DE analysis on scRNA data with poisson GLMM
Chih-Hsuan Wu
Last updated: 2023-11-29
Checks: 6 1
Knit directory: DEanalysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230508) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 91b404e. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Untracked files:
Untracked: .DS_Store
Untracked: .Rhistory
Untracked: analysis/.DS_Store
Untracked: analysis/.Rapp.history
Untracked: analysis/.Rhistory
Untracked: analysis/Bcells.Rmd
Untracked: analysis/CD14+ Monocytes.Rmd
Untracked: analysis/FD_analysis.Rmd
Untracked: analysis/analysis on Kang.Rmd
Untracked: analysis/data2.Rmd
Untracked: analysis/data_clusters.Rmd
Untracked: analysis/figure/
Untracked: analysis/group12_13.Rmd
Untracked: analysis/group12_19.Rmd
Untracked: analysis/group2_19.Rmd
Untracked: analysis/group8_17-2_19.Rmd
Untracked: analysis/group8_17.Rmd
Untracked: analysis/methods_details.Rmd
Untracked: analysis/new_criteria.Rmd
Untracked: data/.Rhistory
Untracked: data/10X_Kang_DEresult.RData
Untracked: data/10X_inputdata.RData
Untracked: data/10X_inputdata_DEresult.RData
Untracked: data/10X_inputdata_cpm.RData
Untracked: data/10X_inputdata_integrated.RData
Untracked: data/10X_inputdata_lognorm.RData
Untracked: data/10Xdata_annotate.rds
Untracked: data/Bcells.Rmd
Untracked: data/Bcellsce.rds
Untracked: data/data2sce.RData
Untracked: data/permutation.RData
Untracked: data/vstcounts.Rdata
Unstaged changes:
Modified: analysis/index.Rmd
Modified: code/DE_methods.R
Modified: code/functions_in_rmd.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/index.Rmd) and HTML
(docs/index.html) files. If you’ve configured a remote Git
repository (see ?wflow_git_remote), click on the hyperlinks
in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | e5cde05 | C-HW | 2023-11-13 | update index page |
| html | fc4ab47 | C-HW | 2023-08-03 | data2 results |
| html | 5ee9460 | C-HW | 2023-07-27 | update index.html |
| html | 949c760 | C-HW | 2023-07-27 | data2 |
| html | b688ebf | C-HW | 2023-07-13 | updatd index.html |
| html | 4da4a48 | C-HW | 2023-07-13 | update index.html |
| html | 35f04ca | C-HW | 2023-07-12 | add 12_19 |
| Rmd | 366cd53 | C-HW | 2023-06-06 | add group8_17&2_19 |
| html | 366cd53 | C-HW | 2023-06-06 | add group8_17&2_19 |
| html | 3b1c8ca | C-HW | 2023-05-25 | update index page |
