Backfits
Jason Willwerscheid
8/17/2019
Last updated: 2019-08-17
Checks: 6 0
Knit directory: scFLASH/
This reproducible R Markdown analysis was created with workflowr (version 1.2.0). The Report tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20181103) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: output/backfit/
Ignored: output/var_type/
Untracked files:
Untracked: analysis/NBapprox.Rmd
Untracked: analysis/pseudocount2.Rmd
Untracked: analysis/trachea4.Rmd
Untracked: code/missing_data.R
Untracked: code/pseudocounts.R
Untracked: code/trachea4.R
Untracked: data/Ensembl2Reactome.txt
Untracked: data/droplet.rds
Untracked: data/hard_bimodal1.txt
Untracked: data/hard_bimodal2.txt
Untracked: data/hard_bimodal3.txt
Untracked: data/mus_pathways.rds
Untracked: docs/figure/pseudocount2.Rmd/
Untracked: output/pseudocount/
Unstaged changes:
Modified: analysis/index.Rmd
Modified: analysis/pseudocount.Rmd
Modified: code/sc_comparisons.R
Modified: code/utils.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 2c164cb | Jason Willwerscheid | 2019-08-17 | wflow_publish(“analysis/backfit.Rmd”) |
Introduction
Although I’ve done much to speed up backfits, they remain slow relative to greedy fits. The question, then, is whether they’re worth the wait. Here, I’ll argue that while the quantitative improvements are relatively small — one can obtain a similar increase in ELBO in a fraction of the time by greedily adding a few additional factors — the qualitative improvements can be dramatic.
