Analises Modelos Mistos
LucianoRogerio e HenriqueBernardino
2021-10-12
Last updated: 2022-04-05
Checks: 6 1
Knit directory: HenriqueDGen/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20211012) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version e020351. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/.DS_Store
Untracked files:
Untracked: output/BLUPsDisease.RDS
Unstaged changes:
Modified: analysis/AnalisesModelosMistos.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/AnalisesModelosMistos.Rmd) and HTML (docs/AnalisesModelosMistos.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | e020351 | LucianoRogerio | 2022-03-29 | Update Henrique Analysis |
| html | e020351 | LucianoRogerio | 2022-03-29 | Update Henrique Analysis |
| Rmd | 350ad46 | LucianoRogerio | 2022-03-23 | Names updates from Data files |
| Rmd | 90b66eb | LucianoRogerio | 2022-03-08 | Update analysis Mixed models Henrique |
| html | 90b66eb | LucianoRogerio | 2022-03-08 | Update analysis Mixed models Henrique |
| Rmd | 2abb2a7 | LucianoRogerio | 2021-12-07 | Small english changes at the website |
| html | 2abb2a7 | LucianoRogerio | 2021-12-07 | Small english changes at the website |
| Rmd | 73653b1 | LucianoRogerio | 2021-12-07 | fix the buttons at the final of each of the webpages |
| html | 73653b1 | LucianoRogerio | 2021-12-07 | fix the buttons at the final of each of the webpages |
| Rmd | f272038 | LucianoRogerio | 2021-12-07 | Update of the analysis and website layout |
| html | f272038 | LucianoRogerio | 2021-12-07 | Update of the analysis and website layout |
| html | f51cdc6 | LucianoRogerio | 2021-11-18 | Add the Dendrogram analysis |
| Rmd | 97d638d | LucianoRogerio | 2021-11-02 | Update of html links |
| html | 97d638d | LucianoRogerio | 2021-11-02 | Update of html links |
| html | b9ef481 | LucianoRogerio | 2021-11-02 | Build site and add new Data |
| Rmd | 7286357 | LucianoRogerio | 2021-11-02 | Insercao do caractere Area de Antracnose e PCA |
| html | 9f9ffff | LucianoRogerio | 2021-10-26 | Build site. |
| Rmd | 170ea91 | LucianoRogerio | 2021-10-26 | BLUPs + Control Means for Diversity analysis |
| Rmd | 60db375 | LucianoRogerio | 2021-10-26 | BLUPS and Means effects estimated for Diversity analysis |
| html | b8ca347 | LucianoRogerio | 2021-10-19 | Build site. |
| Rmd | b87ceea | LucianoRogerio | 2021-10-19 | Add Mixed Models analysis for the repository |
| Rmd | 106f55c | LucianoRogerio | 2021-10-19 | Update Website |
| html | 106f55c | LucianoRogerio | 2021-10-19 | Update Website |
