Pipeline
Last updated: 2024-06-11
Checks: 7 0
Knit directory: PPP/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20240521) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 589e25d. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .RData
Ignored: .Rhistory
Ignored: code/.DS_Store
Ignored: code/TieDIE-devel/.DS_Store
Ignored: code/TieDIE-devel/examples/.DS_Store
Ignored: code/TieDIE-devel/examples/hnsc/.DS_Store
Ignored: code/TieDIE-tiedie2/.DS_Store
Ignored: code/TieDIE-tiedie2/examples/.DS_Store
Ignored: data/.DS_Store
Ignored: data/Phosphoproteome_BCM_GENCODE_v34_harmonized_v1/.DS_Store
Ignored: data/Phosphoproteome_BCM_GENCODE_v34_harmonized_v1/README/.DS_Store
Ignored: data/Proteome_BCM_GENCODE_v34_harmonized_v1/.DS_Store
Ignored: data/Proteome_BCM_GENCODE_v34_harmonized_v1/README/.DS_Store
Ignored: output/.DS_Store
Ignored: output/cnv/.DS_Store
Ignored: output/phos_processed/
Ignored: output/phos_regul_object/
Ignored: output/regulon/.DS_Store
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/Pipeline.Rmd) and HTML
(docs/Pipeline.html) files. If you’ve configured a remote
Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | 589e25d | Zhen Zuo | 2024-06-11 | wflow_publish("*", all = TRUE) |
| html | 6336caf | Zhen Zuo | 2024-06-11 | Update Pipeline.html |
| Rmd | 81703bb | Zhen Zuo | 2024-06-11 | . |
| html | 81703bb | Zhen Zuo | 2024-06-11 | . |
| html | c334119 | Zhen Zuo | 2024-06-11 | Build site. |
| Rmd | b09c43b | Zhen Zuo | 2024-06-11 | Publish files |
| html | 6910e57 | Zhen Zuo | 2024-06-11 | Build site. |
| Rmd | 96dfc6d | Zhen Zuo | 2024-06-11 | Publish files |
library(png)
img <- readPNG("data/Figure2.png")
grid::grid.raster(img)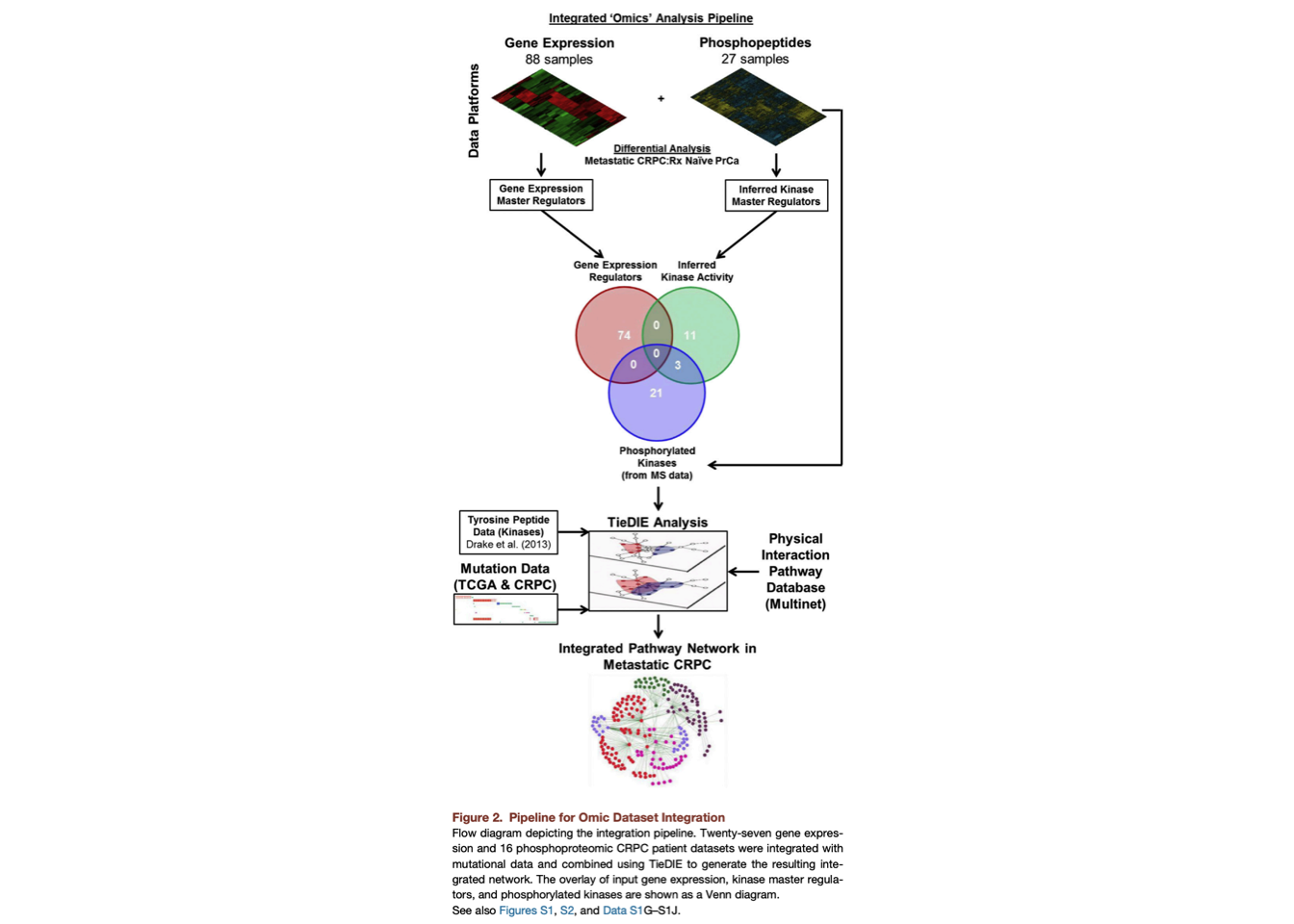
| Version | Author | Date |
|---|---|---|
| 81703bb | Zhen Zuo | 2024-06-11 |
1 Pipeline for Omic Dataset Integration
1.1 Find Differentially Phosphorylated Peptides
- Get phosphorylation data.