In this analysis, I take the best fit from my analysis of variance structures (the fit that “pre-scales” genes and cells) and compare it against a backfitted version of the same. The code used to produce the fits can be viewed here.
source("./code/utils.R")
droplet <- readRDS("./data/droplet.rds")
processed <- preprocess.droplet(droplet)
res <- readRDS("./output/backfit/flashier_fits.rds")
progress.g <- data.table::fread("./output/backfit/greedy_output.txt")
progress.b <- data.table::fread("./output/backfit/backfit_output.txt")Improvement in ELBO
As a quantitative measure of how much backfitting improves the fit, I plot the ELBO attained after each of the last five greedy factors have been added and after each of the first 50 backfitting iterations (backfitting continues for a total of 128 iterations, but the ELBO remains level after the first 30 or so iterations).
From this perspective, backfitting offers about as much improvement as the greedy addition of 3-4 factors. However, a single backfitting iteration takes almost 30 seconds, whereas the entire greedy fit can be performed in less than a minute.
g.iter <- nrow(progress.g)
b.iter <- nrow(progress.b)
progress.g$elapsed.time <- (res$t$greedy * 1:g.iter / g.iter) / 60
progress.b$elapsed.time <- (res$t$greedy + res$t$backfit * 1:b.iter / b.iter) / 60
# Only the final iteration for each greedy factor is needed.
progress.g <- subset(progress.g, c((progress.g$Factor[1:(nrow(progress.g) - 1)]