| Rmd | b9ece4f | LucianoRogerio | 2021-10-12 | Second Commit |
| html | b9ece4f | LucianoRogerio | 2021-10-12 | Second Commit |
| Rmd | d2d70ff | LucianoRogerio | 2021-10-12 | Second Commit |
| html | d2d70ff | LucianoRogerio | 2021-10-12 | Second Commit |
Mixed Models Foliar Disease
library(here)here() starts at /Users/lbd54/Documents/GitHub/HenriqueDGensuppressMessages(library(tidyverse))
suppressMessages(library(plyr))
library(reactable)
suppressMessages(library(data.table))
suppressMessages(source(here::here("code", "MixedModelsFunctions.R")))
PhenoData <- readRDS(here::here("output", "DadosFenotipicosv2.RDS"))
PhenoData$block_number <- as.character(PhenoData$block_number)
PhenoData2 <- PhenoData %>% filter(!is.na(Y))
traits <- unique(PhenoData2$traits)
fmfit <- PhenoData2 %>% dlply(.variables = c("traits"),
.fun = analyzeTrial.lme4FD)
ResFixEffect <- lapply(fmfit, FUN = as.data.frame(anova))
ResAnInt <- matrix(unlist(ResFixEffect,use.names = T),
nrow = 2, byrow = F)
ResAnFin <- rbind(ResAnInt[,1:4],
ResAnInt[,5:8],
ResAnInt[,9:12],
ResAnInt[,13:16])
colnames(ResAnFin) <- c("DF", "SumSq", "MeanSq", "Fvalue")
ResAnovaFinal <- data.frame(Trait = rep(traits, each = 2),
Factor = rep(c("Control", "Block"),
times = 4),
ResAnFin)Table 1. Anova of the fixed effects of Cassava foliar diseases
rdfmfit <- PhenoData2 %>% dlply(.variables = c("traits"),
.fun = analyzeTrialrdMod.lme4)
Deviances <- NULL
for(i in traits){
Deviances[[i]] <- data.frame(Deviance.MM(fmfit[[i]], rdfmfit[[i]]))[2,6:8]
rownames(Deviances[[i]]) <- i
}refitting model(s) with ML (instead of REML)
refitting model(s) with ML (instead of REML)
refitting model(s) with ML (instead of REML)
refitting model(s) with ML (instead of REML)ResDeviances <- data.frame(t(sapply(Deviances, FUN = rbind)))Table 2. Deviance Analysis for cassava foliar disease
H2 <- sapply(fmfit, FUN = getVarComp.lme4) %>% t() %>% as.data.frame()
colnames(H2) <- c("VarClone", "VarRes")
MediasFix <- as.matrix(sapply(fmfit, FUN = (fixef))) %>%
.[rownames(.) == "(Intercept)"] %>% data.frame(Mean = .)
H2 <- cbind(H2, MediasFix)
H2 <- H2 %>% mutate(VarClone = as.numeric(VarClone),
VarRes = as.numeric(VarRes),
VarFen = VarClone + VarRes,
H2 = VarClone/VarFen,
CVg = sqrt(VarClone)/Mean,
CVe = sqrt(VarRes)/Mean)
H2[,"Mean"] <- NULLTable 3. Heritabilities of cassava foliar disease
Obtain the Mean + BLUPs of the clones
MediasFix <- as.matrix(sapply(fmfit, FUN = (fixef)))
MediasFix[2:32, ] <- MediasFix[2:32,] +
matrix(rep(MediasFix[1,], each = 31), nrow = 31, ncol = 4,
byrow = F)