| Rmd | 95be122 | C-HW | 2023-05-25 | update 2_19 |
| html | 95be122 | C-HW | 2023-05-25 | update 2_19 |
| Rmd | 2107af3 | C-HW | 2023-05-24 | add methods_details |
| Rmd | 9a7a569 | C-HW | 2023-05-18 | correct link |
| html | 9a7a569 | C-HW | 2023-05-18 | correct link |
| Rmd | 641a5d8 | C-HW | 2023-05-18 | add link to index |
| html | 641a5d8 | C-HW | 2023-05-18 | add link to index |
| Rmd | 13d726d | C-HW | 2023-05-18 | add DE results on different groups |
| html | 13d726d | C-HW | 2023-05-18 | add DE results on different groups |
| Rmd | fc9f4b6 | C-HW | 2023-05-18 | add new_criteria |
| html | fc9f4b6 | C-HW | 2023-05-18 | add new_criteria |
| html | 7586953 | C-HW | 2023-05-11 | add data_clusters |
| Rmd | 703eb54 | C-HW | 2023-05-09 | create workflowr |
| html | 703eb54 | C-HW | 2023-05-09 | create workflowr |
Introduction
Motivation
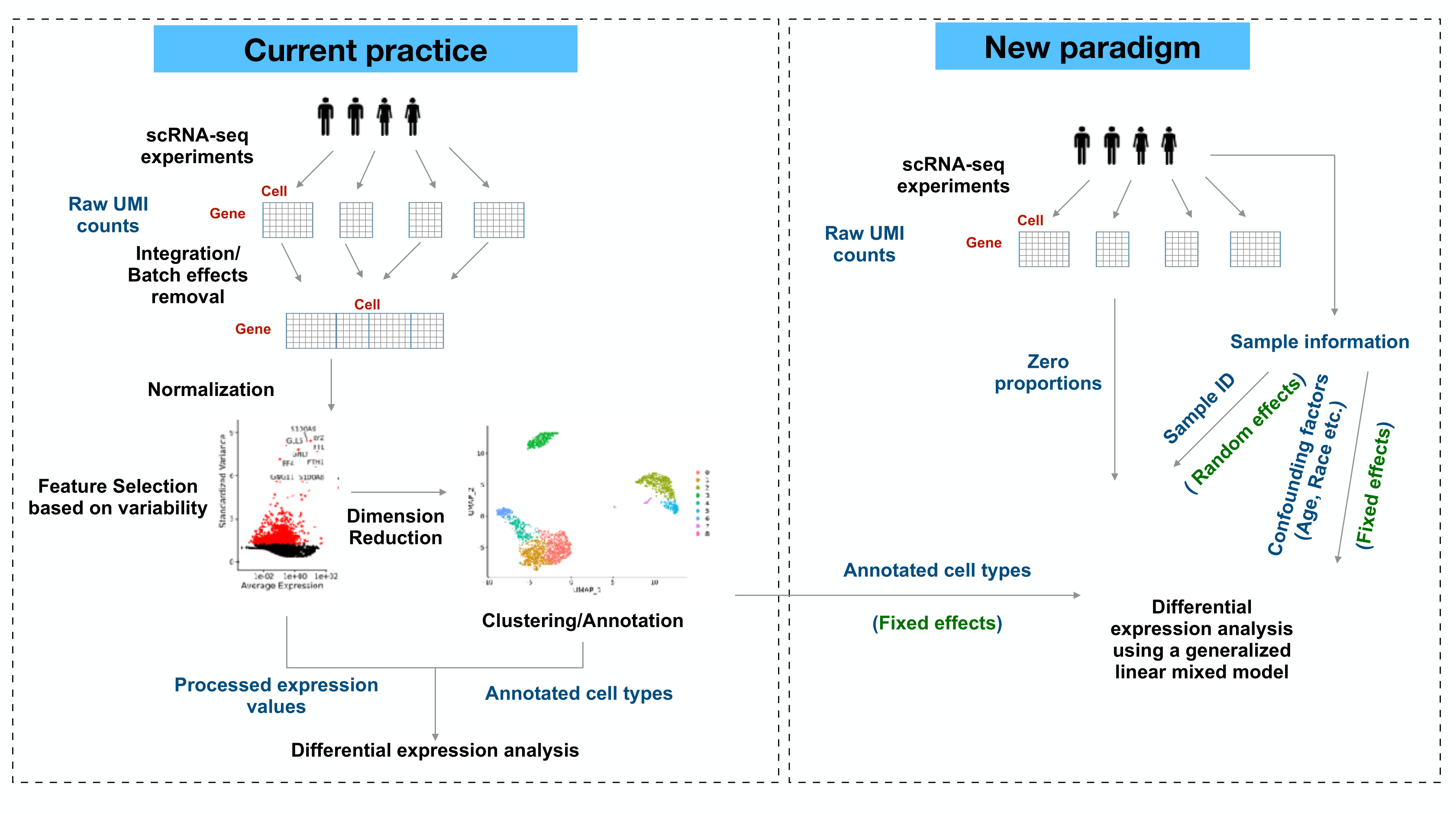
Differential expression (DE) analysis in single-cell transcriptomics reveals cell-type-specific responses. Recent studies have raised concerns about current methods used to identify differentially expressed genes in this context.
scRNA sequencing provides the absolute abundances of RNA molecules in single cells, but normalization - a pre-processing step inherited from the bulk RNA-seq era - reduces this information and returns data as relative abundances, which may mask important differences among cell types
The majority of single-cell DE analysis methods are susceptible to generating false discoveries. This is mainly due to the lack of accounting for variations between biological replicates, commonly referred to as “donor effects”.
- Batch effects are often estimated from leading principal components, representing a consensus from most genes.
- Pseudo-bulk analysis ignores within-sample heterogeneity by treating donor effects as a fixed effect and assuming that each cell from the same donor is equally affected.
Clustering and DE analysis are different problems. The current commonly workflow works well in clustering, but cannot guarantee success in downstream analysis.
Excessive zeros are usually considered as “drop-outs”, while they are actually informative in cell-type heterogeneity. Ignoring zeros in single-cell gene expression data discards valuable information for any analysis.
