Home
Last updated: 2022-05-05
Checks: 6 1
Knit directory: EMBRAPAImputation2022/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20220303) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 8204f0e. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Untracked files:
Untracked: .DS_Store
Untracked: analysis/output.vcf.gz
Untracked: bgzip
Untracked: data/.DS_Store
Untracked: data/DArT2018/
Untracked: data/DArT2020/
Untracked: data/DArT2022/
Untracked: data/GBS/
Untracked: data/data/
Untracked: out.log
Untracked: out.recode.vcf
Untracked: output.log
Untracked: output.recode.vcf
Untracked: output.vcf.gz
Untracked: output/.DS_Store
Untracked: output/DArT2022/
Untracked: output/DCas22_DArt_ReadyForGP_Dos.rds
Untracked: output/out.log
Unstaged changes:
Modified: analysis/index.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/index.Rmd) and HTML (docs/index.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 8204f0e | LucianoRogerio | 2022-03-03 | Start workflowr project. |
New Imputation for EMBRAPA with DArT Marker only and DArT plus GBS Markers
Diversity Array Technology LTDA joint all the genotyping data from the four Genotyping Orders that EMBRAPA requested during these six or seven years in one huge file.
So let’s what we got at the DArT report for EMBRAPA DArT genotyping of 2022
library(genomicMateSelectR)
dir("data/DArT2022")
nskipvcf <- 2
nskipcounts <- 2
VCF2022 <- read.table(here::here("data", "Report_6902_VCF_Ref_Version6.txt"),
sep = "\t", header = T, skip = nskipvcf, comment.char = "")
Counts2022 <- read.table(here::here("data", "SEQ_SNPs_counts_0_Target_extend_Ref.csv"),
sep = ",", header = T, skip = nskipcounts)
Counts2022[1:10,1:30]
VCF2022[1:10,1:30]
genomicMateSelectR::convertDart2vcf(dartvcfInput = here::here("data", "Report_6902_VCF_Ref_Version6.txt"),
dartcountsInput = here::here("data", "SEQ_SNPs_counts_0_Target_extend_Ref.csv"),
nskipvcf = 2, nskipcounts = 2,
outName = "output/DCas22_6902", ncores = 20)Filtering the DArT markers previously the Beagle imputation
library(here); library(tidyverse)
library(magrittr); library(dplyr)
## Parameters for the Filter function
inPath <- "output/"
inName <- "DCas22_6902_DArTseqLD_AllSites_AllChrom_raw"
outPath <- "output/"
outName <- "DCas22_6902_DArTseqLD_AllSites_AllChrom_rawFiltered"
FilterLuc <- function(inPath = NULL, inName, outPath = NULL, outName, CRthresh = 0.6){
system(paste0("vcftools --gzvcf ", inPath, inName, ".vcf.gz --freq2 --out ",
outPath, inName))
system(paste0("vcftools --gzvcf ", inPath, inName, ".vcf.gz --missing-site --out ",
outPath, inName))
INFO <- read.table(paste0(outPath, inName, ".frq"), stringsAsFactors = F,
header = F, skip = 1) %>%
rename(CHROM = V1, POS = V2, N_ALLELES = V3,
N_CHR = V4, FREQ1 = V5, FREQ2 = V6)
callrate <- read.table(paste0(outPath, inName, ".lmiss"), stringsAsFactors = F,
header = T) %>% dplyr::select(CHR, POS, N_DATA, F_MISS) %>%
mutate(CHROM = CHR,
CR = 1 - F_MISS,
.keep = "unused")
stats2filterOn <- left_join(INFO, callrate)
stats2filterOn %<>% dplyr::mutate(FREQ2 = as.numeric(FREQ2)) %>%
dplyr::mutate(MAF = ifelse(FREQ2 > 0.5,
yes = 1 - FREQ2, no = FREQ2)) %>%
dplyr::select(-FREQ1, -FREQ2)
MAFthresh <- (1/max(stats2filterOn$N_DATA, na.rm = T))**2
sitesPassingFilters <- stats2filterOn %>%
dplyr::filter(MAF >= MAFthresh, CR >= CRthresh) %>%
dplyr::select(CHROM, POS)
print(paste0(nrow(sitesPassingFilters), " sites passing filter"))
write.table(sitesPassingFilters, file = paste0(outPath, inName,
".sitesPassing"), row.names = F, col.names = F, quote = F)
system(paste0("vcftools --gzvcf ", inPath, inName, ".vcf.gz",
" ", "--positions ", outPath, inName, ".sitesPassing",
" ", "--recode --stdout | bgzip -c -@ 24 > ", outPath,
outName, ".vcf.gz"))
print(paste0("Filtering Complete: ", outName))
}
FilterLuc(inPath=inPath, inName=inName,
outPath=outPath, outName=outName,
CRthresh = 0.6)Download of the Raw Filtered VCF
cd output/DArT2022
scp lbraatz@cbsulm35.biohpc.cornell.edu:/workdir/lbraatz/DCas22_6902/output/DCas22_6902_DArTseqLD_AllSites_AllChrom_rawFiltered.vcf.gz .