MediasFix <- as.data.frame(MediasFix)
rownames(MediasFix)[1] <- "controlClones"
MediasFix$CLONE <- rownames(MediasFix)
MediasFix %<>% filter(CLONE %like% "control") %>%
as.data.frame() %>% dplyr::select(CLONE, everything())
rownames(MediasFix) <- NULL
MediasFix$CLONE <- gsub(pattern = "control", replacement = "", x = MediasFix$CLONE)
MediasFix %>% filter(CLONE != "Clones") -> MediasFixObtain the Clones BLUPS
BLUPsAle <- lapply(fmfit, FUN = getBLUPs.lme4)
BLUPSDisea <- data.frame(CLONE = rownames(BLUPsAle[1]$Anth))
for(i in names(BLUPsAle)){
drg<-data.frame(CLONE = rownames(BLUPsAle[[i]]), stringsAsFactors=F)
drg[,i] <-BLUPsAle[[i]]
BLUPSDisea<-merge(BLUPSDisea,drg,by="CLONE",all.x=T)
}
BLUPSDisea <- BLUPSDisea %>% filter(CLONE %like% ":1")
BLUPSDisea$CLONE <- gsub(pattern = ":1", replacement = "", BLUPSDisea$CLONE)
BLUPS <- rbind(BLUPSDisea, MediasFix)
saveRDS(BLUPS, here::here("output", "BLUPsDisease.RDS"))Table 4. Blups of the accessions for cassava foliar diseases
Fig 1. Distribution of the BLUPs estimated for cassava foliar diseases
Warning: Removed 72 rows containing non-finite values (stat_density).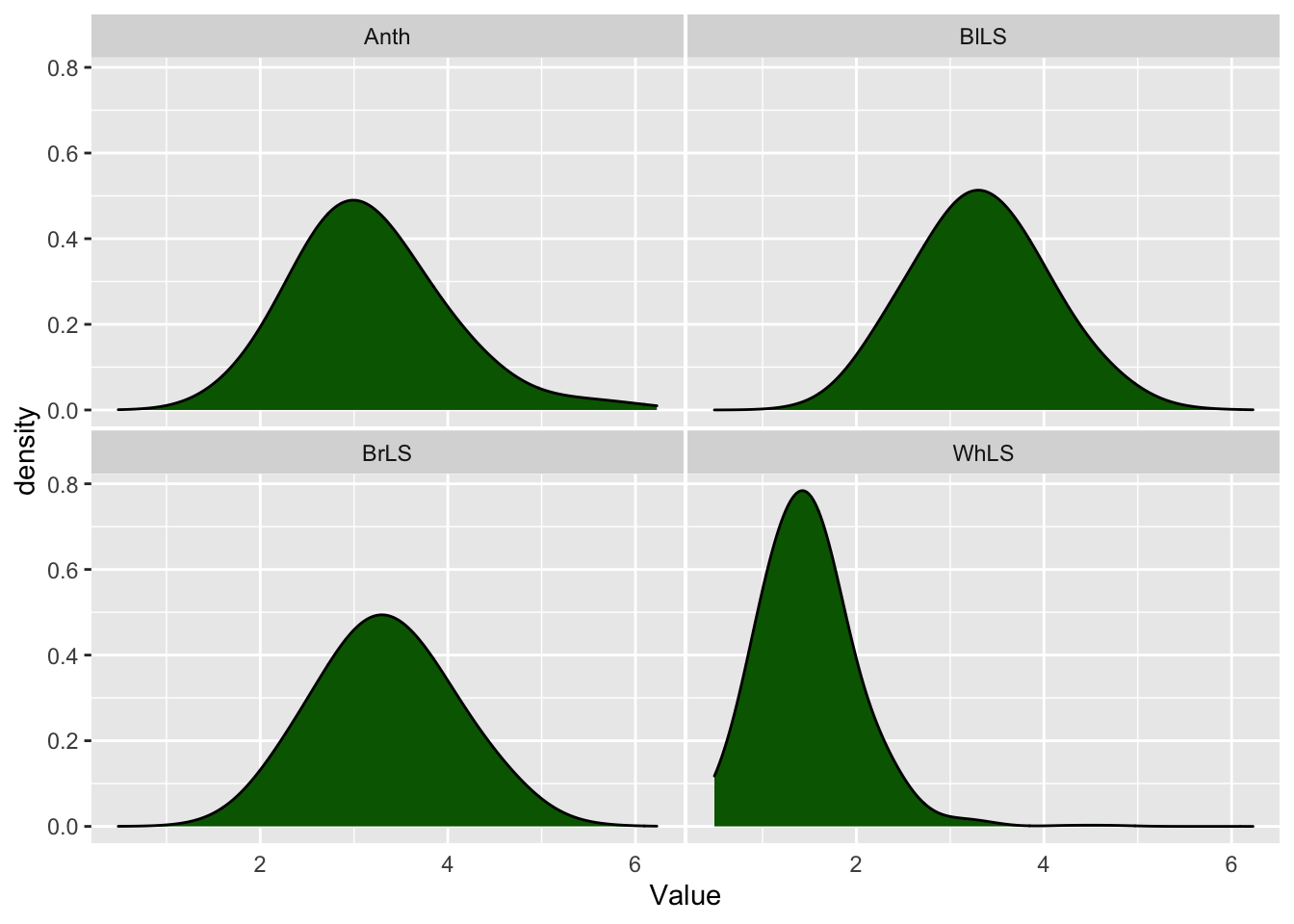
| Version | Author | Date |
|---|---|---|
| 90b66eb | LucianoRogerio | 2022-03-08 |
Mixed Models
Yield Traits
suppressMessages(library(lme4)); suppressMessages(library(tidyverse))
library(reactable); library(here)
AgroData <- readRDS(file = here::here("output", "DadosFenotipicos.rds"))
head(AgroData) Ano Campo Fazenda Local Linha Coluna Stand Trait trial studyDesign
1 2011 Agroverde1 CNPMF CruzAlmas 27 7 18 DMC 1 DBC
2 2011 Agroverde1 CNPMF CruzAlmas 25 14 17 DMC 1 DBC
3 2011 Agroverde1 CNPMF CruzAlmas 16 24 20 DMC 1 DBC
4 2011 Agroverde1 CNPMF CruzAlmas 7 2 16 DMC 1 DBC
5 2011 Agroverde1 CNPMF CruzAlmas 4 11 17 DMC 1 DBC
6 2011 Agroverde1 CNPMF CruzAlmas 3 20 17 DMC 1 DBC
clone rep check new y
1 BGM-0023 1 999 1 29.91
2 BGM-0023 2 999 1 29.93
3 BGM-0023 3 999 1 31.52
4 BGM-0025 1 999 1 35.34
5 BGM-0025 2 999 1 26.68
6 BGM-0025 3 999 1 27.92Table 5. Data entry to perform the mxed model analysis for Agronomic Traits
Trials <- unique(AgroData$trial)
Results <- tibble()
for(i in Trials){
traits <- AgroData %>% filter(trial %in% i) %>% .$Trait %>% unique %>% as.character
results <- tibble()
for(j in traits) {
try(MixedModels <- analyzeTrial.lme4(AgroData %>% filter(trial %in% i & Trait %in% j)))
try(result <- tibble(Trial = i,
Trait = j,
VarG = as.data.frame(VarCorr(MixedModels))[,c("grp","vcov")] %>% .[1,2],
VarE = as.data.frame(VarCorr(MixedModels))[,c("grp","vcov")] %>% .[2,2],