!= progress.g$Factor[2:nrow(progress.g)]), TRUE))
progress.df <- rbind(progress.g, progress.b)
ggplot(subset(progress.df, !(Factor %in% as.character(1:15)) & Iter < 50),
aes(x = elapsed.time, y = Obj, color = Type)) +
geom_point() +
labs(x = "elapsed time (min)", y = "ELBO (unadjusted)",
title = "Greedy factors 16-20 and first 50 backfitting iterations")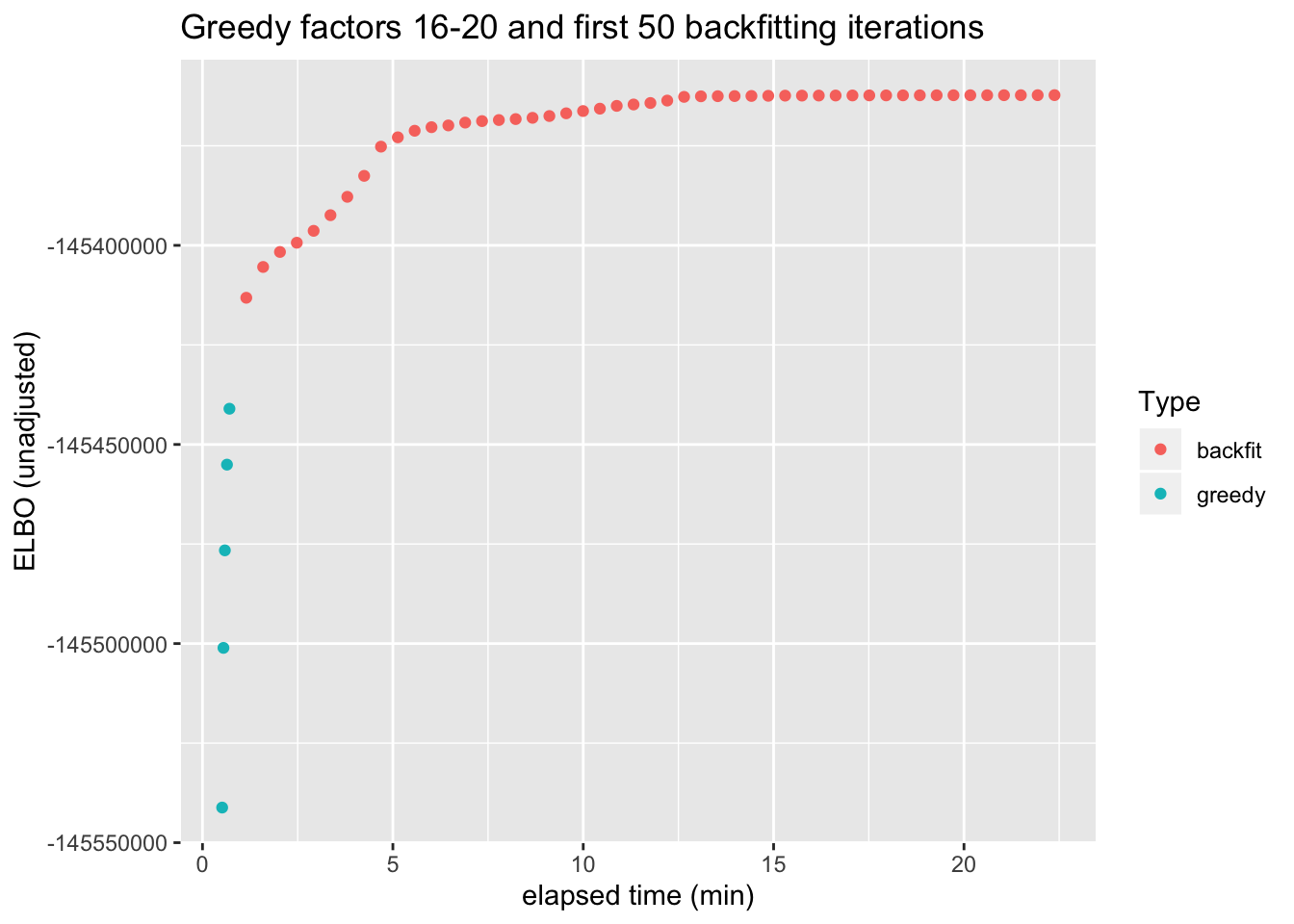
Factor comparison
The ELBO does not tell the whole story. Plotting the factors reveals clear improvements in the backfitted factors. Most obviously, factors 3, 6-8, 11-12, and 17-19 are much sparser and are easily readable as cell-type specific factors. In particular, the unfortunate case of the goblet cells (discussed here) has been cleared up. Factor 2 is also more easily interpretable as indexing a basal-club trajectory without ciliated cells mucking up the situation.
plot.factors(res$fl$greedy, processed$cell.type, title = "Greedy")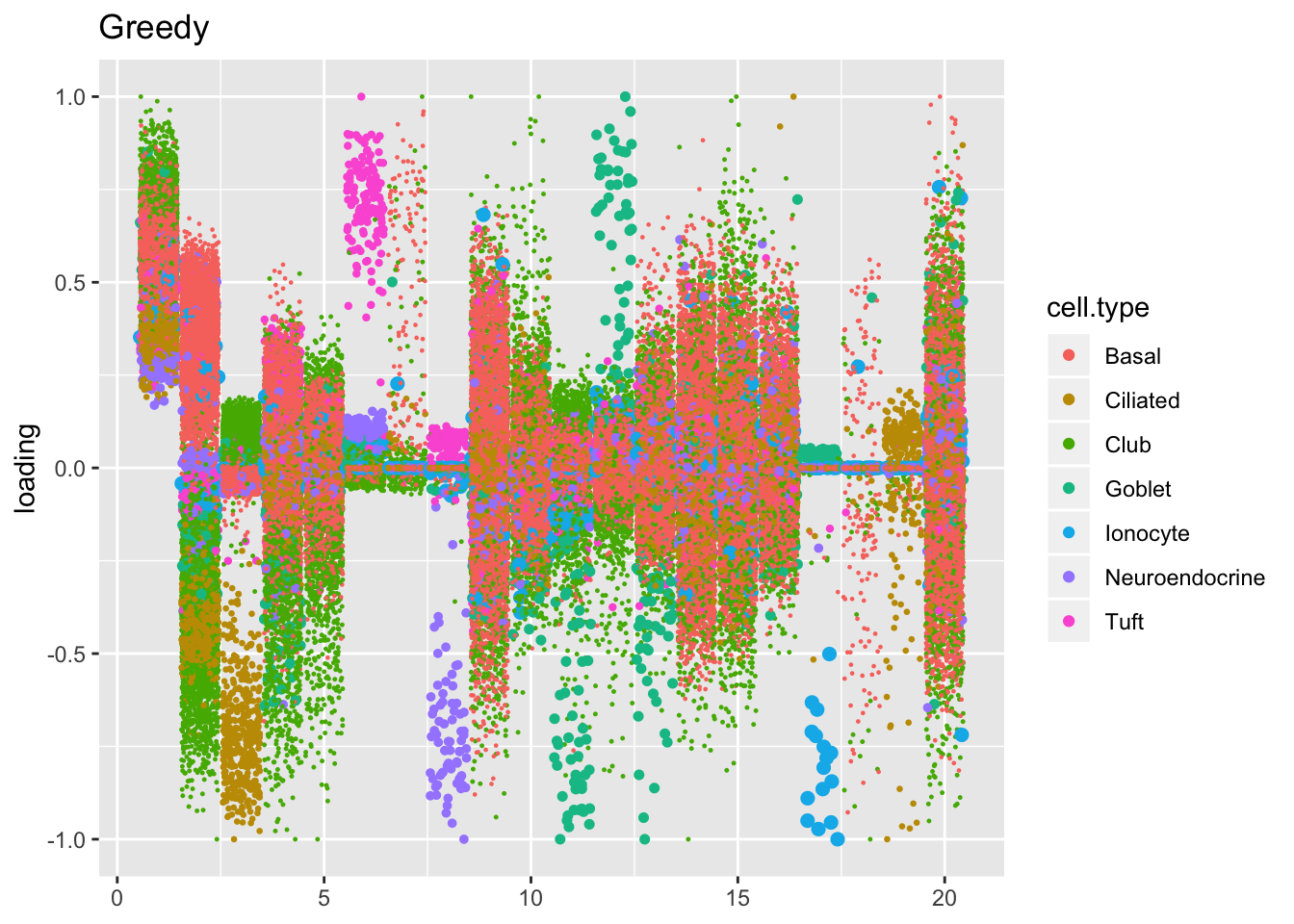
plot.factors(res$fl$backfit, processed$cell.type, title = "Backfit")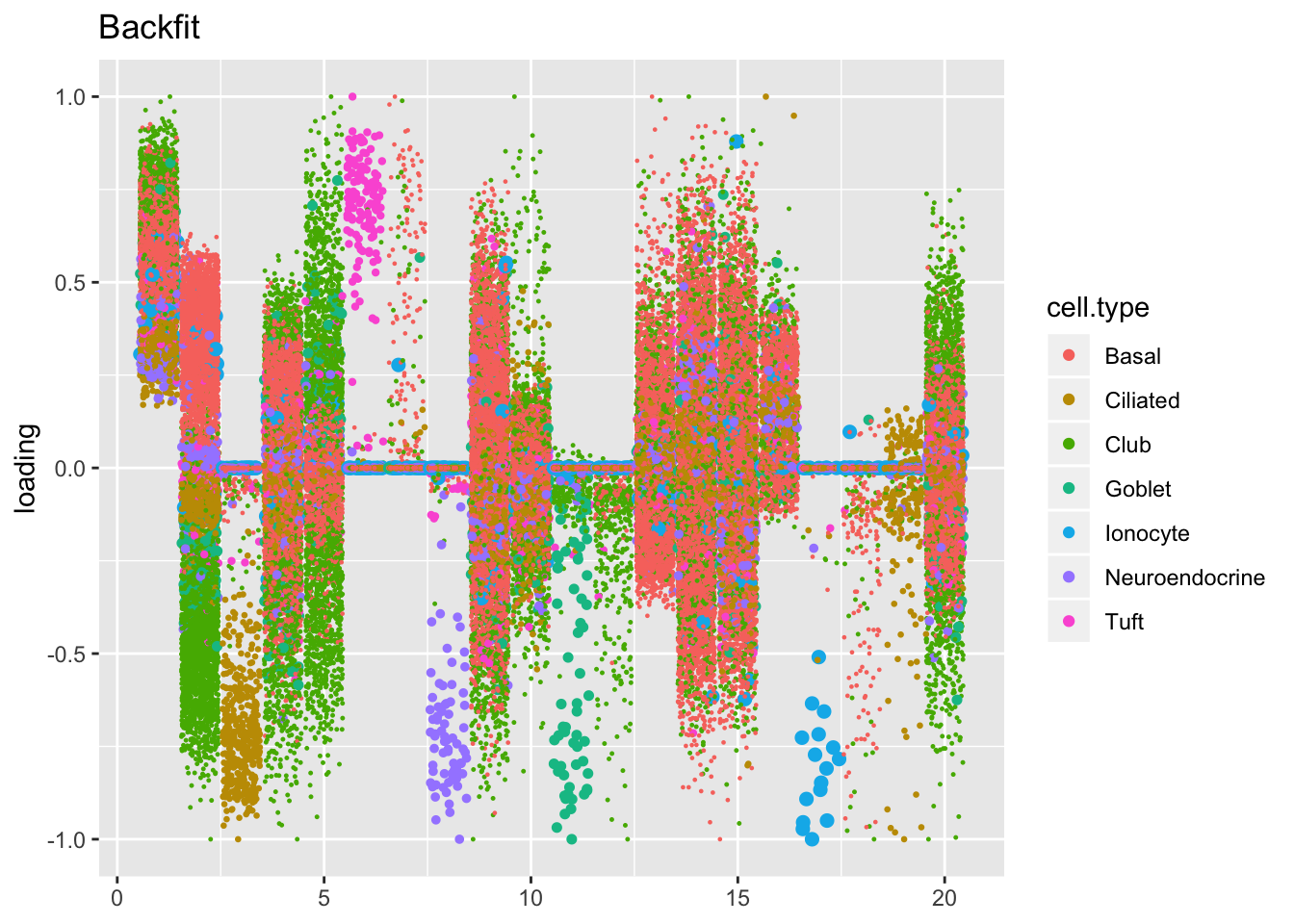
Factors 12 and 13 have increased in importance, while factor 5 (the hillock factor) has become less important. These changes are related: see below. Factors 16, 18, and 20 have also increased in importance (and have possibly borrowed from factor 9), but the significance of these factors is not clear to me.
