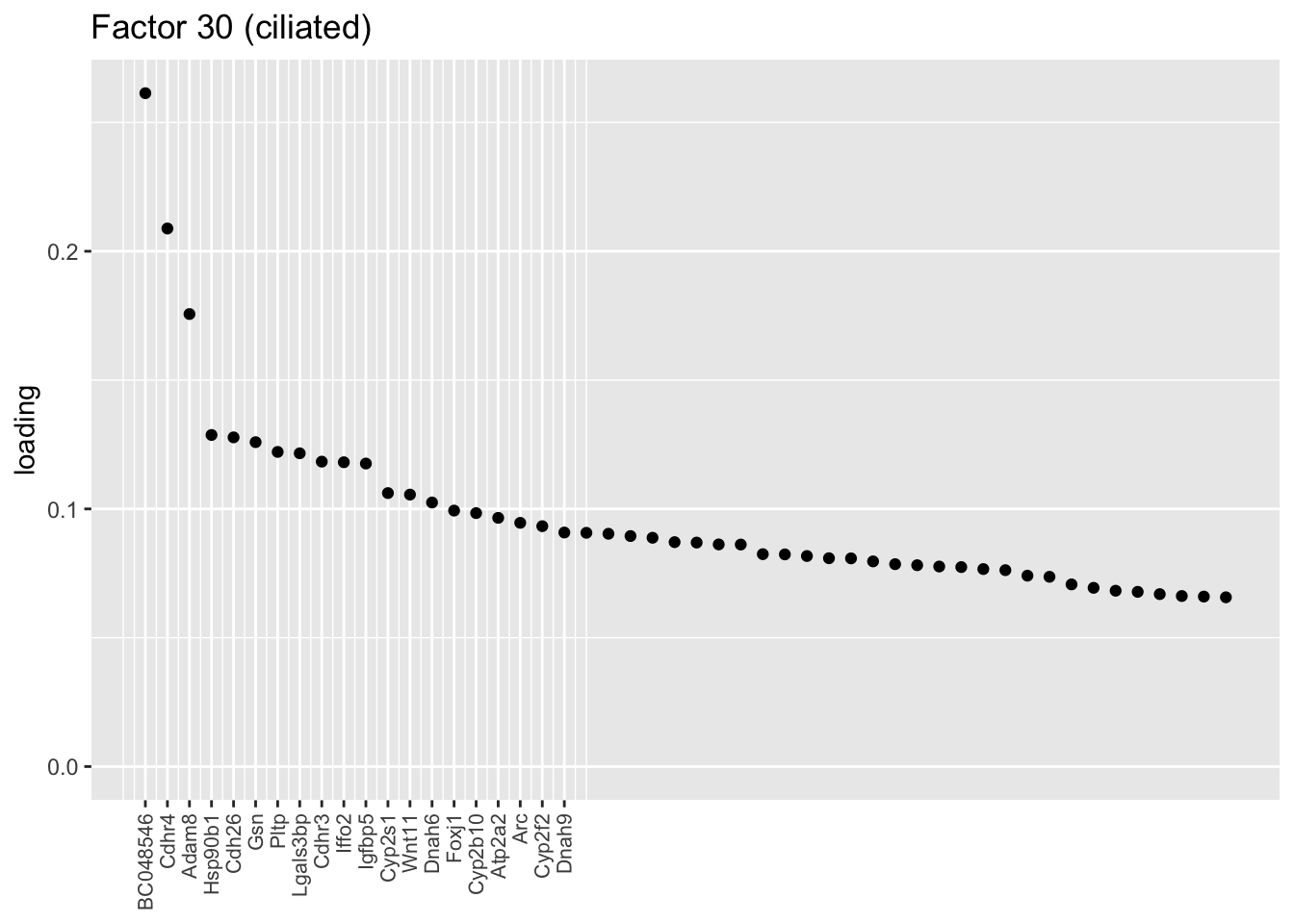
A third ciliated factor (Factor 10) doesn’t cluster with the other two, and indexes yet another ciliated-specific gene set.