scp lbraatz@cbsulm35.biohpc.cornell.edu:/workdir/lbraatz/DCas22_6902/output/DCas22_6902_DArTseqLD_AllSites_AllChrom_raw.lmiss .
scp lbraatz@cbsulm35.biohpc.cornell.edu:/workdir/lbraatz/DCas22_6902/output/DCas22_6902_DArTseqLD_AllSites_AllChrom_raw.frq .
cd ../..MAF and Call Rate of the Raw data
library(tidyverse); library(here)── Attaching packages ─────────────────────────────────────── tidyverse 1.3.1 ──✔ ggplot2 3.3.6 ✔ purrr 0.3.4
✔ tibble 3.1.7 ✔ dplyr 1.0.9
✔ tidyr 1.2.0 ✔ stringr 1.4.0
✔ readr 2.1.2 ✔ forcats 0.5.1── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()here() starts at /Users/lbd54/Documents/GitHub/EMBRAPAImputation2022library(reactable)
INFO <- read.table(here::here("output", "DArT2022", "DCas22_6902_DArTseqLD_AllSites_AllChrom_raw.frq"),
stringsAsFactors = F,
header = F, skip = 1) %>%
rename(CHROM = V1, POS = V2, N_ALLELES = V3,
N_CHR = V4, FREQ1 = V5, FREQ2 = V6)
callrate <- read.table(here::here("output", "DArT2022", "DCas22_6902_DArTseqLD_AllSites_AllChrom_raw.lmiss"),
stringsAsFactors = F,
header = T) %>% dplyr::select(CHR, POS, N_DATA, F_MISS) %>%
mutate(CHROM = CHR,
CR = 1 - F_MISS,
.keep = "unused")
stats2filterOn <- left_join(INFO, callrate)Joining, by = c("CHROM", "POS")stats2filterOn %<>% dplyr::mutate(FREQ2 = as.numeric(FREQ2)) %>%
dplyr::mutate(MAF = ifelse(FREQ2 > 0.5,
yes = 1 - FREQ2, no = FREQ2)) %>%
dplyr::select(-FREQ1, -FREQ2)
MAFthresh <- (1/max(stats2filterOn$N_DATA, na.rm = T))**2
stats2filterOn %<>% filter(!is.na(CHROM), CR >= 0.6, MAF >= MAFthresh) %>%
select(CR, MAF) %>% rename(CallRate = CR) %>% reshape2::melt(.)No id variables; using all as measure variablesstats2filterOn %>% ggplot(aes(x= value)) +
geom_density() + facet_grid(~variable, scales = "free_x") + theme_minimal()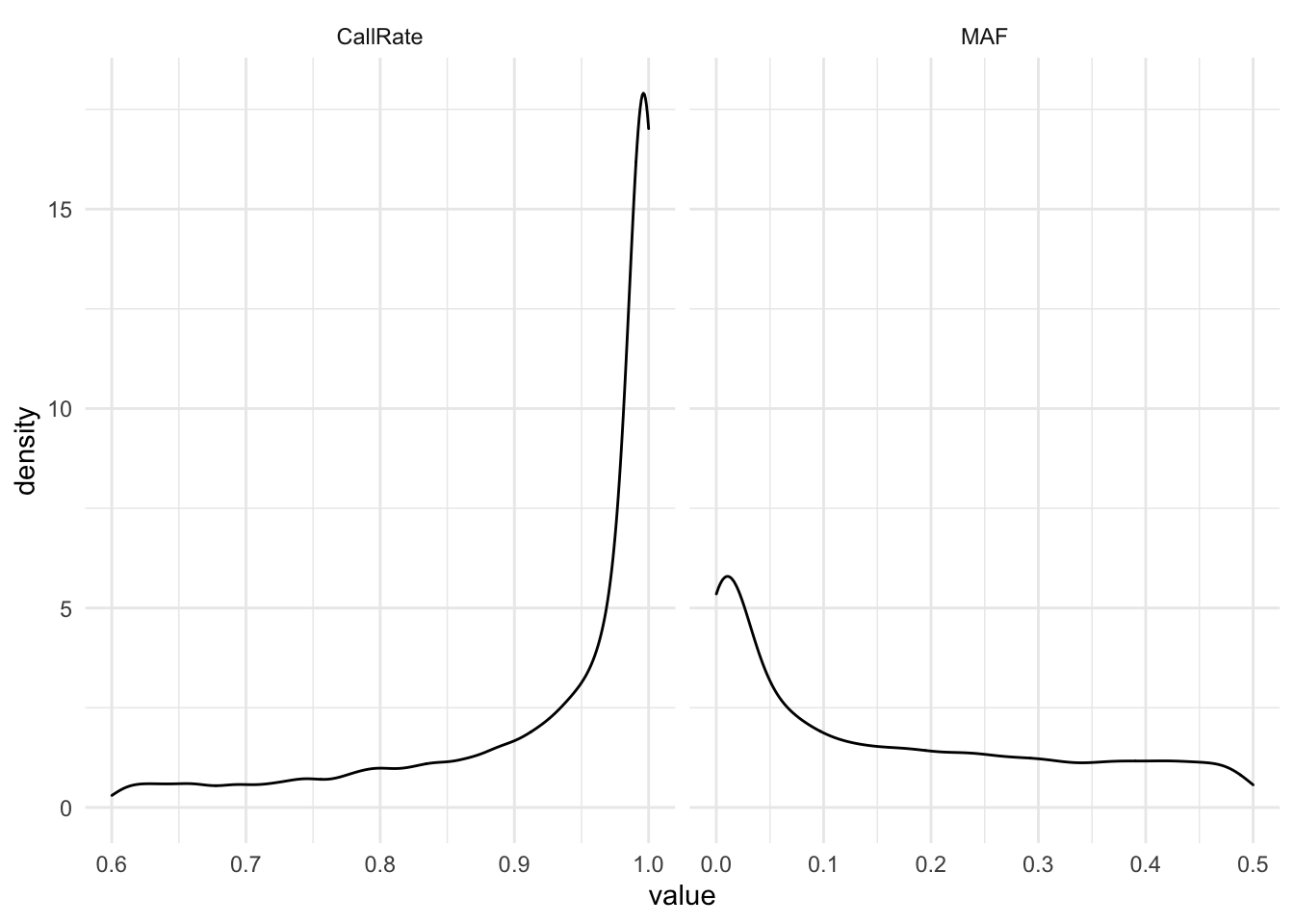
Split the VCF per chromosome