H2 = VarG/(VarG + VarE),
Real = suppressWarnings(MuMIn::r.squaredGLMM(MixedModels)[2])))
try(results <- rbind(results, result))
rm(MixedModels); rm(result)
}
Results <- rbind(Results, results)
rm(traits); rm(results)
}boundary (singular) fit: see help('isSingular')
boundary (singular) fit: see help('isSingular')
boundary (singular) fit: see help('isSingular')
boundary (singular) fit: see help('isSingular')
boundary (singular) fit: see help('isSingular')Error in `contrasts<-`(`*tmp*`, value = contr.funs[1 + isOF[nn]]) :
contrasts can be applied only to factors with 2 or more levels
Error in VarCorr(MixedModels) : object 'MixedModels' not found
Error in rbind(results, result) : object 'result' not foundWarning in rm(MixedModels): object 'MixedModels' not foundWarning in rm(result): object 'result' not foundboundary (singular) fit: see help('isSingular')fixed-effect model matrix is rank deficient so dropping 1 column / coefficient
fixed-effect model matrix is rank deficient so dropping 1 column / coefficient
fixed-effect model matrix is rank deficient so dropping 1 column / coefficient
fixed-effect model matrix is rank deficient so dropping 1 column / coefficient
fixed-effect model matrix is rank deficient so dropping 1 column / coefficientYield data selection
Table 6. Heritability and reliability of all trials for yield traits
Estimação BLUPS e obtenção de médias corrigidas
library(here)
library(furrr)Loading required package: futurelibrary(tidyverse)
source(here::here("code", "MixedModelsFunctions.R"))
AgroDataSel <- readRDS(here::here("data", "DadosFenotipicosSel.rds")) %>%
mutate(trial = as.character(trial),
rep = as.character(rep),
repTrial = as.factor(paste(trial, rep, sep =":")))
#NCT <- 4
#plan(sequential)
#RhpcBLASctl::blas_set_num_threads(NCT)
traits <- table(AgroDataSel$Trait) %>% .[order(.)] %>% names
for(i in traits){
print(paste("Trait", i, sep = " "))
DataMM <- AgroDataSel %>% filter(Trait == i)
MM <- analyzeTrial.lme4Conj(DataMM)
blups <- ranef(MM)$clone + fixef(MM)[1]
Blups <- tibble(id = rownames(blups),
blups = blups$`(Intercept)`)
colnames(Blups)[2] <- i
file <- here::here("output", "MixedModels",
paste("Blups_", i, ".rds", sep = ""))
saveRDS(object = Blups, file = file)
rm(DataMM); rm(MM); rm(blups); rm(Blups); rm(file)
}[1] "Trait Vigor12M"
[1] "Trait NR"
[1] "Trait DRY"
[1] "Trait DMC"
[1] "Trait PTR"
[1] "Trait PPA"BlupsTraits <- readRDS(here::here("output", "MixedModels", "Blups_Vigor12M.rds"))
IDClones <- tibble(id = unique(AgroDataSel$clone) %>% .[order(.)])
