Pre-copulatory sexual selection predicts sexual size dimorphism: a meta-analysis of comparative studies
Main analyses
Lennart Winkler1, Robert P
Freckleton2, Tamas Szekely3, 4 &
Tim Janicke1,5
1Applied Zoology,
Technical University Dresden 2Department of
Zoology, University of Oxford, South Parks Road, Oxford OX1 3PS,
UK
3Milner Centre for Evolution, University of Bath,
Bath, UK84Department of Evolutionary Zoology and
Human Behaviour, University of Debrecen, Debrecen, Hungary
5Centre d’Écologie Fonctionnelle et Évolutive, UMR 5175,
CNRS, Université de Montpellier
Last updated: 2024-05-01
Checks: 6 1
Knit directory:
SSD_and_sexual_selection_2023/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230430) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version b028b7d. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Untracked files:
Untracked: data/Data_SexSelSSD.csv
Untracked: data/Pyologeny_SexSelSSD.txt
Unstaged changes:
Modified: .Rprofile
Modified: .gitattributes
Modified: .gitignore
Modified: README.md
Modified: SSD_and_sexual_selection_2023.Rproj
Modified: _workflowr.yml
Modified: analysis/_site.yml
Modified: analysis/about.Rmd
Modified: analysis/index.Rmd
Modified: analysis/index2.Rmd
Modified: analysis/license.Rmd
Modified: code/README.md
Modified: data/README.md
Deleted: data/Supplement4_SexSelSSD_V01.csv
Deleted: data/Supplement6_SexSelSSD_V01.txt
Modified: output/README.md
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/index.Rmd) and HTML
(docs/index.html) files. If you’ve configured a remote Git
repository (see ?wflow_git_remote), click on the hyperlinks
in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | b028b7d | LennartWinkler | 2023-05-24 | Build site. |
| Rmd | c1d3311 | LennartWinkler | 2023-05-24 | wflow_publish(all = T) |
| html | 71d667c | LennartWinkler | 2023-05-03 | Build site. |
| Rmd | 378a58c | LennartWinkler | 2023-05-03 | wflow_publish(republish = TRUE, all = T) |
| html | 085f45f | LennartWinkler | 2023-05-03 | Build site. |
| Rmd | 85c143e | LennartWinkler | 2023-05-03 | update |
| html | 8edd942 | LennartWinkler | 2023-05-01 | Build site. |
| Rmd | d131cd9 | LennartWinkler | 2023-05-01 | wflow_publish(republish = TRUE, all = T) |
| Rmd | 57ca562 | LennartWinkler | 2023-04-30 | update |
| html | 054a12b | LennartWinkler | 2023-04-30 | Build site. |
| Rmd | 1a5561c | LennartWinkler | 2023-04-30 | wflow_publish(all = T) |
| Rmd | 4482c88 | LennartWinkler | 2023-04-30 | Start workflowr project. |
Supplementary material reporting R code for the manuscript ‘Pre-copulatory sexual selection predicts sexual size dimorphism: a meta-analysis of comparative studies’. Additional analyses excluding studies that did not control for phylogenetic non-independence (Supplement 2) can be found at: https://https://lennartwinkler.github.io/SSD_and_sexual_selection_2023/index2.html # Load and prepare data Before we started the analyses, we loaded all necessary packages and data.
rm(list = ls()) # Clear work environment
# Load R-packages ####
list_of_packages=cbind('ape','matrixcalc','metafor','Matrix','MASS','pwr','psych','multcomp','data.table','ggplot2','RColorBrewer','MCMCglmm','ggdist','cowplot','PupillometryR','dplyr','wesanderson','gridExtra')
lapply(list_of_packages, require, character.only = TRUE)
# Load data set ####
MetaData <- read.csv("./data/Data_SexSelSSD.csv", sep=";", header=TRUE) # Load data set
N_Studies <- length(summary(as.factor(MetaData$Study_ID))) # Number of included primary studies
Tree<- read.tree("./data/Pyologeny_SexSelSSD.txt") # Load phylogenetic tree
# Prune phylogenetic tree
MetaData_Class_Data <- unique(MetaData$Class)
Tree_Class<-drop.tip(Tree, Tree$tip.label[-na.omit(match(MetaData_Class_Data, Tree$tip.label))])
forcedC_Moderators <- as.matrix(forceSymmetric(vcv(Tree_Class, corr=TRUE)))
# Order moderator levels
MetaData$SexSel_Episode=as.factor(MetaData$SexSel_Episode)
MetaData$SexSel_Episode=relevel(MetaData$SexSel_Episode,c("post-copulatory"))
MetaData$SexSel_Episode=relevel(MetaData$SexSel_Episode,c("pre-copulatory"))
MetaData$Class=as.factor(MetaData$Class)
MetaData$z=as.numeric(MetaData$z)
# Set figure theme and colors
theme=theme(panel.border = element_blank(),
panel.background = element_blank(),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
legend.position = c(0.2,0.5),
legend.title = element_blank(),
legend.text = element_text(colour="black", size=12),
axis.line.x = element_line(colour = "black", linewidth = 1),
axis.line.y = element_line(colour = "black", linewidth = 1),
axis.text.x = element_text(face="plain", color="black", size=12, angle=0),
axis.text.y = element_text(face="plain", color="black", size=12, angle=0),
axis.title.x = element_text(size=14,face="plain", margin = margin(r=0,10,0,0)),
axis.title.y = element_text(size=14,face="plain", margin = margin(r=10,0,0,0)),
axis.ticks = element_line(linewidth = 1),
axis.ticks.length = unit(.3, "cm"))
colpal=c('#003d6cff','#0c7accff','#5a9c97ff')
colpal2=brewer.pal(7, 'Dark2')
colpal3=brewer.pal(10, 'BrBG')
colpal4=c("grey50","grey65")
colpal5=c("#8C2D04","#CC4C02", "#EC7014", "#FE9929", "#FEC44F","#A8DDB5","#A8DDB5", "#7BCCC4", "#4EB3D3", "#2B8CBE", "#08589E")
colpal6=c("#8C2D04","#CC4C02", "#EC7014", "#FE9929", "#FEC44F","#A8DDB5", "#7BCCC4", "#4EB3D3", "#2B8CBE", "#08589E")
Meta_col=c('#5a9c97ff','#0c7accff','#003d6cff','black')Global models
We addressed the question if increasing sexual selection correlated with an increasingly male-biased SSD. For this we ran a global model including an observation-level index and the study identifier as random termson correlation coefficients that were positive if increasing sexual selection correlated with an increasingly male-biased SSD, but negative if increasing sexual selection correlated with an increasingly female-biased SSD. First, we ran a global model including the phylogeny:
Model_REML_Null = rma.mv(r ~ 1, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_Null)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-23.7816 47.5632 55.5632 66.7464 55.9080
Variance Components:
estim sqrt nlvls fixed factor R
sigma^2.1 0.0205 0.1431 73 no Study_ID no
sigma^2.2 0.0484 0.2199 122 no Index no
sigma^2.3 0.0130 0.1140 11 no Class yes
Test for Heterogeneity:
Q(df = 121) = 1559.2739, p-val < .0001
Model Results:
estimate se zval pval ci.lb ci.ub
0.2498 0.0733 3.4095 0.0007 0.1062 0.3934 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Second, we ran a global model without the phylogeny:
Model_cREML_Null = rma.mv(r ~ 1, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_Null)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-25.1523 50.3047 56.3047 64.6921 56.5098
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0316 0.1777 73 no Study_ID
sigma^2.2 0.0460 0.2144 122 no Index
Test for Heterogeneity:
Q(df = 121) = 1559.2739, p-val < .0001
Model Results:
estimate se zval pval ci.lb ci.ub
0.2670 0.0321 8.3255 <.0001 0.2042 0.3299 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Moderator tests for phylogenetic models
Next, we ran a series of models that test the effect of different moderators. Again we started with models including the phylogeny.
Sexual selection episode
The first model explores the effect of the different episodes of sexual selection (i.e. pre-copulatory, post-copulatory or both):
MetaData$SexSel_Episode=relevel(MetaData$SexSel_Episode,c("pre-copulatory"))
Model_REML_by_SexSelEpisode = rma.mv(r ~ factor(SexSel_Episode), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SexSelEpisode)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-8.4861 16.9722 28.9722 45.6469 29.7222
Variance Components:
estim sqrt nlvls fixed factor R
sigma^2.1 0.0167 0.1293 73 no Study_ID no
sigma^2.2 0.0338 0.1839 122 no Index no
sigma^2.3 0.0125 0.1119 11 no Class yes
Test for Residual Heterogeneity:
QE(df = 119) = 1337.5244, p-val < .0001
Test of Moderators (coefficients 2:3):
QM(df = 2) = 35.8143, p-val < .0001
Model Results:
estimate se zval pval
intrcpt 0.2494 0.0755 3.3009 0.0010
factor(SexSel_Episode)post-copulatory -0.3476 0.0816 -4.2614 <.0001
factor(SexSel_Episode)both 0.1415 0.0531 2.6632 0.0077
ci.lb ci.ub
intrcpt 0.1013 0.3974 ***
factor(SexSel_Episode)post-copulatory -0.5075 -0.1877 ***
factor(SexSel_Episode)both 0.0374 0.2456 **
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We then re-leveled the model for post-hoc comparisons:
MetaData$SexSel_Episode=relevel(MetaData$SexSel_Episode,c("post-copulatory"))
Model_REML_by_SexSelEpisode2 = rma.mv(r ~ factor(SexSel_Episode), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SexSelEpisode2)
MetaData$SexSel_Episode=relevel(MetaData$SexSel_Episode,c("both"))
Model_REML_by_SexSelEpisode3 = rma.mv(r ~ factor(SexSel_Episode), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SexSelEpisode3)Finally, we computed FDR corrected p-values:
tab1=as.data.frame(round(p.adjust(c(.0001, 0.3004, .0001), method = 'fdr'),digit=3),row.names=cbind("Pre-copulatory","Post-copulatory","Both"))
colnames(tab1)<-cbind('P-value')
tab1 P-value
Pre-copulatory 0.0
Post-copulatory 0.3
Both 0.0Plot sexual selection mode (Figure 3)
Here we plot the sexual selection mode moderator:
MetaData$SexSel_Episode=factor(MetaData$SexSel_Episode, levels = c("both","post-copulatory" ,"pre-copulatory"))
ggplot(MetaData, aes(x=SexSel_Episode, y=r, fill = SexSel_Episode, colour = SexSel_Episode)) +
geom_hline(yintercept=0, linetype="longdash", color = "black", linewidth=1)+
geom_flat_violin(position = position_nudge(x = 0.25, y = 0),adjust =1, trim = F,alpha=0.6)+
geom_point(position = position_jitter(width = .1), size = 2.5,alpha=0.6,stroke=0,shape=19)+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelEpisode$ci.lb[1], x=3.25, xend= 3.25, yend= Model_REML_by_SexSelEpisode$ci.ub[1]), alpha=1,linewidth=1,color='black')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelEpisode2$ci.lb[1], x=2.25, xend= 2.25, yend= Model_REML_by_SexSelEpisode2$ci.ub[1]), alpha=1,linewidth=1,color='black')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelEpisode3$ci.lb[1], x=1.25, xend= 1.25, yend= Model_REML_by_SexSelEpisode3$ci.ub[1]), alpha=1,linewidth=1,color='black')+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelEpisode$b[1,1], x=3.25), size = 3.5,alpha=1,stroke=1,shape=21,fill='grey50',color='black')+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelEpisode2$b[1,1], x=2.25), size = 3.5,alpha=1,stroke=1,shape=21,fill='white',color='black')+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelEpisode3$b[1,1], x=1.25), size = 3.5,alpha=1,stroke=1,shape=21,fill='black',color='black')+
ylab(expression(paste("Effect size (", italic("r"),')')))+xlab('Sexual selection episode')+coord_flip()+guides(fill = FALSE, colour = FALSE) +
scale_color_manual(values =colpal)+
scale_fill_manual(values =colpal)+
scale_x_discrete(labels=c("Pre- and \n post-copulatory","Post-copulatory" ,"Pre-copulatory"),expand=c(.1,0))+
annotate("text", x=1, y=1.2, label= "n = 48",size=4.5) +
annotate("text", x=2, y=1.2, label= "n = 19",size=4.5) +
annotate("text", x=3, y=1.2, label= "n = 55",size=4.5) + theme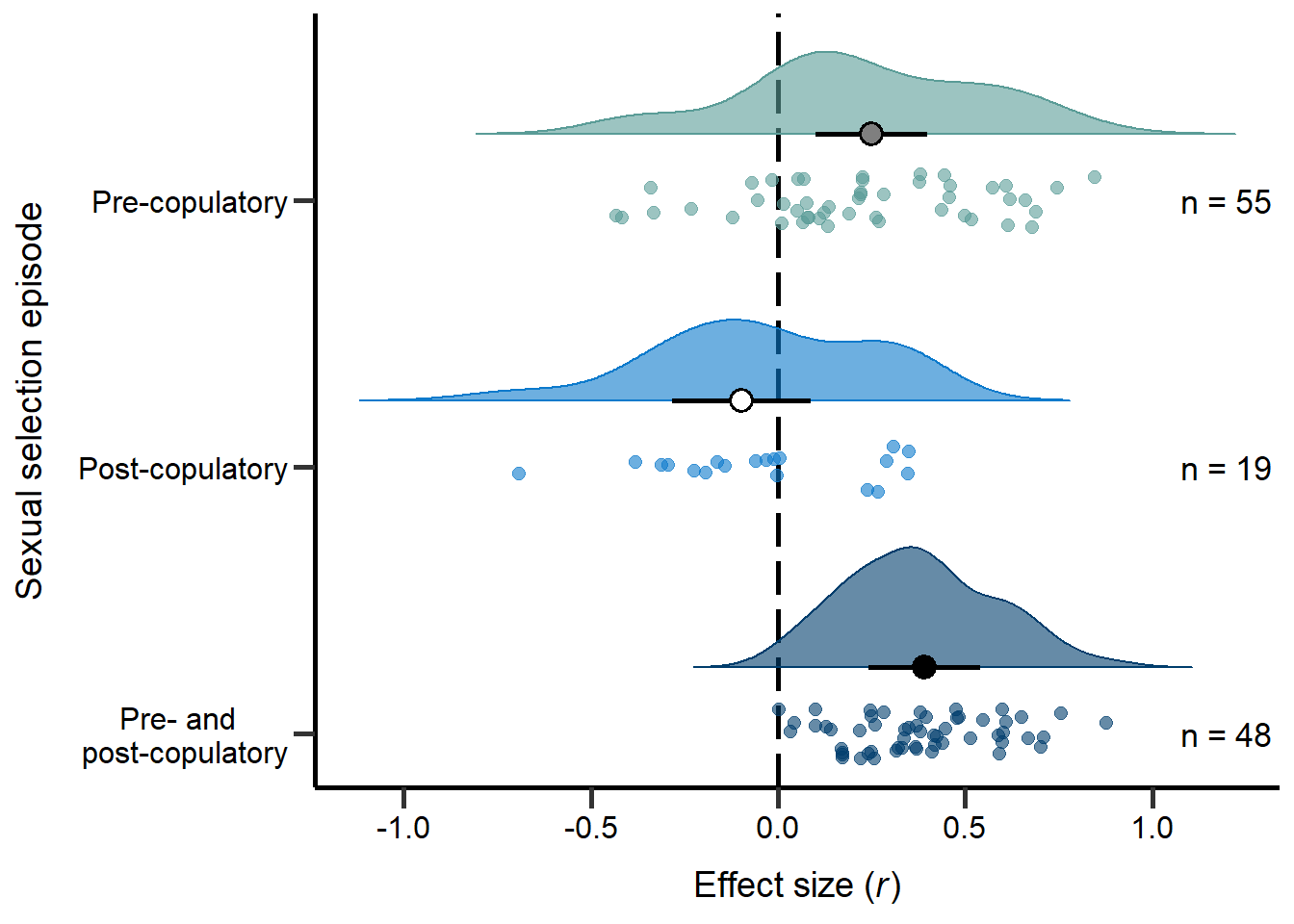
Figure 3: Raincloud plot of correlation coefficients between SSD and the modes of sexual selection proxies (i.e. pre-copulatory, post-copulatory or both) including sample sizes and estimates with 95%CI from phylogenetic model.