plot.one.factor(res$fl, 10, NULL,
title = "Factor 10 (ciliated)",
top.n = top.n, label.top.n = label.top.n)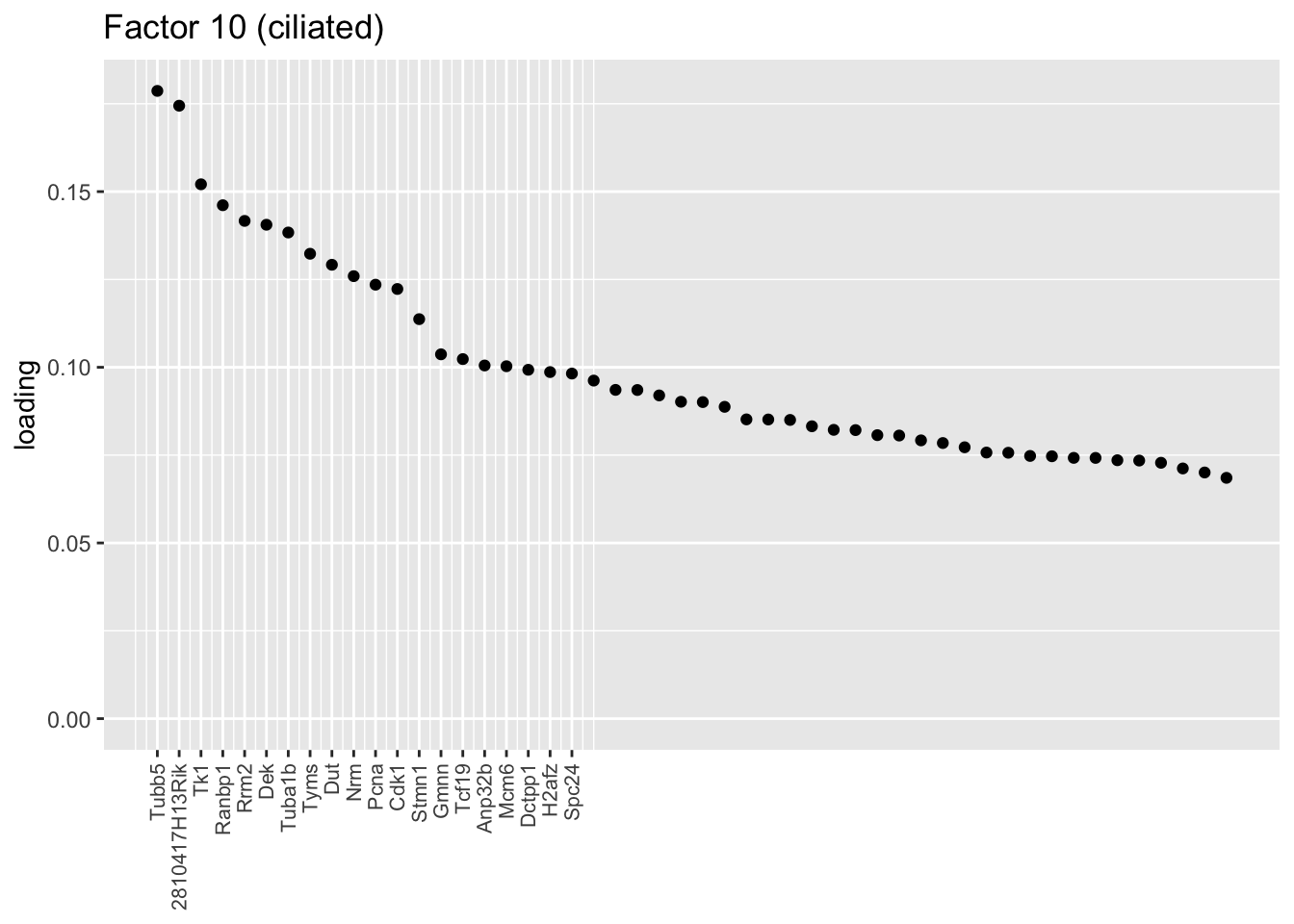
Hillock factors
One feature that is identified in the pulse-seq dataset but not the droplet-based dataset is a subpopulation of transitional cells along the basal-to-club trajectory organized in “hillocks.” Factor 8 turns out to be the factor with largest mean loading among genes that Montoro et al. identify as particularly highly expressed in hillock cells (indicated below