BlupsTraits <- IDClones %>% left_join(BlupsTraits, by = "id")
for(i in traits[-1]){
filename <- paste("Blups_", i, ".rds", sep = "")
BlupsTraits <- BlupsTraits %>%
left_join(readRDS(here::here("output", "MixedModels", filename)))
colnames(BlupsTraits)[colnames(BlupsTraits) == "blups"] <- i
}Joining, by = "id"Joining, by = "id"
Joining, by = "id"
Joining, by = "id"
Joining, by = "id"saveRDS(object = BlupsTraits,
file = here::here("output", "BlupsFenHen.rds"))Fig 2. Distribution of the BLUPs estimated for cassava agronomic traits
Warning: Removed 681 rows containing non-finite values (stat_density).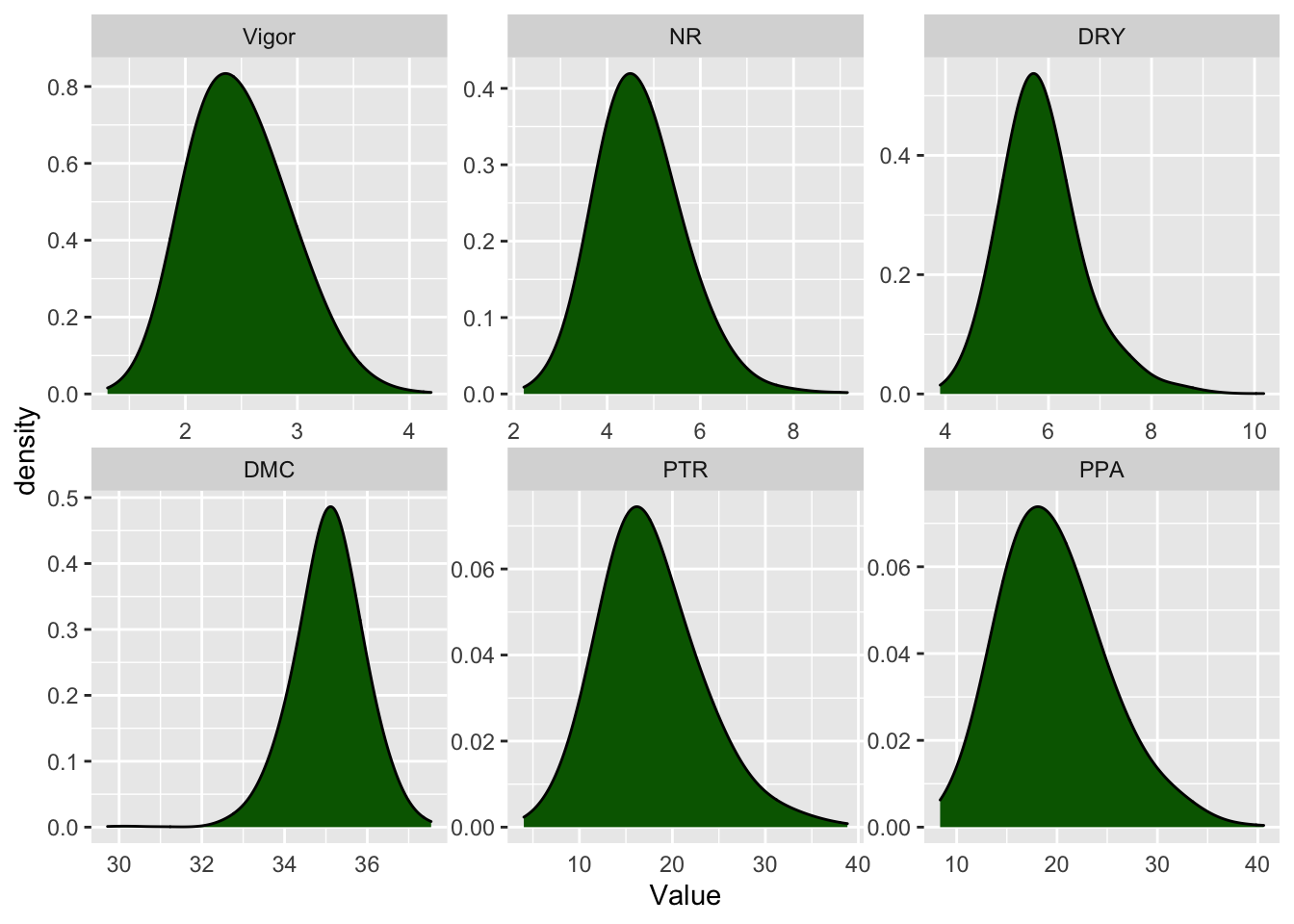
Joint BLUPS from Disease and Yield traits
AllBlups <- BLUPS %>% left_join(BlupsTraits, by = c("CLONE" = "id")) %>%
dplyr::rename(Vigor = Vigor12M)
saveRDS(AllBlups, file = here::here("output", "BLUPsDiseaseAgro.rds"))Table 7. BLUPs plus intercept for all disease and agronomic traits for cassava.