| Version | Author | Date |
|---|---|---|
| 8edd942 | LennartWinkler | 2023-05-01 |
Sexual selection category
Next we explored the effect of the sexual selection category (i.e. density, mating system, operational sex ratio (OSR), post-mating competition, pre-mating competition, trait-based, other):
MetaData$SexSel_Category=as.factor(MetaData$SexSel_Category)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Postmating competition"))
Model_REML_by_SexSelCat = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SexSelCat)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
5.8584 -11.7168 8.2832 35.7326 10.3986
Variance Components:
estim sqrt nlvls fixed factor R
sigma^2.1 0.0209 0.1446 73 no Study_ID no
sigma^2.2 0.0181 0.1345 122 no Index no
sigma^2.3 0.0123 0.1109 11 no Class yes
Test for Residual Heterogeneity:
QE(df = 115) = 916.8832, p-val < .0001
Test of Moderators (coefficients 2:7):
QM(df = 6) = 83.7711, p-val < .0001
Model Results:
estimate se zval pval
intrcpt -0.1218 0.0915 -1.3305 0.1834
factor(SexSel_Category)Density 0.3310 0.1209 2.7376 0.0062
factor(SexSel_Category)Mating system 0.4737 0.0793 5.9704 <.0001
factor(SexSel_Category)OSR 0.7397 0.1285 5.7572 <.0001
factor(SexSel_Category)Other 0.4501 0.0933 4.8255 <.0001
factor(SexSel_Category)Premating competition 0.4601 0.0819 5.6138 <.0001
factor(SexSel_Category)Trait-based 0.1059 0.0891 1.1887 0.2346
ci.lb ci.ub
intrcpt -0.3012 0.0576
factor(SexSel_Category)Density 0.0940 0.5679 **
factor(SexSel_Category)Mating system 0.3182 0.6292 ***
factor(SexSel_Category)OSR 0.4879 0.9915 ***
factor(SexSel_Category)Other 0.2673 0.6329 ***
factor(SexSel_Category)Premating competition 0.2994 0.6207 ***
factor(SexSel_Category)Trait-based -0.0687 0.2805
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We then re-leveled the model for post-hoc comparisons:
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Trait-based"))
Model_REML_by_SexSelCat2 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SexSelCat2)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Density"))
Model_REML_by_SexSelCat3 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SexSelCat3)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Premating competition"))
Model_REML_by_SexSelCat4 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SexSelCat4)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Mating system"))
Model_REML_by_SexSelCat5 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SexSelCat5)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("OSR"))
Model_REML_by_SexSelCat6 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SexSelCat6)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Other"))
Model_REML_by_SexSelCat7 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SexSelCat7)Finally, we computed FDR corrected p-values:
tab2=as.data.frame(round(p.adjust(c(0.1834, 0.8561, 0.0826, .0001, .0001, .0001, 0.0002), method = 'fdr'),digit=3),row.names=cbind("Postmating competition","Trait-based","Density",'Premating competition',"Mating system","OSR","Other"))
colnames(tab2)<-cbind('P-value')
tab2 P-value
Postmating competition 0.214
Trait-based 0.856
Density 0.116
Premating competition 0.000
Mating system 0.000
OSR 0.000
Other 0.000Plot sexual selection category (Figure S1)
Here we plot the sexual selection category moderator:
MetaData$SexSel_Category=factor(MetaData$SexSel_Category, levels = rev(c( "Postmating competition","Trait-based" ,"Density","Premating competition" ,"Mating system" , "OSR", "Other")))
ggplot(MetaData, aes(x=SexSel_Category, y=r, fill = SexSel_Category, colour = SexSel_Category)) +
geom_hline(yintercept=0, linetype="longdash", color = "black", linewidth=1)+
geom_flat_violin(position = position_nudge(x = 0.25, y = 0),adjust =1, trim = F,alpha=0.6)+
geom_point(position = position_jitter(width = .1), size = 2.5,alpha=0.6,stroke=0,shape=19)+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat$b[1,1], x=7.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat2$b[1,1], x=6.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat3$b[1,1], x=5.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat4$b[1,1], x=4.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat5$b[1,1], x=3.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat6$b[1,1], x=2.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat7$b[1,1], x=1.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat$ci.lb[1], x=7.25, xend= 7.25, yend= Model_REML_by_SexSelCat$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat2$ci.lb[1], x=6.25, xend= 6.25, yend= Model_REML_by_SexSelCat2$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat3$ci.lb[1], x=5.25, xend= 5.25, yend= Model_REML_by_SexSelCat3$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat4$ci.lb[1], x=4.25, xend= 4.25, yend= Model_REML_by_SexSelCat4$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat5$ci.lb[1], x=3.25, xend= 3.25, yend= Model_REML_by_SexSelCat5$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat6$ci.lb[1], x=2.25, xend= 2.25, yend= Model_REML_by_SexSelCat6$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SexSelCat7$ci.lb[1], x=1.25, xend= 1.25, yend= Model_REML_by_SexSelCat7$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
ylab(expression(paste("Effect size (", italic("r"),')')))+xlab('Sexual selection category')+coord_flip()+guides(fill = FALSE, colour = FALSE) +
scale_color_manual(values =colpal2)+
scale_fill_manual(values =colpal2)+
scale_x_discrete(labels=rev(c( "Post-mating\ncompetition","Trait-based" ,"Population\ndensity","Pre-mating\ncompetition" ,"Mating\nsystem" , "Sex ratio", "Other")),expand=c(.1,0))+
annotate("text", x=7, y=1.2, label= "n = 19",size=4.5) +
annotate("text", x=6, y=1.2, label= "n = 17",size=4.5) +
annotate("text", x=5, y=1.2, label= "n = 5",size=4.5) +
annotate("text", x=4, y=1.2, label= "n = 22",size=4.5) +
annotate("text", x=3, y=1.2, label= "n = 36",size=4.5) +
annotate("text", x=2, y=1.2, label= "n = 6",size=4.5) +
annotate("text", x=1, y=1.2, label= "n = 17",size=4.5) + theme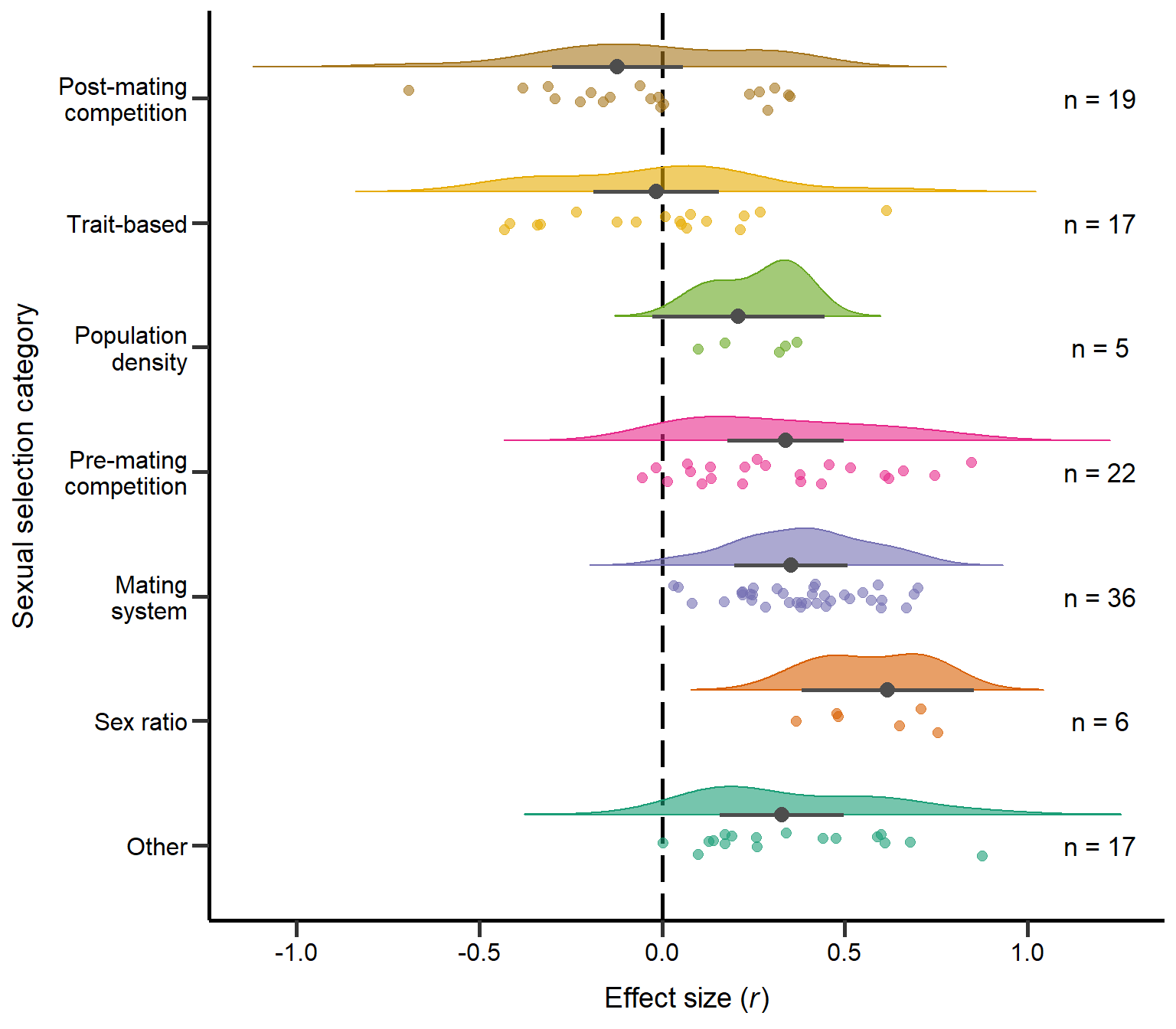
Figure S1: Raincloud plot of correlation coefficients between SSD and different sexual selection categories including sample sizes and estimates with 95%CI from phylogenetic model.