require(furrr); plan(multisession, workers = 18)
options(future.globals.maxSize=+Inf); options(future.rng.onMisuse="ignore")
vcfIn<-here::here("output/","DCas22_6902_DArTseqLD_AllSites_AllChrom_rawFiltered.vcf.gz")
filters<-"--minDP 4 --maxDP 50" # because using GT not PL for impute (Beagle5)
outPath<-here::here("output/")
outSuffix<-"DCas22_6902_DArTseqLD_AllSites_AllChrom_rawFiltered"
future_map(1:18,
~genomicMateSelectR::splitVCFbyChr(Chr=.,
vcfIn=vcfIn,filters=filters,
outPath=outPath,
outSuffix=outSuffix))
plan(sequential)Imputation DArT markers only
Imputation is performed by chromosome
java -Xms2g -Xmx [maxmem] -jar /programs/beagle/beagle.jar gt= [targetVCF] map= [mapFile] out= [outName] nthreads= [nthreads] impute= [impute] ne= [ne]runBeagle5Luc <- function(targetVCF, mapFile, outName, nthreads, maxmem = "500g",
impute = TRUE, ne = 1e+05, samplesToExclude = NULL){
system(paste0("java -Xms2g -Xmx", maxmem, " -jar /programs/beagle/beagle.jar ",
"gt=", targetVCF, " ", "map=", mapFile, " ",
"out=", outName, " ", "nthreads=", nthreads,
" impute=", impute, " ne=", ne,
ifelse(!is.null(samplesToExclude),
paste0(" excludesamples=", samplesToExclude), "")))}
targetVCFpath<-here::here("output/") # location of the targetVCF
mapPath<-here::here("data", "CassavaGeneticMapV6updated/")
outPath<-here::here("output/")
outSuffix<-"DCas22_6902"
library(tidyverse); library(magrittr);
purrr::map(1:18,
~runBeagle5Luc(targetVCF=paste0(targetVCFpath,"chr",.,
"_DCas22_6902_DArTseqLD_AllSites_AllChrom_rawFiltered.vcf.gz"),
mapFile=paste0(mapPath,"chr",.,
"_cassava_cM_pred.v6_91019.map"),
outName=paste0(outPath,"chr",.,
"_DCas22_6902_DArT_imputed"),
nthreads=110))Organize the Beagle logs in a directory
cd ~/Desktop/Genotyping/DArT/EMBRAPA/DCas22_6902/output/
mkdir BeagleLogs
cp *_DCas22_6902_DArT_imputed.log BeagleLogs/.