sessionInfo()R version 4.1.2 (2021-11-01)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS Big Sur 11.6.5
Matrix products: default
LAPACK: /Library/Frameworks/R.framework/Versions/4.1-arm64/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] furrr_0.2.3 future_1.24.0 lme4_1.1-28 sommer_4.1.5
[5] crayon_1.5.1 lattice_0.20-45 MASS_7.3-56 Matrix_1.4-1
[9] data.table_1.14.3 reactable_0.2.3 plyr_1.8.7 forcats_0.5.1
[13] stringr_1.4.0 dplyr_1.0.8 purrr_0.3.4 readr_2.1.2
[17] tidyr_1.2.0 tibble_3.1.6 ggplot2_3.3.5 tidyverse_1.3.1
[21] here_1.0.1
loaded via a namespace (and not attached):
[1] nlme_3.1-157 fs_1.5.2 lubridate_1.8.0 httr_1.4.2
[5] rprojroot_2.0.3 tools_4.1.2 backports_1.4.1 bslib_0.3.1
[9] utf8_1.2.2 R6_2.5.1 DBI_1.1.2 colorspace_2.0-3
[13] withr_2.5.0 tidyselect_1.1.2 compiler_4.1.2 git2r_0.30.1
[17] cli_3.2.0 rvest_1.0.2 xml2_1.3.3 labeling_0.4.2
[21] sass_0.4.1 scales_1.1.1 digest_0.6.29 minqa_1.2.4
[25] rmarkdown_2.13 MuMIn_1.46.0 pkgconfig_2.0.3 htmltools_0.5.2
[29] parallelly_1.30.0 dbplyr_2.1.1 fastmap_1.1.0 highr_0.9
[33] htmlwidgets_1.5.4 rlang_1.0.2 readxl_1.4.0 rstudioapi_0.13
[37] jquerylib_0.1.4 generics_0.1.2 farver_2.1.0 jsonlite_1.8.0
[41] crosstalk_1.2.0 magrittr_2.0.3 Rcpp_1.0.8.3 munsell_0.5.0
[45] fansi_1.0.3 lifecycle_1.0.1 stringi_1.7.6 whisker_0.4
[49] yaml_2.3.5 grid_4.1.2 parallel_4.1.2 listenv_0.8.0
[53] promises_1.2.0.1 haven_2.4.3 splines_4.1.2 hms_1.1.1
[57] knitr_1.38 pillar_1.7.0 boot_1.3-28 codetools_0.2-18
[61] stats4_4.1.2 reshape2_1.4.4 reprex_2.0.1 glue_1.6.2
[65] evaluate_0.15 modelr_0.1.8 vctrs_0.4.0 nloptr_2.0.0
[69] tzdb_0.3.0 httpuv_1.6.5 cellranger_1.1.0 gtable_0.3.0
[73] reactR_0.4.4 assertthat_0.2.1 xfun_0.30 broom_0.7.12
[77] later_1.3.0 workflowr_1.7.0 globals_0.14.0 ellipsis_0.3.2