| Version | Author | Date |
|---|---|---|
| 8edd942 | LennartWinkler | 2023-05-01 |
Type of SSD measure
Next we explored the effect of the type of SSD measure (i.e. body mass or size):
MetaData$SSD_Proxy=as.factor(MetaData$SSD_Proxy)
MetaData$SSD_Proxy=relevel(MetaData$SSD_Proxy,c("Body mass"))
Model_REML_by_SSDMeasure = rma.mv(r ~ SSD_Proxy, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SSDMeasure)
Multivariate Meta-Analysis Model (k = 120; method: REML)
logLik Deviance AIC BIC AICc
-23.6341 47.2682 57.2682 71.1216 57.8039
Variance Components:
estim sqrt nlvls fixed factor R
sigma^2.1 0.0235 0.1534 72 no Study_ID no
sigma^2.2 0.0485 0.2203 120 no Index no
sigma^2.3 0.0047 0.0688 11 no Class yes
Test for Residual Heterogeneity:
QE(df = 118) = 1511.0606, p-val < .0001
Test of Moderators (coefficient 2):
QM(df = 1) = 2.3553, p-val = 0.1249
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.3292 0.0715 4.6056 <.0001 0.1891 0.4693 ***
SSD_ProxyBody size -0.1015 0.0662 -1.5347 0.1249 -0.2312 0.0281
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We then re-leveled the model for post-hoc comparisons:
MetaData$SSD_Proxy=relevel(MetaData$SSD_Proxy,c("Body size"))
Model_REML_by_SSDMeasure2 = rma.mv(r ~ SSD_Proxy, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_SSDMeasure2)Finally, we computed FDR corrected p-values:
tab3=as.data.frame(round(p.adjust(c(.0001, .0001), method = 'fdr'),digit=3),row.names=cbind("Body mass","Body size"))
colnames(tab3)<-cbind('P-value')
tab3 P-value
Body mass 0
Body size 0Plot Type of SSD measure (Figure S2A)
Here we plot the type of SSD measure moderator:
MetaData_NAProxy=MetaData[!is.na(MetaData$SSD_Proxy),]
MetaData_NAProxy$SSD_Proxy=as.factor(MetaData_NAProxy$SSD_Proxy)
MetaData_NAProxy$SSD_Proxy=relevel(MetaData_NAProxy$SSD_Proxy,c("Body size"))
ggplot(MetaData_NAProxy, aes(x=SSD_Proxy, y=r, fill = SSD_Proxy, colour = SSD_Proxy)) +
geom_hline(yintercept=0, linetype="longdash", color = "black", linewidth=1)+
geom_flat_violin(position = position_nudge(x = 0.25, y = 0),adjust =1, trim = F,alpha=0.6)+
geom_point(position = position_jitter(width = .1), size = 2.5,alpha=0.6,stroke=0,shape=19)+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SSDMeasure$b[1,1], x=2.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_SSDMeasure2$b[1,1], x=1.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SSDMeasure$ci.lb[1], x=2.25, xend= 2.25, yend= Model_REML_by_SSDMeasure$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_SSDMeasure2$ci.lb[1], x=1.25, xend= 1.25, yend= Model_REML_by_SSDMeasure2$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
ylab(expression(paste("Effect size (", italic("r"),')')))+xlab('SSD proxy')+coord_flip()+guides(fill = FALSE, colour = FALSE) +
scale_color_manual(values =colpal4)+
scale_fill_manual(values =colpal4)+
scale_x_discrete(labels=rev(c("Body \nmass ","Body \n size ")),expand=c(.15,0))+
annotate("text", x=2, y=1.1, label= "n = 57",size=3.5) +
annotate("text", x=1, y=1.1, label= "n = 63",size=3.5) + theme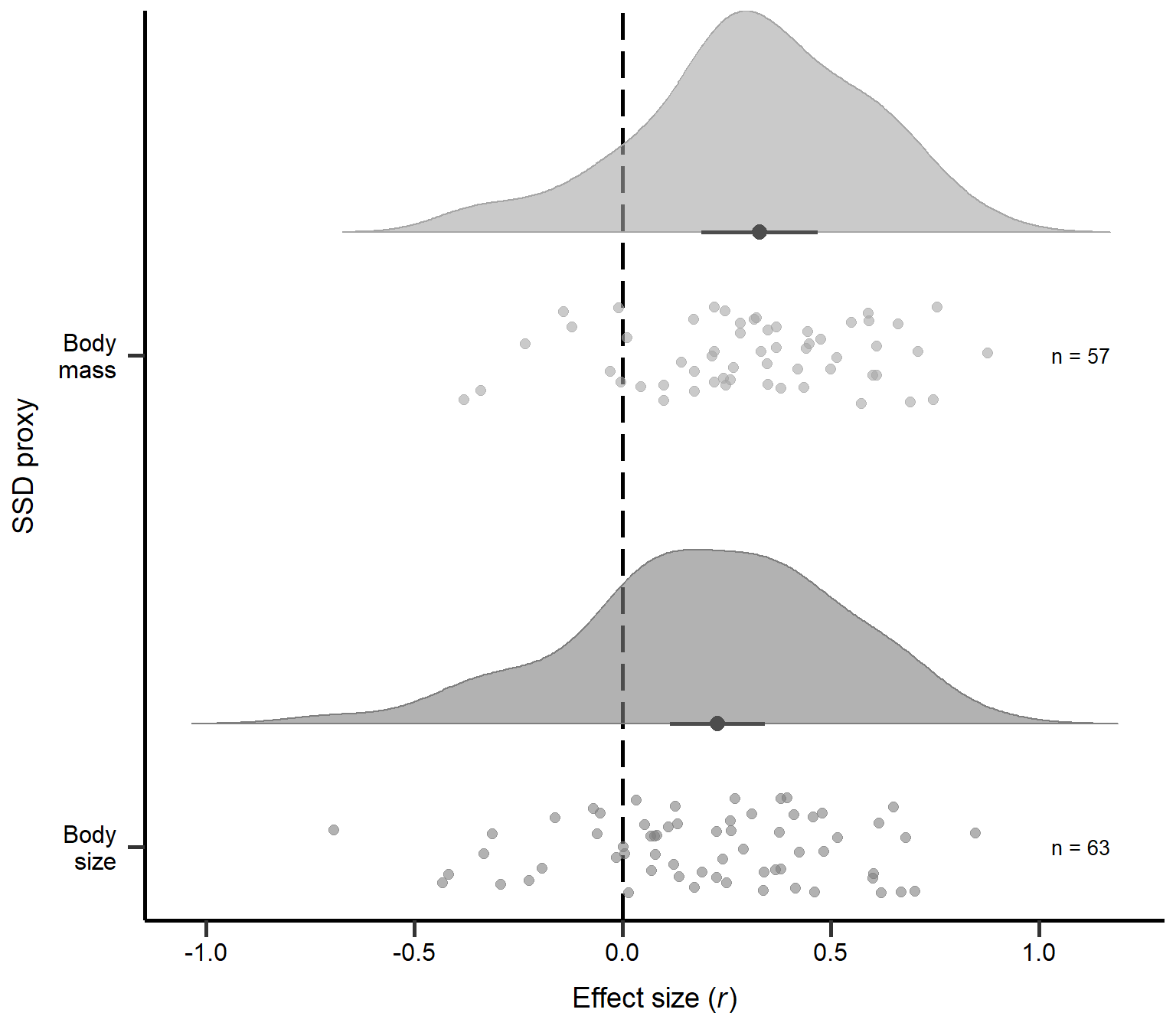
Figure S2A: Raincloud plot of correlation coefficients for different types of SSD measures (i.e. body mass or size) including sample sizes and estimates with 95% CI from phylogenetic model.
SSD measure controlled for body size?
Next we explored the effect if the primary study controlled the SSD for body size (i.e. uncontrolled or controlled):
MetaData$BodySizeControlled=as.factor(MetaData$BodySizeControlled)
MetaData$BodySizeControlled=relevel(MetaData$BodySizeControlled,c("No"))
Model_REML_by_BodySizeCont = rma.mv(r ~ BodySizeControlled, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_BodySizeCont)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-23.6694 47.3387 57.3387 71.2762 57.8650
Variance Components:
estim sqrt nlvls fixed factor R
sigma^2.1 0.0210 0.1449 73 no Study_ID no
sigma^2.2 0.0483 0.2199 122 no Index no
sigma^2.3 0.0120 0.1094 11 no Class yes
Test for Residual Heterogeneity:
QE(df = 120) = 1555.0586, p-val < .0001
Test of Moderators (coefficient 2):
QM(df = 1) = 0.6071, p-val = 0.4359
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.2629 0.0732 3.5909 0.0003 0.1194 0.4063 ***
BodySizeControlledYes -0.0674 0.0865 -0.7792 0.4359 -0.2368 0.1021
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We then re-leveled the model for post-hoc comparisons:
MetaData$BodySizeControlled=relevel(MetaData$BodySizeControlled,c("Yes"))
Model_REML_by_BodySizeCont2 = rma.mv(r ~ BodySizeControlled, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_BodySizeCont2)Finally, we computed FDR corrected p-values:
tab4=as.data.frame(round(p.adjust(c(.0001, 0.0517), method = 'fdr'),digit=3),row.names=cbind("uncontrolled","controlled"))
colnames(tab4)<-cbind('P-value')
tab4 P-value
uncontrolled 0.000
controlled 0.052Plot: SSD measure controlled for body size? (Figure S2B)
Here we plot effect sizes if type of SSD measure controlled for body size:
MetaData$BodySizeControlled=as.factor(MetaData$BodySizeControlled)
MetaData$BodySizeControlled=relevel(MetaData$BodySizeControlled,c("Yes"))
ggplot(MetaData, aes(x=BodySizeControlled, y=r, fill = BodySizeControlled, colour = BodySizeControlled)) +
geom_hline(yintercept=0, linetype="longdash", color = "black", linewidth=1)+
geom_flat_violin(position = position_nudge(x = 0.25, y = 0),adjust =1, trim = F,alpha=0.6)+
geom_point(position = position_jitter(width = .1), size = 2.5,alpha=0.6,stroke=0,shape=19)+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_BodySizeCont$b[1,1], x=2.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_point(inherit.aes = F,mapping = aes(y=Model_REML_by_BodySizeCont2$b[1,1], x=1.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_BodySizeCont$ci.lb[1], x=2.25, xend= 2.25, yend= Model_REML_by_BodySizeCont$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_REML_by_BodySizeCont2$ci.lb[1], x=1.25, xend= 1.25, yend= Model_REML_by_BodySizeCont2$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
ylab(expression(paste("Effect size (", italic("r"),')')))+xlab('Controlling SSD \n for body size or mass')+coord_flip()+guides(fill = FALSE, colour = FALSE) +
scale_color_manual(values =colpal4)+
scale_fill_manual(values =colpal4)+
scale_x_discrete(labels=(c("Controlled","Uncontrolled")),expand=c(.15,0))+
annotate("text", x=2, y=1.1, label= "n = 105",size=3.5) +
annotate("text", x=1, y=1.1, label= "n = 17",size=3.5) + theme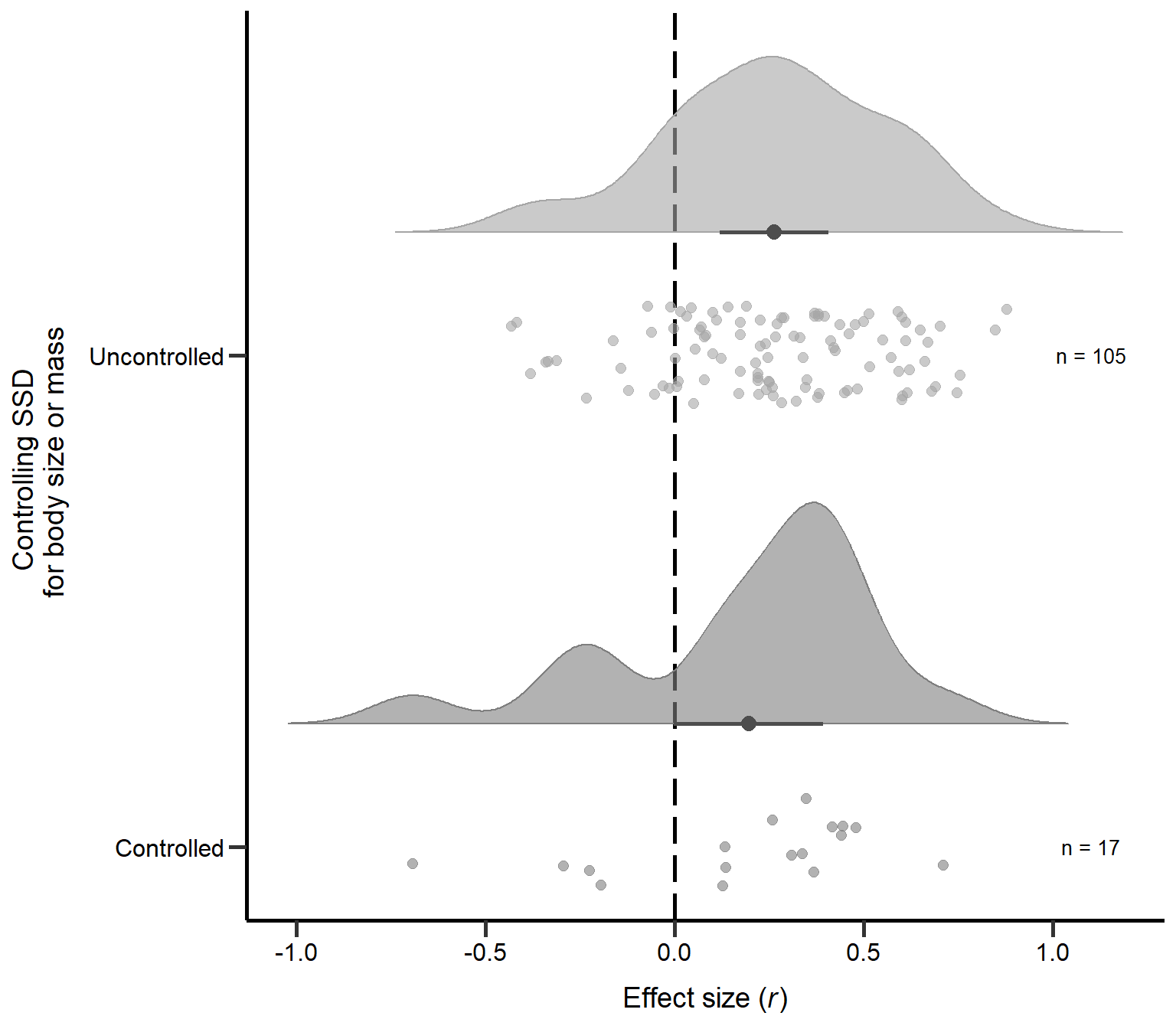
Figure S2B: Raincloud plot of correlation coefficients for primary studies controlling SSD for body size or mass (uncontrolled or controlled) including sample sizes and estimates with 95% CI from phylogenetic model.