in red). Factor 14, included among the basal factors, is also highly loaded on many of these genes. I’d conjecture that Factor 8 (which clusters with Factor 26) picks out cells nearer to the club end of the trajectory, while 14 picks out cells nearer to the basal end.
plot.one.factor(res$fl, 8, droplet$hillock.genes,
title = "Factor 8 (club-hillock)",
top.n = top.n, label.top.n = 10)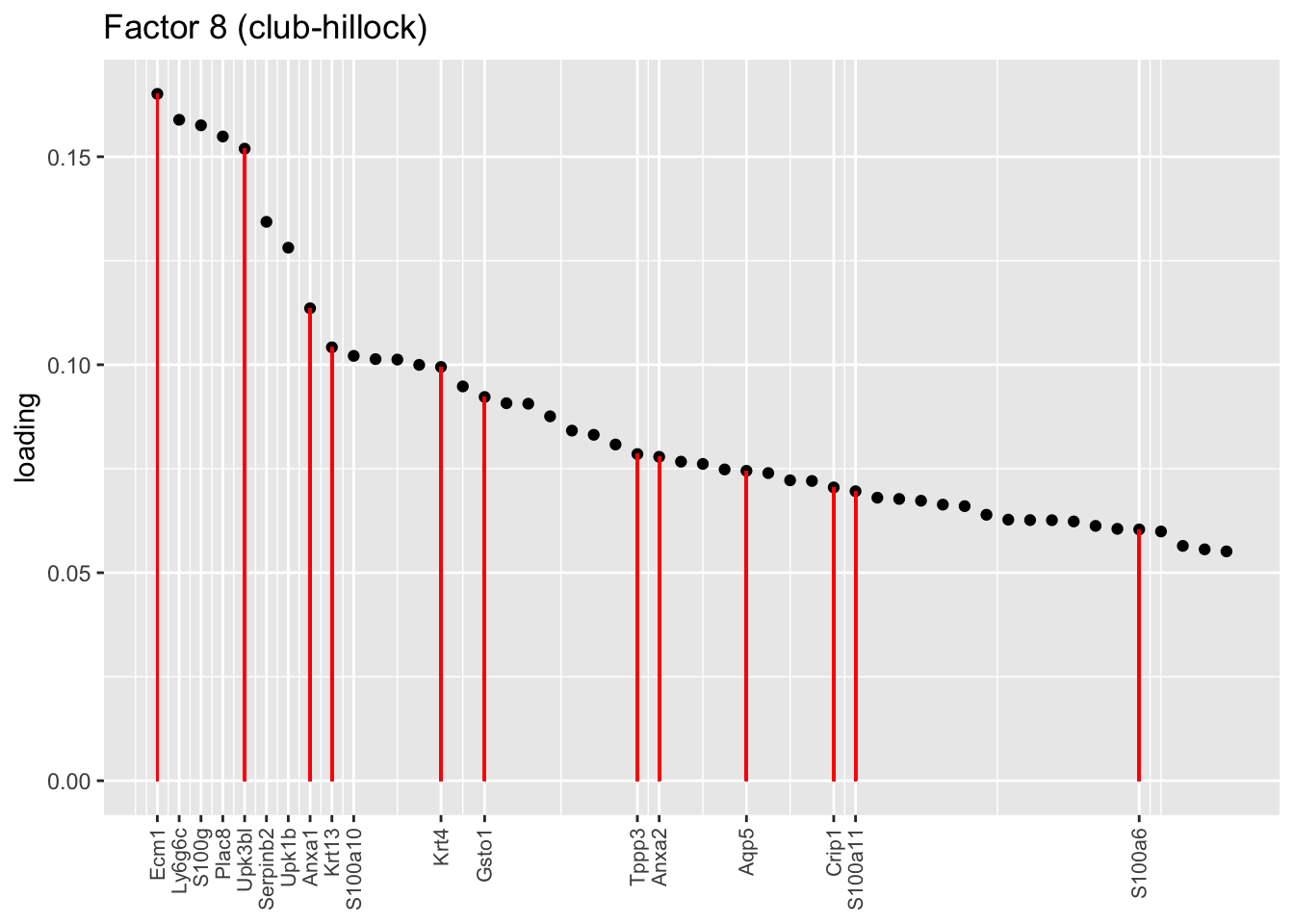
plot.one.factor(res$fl, 26, droplet$hillock.genes,
title = "Factor 26 (club-hillock)",