pve.df <- data.frame(k = 1:res$fl$greedy$n.factors,
greedy = res$fl$greedy$pve,
backfit = res$fl$backfit$pve)
ggplot(pve.df, aes(x = greedy, y = backfit)) +
geom_text(label = pve.df$k, size = 3) +
geom_abline(slope = 1, linetype = "dashed") +
scale_x_log10() + scale_y_log10() +
labs(title = "Proportion of variance explained")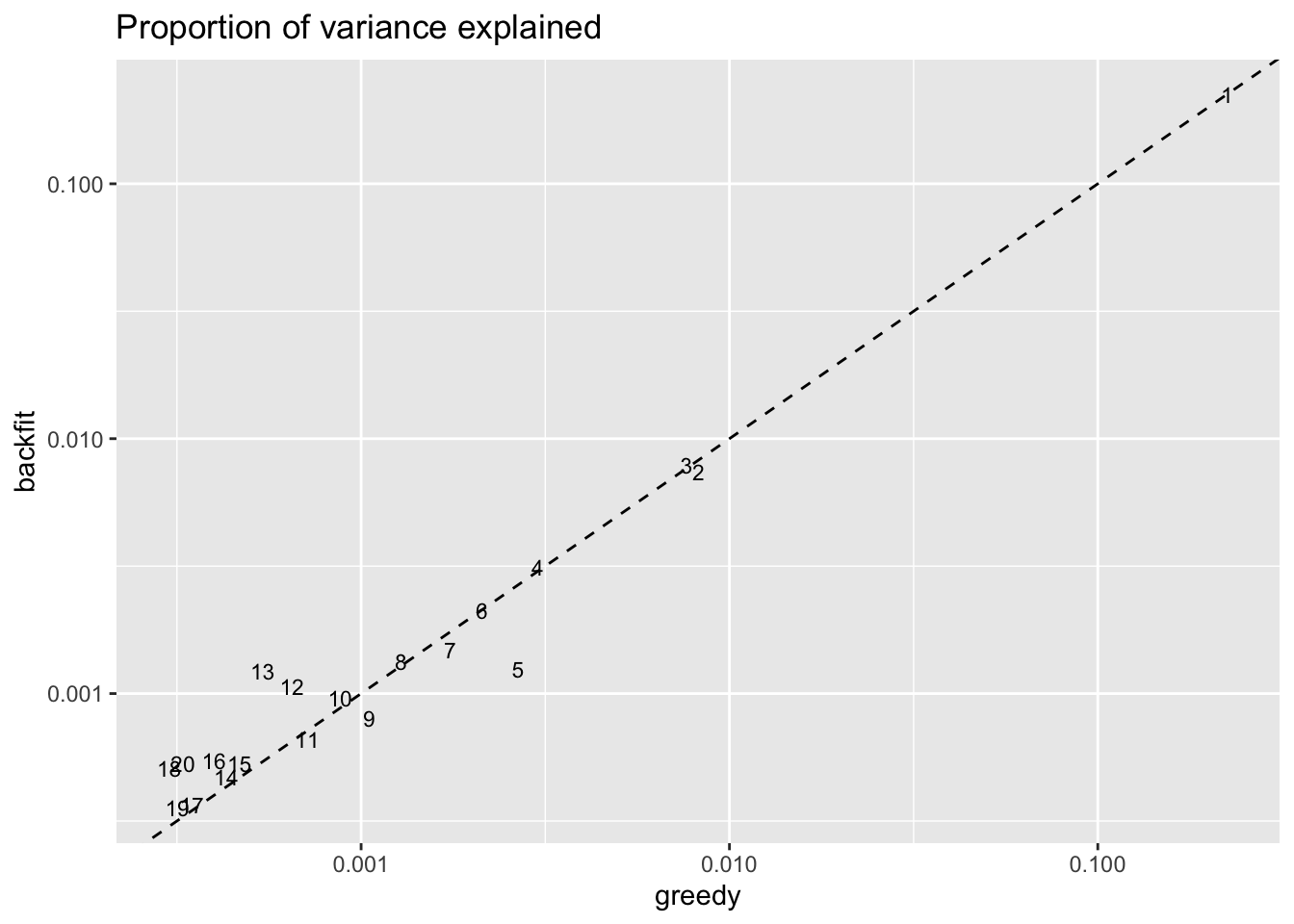
Hillock factor
One of the most interesting changes concerns the “hillock factor.” As far as I can tell, the burden of representing hillock cells is transferred from a single factor (factor 5) to three separate factors (5, 12, and 13). Note, in particular, that Krt13 (a marker gene for hillock cells) has entirely disappeared from the top loadings for factor 5. Instead, the two backfitted factors that are most highly loaded on Krt13 are factors 12 and 13, which are no longer confounded with goblet-specific effects. Intriguingly, factor 12 is almost exclusively loaded on club cells, whereas factor 13 is non-sparse and includes basal and tuft cells.