We provide a generalized linear mixed model framework (GLMM) to detect differentially expressed genes (DEGs) between two given cell types. The model takes donor-specific variations as random effects, and uses raw UMI counts to prevent biases in DE analysis.
| Version | Author | Date |
|---|---|---|
| c347de5 | C-HW | 2023-11-15 |
GLMM for DE analysis
Poisson GLMM
For each count \(X_{cgk}\) sampled from cell \(c\), donor \(k\), and gene \(g\),
\[ \begin{aligned} X_{cgk}|\lambda_{cgk} & \sim Poisson(\lambda_{cgk})\\ \log \lambda_{cgk} & = \mu_g + X_c{\beta_g} + \epsilon_{gk}\\ \end{aligned} \] where \(X_c\) is the indicator for different cell types, and \(\epsilon_{gk}\sim N(0,\sigma_{g}^2)\) represents the random effects for donor \(k\). Our goal is to test \(H_0: \beta_g = 0\). Here \(e^{\beta_g}\) represents the fold change of gene \(g\) between two cell types.
Binomial GLMM
\[ \begin{aligned} \mathbb{1}_{X_{cgk}=0}|p_{cgk} & \sim Bernoulli(p_{cgk})\\ \log \frac{p_{cgk}}{1-p_{cgk}} & = \mu_g + X_c\beta_{g} + \epsilon_{gk}\\ \end{aligned} \] where \(X_c\) is the indicator for different cell types, and \(\epsilon_{gk}\sim N(0,\sigma_{g}^2)\) represents the random effects for donor \(k\). Our goal is to test \(H_0: \beta_g = 0\).
New criteria on determining DEGs
We proposed new criteria that based on the convention and also the gene mean and the difference in mean. If the log2 gene mean in two groups is lower than a certain value (-2.25 in case study 1) and the log2 mean difference is smaller than a threshold (-1 in case study 1), the genes would not be considered as a DEGs. These can also be used as a filter before any DE analysis to speed up the computation. Both of these criteria are adjustable, depending on the dataset’s performance and characteristics. An examination in heatmaps and mean difference against mean plot in advanced can be helpful to determine the thresholds when analyzing a new dataset. More details can be found here.
Benchmarked methods
In this project, we compare a few methods performing the DE analysis results. Our comparison encompassed Poisson-glmm and Binomial-glmm from the new paradigm, as well as pseudo-bulk approaches including DESeq2 and edgeR. Additionally, we assessed the performance of single cell specific tools including MAST, Wilcox in Seurat, and linear mixed models in Muscat. More details can be found here.
Applications
Analysis on case study 1
- About data
In case study 1, a 10X scRNA-seq dataset of post-menopausal fallopian tubes, with 57,182 cells sourced from five donors, covering 29,382 genes is analyzed. The 20 clusters are obtained via HIPPO algorithm. There is no pre-filtering procedure applied on this dataset, except for built-in filtering steps in each method. We use sctransform to get the VST data, and the integration workflow provided by Seurat to obtain the integrated data.
Analysis on case study 2
About data In case study 2, the dataset contains 10X droplet-based scRNA-seq PBCM data from 8 Lupus patients obtained before and after 6h-treatment with IFN-\(\beta\). After removing undetected genes and lowly expressed genes (less than 10 cells expressing more than 1), the dataset consists of 29065 cells and 7661 genes. The integrated data is replaced by log2-transformed normalized expression values obtained via computeLibrarayFactors and logNormCounts functions in Muscat.
Analyses
sessionInfo()R version 4.2.2 (2022-10-31)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur ... 10.16
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
loaded via a namespace (and not attached):
[1] Rcpp_1.0.11 rstudioapi_0.15.0 whisker_0.4.1 knitr_1.27
[5] magrittr_2.0.3 workflowr_1.7.0 R6_2.5.1 rlang_1.1.1
[9] fastmap_1.1.1 fansi_1.0.4 stringr_1.5.0 tools_4.2.2
[13] xfun_0.39 png_0.1-8 utf8_1.2.3 cli_3.6.1
[17] jquerylib_0.1.4 git2r_0.32.0 htmltools_0.5.5 rprojroot_2.0.3
[21] yaml_2.3.7 digest_0.6.33 tibble_3.2.1 lifecycle_1.0.3
[25] later_1.3.1 vctrs_0.6.3 sass_0.4.7 promises_1.2.0.1
[29] fs_1.6.3 glue_1.6.2 cachem_1.0.8 evaluate_0.21
[33] rmarkdown_2.23 stringi_1.7.12 pillar_1.9.0 compiler_4.2.2
[37] bslib_0.5.0 jsonlite_1.8.7 httpuv_1.6.11 pkgconfig_2.0.3