rm *_DCas22_6902_DArT_imputed.logPost Imputation Filter
Standard post-imputation filter: \(CR≥0.6\), \(MAF≥(1/7827)^2\).
Loop to filter all 18 VCF files in parallel
inPath<-here::here("output/")
outPath<-here::here("output/")
require(furrr); plan(multisession, workers = 18)
future_map(1:18,
~FilterLuc(inPath=inPath,
inName=paste0("chr",.,"_DCas22_6902_DArT_imputed"),
outPath=outPath,
outName=paste0("chr",.,"_DCas22_6902_DArT_imputedAndFiltered"),
CRthresh = 0.6))
plan(sequential)Let’s check what we got
purrr::map(1:18,~system(paste0("zcat ",here::here("output/"),"chr",.,"_DCas22_6902_DArT_imputedAndFiltered.vcf.gz | wc -l")))Chr 1 - 1064
Chr 2 - 696
Chr 3 - 682
Chr 4 - 682
Chr 5 - 634
Chr 6 - 656
Chr 7 - 428
Chr 8 - 532
Chr 9 - 520
Chr 10 - 664
Chr 11 - 591
Chr 12 - 468
Chr 13 - 508
Chr 14 - 707
Chr 15 - 516
Chr 16 - 437
Chr 17 - 543
Chr 18 - 480Formats for GS and GWAS Analysis
for(i in 1:18){
system(paste0("gzcat chr", i, "_DCas22_6902_DArT_imputedAndFiltered.vcf.gz",
" > ",
"chr", i, "_DCas22_6902_DArT_imputedAndFiltered.vcf"))
}
VCFFile <- tibble()
inPath <- "output/DArT2022/"
outPath <- "output/DArT2022/"
outName <- "AllChrom_DArT_ReadyForGP_2022May04"
for(i in 1:18){
x <- read.table(file = paste0(inPath, "chr", i, "_DCas22_6902_DArT_imputedAndFiltered.vcf"),
header = T, comment.char = "", skip = 9, sep = "\t")
print(paste0("chr - ", i, " - NSNPs ", nrow(x)))
VCFFile <- rbind(VCFFile, x)
}
VCFFile %<>% rename(`#CHROM` = X.CHROM)
# Header ----------------------------------------
header<-c("##fileformat=VCFv4.2",
"##filedate=20220504",
"##source=\"beagle.28Sep18.793.jar\"",
"##INFO=<ID=AF,Number=A,Type=Float,Description=\"Estimated ALT Allele Frequencies\">",
"##INFO=<ID=DR2,Number=1,Type=Float,Description=\"Dosage R-Squared: estimated squared correlation between estimated REF dose [P(RA) + 2*P(RR)] and true REF dose\">",
"##INFO=<ID=IMP,Number=0,Type=Flag,Description=\"Imputed marker\">",
"##FORMAT=<ID=GT,Number=1,Type=String,Description=\"Genotype\">",
"##FORMAT=<ID=DS,Number=A,Type=Float,Description=\"estimated ALT dose [P(RA) + P(AA)]\">",
"##FORMAT=<ID=GP,Number=G,Type=Float,Description=\"Estimated Genotype Probability\">")
# Write to disk ----------------------------------------
options("scipen"=1000, "digits"=4)
# for a few SNPs, position kept printing in sci notation e.g. 1e3, screws up Beagle etc., this avoids that (I hope)
write_lines(header,
file = paste0(outPath, outName,".vcf"))
write.table(VCFFile,
paste0(outPath, outName,".vcf"),
append = TRUE, sep = "\t", row.names=F, col.names=T, quote=F)
system(paste0("cat ", outPath, outName, ".vcf | bgzip -c > ", outPath, outName, ".vcf.gz"))
## Convert VCF to a Dosage file
DosFile <- VCFFile
rownames(DosFile) <- DosFile$ID
DosFile %<>% dplyr::select(-c(1:9)) %>% t %>% as.data.frame
str(DosFile[,1:10])
for(i in colnames(DosFile)[1:10]){