Phylogentic vs non-phylogenetic studies
Next we explored the effect of studies controlling for phylogeny vs non-phylogenetic studies:
MetaData$PhyloControlled=as.factor(MetaData$PhyloControlled)
MetaData$PhyloControlled=relevel(MetaData$PhyloControlled,c("No"))
Model_cREML_by_Phylo = rma.mv(r ~ PhyloControlled, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_cREML_by_Phylo)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-20.7468 41.4937 51.4937 65.4311 52.0200
Variance Components:
estim sqrt nlvls fixed factor R
sigma^2.1 0.0134 0.1159 73 no Study_ID no
sigma^2.2 0.0503 0.2244 122 no Index no
sigma^2.3 0.0117 0.1083 11 no Class yes
Test for Residual Heterogeneity:
QE(df = 120) = 1350.6816, p-val < .0001
Test of Moderators (coefficient 2):
QM(df = 1) = 6.8605, p-val = 0.0088
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.4194 0.0959 4.3714 <.0001 0.2314 0.6075 ***
PhyloControlledYes -0.2067 0.0789 -2.6192 0.0088 -0.3614 -0.0520 **
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We then re-leveled the model for post-hoc comparisons:
MetaData$PhyloControlled=relevel(MetaData$PhyloControlled,c("Yes"))
Model_cREML_by_Phylo2 = rma.mv(r ~ PhyloControlled, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_cREML_by_Phylo2)Finally, we computed FDR corrected p-values:
tab5=as.data.frame(round(p.adjust(c(0.0001, 0.0027), method = 'fdr'),digit=3),row.names=cbind("Non-phylogenetic","With phylogeny"))
colnames(tab5)<-cbind('P-value')
tab5 P-value
Non-phylogenetic 0.000
With phylogeny 0.003Plot Phylogentic vs non-phylogenetic studies (Figure S2C)
Here we plot the type of SSD measure moderator:
MetaData$PhyloControlled=as.factor(MetaData$PhyloControlled)
MetaData$PhyloControlled=relevel(MetaData$PhyloControlled,c("No"))
ggplot(MetaData, aes(x=PhyloControlled, y=r, fill = PhyloControlled, colour = PhyloControlled)) +
geom_hline(yintercept=0, linetype="longdash", color = "black", linewidth=1)+
geom_flat_violin(position = position_nudge(x = 0.25, y = 0),adjust =1, trim = F,alpha=0.6)+
geom_point(position = position_jitter(width = .1), size = 2.5,alpha=0.6,stroke=0,shape=19)+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Phylo2$b[1,1], x=2.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Phylo$b[1,1], x=1.25), size = 3.5,alpha=1,stroke=0,shape=19,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Phylo2$ci.lb[1], x=2.25, xend= 2.25, yend= Model_cREML_by_Phylo2$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Phylo$ci.lb[1], x=1.25, xend= 1.25, yend= Model_cREML_by_Phylo$ci.ub[1]), alpha=1,linewidth=1,color='grey30')+
ylab(expression(paste("Effect size (", italic("r"),')')))+xlab('Controlling for phylogeny')+coord_flip()+guides(fill = FALSE, colour = FALSE) +
scale_color_manual(values =colpal4)+
scale_fill_manual(values =colpal4)+
scale_x_discrete(labels=(c("Phylogenetically \n uncorrected ","Phylogenetically \n corrected ")),expand=c(.15,0))+
annotate("text", x=1, y=1.1, label= "n = 20",size=3.5) +
annotate("text", x=2, y=1.1, label= "n = 102",size=3.5) + theme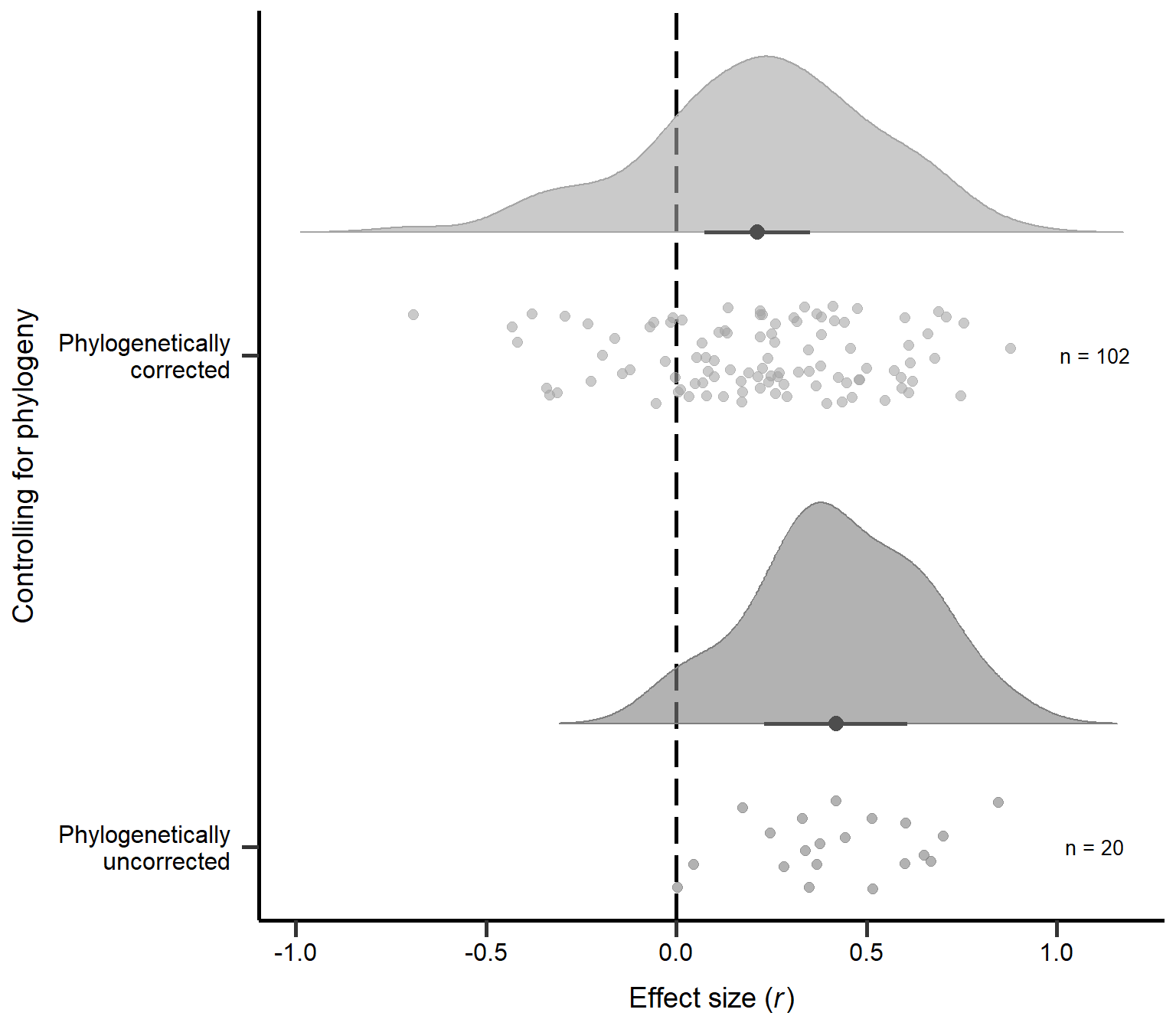
Figure S2C: Raincloud plot of correlation coefficients for non-phylogenetic and phylogenetic analyses in primary studies including sample sizes and estimates with 95% CI from phylogenetic model (Table 4).
Percentage of species with female-biased SSD
There was variation in the primary studies regarding the typical SSD in the studied taxa (i.e. some studies focused on taxa with more male-biased SSD, while others on taxa with more female-biased SSD). Still, there was no significant relationship between the percentage of species with a female-biased SSD and effect sizes.
MetaData_SSDbias=MetaData
MetaData_SSDbias=MetaData_SSDbias[!is.na(MetaData_SSDbias$SSD_SexBias_in_perc_F),]
Model_REML_SSbias = rma.mv(r ~ SSD_SexBias_in_perc_F, V=Var_r, data = MetaData_SSDbias, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_SSbias)
Multivariate Meta-Analysis Model (k = 104; method: REML)
logLik Deviance AIC BIC AICc
-20.8873 41.7746 51.7746 64.8995 52.3996
Variance Components:
estim sqrt nlvls fixed factor R
sigma^2.1 0.0280 0.1673 64 no Study_ID no
sigma^2.2 0.0471 0.2170 104 no Index no
sigma^2.3 0.0051 0.0716 11 no Class yes
Test for Residual Heterogeneity:
QE(df = 102) = 1443.6764, p-val < .0001
Test of Moderators (coefficient 2):
QM(df = 1) = 3.2778, p-val = 0.0702
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.3765 0.0807 4.6657 <.0001 0.2183 0.5346 ***
SSD_SexBias_in_perc_F -0.0021 0.0012 -1.8105 0.0702 -0.0044 0.0002 .
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Figure S3A
model_p1 <- predict(Model_REML_SSbias) # Extract model predictions
ggplot(MetaData_SSDbias, aes(x=as.numeric(SSD_SexBias_in_perc_F), y=r)) +
theme(panel.grid.major = element_blank(), panel.grid.minor = element_blank(),panel.background = element_blank(), axis.line = element_line(colour = "black"))+
theme(axis.text=element_text(size=13),
axis.title=element_text(size=14))+ theme(legend.position="none")+
geom_point(shape=16, size = 3,alpha=0.5)+xlab('% of species with \n female-biased SSD')+ylab(expression(paste("Effect size (", italic("r"),')')))+
geom_hline(yintercept=0, linetype="dashed", color = "black", linewidth=1)+
geom_line( aes(y = model_p1$pred), size = 1)+
geom_ribbon( aes(ymin = model_p1$ci.lb, ymax = model_p1$ci.ub, color = 'black',linetype=NA), alpha = .15,show.legend = F, outline.type = "both") +
theme
Figure S3A: Scatter plot of the sPercentage of species with a female-biased SSD. Dashed line marks a correlation coefficient of zero, black line represents predictions from REML model (controlling for phylogeny) and grey area represents 95% CI on model predictions.
Test for publication bias
To test for publication bias, we transformed r into z scores and ran multilevel mixed-effects models (restricted maximum likelihood) with z as the predictor and its standard error as the response with study ID and an observation level random effect. Models were weight by the mean standard error of z across all studies. While the variance in r depends on the effect size and the sample size, the variance in z is only dependent on the sample size. Hence, if z values correlate with the variance in z, this indicates that small studies were only published, if the effect was large, suggesting publication bias.
Model_REML_PublBias = rma.mv(z ~ SE_z, V=rep((mean(SE_z)*mean(SE_z))*N,length(SE_z)), data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML",control=list(rel.tol=1e-8))
summary(Model_REML_PublBias)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-145.5101 291.0202 301.0202 314.9577 301.5465
Variance Components:
estim sqrt nlvls fixed factor R
sigma^2.1 0.0000 0.0000 73 no Study_ID no
sigma^2.2 0.0000 0.0000 122 no Index no
sigma^2.3 0.0000 0.0002 11 no Class yes
Test for Residual Heterogeneity:
QE(df = 120) = 19.2579, p-val = 1.0000
Test of Moderators (coefficient 2):
QM(df = 1) = 1.5648, p-val = 0.2110
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.1715 0.1977 0.8678 0.3855 -0.2159 0.5590
SE_z 0.8181 0.6540 1.2509 0.2110 -0.4637 2.0999
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Figure S3B
# Extract model predictions
model_p2 <- predict(Model_REML_PublBias)
ggplot(MetaData, aes(x=SE_z, y=z)) +
theme(panel.grid.major = element_blank(), panel.grid.minor = element_blank(),panel.background = element_blank(), axis.line = element_line(colour = "black"))+
theme(axis.text=element_text(size=13),
axis.title=element_text(size=14))+ theme(legend.position="none")+ylab(expression(paste("Effect size (", italic("z"),')')))+
geom_point(shape=16, size = 3,alpha=0.5)+xlab('Standard error')+
geom_hline(yintercept=0, linetype="dashed", color = "black", linewidth=1)+
geom_line( aes(y = model_p2$pred), size = 1)+
geom_ribbon( aes(ymin = model_p2$ci.lb, ymax = model_p2$ci.ub, color = 'black',linetype=NA), alpha = .15,show.legend = F, outline.type = "both") +
theme
Figure S3B: Scatter plot of the standard error in z scores of each study against the z score of each primary study (transformed correlation coefficients). Dashed line marks a correlation coefficient of zero, black line represents predictions from REML model (controlling for phylogeny) and grey area represents 95% CI on model predictions.