top.n = 60, label.top.n = 10)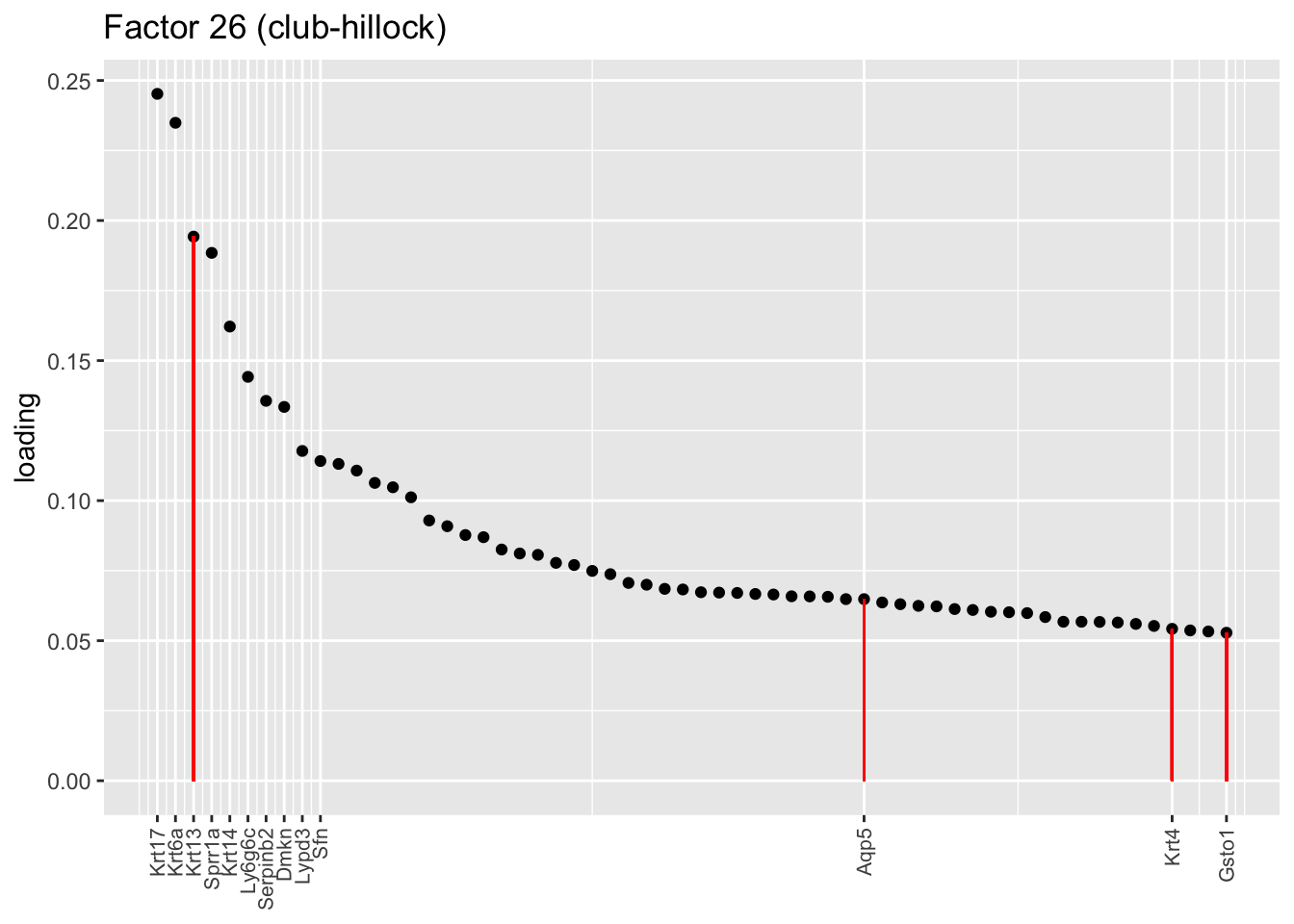
plot.one.factor(res$fl, 14, droplet$hillock.genes,
title = "Factor 14 (basal-hillock)",
top.n = top.n, label.top.n = 10)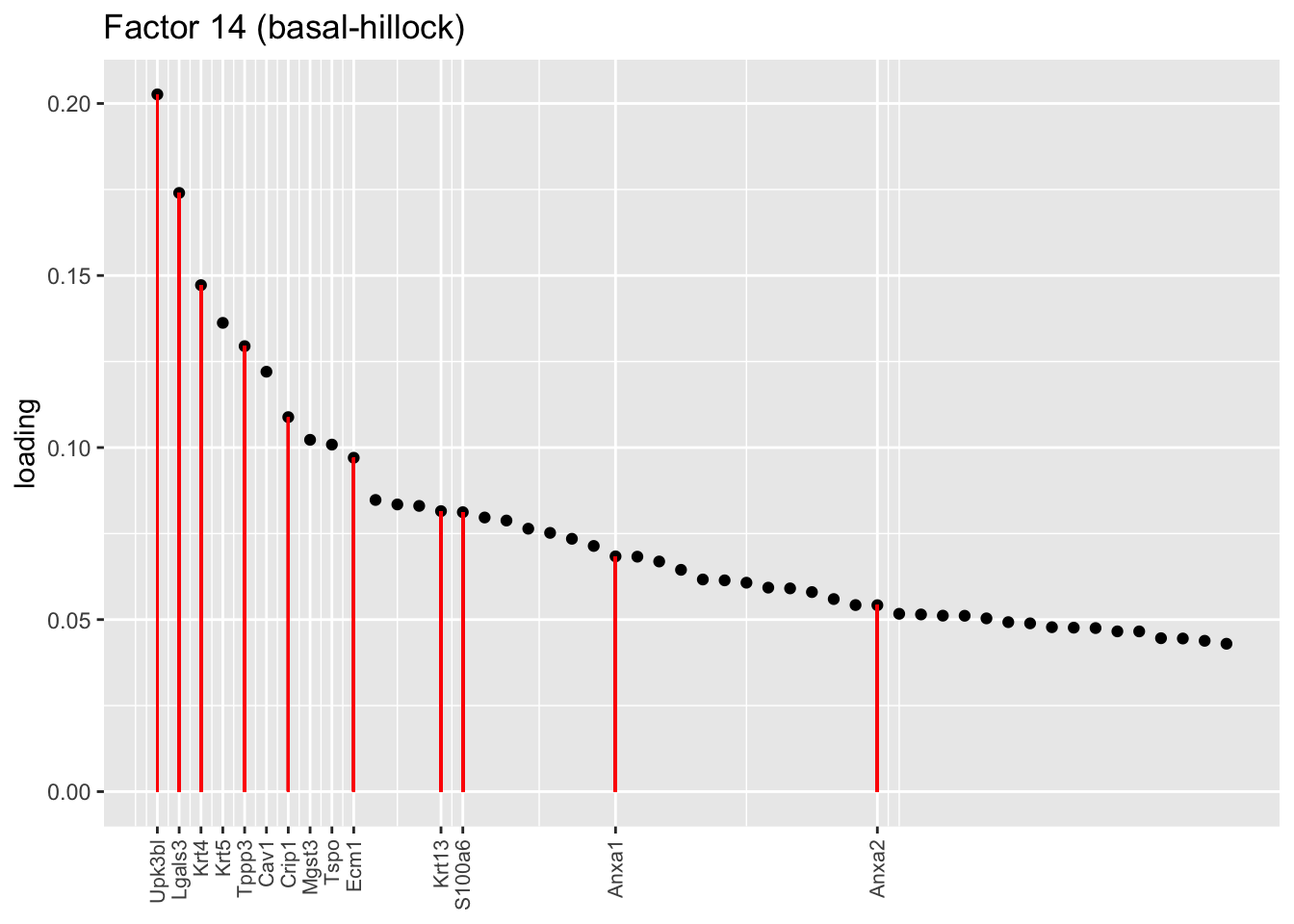
Sparse basal factors
The next cluster is comprised of a small subset of basal cells. I don’t have much to say about it, except that many of the top genes are also highly expressed in ciliated cells, so it’s possible that these are basal cells that have begun to differentiate into ciliated cells.