DosFile[,i] <- ifelse(test = DosFile[,i] == "0|0",
yes = "0",
no = ifelse(test = DosFile[,i] == "1|1",
yes = "2",
no = "1")) %>%
as.numeric
}
DArTClones <- rownames(DosFile) %>% gsub(pattern = "BGM.", replacement = "BGM-") %>%
gsub(pattern = ".TB", replacement = "-TB") %>%
gsub(pattern = ".T.Bco", replacement = "-TBco") %>%
gsub(pattern = ".TR", replacement = "-TR") %>%
gsub(pattern = ".T.Rx", replacement = "-TRx") %>%
gsub(pattern = "BR.([0-9])+.DArT.PL([0-9])+_([A-Z])([0-9])+...", replacement = "") %>%
gsub(pattern = "BR.SET([0-9])+.18.", replacement = "") %>%
gsub(pattern = "X0", replacement = "0") %>%
gsub(pattern = "X1", replacement = "1") %>%
gsub(pattern = "X201", replacement = "201") %>%
gsub(pattern = "X3", replacement = "3") %>%
gsub(pattern = "X4", replacement = "4") %>%
gsub(pattern = "X5", replacement = "5") %>%
gsub(pattern = "X7", replacement = "7") %>%
gsub(pattern = "X9", replacement = "9")
DArTClones <- cbind(as.matrix(rownames(DosFile)), as.matrix(DArTClones))
dup <- DArTClones[duplicated(DArTClones[,2]),1]
dup[dup == "TMEB14"] <- "BR.20.DArT.PL23_H01...TMEB14"
for(i in 1:nrow(DArTClones)){
DArTClones[i,2] <- base::ifelse(test = any(DArTClones[i,1] == dup),
yes = DArTClones[i,1],
no = DArTClones[i,2])
}
rownames(DosFile) <- DArTClones[,2]
### It still needs to correct the names
saveRDS(as.matrix(DosFile), file = "output/DCas22_DArt_ReadyForGP_Dos.rds")
sessionInfo()R version 4.1.2 (2021-11-01)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS Big Sur 11.6.5
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.1-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.1-arm64/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] reactable_0.2.3 here_1.0.1 forcats_0.5.1 stringr_1.4.0
[5] dplyr_1.0.9 purrr_0.3.4 readr_2.1.2 tidyr_1.2.0
[9] tibble_3.1.7 ggplot2_3.3.6 tidyverse_1.3.1
loaded via a namespace (and not attached):
[1] Rcpp_1.0.8.3 lubridate_1.8.0 assertthat_0.2.1 rprojroot_2.0.3
[5] digest_0.6.29 utf8_1.2.2 plyr_1.8.7 R6_2.5.1
[9] cellranger_1.1.0 backports_1.4.1 reprex_2.0.1 evaluate_0.15
[13] highr_0.9 httr_1.4.3 pillar_1.7.0 rlang_1.0.2
[17] readxl_1.4.0 rstudioapi_0.13 whisker_0.4 jquerylib_0.1.4
[21] rmarkdown_2.14 labeling_0.4.2 htmlwidgets_1.5.4 munsell_0.5.0
[25] broom_0.8.0 compiler_4.1.2 httpuv_1.6.5 modelr_0.1.8
[29] xfun_0.30 pkgconfig_2.0.3 htmltools_0.5.2 tidyselect_1.1.2
[33] workflowr_1.7.0 fansi_1.0.3 crayon_1.5.1 tzdb_0.3.0
[37] dbplyr_2.1.1 withr_2.5.0 later_1.3.0 grid_4.1.2
[41] jsonlite_1.8.0 gtable_0.3.0 lifecycle_1.0.1 DBI_1.1.2
[45] git2r_0.30.1 magrittr_2.0.3 scales_1.2.0 cli_3.3.0
[49] stringi_1.7.6 farver_2.1.0 reshape2_1.4.4 fs_1.5.2
[53] promises_1.2.0.1 xml2_1.3.3 bslib_0.3.1 ellipsis_0.3.2
[57] generics_0.1.2 vctrs_0.4.1 tools_4.1.2 glue_1.6.2
[61] hms_1.1.1 fastmap_1.1.0 yaml_2.3.5 colorspace_2.0-3
[65] rvest_1.0.2 knitr_1.38 haven_2.5.0 sass_0.4.1