Publication year
Next we explored the effect of the publication year of each study:
Model_REML_by_Year = rma.mv(r ~ Year, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
summary(Model_REML_by_Year)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-21.5680 43.1360 53.1360 67.0735 53.6623
Variance Components:
estim sqrt nlvls fixed factor R
sigma^2.1 0.0177 0.1332 73 no Study_ID no
sigma^2.2 0.0476 0.2183 122 no Index no
sigma^2.3 0.0118 0.1085 11 no Class yes
Test for Residual Heterogeneity:
QE(df = 120) = 1275.7703, p-val < .0001
Test of Moderators (coefficient 2):
QM(df = 1) = 5.0277, p-val = 0.0249
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 11.7151 5.1141 2.2907 0.0220 1.6917 21.7385 *
Year -0.0057 0.0025 -2.2423 0.0249 -0.0107 -0.0007 *
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Plot publication year (Figure S3C)
Here we plot the publication year:
# Extract model predictions
Model_REML_by_Year = rma.mv(r ~ Year, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index, ~ 1 | Class), R = list(Class = forcedC_Moderators), method = "REML")
model_p4 <- predict(Model_REML_by_Year)
ggplot(MetaData, aes(x=as.numeric(Year), y=r)) +
theme(panel.grid.major = element_blank(), panel.grid.minor = element_blank(),panel.background = element_blank(), axis.line = element_line(colour = "black"))+
theme(axis.text=element_text(size=13),
axis.title=element_text(size=14))+ theme(legend.position="none")+
geom_point(shape=16, size = 3,alpha=0.5)+xlab('Publication year')+ylab(expression(paste("Effect size (", italic("r"),')')))+
geom_hline(yintercept=0, linetype="dashed", color = "black", linewidth=1)+
geom_line( aes(y = model_p4$pred), size = 1)+
geom_ribbon( aes(ymin = model_p4$ci.lb, ymax = model_p4$ci.ub, color = 'black',linetype=NA), alpha = .15,show.legend = F, outline.type = "both") +
theme
Figure S3C: Scatter plot of correlation coefficients against the publication year of each study. Dashed line marks a correlation coefficient of zero, black line represents predictions from REML model (controlling for phylogeny) and grey area represents 95% CI on model predictions.
Moderator tests for non-phylogenetic models
Here we ran all models without the phylogeny.
Sexual selection episode
The first model explores the effect of the sexual selection episode (i.e. pre-copulatory, post-copulatory or both):
MetaData$SexSel_Episode=relevel(MetaData$SexSel_Episode,c("pre-copulatory"))
Model_REML_by_cSexSelEpisode = rma.mv(r ~ factor(SexSel_Episode), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSexSelEpisode)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-10.3162 20.6324 30.6324 44.5280 31.1634
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0243 0.1559 73 no Study_ID
sigma^2.2 0.0337 0.1836 122 no Index
Test for Residual Heterogeneity:
QE(df = 119) = 1337.5244, p-val < .0001
Test of Moderators (coefficients 2:3):
QM(df = 2) = 34.5137, p-val < .0001
Model Results:
estimate se zval pval
intrcpt 0.2379 0.0407 5.8383 <.0001
factor(SexSel_Episode)both 0.1673 0.0543 3.0825 0.0021
factor(SexSel_Episode)post-copulatory -0.3150 0.0833 -3.7828 0.0002
ci.lb ci.ub
intrcpt 0.1580 0.3177 ***
factor(SexSel_Episode)both 0.0609 0.2737 **
factor(SexSel_Episode)post-copulatory -0.4783 -0.1518 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We then re-leveled the model for post-hoc comparisons:
MetaData$SexSel_Episode=relevel(MetaData$SexSel_Episode,c("post-copulatory"))
Model_REML_by_cSexSelEpisode2 = rma.mv(r ~ factor(SexSel_Episode), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSexSelEpisode2)
MetaData$SexSel_Episode=relevel(MetaData$SexSel_Episode,c("both"))
Model_REML_by_cSexSelEpisode3 = rma.mv(r ~ factor(SexSel_Episode), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSexSelEpisode3)Finally, we computed FDR corrected p-values:
tab1=as.data.frame(round(p.adjust(c(0.0001, 0.2970, .0001), method = 'fdr'),digit=3),row.names=cbind("Pre-copulatory","Post-copulatory","Both"))
colnames(tab1)<-cbind('P-value')
tab1 P-value
Pre-copulatory 0.000
Post-copulatory 0.297
Both 0.000Sexual selection category
Next we explored the effect of the sexual selection category (i.e. density, mating system, operational sex ratio (OSR), post-mating competition, pre-mating competition, trait-based, other):
MetaData$SexSel_Category=as.factor(MetaData$SexSel_Category)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Postmating competition"))
Model_REML_by_cSexSelCat = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSexSelCat)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
4.7833 -9.5666 8.4334 33.1378 10.1477
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0268 0.1636 73 no Study_ID
sigma^2.2 0.0181 0.1346 122 no Index
Test for Residual Heterogeneity:
QE(df = 115) = 916.8832, p-val < .0001
Test of Moderators (coefficients 2:7):
QM(df = 6) = 83.7704, p-val < .0001
Model Results:
estimate se zval pval
intrcpt -0.0721 0.0685 -1.0516 0.2930
factor(SexSel_Category)Other 0.4433 0.0942 4.7044 <.0001
factor(SexSel_Category)OSR 0.7141 0.1285 5.5581 <.0001
factor(SexSel_Category)Mating system 0.4713 0.0792 5.9503 <.0001
factor(SexSel_Category)Premating competition 0.4308 0.0837 5.1490 <.0001
factor(SexSel_Category)Density 0.3274 0.1226 2.6695 0.0076
factor(SexSel_Category)Trait-based 0.0776 0.0880 0.8815 0.3780
ci.lb ci.ub
intrcpt -0.2064 0.0622
factor(SexSel_Category)Other 0.2586 0.6280 ***
factor(SexSel_Category)OSR 0.4623 0.9659 ***
factor(SexSel_Category)Mating system 0.3161 0.6266 ***
factor(SexSel_Category)Premating competition 0.2668 0.5948 ***
factor(SexSel_Category)Density 0.0870 0.5678 **
factor(SexSel_Category)Trait-based -0.0949 0.2500
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We then re-leveled the model for post-hoc comparisons:
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Trait-based"))
Model_REML_by_cSexSelCat2 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSexSelCat2)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Density"))
Model_REML_by_cSexSelCat3 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSexSelCat3)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Premating competition"))
Model_REML_by_cSexSelCat4 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSexSelCat4)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Mating system"))
Model_REML_by_cSexSelCat5 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSexSelCat5)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("OSR"))
Model_REML_by_cSexSelCat6 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSexSelCat6)
MetaData$SexSel_Category=relevel(MetaData$SexSel_Category,c("Other"))
Model_REML_by_cSexSelCat7 = rma.mv(r ~ factor(SexSel_Category), V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSexSelCat7)Finally, we computed FDR corrected p-values:
tab2=as.data.frame(round(p.adjust(c(0.2930, 0.9215, 0.0124, .0001, .0001, .0001, .0001), method = 'fdr'),digit=3),row.names=cbind("Postmating competition","Trait-based","Density",'Premating competition',"Mating system","OSR","Other"))
colnames(tab2)<-cbind('P-value')
tab2 P-value
Postmating competition 0.342
Trait-based 0.921
Density 0.017
Premating competition 0.000
Mating system 0.000
OSR 0.000
Other 0.000Phylogenetic classes
Next we explored the effect of the phylogenetic classes:
MetaData$Class=relevel(MetaData$Class,c("Actinopterygii"))
Model_cREML_by_Class5 = rma.mv(r ~ Class, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Class5)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-19.8965 39.7930 65.7930 101.0169 69.5455
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0183 0.1352 73 no Study_ID
sigma^2.2 0.0512 0.2262 122 no Index
Test for Residual Heterogeneity:
QE(df = 111) = 1222.6024, p-val < .0001
Test of Moderators (coefficients 2:11):
QM(df = 10) = 16.3649, p-val = 0.0897
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.2199 0.1099 2.0012 0.0454 0.0045 0.4352 *
ClassAmphibia -0.1432 0.1483 -0.9659 0.3341 -0.4338 0.1474
ClassAnimalia 0.0060 0.2469 0.0244 0.9805 -0.4780 0.4900
ClassAves -0.0023 0.1223 -0.0187 0.9851 -0.2419 0.2373
ClassInsecta 0.1621 0.1586 1.0222 0.3067 -0.1487 0.4729
ClassMalacostraca -0.2177 0.3535 -0.6157 0.5381 -0.9106 0.4753
ClassMammalia 0.1666 0.1195 1.3939 0.1633 -0.0677 0.4009
ClassNematoda 0.2021 0.2646 0.7637 0.4450 -0.3165 0.7208
ClassPisces 0.0384 0.2897 0.1327 0.8944 -0.5293 0.6062
ClassReptilia -0.0980 0.1426 -0.6870 0.4921 -0.3775 0.1816
ClassTrematoda -0.1598 0.2298 -0.6954 0.4868 -0.6102 0.2906
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1MetaData$Class=relevel(MetaData$Class,c("Amphibia"))
Model_cREML_by_Class4 = rma.mv(r ~ Class, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Class4)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-19.8965 39.7930 65.7930 101.0169 69.5455
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0183 0.1352 73 no Study_ID
sigma^2.2 0.0512 0.2262 122 no Index
Test for Residual Heterogeneity:
QE(df = 111) = 1222.6024, p-val < .0001
Test of Moderators (coefficients 2:11):
QM(df = 10) = 16.3649, p-val = 0.0897
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.0766 0.0996 0.7697 0.4415 -0.1185 0.2718
ClassActinopterygii 0.1432 0.1483 0.9659 0.3341 -0.1474 0.4338
ClassAnimalia 0.1492 0.2425 0.6153 0.5383 -0.3261 0.6246
ClassAves 0.1409 0.1137 1.2394 0.2152 -0.0819 0.3638
ClassInsecta 0.3053 0.1524 2.0028 0.0452 0.0065 0.6041 *
ClassMalacostraca -0.0744 0.3505 -0.2124 0.8318 -0.7614 0.6125
ClassMammalia 0.3098 0.1112 2.7860 0.0053 0.0919 0.5278 **
ClassNematoda 0.3453 0.2605 1.3255 0.1850 -0.1653 0.8559
ClassPisces 0.1817 0.2859 0.6353 0.5252 -0.3788 0.7421
ClassReptilia 0.0452 0.1349 0.3353 0.7374 -0.2192 0.3096
ClassTrematoda -0.0166 0.2291 -0.0724 0.9423 -0.4656 0.4324
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1MetaData$Class=relevel(MetaData$Class,c("Animalia"))
Model_cREML_by_Class11 = rma.mv(r ~ Class, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Class11)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-19.8965 39.7930 65.7930 101.0169 69.5455
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0183 0.1352 73 no Study_ID
sigma^2.2 0.0512 0.2262 122 no Index
Test for Residual Heterogeneity:
QE(df = 111) = 1222.6024, p-val < .0001
Test of Moderators (coefficients 2:11):
QM(df = 10) = 16.3649, p-val = 0.0897
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.2259 0.2212 1.0214 0.3071 -0.2076 0.6594
ClassAmphibia -0.1492 0.2425 -0.6153 0.5383 -0.6246 0.3261
ClassActinopterygii -0.0060 0.2469 -0.0244 0.9805 -0.4900 0.4780
ClassAves -0.0083 0.2279 -0.0365 0.9709 -0.4549 0.4383
ClassInsecta 0.1561 0.2495 0.6256 0.5316 -0.3329 0.6450
ClassMalacostraca -0.2237 0.4023 -0.5560 0.5782 -1.0121 0.5648
ClassMammalia 0.1606 0.2266 0.7085 0.4786 -0.2836 0.6048
ClassNematoda 0.1961 0.3269 0.5998 0.5486 -0.4446 0.8368
ClassPisces 0.0324 0.3475 0.0933 0.9257 -0.6487 0.7135
ClassReptilia -0.1040 0.2392 -0.4349 0.6636 -0.5728 0.3647
ClassTrematoda -0.1658 0.3025 -0.5483 0.5835 -0.7586 0.4270
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1MetaData$Class=relevel(MetaData$Class,c("Aves"))
Model_cREML_by_Class = rma.mv(r ~ Class, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Class)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-19.8965 39.7930 65.7930 101.0169 69.5455
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0183 0.1352 73 no Study_ID
sigma^2.2 0.0512 0.2262 122 no Index
Test for Residual Heterogeneity:
QE(df = 111) = 1222.6024, p-val < .0001
Test of Moderators (coefficients 2:11):
QM(df = 10) = 16.3649, p-val = 0.0897
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.2176 0.0549 3.9626 <.0001 0.1100 0.3252 ***
ClassAnimalia 0.0083 0.2279 0.0365 0.9709 -0.4383 0.4549
ClassAmphibia -0.1409 0.1137 -1.2394 0.2152 -0.3638 0.0819
ClassActinopterygii 0.0023 0.1223 0.0187 0.9851 -0.2373 0.2419
ClassInsecta 0.1644 0.1276 1.2880 0.1977 -0.0858 0.4145
ClassMalacostraca -0.2154 0.3405 -0.6325 0.5270 -0.8827 0.4520
ClassMammalia 0.1689 0.0731 2.3113 0.0208 0.0257 0.3121 *
ClassNematoda 0.2044 0.2469 0.8277 0.4078 -0.2796 0.6883
ClassPisces 0.0407 0.2736 0.1489 0.8817 -0.4955 0.5770
ClassReptilia -0.0957 0.1056 -0.9064 0.3647 -0.3026 0.1112
ClassTrematoda -0.1575 0.2126 -0.7409 0.4588 -0.5742 0.2592
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1MetaData$Class=relevel(MetaData$Class,c("Insecta"))
Model_cREML_by_Class8 = rma.mv(r ~ Class, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Class8)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-19.8965 39.7930 65.7930 101.0169 69.5455
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0183 0.1352 73 no Study_ID
sigma^2.2 0.0512 0.2262 122 no Index
Test for Residual Heterogeneity:
QE(df = 111) = 1222.6024, p-val < .0001
Test of Moderators (coefficients 2:11):
QM(df = 10) = 16.3649, p-val = 0.0897
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.3819 0.1154 3.3090 0.0009 0.1557 0.6082 ***
ClassAves -0.1644 0.1276 -1.2880 0.1977 -0.4145 0.0858
ClassAnimalia -0.1561 0.2495 -0.6256 0.5316 -0.6450 0.3329
ClassAmphibia -0.3053 0.1524 -2.0028 0.0452 -0.6041 -0.0065 *
ClassActinopterygii -0.1621 0.1586 -1.0222 0.3067 -0.4729 0.1487
ClassMalacostraca -0.3797 0.3553 -1.0688 0.2852 -1.0761 0.3166
ClassMammalia 0.0045 0.1252 0.0361 0.9712 -0.2409 0.2500
ClassNematoda 0.0400 0.2670 0.1499 0.8808 -0.4833 0.5633
ClassPisces -0.1236 0.2918 -0.4237 0.6718 -0.6956 0.4484
ClassReptilia -0.2601 0.1470 -1.7694 0.0768 -0.5481 0.0280 .