hillock.genes <- c("Krt13", "Krt4", "Ecm1", "S100a11", "Cldn3", "Lgals3", "Anxa1",
"S100a6", "Upk3bl", "Aqp5", "Anxa2", "Crip1", "Gsto1", "Tppp3")
plot.one.factor(res$fl$greedy, 5, hillock.genes, "Factor 5 (greedy)", invert = TRUE)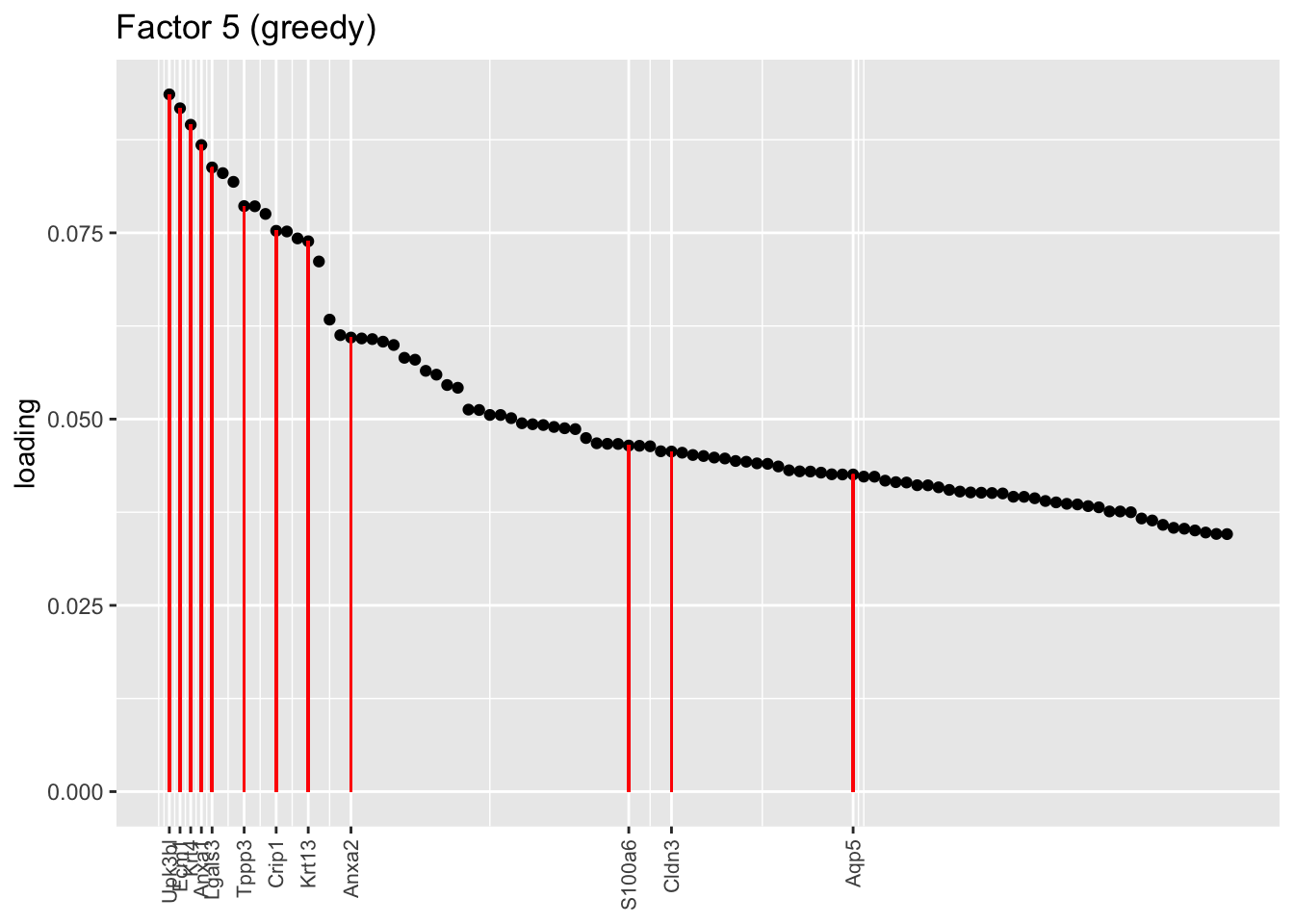
plot.one.factor(res$fl$backfit, 5, hillock.genes, "Factor 5 (backfit)", invert = TRUE)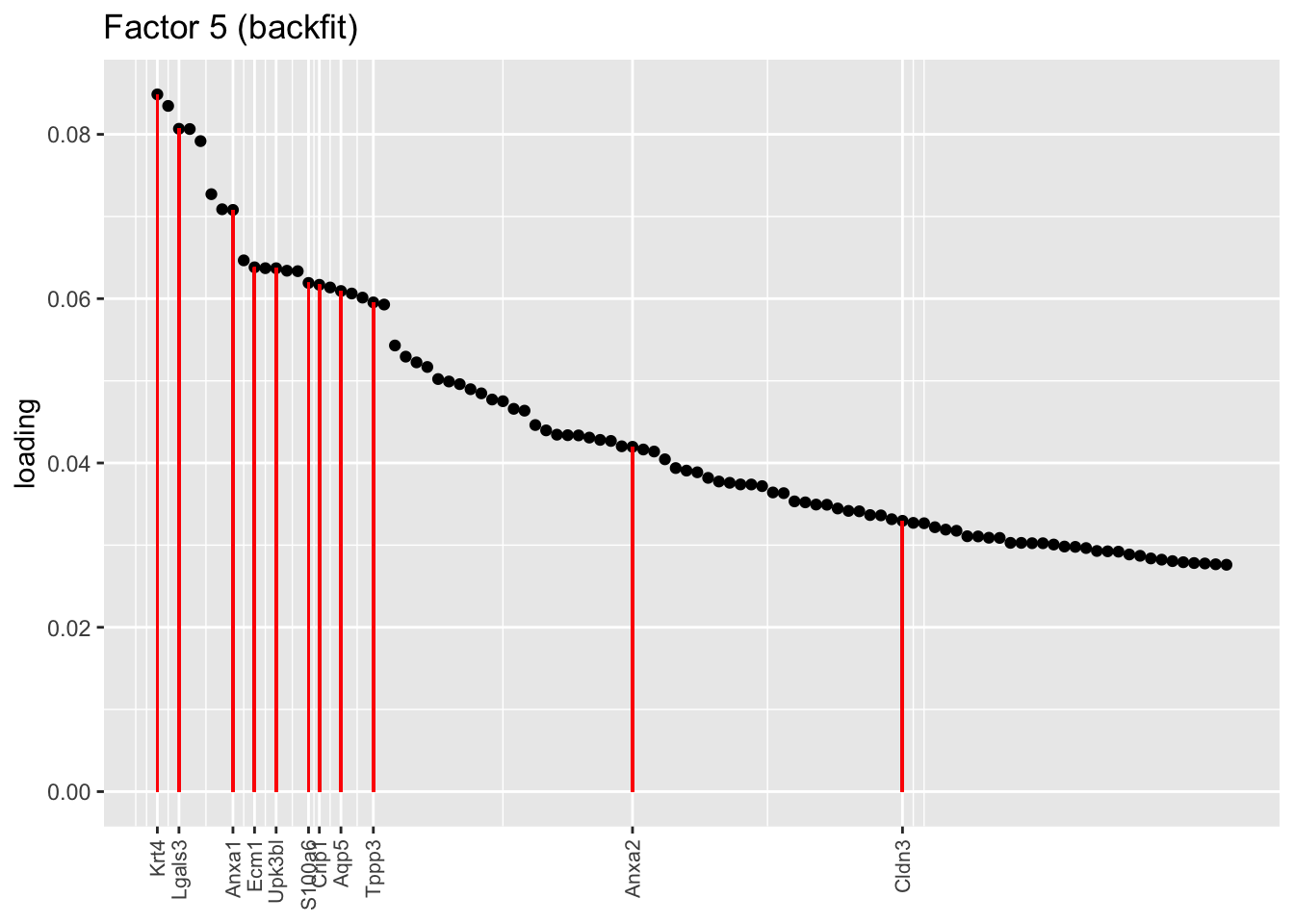