plot.one.factor(res$fl, 10, NULL,
title = "Factor 10 (basal)",
top.n = top.n, label.top.n = label.top.n)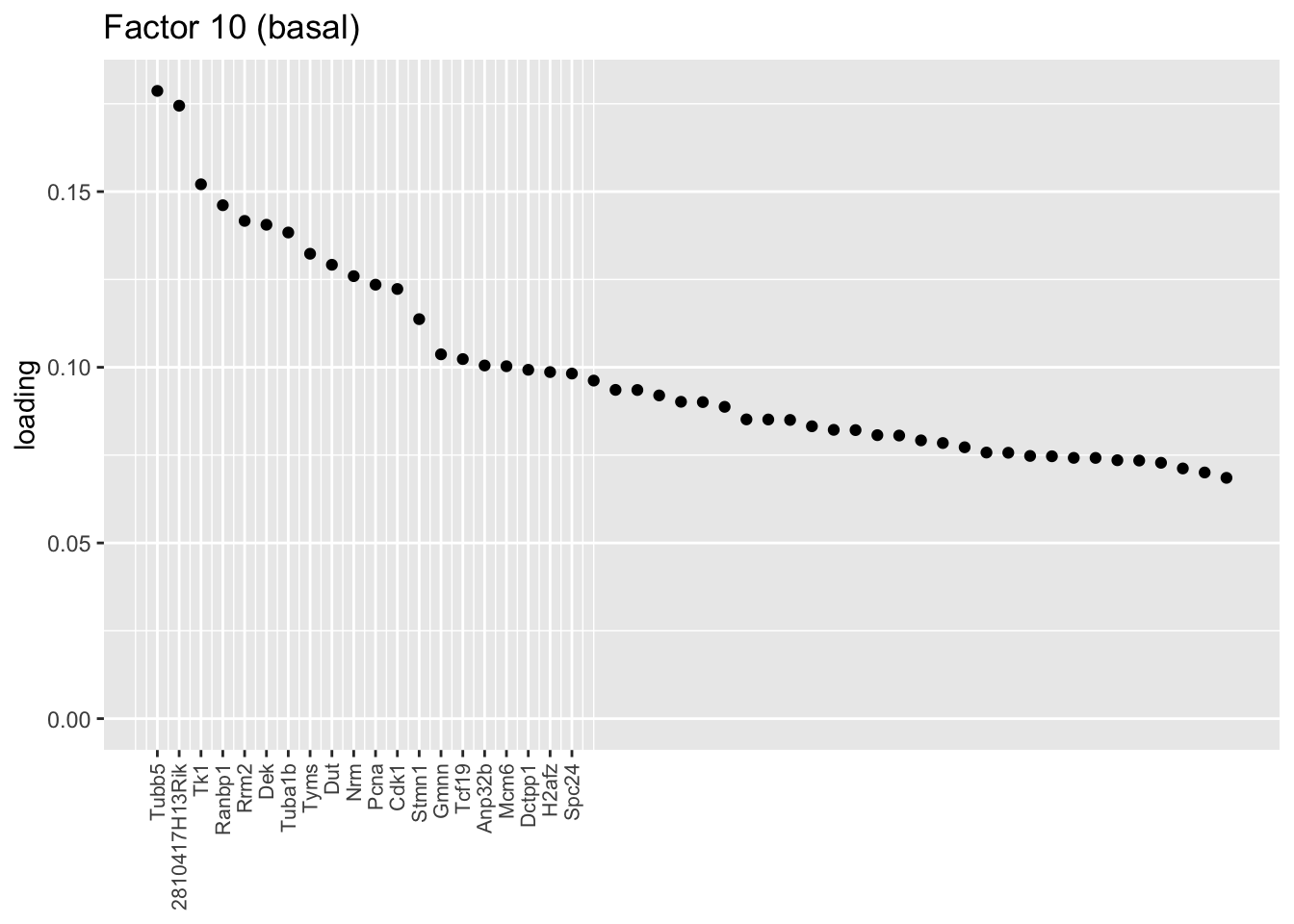
plot.one.factor(res$fl, 11, NULL,
title = "Factor 11 (basal)",
top.n = top.n, label.top.n = label.top.n)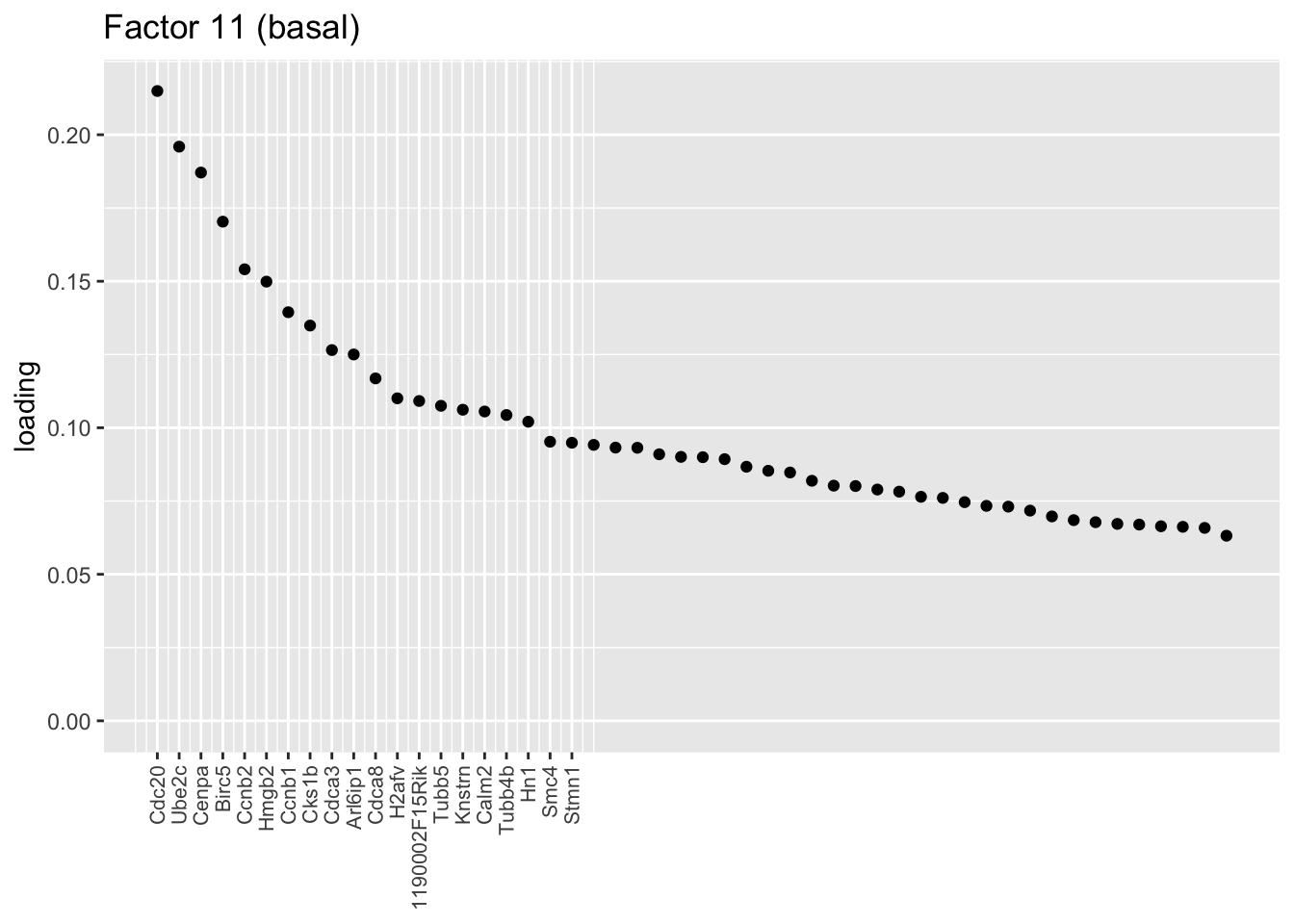
Ionocytes, neuroendocrine cells, tuft cells
These are each relatively clean factors that are specific to a single cell type. The tuft factor does not distinguish between tuft-1 (blue) and tuft-2 (red) subpopulations.