ClassTrematoda -0.3219 0.2349 -1.3702 0.1706 -0.7823 0.1386
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1MetaData$Class=relevel(MetaData$Class,c("Malacostraca"))
Model_cREML_by_Class7 = rma.mv(r ~ Class, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Class7)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-19.8965 39.7930 65.7930 101.0169 69.5455
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0183 0.1352 73 no Study_ID
sigma^2.2 0.0512 0.2262 122 no Index
Test for Residual Heterogeneity:
QE(df = 111) = 1222.6024, p-val < .0001
Test of Moderators (coefficients 2:11):
QM(df = 10) = 16.3649, p-val = 0.0897
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.0022 0.3360 0.0065 0.9948 -0.6564 0.6608
ClassInsecta 0.3797 0.3553 1.0688 0.2852 -0.3166 1.0761
ClassAves 0.2154 0.3405 0.6325 0.5270 -0.4520 0.8827
ClassAnimalia 0.2237 0.4023 0.5560 0.5782 -0.5648 1.0121
ClassAmphibia 0.0744 0.3505 0.2124 0.8318 -0.6125 0.7614
ClassActinopterygii 0.2177 0.3535 0.6157 0.5381 -0.4753 0.9106
ClassMammalia 0.3843 0.3397 1.1313 0.2579 -0.2815 1.0500
ClassNematoda 0.4198 0.4134 1.0155 0.3099 -0.3904 1.2300
ClassPisces 0.2561 0.4298 0.5958 0.5513 -0.5864 1.0986
ClassReptilia 0.1197 0.3481 0.3437 0.7310 -0.5627 0.8020
ClassTrematoda 0.0578 0.3943 0.1467 0.8834 -0.7150 0.8307
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1MetaData$Class=relevel(MetaData$Class,c("Mammalia"))
Model_cREML_by_Class3 = rma.mv(r ~ Class, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Class3)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-19.8965 39.7930 65.7930 101.0169 69.5455
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0183 0.1352 73 no Study_ID
sigma^2.2 0.0512 0.2262 122 no Index
Test for Residual Heterogeneity:
QE(df = 111) = 1222.6024, p-val < .0001
Test of Moderators (coefficients 2:11):
QM(df = 10) = 16.3649, p-val = 0.0897
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.3865 0.0495 7.8052 <.0001 0.2894 0.4835 ***
ClassMalacostraca -0.3843 0.3397 -1.1313 0.2579 -1.0500 0.2815
ClassInsecta -0.0045 0.1252 -0.0361 0.9712 -0.2500 0.2409
ClassAves -0.1689 0.0731 -2.3113 0.0208 -0.3121 -0.0257 *
ClassAnimalia -0.1606 0.2266 -0.7085 0.4786 -0.6048 0.2836
ClassAmphibia -0.3098 0.1112 -2.7860 0.0053 -0.5278 -0.0919 **
ClassActinopterygii -0.1666 0.1195 -1.3939 0.1633 -0.4009 0.0677
ClassNematoda 0.0355 0.2458 0.1444 0.8851 -0.4462 0.5172
ClassPisces -0.1282 0.2726 -0.4702 0.6382 -0.6624 0.4061
ClassReptilia -0.2646 0.1031 -2.5675 0.0102 -0.4666 -0.0626 *
ClassTrematoda -0.3264 0.2106 -1.5498 0.1212 -0.7392 0.0864
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1MetaData$Class=relevel(MetaData$Class,c("Nematoda"))
Model_cREML_by_Class10 = rma.mv(r ~ Class, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Class10)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-19.8965 39.7930 65.7930 101.0169 69.5455
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0183 0.1352 73 no Study_ID
sigma^2.2 0.0512 0.2262 122 no Index
Test for Residual Heterogeneity:
QE(df = 111) = 1222.6024, p-val < .0001
Test of Moderators (coefficients 2:11):
QM(df = 10) = 16.3649, p-val = 0.0897
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.4220 0.2407 1.7528 0.0796 -0.0499 0.8938 .
ClassMammalia -0.0355 0.2458 -0.1444 0.8851 -0.5172 0.4462
ClassMalacostraca -0.4198 0.4134 -1.0155 0.3099 -1.2300 0.3904
ClassInsecta -0.0400 0.2670 -0.1499 0.8808 -0.5633 0.4833
ClassAves -0.2044 0.2469 -0.8277 0.4078 -0.6883 0.2796
ClassAnimalia -0.1961 0.3269 -0.5998 0.5486 -0.8368 0.4446
ClassAmphibia -0.3453 0.2605 -1.3255 0.1850 -0.8559 0.1653
ClassActinopterygii -0.2021 0.2646 -0.7637 0.4450 -0.7208 0.3165
ClassPisces -0.1637 0.3603 -0.4543 0.6496 -0.8698 0.5425
ClassReptilia -0.3001 0.2574 -1.1660 0.2436 -0.8045 0.2044
ClassTrematoda -0.3619 0.3171 -1.1415 0.2537 -0.9833 0.2595
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1MetaData$Class=relevel(MetaData$Class,c("Pisces"))
Model_cREML_by_Class6 = rma.mv(r ~ Class, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Class6)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-19.8965 39.7930 65.7930 101.0169 69.5455
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0183 0.1352 73 no Study_ID
sigma^2.2 0.0512 0.2262 122 no Index
Test for Residual Heterogeneity:
QE(df = 111) = 1222.6024, p-val < .0001
Test of Moderators (coefficients 2:11):
QM(df = 10) = 16.3649, p-val = 0.0897
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.2583 0.2680 0.9636 0.3352 -0.2671 0.7837
ClassNematoda 0.1637 0.3603 0.4543 0.6496 -0.5425 0.8698
ClassMammalia 0.1282 0.2726 0.4702 0.6382 -0.4061 0.6624
ClassMalacostraca -0.2561 0.4298 -0.5958 0.5513 -1.0986 0.5864
ClassInsecta 0.1236 0.2918 0.4237 0.6718 -0.4484 0.6956
ClassAves -0.0407 0.2736 -0.1489 0.8817 -0.5770 0.4955
ClassAnimalia -0.0324 0.3475 -0.0933 0.9257 -0.7135 0.6487
ClassAmphibia -0.1817 0.2859 -0.6353 0.5252 -0.7421 0.3788
ClassActinopterygii -0.0384 0.2897 -0.1327 0.8944 -0.6062 0.5293
ClassReptilia -0.1364 0.2831 -0.4819 0.6298 -0.6912 0.4184
ClassTrematoda -0.1983 0.3383 -0.5861 0.5578 -0.8612 0.4647
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1MetaData$Class=relevel(MetaData$Class,c("Reptilia"))
Model_cREML_by_Class2 = rma.mv(r ~ Class, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Class2)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-19.8965 39.7930 65.7930 101.0169 69.5455
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0183 0.1352 73 no Study_ID
sigma^2.2 0.0512 0.2262 122 no Index
Test for Residual Heterogeneity:
QE(df = 111) = 1222.6024, p-val < .0001
Test of Moderators (coefficients 2:11):
QM(df = 10) = 16.3649, p-val = 0.0897
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.1219 0.0910 1.3390 0.1806 -0.0565 0.3003
ClassPisces 0.1364 0.2831 0.4819 0.6298 -0.4184 0.6912
ClassNematoda 0.3001 0.2574 1.1660 0.2436 -0.2044 0.8045
ClassMammalia 0.2646 0.1031 2.5675 0.0102 0.0626 0.4666 *
ClassMalacostraca -0.1197 0.3481 -0.3437 0.7310 -0.8020 0.5627
ClassInsecta 0.2601 0.1470 1.7694 0.0768 -0.0280 0.5481 .