plot.one.factor(res$fl$backfit, 12, hillock.genes, "Factor 12 (backfit)", invert = TRUE)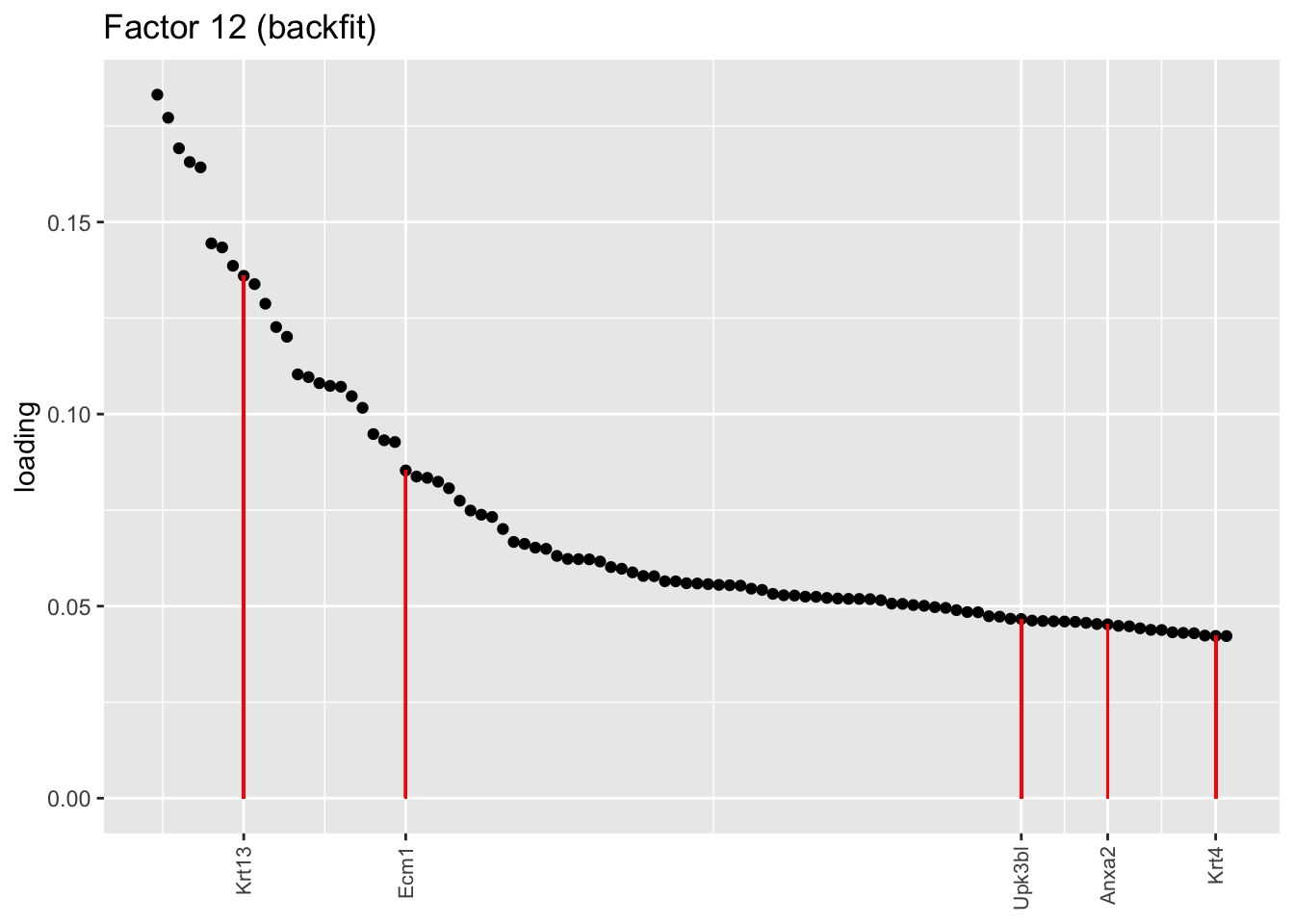
plot.one.factor(res$fl$backfit, 13, hillock.genes, "Factor 13 (backfit)", invert = FALSE)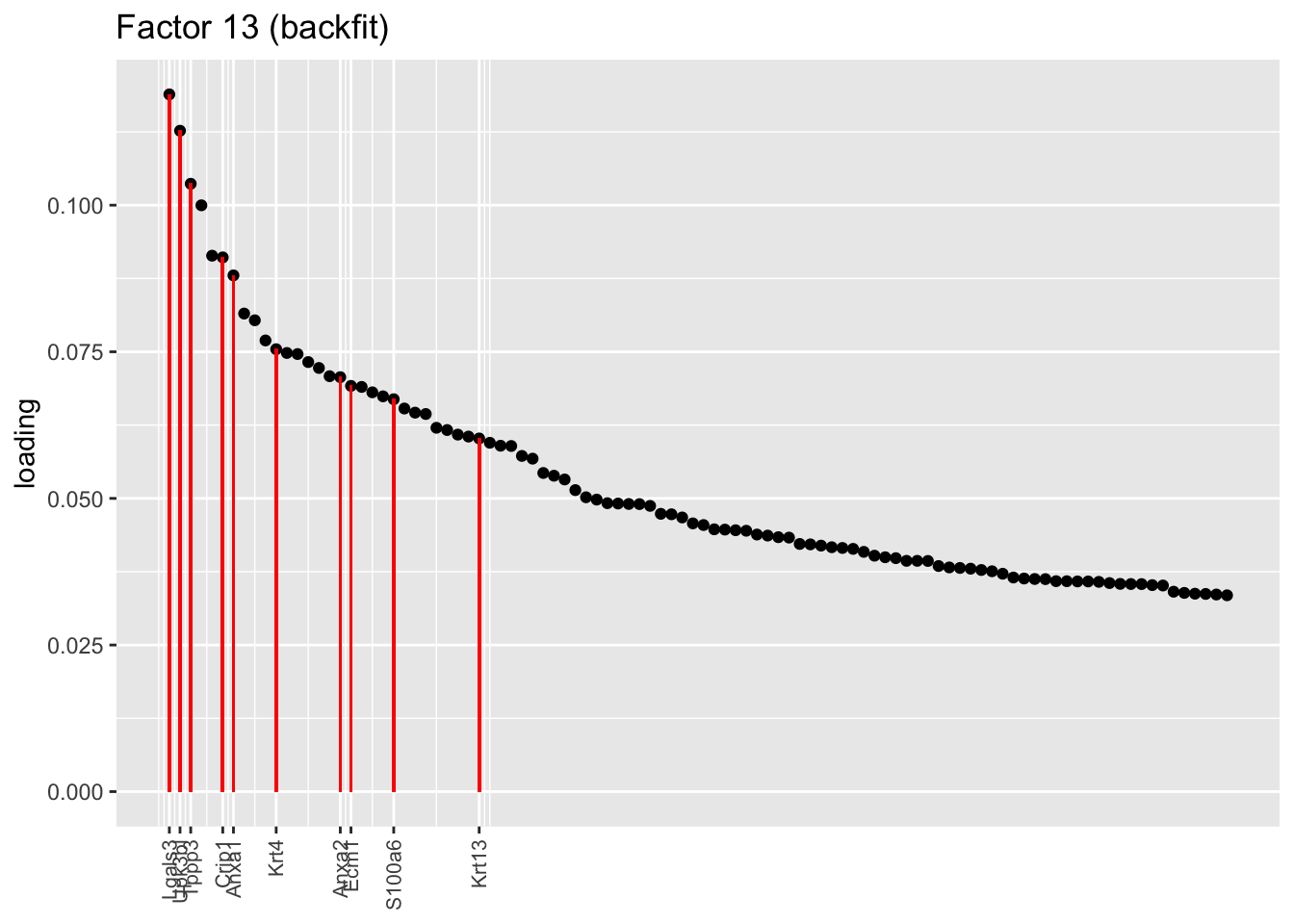
sessionInfo()R version 3.5.3 (2019-03-11)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Mojave 10.14.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] flashier_0.1.11 ggplot2_3.2.0 Matrix_1.2-15