plot.one.factor(res$fl, 20, droplet$ionocyte.genes,
title = "Factor 20 (ionocytes)",
top.n = 70, label.top.n = 9)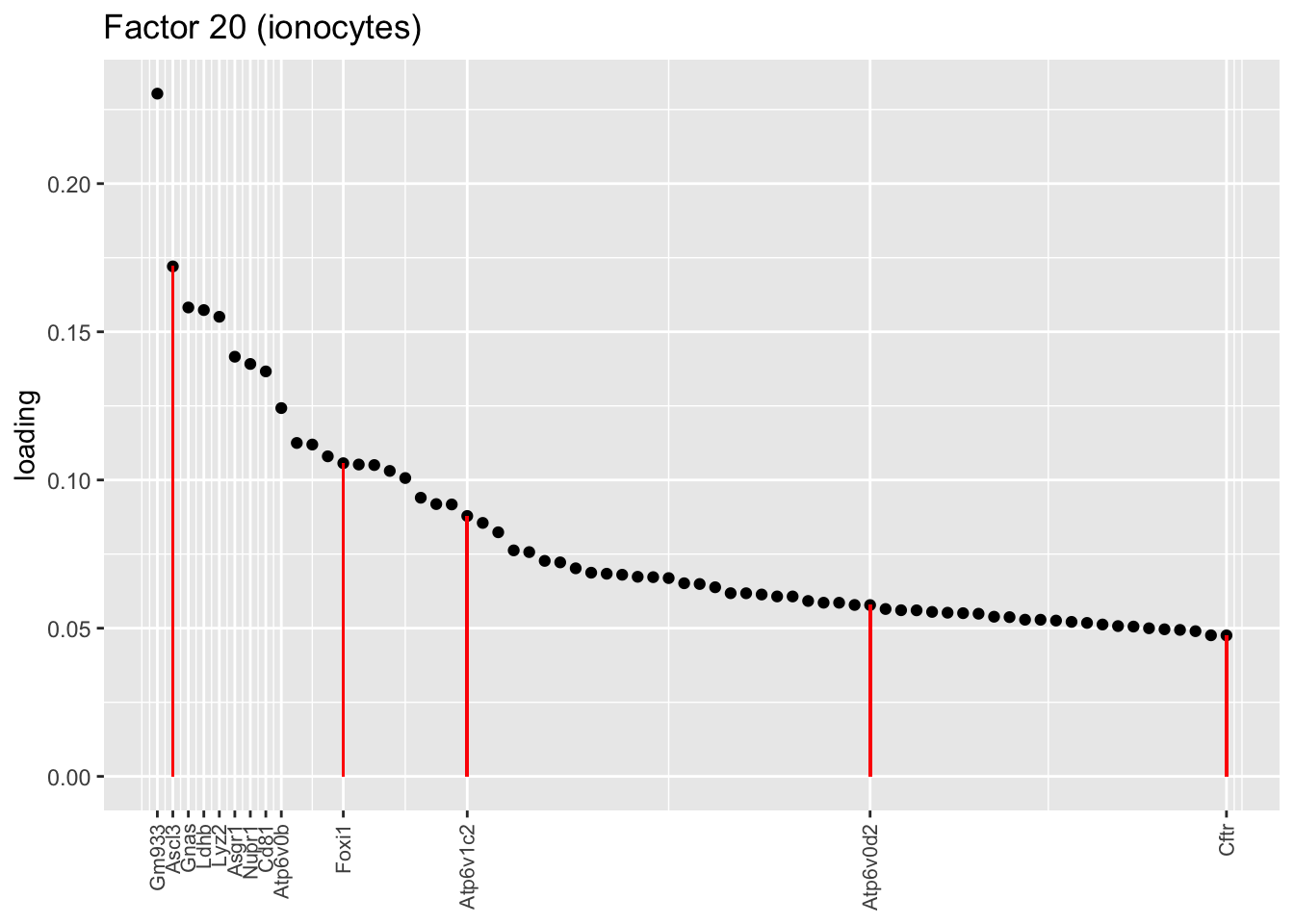
plot.one.factor(res$fl, 12, NULL,
title = "Factor 12 (neuroendocrine)",
top.n = top.n, label.top.n = label.top.n)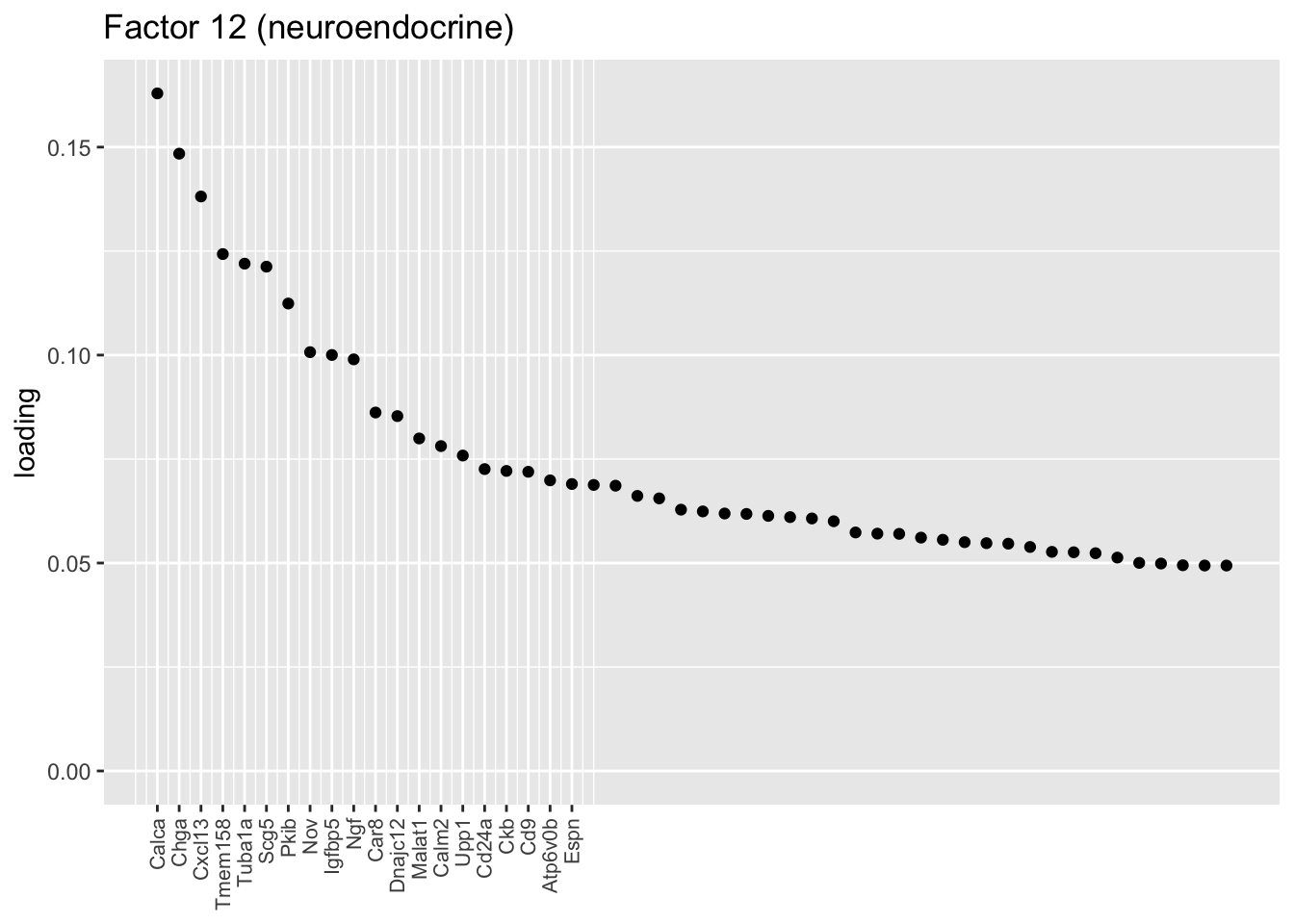
plot.one.factor(res$fl, 6, droplet$all.tuft.genes,
title = "Factor 6 (tuft)", gene.colors = droplet$all.tuft.colors,
top.n = 100, label.top.n = 4)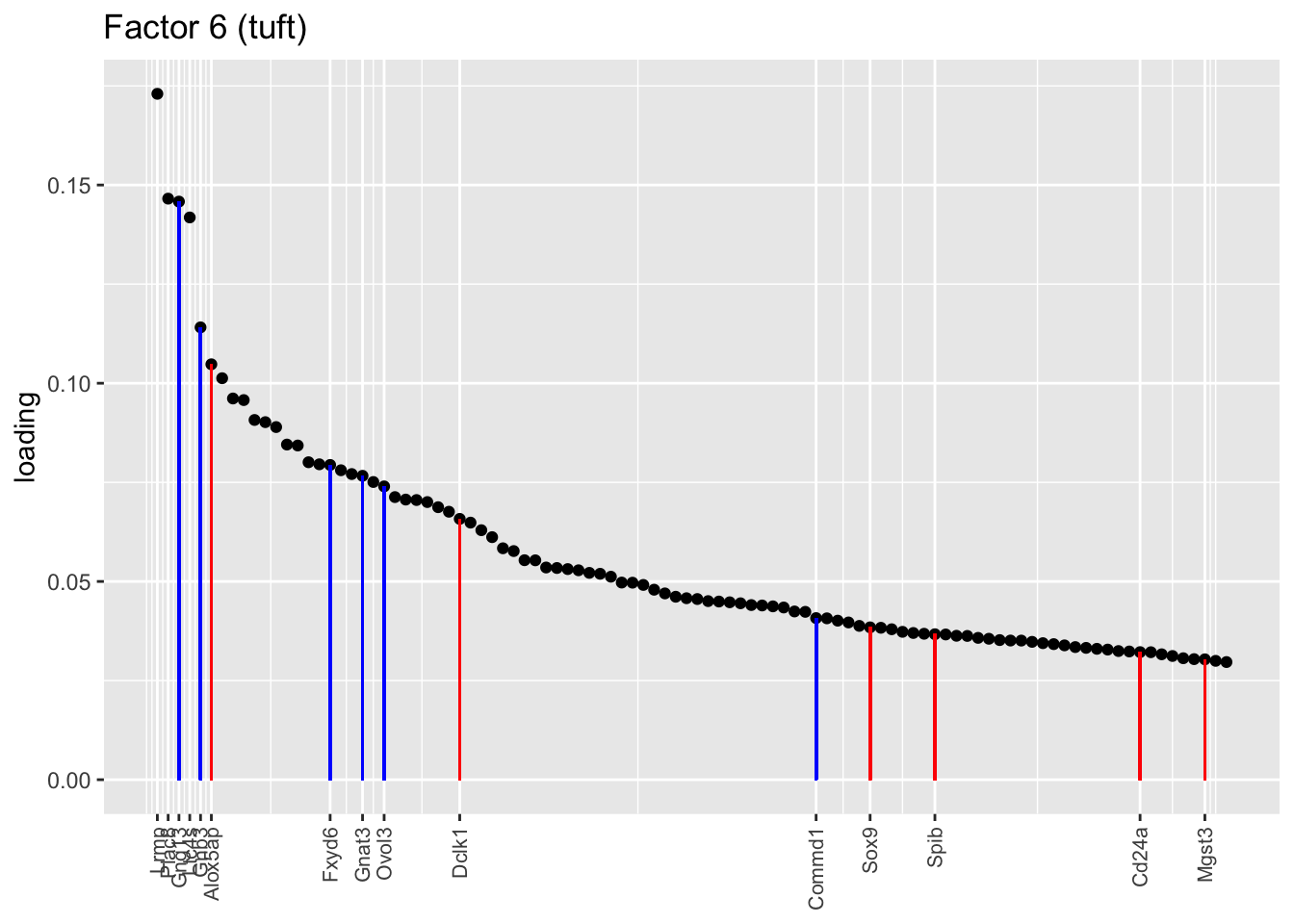