- Replicates were averaged.
- Run t-test on naive and benign samples to identify differentially phosphorylated peptide (FDA < 0.1).
- Map differentially phosphorylated peptide to known kinases. Save it
and pass to the next step.
Please double check. I cannot find details about how this map works, I think it uses the predicted kinase-substrate interactions you shared with me and map sep_15 to kinase. .
1.2 Integration of Transcriptomic and Phosphoproteomic Datasets Using TieDIE
1.2.1 Apply the master regulator inference algorithm (MARINa)
We first applied the master regulator inference algorithm (MARINa) (Alvarez et al., 2015), a method to infer the activity of a given protein based on the differential expression/phosphorylation of the targets it regulates.
I am confused about this part. Can you infer the activity of a given protein based on protein abundance? I do not understand why people infer protein activity based on expression/phosphorylation data instead of protein data itself.
1.2.1.1 Input
- Known kinases from mapped differentially phosphorylated peptide (from last step)
- Gene Expression data for normal and tumor
- Gene level pathway network (Object of class regulon with XXX regulators, XXX targets and XXX interactions)
- Phosphorylation data for normal and tumor
- Phosphorylation level pathway network (Object of class regulon with XXX regulators, XXX targets and XXX interactions)
1.2.1.2 Output
- Transcription factors with differential activity (repression/activation).
- Differentially activated kinase regulator (only higher in tumor, based on the predicted upstream kinases for each phosphopeptide).
I think it dependents on the pathway network we give as input, it does not have to be transcription factors based on my observation. It can be a regulator in general.
This allowed us to identify transcription factors with differential activity (repression/activation) as well as differentially activated kinase regulators (based on the predicted upstream kinases for each phosphopeptide) in metastatic CRPC samples as compared with treatment naive prostate cancers (Data S1G and S1H)..
In addition, kinases directly identified by the mass spectrometer in our phosphoproteomic dataset (phosphorylated kinases) were merged with the kinase regulators before input to TieDIE.
1.2.1.3 Mechanisms and details
Given a data matrix and a regulon, by running
vpres <- viper(dset, regulon, verbose = FALSE), it will
generate regulator’s activity matrix for each sample and regulator. This
is running for each sample,
but it can also run on multiple samples together (similar to MARINa),
mrs <- msviper(signature, regulon, nullmodel, verbose = FALSE)
to get top regulators with Normalized Enrichment Score (NES) and
p-value.
In this section, msviper was used to get the output.
In other words, it performs a KS test for each regulator using as
input the differential activity (in metastatic CRPC vs treatment naïve
prostate cancer) of its targets. It applies a test based on the
Kolmogorov-Smirnov test to assess whether the differential activity of
activated targets have higher levels, and/or the differential activity
of inhibited targets have lower levels, than expected by chance based on
a uniform distribution. In the case of TF regulators, the expression
levels of the targets are used for this inference; in the case of kinase
regulators, the phosphorylation levels are used for the target levels.
To facilitate this analysis, we combined multiple databases of predicted
kinase-substrate predicted interactions to produce a comprehensive
‘regulome’ of candidate regulator kinases that are predicted to
phosphorylate at least 25 proteins on at least one site (Drake et al.,
2012; Lachmann and Ma’ayan, 2009). We ran the MARINa algorithm (Alvarez
et al., 2015), to find ‘kinase regulators’ with significantly higher
activity–as inferred from the peptides they are predicted to
phosphorylate–in CRPC samples compared to the control primary and benign
tumor samples. For the TF targets we used a predetermined interactome of
transcription factor-to-target regulatory edges, inferred using a
diverse sample of normal, primary and metastatic prostate cancer samples
as well as cell lines (Aytes et al., 2014a).
sessionInfo()R version 4.4.0 (2024-04-24)
Platform: aarch64-apple-darwin20
Running under: macOS Sonoma 14.5
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: America/New_York
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] png_0.1-8
loaded via a namespace (and not attached):
[1] vctrs_0.6.5 cli_3.6.2 knitr_1.47 rlang_1.1.4
[5] xfun_0.44 highr_0.11 stringi_1.8.4 promises_1.3.0
[9] jsonlite_1.8.8 workflowr_1.7.1 glue_1.7.0 rprojroot_2.0.4
[13] git2r_0.33.0 htmltools_0.5.8.1 httpuv_1.6.15 sass_0.4.9
[17] fansi_1.0.6 rmarkdown_2.27 grid_4.4.0 jquerylib_0.1.4
[21] evaluate_0.24.0 tibble_3.2.1 fastmap_1.2.0 yaml_2.3.8
[25] lifecycle_1.0.4 whisker_0.4.1 stringr_1.5.1 compiler_4.4.0
[29] fs_1.6.4 Rcpp_1.0.12 pkgconfig_2.0.3 rstudioapi_0.16.0
[33] later_1.3.2 digest_0.6.35 R6_2.5.1 utf8_1.2.4
[37] pillar_1.9.0 magrittr_2.0.3 bslib_0.7.0 tools_4.4.0
[41] cachem_1.1.0