ClassAves 0.0957 0.1056 0.9064 0.3647 -0.1112 0.3026
ClassAnimalia 0.1040 0.2392 0.4349 0.6636 -0.3647 0.5728
ClassAmphibia -0.0452 0.1349 -0.3353 0.7374 -0.3096 0.2192
ClassActinopterygii 0.0980 0.1426 0.6870 0.4921 -0.1816 0.3775
ClassTrematoda -0.0618 0.2255 -0.2742 0.7839 -0.5037 0.3801
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1MetaData$Class=relevel(MetaData$Class,c("Trematoda"))
Model_cREML_by_Class9 = rma.mv(r ~ Class, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Class9)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-19.8965 39.7930 65.7930 101.0169 69.5455
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0183 0.1352 73 no Study_ID
sigma^2.2 0.0512 0.2262 122 no Index
Test for Residual Heterogeneity:
QE(df = 111) = 1222.6024, p-val < .0001
Test of Moderators (coefficients 2:11):
QM(df = 10) = 16.3649, p-val = 0.0897
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.0600 0.2063 0.2910 0.7710 -0.3443 0.4644
ClassReptilia 0.0618 0.2255 0.2742 0.7839 -0.3801 0.5037
ClassPisces 0.1983 0.3383 0.5861 0.5578 -0.4647 0.8612
ClassNematoda 0.3619 0.3171 1.1415 0.2537 -0.2595 0.9833
ClassMammalia 0.3264 0.2106 1.5498 0.1212 -0.0864 0.7392
ClassMalacostraca -0.0578 0.3943 -0.1467 0.8834 -0.8307 0.7150
ClassInsecta 0.3219 0.2349 1.3702 0.1706 -0.1386 0.7823
ClassAves 0.1575 0.2126 0.7409 0.4588 -0.2592 0.5742
ClassAnimalia 0.1658 0.3025 0.5483 0.5835 -0.4270 0.7586
ClassAmphibia 0.0166 0.2291 0.0724 0.9423 -0.4324 0.4656
ClassActinopterygii 0.1598 0.2298 0.6954 0.4868 -0.2906 0.6102
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Finally, we computed FDR corrected p-values:
tab2=as.data.frame(round(p.adjust(c(.0001, 0.1806, .0001, 0.4415, 0.0454, 0.3352, 0.9948, 0.0009, 0.7710, 0.0796, 0.3071), method = 'fdr'),digit=3),row.names=cbind( "Animalia","Nematoda","Trematoda","Insecta","Malacostraca","Pisces" ,"Actinopterygii","Amphibia","Mammalia","Reptilia","Aves"))
colnames(tab2)<-cbind('P-value')
tab2 P-value
Animalia 0.001
Nematoda 0.331
Trematoda 0.001
Insecta 0.540
Malacostraca 0.125
Pisces 0.461
Actinopterygii 0.995
Amphibia 0.003
Mammalia 0.848
Reptilia 0.175
Aves 0.461Plot phylogenetic classes (Figure 1)
Here we plot the phylogenetic classes:
MetaData$Class=as.factor(MetaData$Class)
MetaData$Class=factor(MetaData$Class, levels = (c("Animalia","Nematoda","Trematoda","Insecta","Malacostraca","Pisces" ,"Actinopterygii","Amphibia","Mammalia","Reptilia","Aves")))
MetaData_sorted=MetaData
MetaData_sorted$Class[MetaData_sorted$Class=='Pisces']='Actinopterygii'
ggplot() +
geom_hline(yintercept=0, linetype="longdash", color = "black", linewidth=1)+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class$ci.lb[1], x=11.35, xend= 11.35, yend= Model_cREML_by_Class$ci.ub[1]),linewidth=1,color=colpal5[11], alpha=1)+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class2$ci.lb[1], x=10.35, xend= 10.35, yend= Model_cREML_by_Class2$ci.ub[1]), alpha=1,linewidth=1,color=colpal5[10])+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class3$ci.lb[1], x=9.35, xend= 9.35, yend= Model_cREML_by_Class3$ci.ub[1]), alpha=1,linewidth=1,color=colpal5[9])+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class4$ci.lb[1], x=8.35, xend= 8.35, yend= Model_cREML_by_Class4$ci.ub[1]), alpha=1,linewidth=1,color=colpal5[8])+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class5$ci.lb[1], x=7.35, xend= 7.35, yend= Model_cREML_by_Class5$ci.ub[1]), alpha=1,linewidth=1,color=colpal5[7])+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class6$ci.lb[1], x=6.35, xend= 6.35, yend= Model_cREML_by_Class6$ci.ub[1]), alpha=1,linewidth=1,color=colpal5[6])+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class7$ci.lb[1], x=5.35, xend= 5.35, yend= Model_cREML_by_Class7$ci.ub[1]), alpha=1,linewidth=1,color=colpal5[5])+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class8$ci.lb[1], x=4.35, xend= 4.35, yend= Model_cREML_by_Class8$ci.ub[1]), alpha=1,linewidth=1,color=colpal5[4])+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class9$ci.lb[1], x=3.35, xend= 3.35, yend= Model_cREML_by_Class9$ci.ub[1]), alpha=1,linewidth=1,color=colpal5[3])+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class10$ci.lb[1], x=2.35, xend= 2.35, yend= Model_cREML_by_Class10$ci.ub[1]), alpha=1,linewidth=1,color=colpal5[2])+
geom_segment(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class11$ci.lb[1], x=1.35, xend= 1.35, yend= Model_cREML_by_Class11$ci.ub[1]), alpha=1,linewidth=1,color=colpal5[1])+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class$b[1,1], x=11.35), size = 3.5,alpha=1,stroke=0,shape=19,color='white')+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class2$b[1,1], x=10.35), size = 3.5,alpha=1,stroke=0,shape=19,color='white')+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class3$b[1,1], x=9.35), size = 3.5,alpha=1,stroke=0,shape=19,color='white')+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class4$b[1,1], x=8.35), size = 3.5,alpha=1,stroke=0,shape=19,color='white')+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class5$b[1,1], x=7.35), size = 3.5,alpha=1,stroke=0,shape=19,color='white')+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class6$b[1,1], x=6.35), size = 3.5,alpha=1,stroke=0,shape=19,color='white')+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class7$b[1,1], x=5.35), size = 3.5,alpha=1,stroke=0,shape=19,color='white')+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class8$b[1,1], x=4.35), size = 3.5,alpha=1,stroke=0,shape=19,color='white')+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class9$b[1,1], x=3.35), size = 3.5,alpha=1,stroke=0,shape=19,color='white')+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class10$b[1,1], x=2.35), size = 3.5,alpha=1,stroke=0,shape=19,color='white')+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class11$b[1,1], x=1.35), size = 3.5,alpha=1,stroke=0,shape=19,color='white')+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class$b[1,1], x=11.35), size = 3.5,alpha=1,stroke=0,shape=19,color=colpal5[11])+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class2$b[1,1], x=10.35), size = 3.5,alpha=1,stroke=0,shape=19,color=colpal5[10])+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class3$b[1,1], x=9.35), size = 3.5,alpha=1,stroke=0,shape=19,color=colpal5[9])+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class4$b[1,1], x=8.35), size = 3.5,alpha=1,stroke=0,shape=19,color=colpal5[8])+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class5$b[1,1], x=7.35), size = 3.5,alpha=1,stroke=0,shape=19,color=colpal5[7])+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class6$b[1,1], x=6.35), size = 3.5,alpha=1,stroke=0,shape=19,color=colpal5[6])+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class7$b[1,1], x=5.35), size = 3.5,alpha=1,stroke=0,shape=19,color=colpal5[5])+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class8$b[1,1], x=4.35), size = 3.5,alpha=1,stroke=0,shape=19,color=colpal5[4])+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class9$b[1,1], x=3.35), size = 3.5,alpha=1,stroke=0,shape=19,color=colpal5[3])+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class10$b[1,1], x=2.35), size = 3.5,alpha=1,stroke=0,shape=19,color=colpal5[2])+
geom_point(inherit.aes = F,mapping = aes(y=Model_cREML_by_Class11$b[1,1], x=1.35), size = 3.5,alpha=1,stroke=0,shape=19,color=colpal5[1])+
ylab(expression(paste("Effect size (", italic("r"),')')))+xlab('Class')+coord_flip()+guides(fill = FALSE, colour = FALSE) +
scale_x_discrete(labels=(c("Animalia","Nematoda","Trematoda","Insecta","Malacostraca","Pisces" ,"Actinopterygii","Amphibia","Mammalia","Reptilia","Aves")),expand=c(.1,0))+
geom_point(data=MetaData,aes(x=Class, y=r, fill = Class, colour = Class),position = position_jitter(width = .1), size = 2.5,alpha=0.6,stroke=0,shape=19)+
scale_color_manual(values =colpal5)+
scale_fill_manual(values =colpal5)+
theme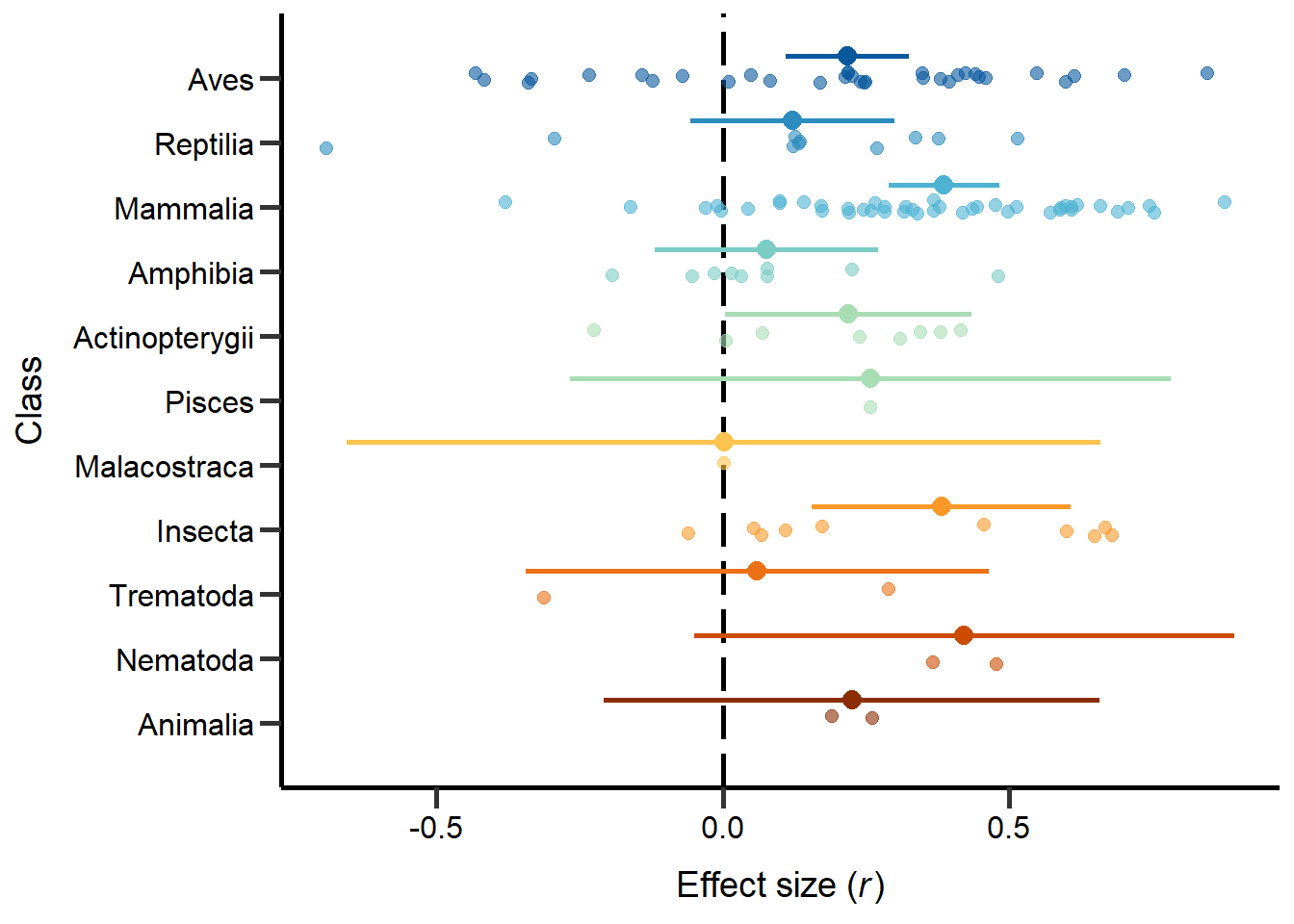
Figure 1: Phylogeny based on classes including sample sizes for the number of studies and number of effect sizes, respectively, and estimates with 95%CI from non-phylogenetic model.
| Version | Author | Date |
|---|---|---|
| 8edd942 | LennartWinkler | 2023-05-01 |
Type of SSD measure
Next we explored the effect of the type of SSD measure (i.e. body mass or size):
MetaData$SSD_Proxy=as.factor(MetaData$SSD_Proxy)
MetaData$SSD_Proxy=relevel(MetaData$SSD_Proxy,c("Body mass"))
Model_REML_by_cSSDMeasure = rma.mv(r ~ SSD_Proxy, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSSDMeasure)
Multivariate Meta-Analysis Model (k = 120; method: REML)
logLik Deviance AIC BIC AICc
-23.8130 47.6260 55.6260 66.7088 55.9800
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0266 0.1632 72 no Study_ID
sigma^2.2 0.0478 0.2187 120 no Index
Test for Residual Heterogeneity:
QE(df = 118) = 1511.0606, p-val < .0001
Test of Moderators (coefficient 2):
QM(df = 1) = 4.1937, p-val = 0.0406
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.3357 0.0455 7.3733 <.0001 0.2464 0.4249 ***
SSD_ProxyBody size -0.1221 0.0596 -2.0478 0.0406 -0.2390 -0.0052 *
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We then re-leveled the model for post-hoc comparisons:
MetaData$SSD_Proxy=relevel(MetaData$SSD_Proxy,c("Body size"))
Model_REML_by_cSSDMeasure2 = rma.mv(r ~ SSD_Proxy, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cSSDMeasure2)Finally, we computed FDR corrected p-values:
tab3=as.data.frame(round(p.adjust(c(.0001, .0001), method = 'fdr'),digit=3),row.names=cbind("Body mass","Body size"))
colnames(tab3)<-cbind('P-value')
tab3 P-value
Body mass 0
Body size 0SSD measure controlled for body size?