loaded via a namespace (and not attached):
[1] Rcpp_1.0.1 plyr_1.8.4 compiler_3.5.3
[4] pillar_1.3.1 git2r_0.25.2 workflowr_1.2.0
[7] iterators_1.0.10 tools_3.5.3 digest_0.6.18
[10] evaluate_0.13 tibble_2.1.1 gtable_0.3.0
[13] lattice_0.20-38 pkgconfig_2.0.2 rlang_0.3.1
[16] foreach_1.4.4 parallel_3.5.3 yaml_2.2.0
[19] ebnm_0.1-24 xfun_0.6 withr_2.1.2
[22] stringr_1.4.0 dplyr_0.8.0.1 knitr_1.22
[25] fs_1.2.7 rprojroot_1.3-2 grid_3.5.3
[28] tidyselect_0.2.5 data.table_1.12.2 glue_1.3.1
[31] R6_2.4.0 rmarkdown_1.12 mixsqp_0.1-119
[34] reshape2_1.4.3 ashr_2.2-38 purrr_0.3.2
[37] magrittr_1.5 whisker_0.3-2 MASS_7.3-51.1
[40] codetools_0.2-16 backports_1.1.3 scales_1.0.0
[43] htmltools_0.3.6 assertthat_0.2.1 colorspace_1.4-1
[46] labeling_0.3 stringi_1.4.3 pscl_1.5.2
[49] doParallel_1.0.14 lazyeval_0.2.2 munsell_0.5.0
[52] truncnorm_1.0-8 SQUAREM_2017.10-1 crayon_1.3.4