sessionInfo()R version 3.5.3 (2019-03-11)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Mojave 10.14.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] flashier_0.1.17 ggplot2_3.2.0 Matrix_1.2-15
loaded via a namespace (and not attached):
[1] Rcpp_1.0.1 plyr_1.8.4 compiler_3.5.3
[4] pillar_1.3.1 git2r_0.25.2 workflowr_1.2.0
[7] iterators_1.0.10 tools_3.5.3 digest_0.6.18
[10] evaluate_0.13 tibble_2.1.1 gtable_0.3.0
[13] lattice_0.20-38 pkgconfig_2.0.2 rlang_0.3.1
[16] foreach_1.4.4 parallel_3.5.3 yaml_2.2.0
[19] ebnm_0.1-24 xfun_0.6 withr_2.1.2
[22] stringr_1.4.0 dplyr_0.8.0.1 knitr_1.22
[25] fs_1.2.7 rprojroot_1.3-2 grid_3.5.3
[28] tidyselect_0.2.5 glue_1.3.1 R6_2.4.0
[31] rmarkdown_1.12 mixsqp_0.1-119 reshape2_1.4.3
[34] ashr_2.2-38 purrr_0.3.2 magrittr_1.5
[37] whisker_0.3-2 MASS_7.3-51.1 codetools_0.2-16
[40] backports_1.1.3 scales_1.0.0 htmltools_0.3.6
[43] assertthat_0.2.1 colorspace_1.4-1 labeling_0.3
[46] stringi_1.4.3 pscl_1.5.2 doParallel_1.0.14
[49] lazyeval_0.2.2 munsell_0.5.0 truncnorm_1.0-8
[52] SQUAREM_2017.10-1 crayon_1.3.4