Next we explored the effect if the primary study controlled the SSD for body size (i.e. uncontrolled or controlled):
MetaData$BodySizeControlled=as.factor(MetaData$BodySizeControlled)
MetaData$BodySizeControlled=relevel(MetaData$BodySizeControlled,c("No"))
Model_REML_by_cBodySizeCont = rma.mv(r ~ BodySizeControlled, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cBodySizeCont)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-24.8368 49.6736 57.6736 68.8236 58.0214
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0309 0.1759 73 no Study_ID
sigma^2.2 0.0463 0.2152 122 no Index
Test for Residual Heterogeneity:
QE(df = 120) = 1555.0586, p-val < .0001
Test of Moderators (coefficient 2):
QM(df = 1) = 0.9645, p-val = 0.3261
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.2803 0.0348 8.0595 <.0001 0.2121 0.3485 ***
BodySizeControlledYes -0.0868 0.0884 -0.9821 0.3261 -0.2600 0.0864
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We then re-leveled the model for post-hoc comparisons:
MetaData$BodySizeControlled=relevel(MetaData$BodySizeControlled,c("Yes"))
Model_REML_by_cBodySizeCont2 = rma.mv(r ~ BodySizeControlled, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_REML_by_cBodySizeCont2)Finally, we computed FDR corrected p-values:
tab4=as.data.frame(round(p.adjust(c(.0001, 0.0172), method = 'fdr'),digit=3),row.names=cbind("uncontrolled","controlled"))
colnames(tab4)<-cbind('P-value')
tab4 P-value
uncontrolled 0.000
controlled 0.017Phylogentic vs non-phylogenetic studies
Next we explored the effect of studies controlling for phylogeny vs non-phylogenetic studies:
MetaData$PhyloControlled=as.factor(MetaData$PhyloControlled)
MetaData$PhyloControlled=relevel(MetaData$PhyloControlled,c("No"))
Model_cREML_by_cPhylo = rma.mv(r ~ PhyloControlled, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_cPhylo)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-22.1062 44.2124 52.2124 63.3624 52.5603
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0230 0.1515 73 no Study_ID
sigma^2.2 0.0485 0.2202 122 no Index
Test for Residual Heterogeneity:
QE(df = 120) = 1350.6816, p-val < .0001
Test of Moderators (coefficient 2):
QM(df = 1) = 6.8163, p-val = 0.0090
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.4465 0.0759 5.8802 <.0001 0.2977 0.5953 ***
PhyloControlledYes -0.2163 0.0828 -2.6108 0.0090 -0.3786 -0.0539 **
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We then re-leveled the model for post-hoc comparisons:
MetaData$PhyloControlled=relevel(MetaData$PhyloControlled,c("Yes"))
Model_cREML_by_cPhylo2 = rma.mv(r ~ PhyloControlled, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_cPhylo2)Finally, we computed FDR corrected p-values:
tab5=as.data.frame(round(p.adjust(c(0.0001, 0.0001), method = 'fdr'),digit=3),row.names=cbind("Non-phylogenetic","With phylogeny"))
colnames(tab5)<-cbind('P-value')
tab5 P-value
Non-phylogenetic 0
With phylogeny 0Percentage of species with female-biased SSD
Model_cREML_SSbias = rma.mv(r ~ SSD_SexBias_in_perc_F, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_SSbias)
Multivariate Meta-Analysis Model (k = 104; method: REML)
logLik Deviance AIC BIC AICc
-21.0804 42.1607 50.1607 60.6606 50.5731
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0315 0.1775 64 no Study_ID
sigma^2.2 0.0465 0.2157 104 no Index
Test for Residual Heterogeneity:
QE(df = 102) = 1443.6764, p-val < .0001
Test of Moderators (coefficient 2):
QM(df = 1) = 4.8150, p-val = 0.0282
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.3818 0.0550 6.9477 <.0001 0.2741 0.4895 ***
SSD_SexBias_in_perc_F -0.0023 0.0011 -2.1943 0.0282 -0.0044 -0.0003 *
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Test for publication bias
Model_cREML_PublBias = rma.mv(z ~ SE_z, V=rep(((mean(SE_z)*mean(SE_z))*N),length(SE_z)), data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_PublBias)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-145.5101 291.0202 299.0202 310.1702 299.3680
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0000 0.0000 73 no Study_ID
sigma^2.2 0.0000 0.0000 122 no Index
Test for Residual Heterogeneity:
QE(df = 120) = 19.2579, p-val = 1.0000
Test of Moderators (coefficient 2):
QM(df = 1) = 1.5648, p-val = 0.2110
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 0.1715 0.1977 0.8678 0.3855 -0.2159 0.5590
SE_z 0.8181 0.6540 1.2509 0.2110 -0.4637 2.0999
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Publication year
Next we explored the effect of the publication year of each study:
Model_cREML_by_Year = rma.mv(r ~ Year, V=Var_r, data = MetaData, random = c(~ 1 | Study_ID,~ 1 | Index), method = "REML")
summary(Model_cREML_by_Year)
Multivariate Meta-Analysis Model (k = 122; method: REML)
logLik Deviance AIC BIC AICc
-22.8685 45.7371 53.7371 64.8871 54.0849
Variance Components:
estim sqrt nlvls fixed factor
sigma^2.1 0.0281 0.1677 73 no Study_ID
sigma^2.2 0.0453 0.2129 122 no Index
Test for Residual Heterogeneity:
QE(df = 120) = 1275.7703, p-val < .0001
Test of Moderators (coefficient 2):
QM(df = 1) = 5.0616, p-val = 0.0245
Model Results:
estimate se zval pval ci.lb ci.ub
intrcpt 12.3544 5.3730 2.2994 0.0215 1.8236 22.8853 *
Year -0.0060 0.0027 -2.2498 0.0245 -0.0113 -0.0008 *
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Forest plot
# Sort data by Class
MetaData$Class=as.factor(MetaData$Class)
MetaData$Class=factor(MetaData$Class, levels = c("Animalia","Nematoda","Trematoda","Insecta","Malacostraca","Pisces" ,"Actinopterygii","Amphibia","Mammalia","Reptilia","Aves"))
MetaData_sorted <-MetaData[order(MetaData$r,decreasing = T),]
MetaData_sorted$Class[MetaData_sorted$Class=='Pisces']='Actinopterygii'
MetaData_sorted <-MetaData_sorted[order(MetaData_sorted$Class),]
# Add global effect size to data
forestData=MetaData_sorted[,c(3,5,10,15,18,19)]
forestData[,c(4:6)]=lapply(forestData[,c(4:6)],as.numeric)
forestData$SexSel_Episode=as.factor(forestData$SexSel_Episode)
forestData$SexSel_Episode=factor(forestData$SexSel_Episode, levels = c("pre-copulatory","post-copulatory" ,"both"))Figure 2
ggplot(data=forestData, aes(y=1:nrow(forestData), x=r, xmin=lCI, xmax=uCI,color=Class,shape=SexSel_Episode,fill=SexSel_Episode)) +
geom_vline(xintercept=Model_REML_Null$b[1,1], color='black', linetype='solid', alpha=.9,linewidth=0.75)+
annotate("rect", xmin = Model_REML_Null$ci.lb[1], xmax = Model_REML_Null$ci.ub[1], ymin = 1, ymax = nrow(forestData),alpha = .25)+
geom_vline(xintercept=0, color='black', linetype='dashed', alpha=.5,linewidth=0.8) +
geom_errorbarh(height=0,size = ifelse(1:nrow(forestData) == c(1), 3.3, 3.3), alpha=1,colour='white')+
geom_errorbarh(height=0,size = ifelse(1:nrow(forestData) == c(1), 3.3, 3.3), alpha=1)+
geom_point(size = ifelse(1:nrow(forestData) == c(1), 2, 2), alpha=1,stroke=1,colour='white')+
geom_point(size = ifelse(1:nrow(forestData) == c(1), 2, 2), alpha=1,stroke=1,colour='black')+
scale_y_continuous(breaks=seq(1, nrow(forestData), by=1), labels=forestData$AuthorsAndYear,limits=c(1,length(forestData$AuthorsAndYear)),expand=c(0.015,0.015)) +
labs(title='', x=expression(paste(italic("r "),'(95% CI)')), y = 'Study') +
theme_classic()+theme(axis.text.x=element_text(size=12),
axis.title=element_text(size=12))+ labs(fill = c("Sexual selection episode"),shape = c("Sexual selection mode"))+
scale_colour_manual(values=colpal6, labels=NULL,guide = "none")+
scale_shape_manual(values=c(21,21,21) ,labels=c("Pre-copulatory","Post-copulatory" ,"Both"))+
scale_fill_manual(values=c('grey50','white','black'),labels=c("Pre-copulatory","Post-copulatory" ,"Both"))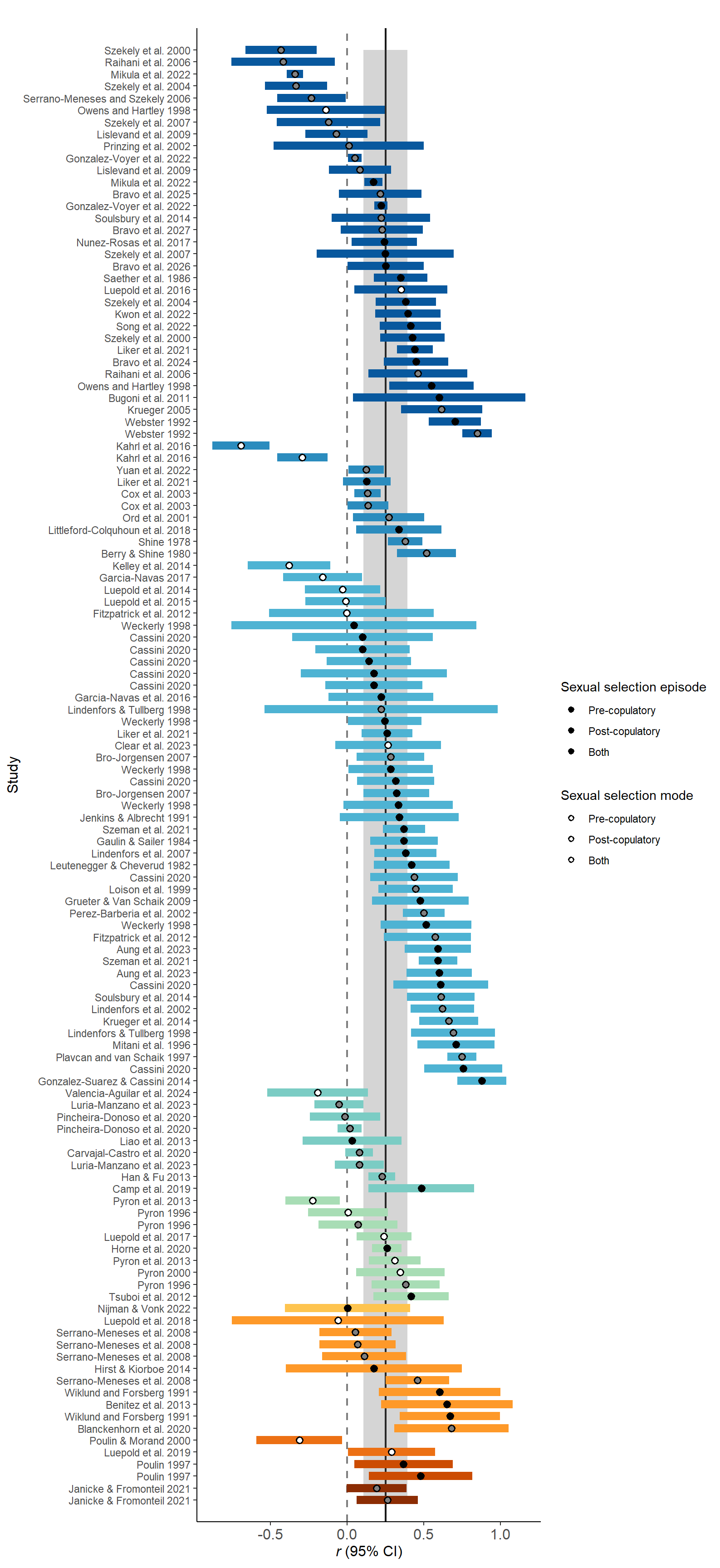
Figure 2: Forest plot including correlation coefficients (±95% CI), as well as the global effect size (in grey) with and without controlling for phylogeny. Different animal classes highlighted by background colors (see Figure 1 for class names) and sexual selection modes pre- (red), post-copulatory (blue) and both (green) by color of effect size.
| Version | Author | Date |
|---|---|---|
| 054a12b | LennartWinkler | 2023-04-30 |
R version 4.3.3 (2024-02-29 ucrt)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 19045)
Matrix products: default
locale:
[1] LC_COLLATE=German_Germany.utf8 LC_CTYPE=German_Germany.utf8
[3] LC_MONETARY=German_Germany.utf8 LC_NUMERIC=C
[5] LC_TIME=German_Germany.utf8
time zone: Europe/Berlin
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] gridExtra_2.3 wesanderson_0.3.7 PupillometryR_0.0.5
[4] rlang_1.1.3 dplyr_1.1.4 cowplot_1.1.3
[7] ggdist_3.3.2 MCMCglmm_2.35 coda_0.19-4.1
[10] RColorBrewer_1.1-3 ggplot2_3.5.0 data.table_1.15.2
[13] multcomp_1.4-25 TH.data_1.1-2 survival_3.5-8
[16] mvtnorm_1.2-4 psych_2.4.3 pwr_1.3-0
[19] MASS_7.3-60.0.1 metafor_4.4-0 numDeriv_2016.8-1.1
[22] metadat_1.2-0 Matrix_1.6-5 matrixcalc_1.0-6
[25] ape_5.7-1 workflowr_1.7.1
loaded via a namespace (and not attached):
[1] tidyselect_1.2.1 farver_2.1.1 fastmap_1.1.1
[4] tensorA_0.36.2.1 mathjaxr_1.6-0 promises_1.2.1
[7] digest_0.6.35 lifecycle_1.0.4 processx_3.8.4
[10] magrittr_2.0.3 compiler_4.3.3 sass_0.4.9
[13] tools_4.3.3 utf8_1.2.4 yaml_2.3.8
[16] knitr_1.45 labeling_0.4.3 mnormt_2.1.1
[19] withr_3.0.0 purrr_1.0.2 grid_4.3.3
[22] fansi_1.0.6 git2r_0.33.0 colorspace_2.1-0
[25] scales_1.3.0 cli_3.6.1 rmarkdown_2.26
[28] generics_0.1.3 rstudioapi_0.16.0 httr_1.4.7
[31] cachem_1.0.8 stringr_1.5.1 splines_4.3.3
[34] parallel_4.3.3 vctrs_0.6.5 sandwich_3.1-0
[37] jsonlite_1.8.8 callr_3.7.5 jquerylib_0.1.4
[40] tidyr_1.3.1 glue_1.7.0 codetools_0.2-19
[43] ps_1.7.6 distributional_0.4.0 stringi_1.8.3
[46] cubature_2.1.0 gtable_0.3.4 later_1.3.2
[49] munsell_0.5.0 tibble_3.2.1 pillar_1.9.0
[52] htmltools_0.5.7 R6_2.5.1 rprojroot_2.0.4
[55] evaluate_0.23 lattice_0.22-5 highr_0.10
[58] corpcor_1.6.10 httpuv_1.6.14 bslib_0.6.1
[61] Rcpp_1.0.12 nlme_3.1-164 whisker_0.4.1
[64] xfun_0.42 fs_1.6.3 zoo_1.8-12
[67] getPass_0.2-4 pkgconfig_2.0.3