Preparations needed for launching the analysis
Maeva Techer
2023-12-18
Last updated: 2023-12-18
Checks: 7 0
Knit directory:
locust-comparative-genomics/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20221025) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 9cf4d78. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: data/.DS_Store
Ignored: data/.Rhistory
Ignored: figures/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/1_workenvs-prep.Rmd) and
HTML (docs/1_workenvs-prep.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 53877fa | Maeva A. TECHER | 2023-12-18 | add pages |
Work environment
Environment
- Grace Cluster at Texas A&M University, College Station, Texas, USA
- Local Mac: macOS Monterey Version 12.6
- R version 4.2.1
Directories
| Path | Purpose |
|---|---|
/scratch/user/XXXX/locust-phase |
Working repository |
/scratch/user/XXXX/locust-phase/workflow |
Location of the Snakemake pipeline |
/scratch/user/XXXX/refgenomes/locust-complete |
Reference genome, annotation and index |
/scratch/user/XXXX/locust-phase/data/reads |
Raw FASTQ.GZ files |
/scratch/user/XXXX/locust-phase/data/kaiju |
Kaiju database and output |
/scratch/user/XXXX/locust-phase/data/trimming |
Trimmed FASTQ files |
/scratch/user/XXXX/locust-phase/data/alignment/STAR |
Output from STAR |
/scratch/user/XXXX/locust-phase/data/alignment/STAR_SALMON |
Output from STAR-Salmon |
/scratch/user/XXXX/locust-phase/data/alignment/STAR_RSEM |
Output from STAR-Salmon |
/scratch/user/XXXX/locust-phase/data/analysis |
R analysis (DGE) |
Snakemake pipeline
We use a Snakemake pipeline for each species. Therefore, it is essential to verify that each software is installed 1) locally or via a conda environment or 2) as a module or cloned via Github on the Grace cluster at Texas A&M University is essential.
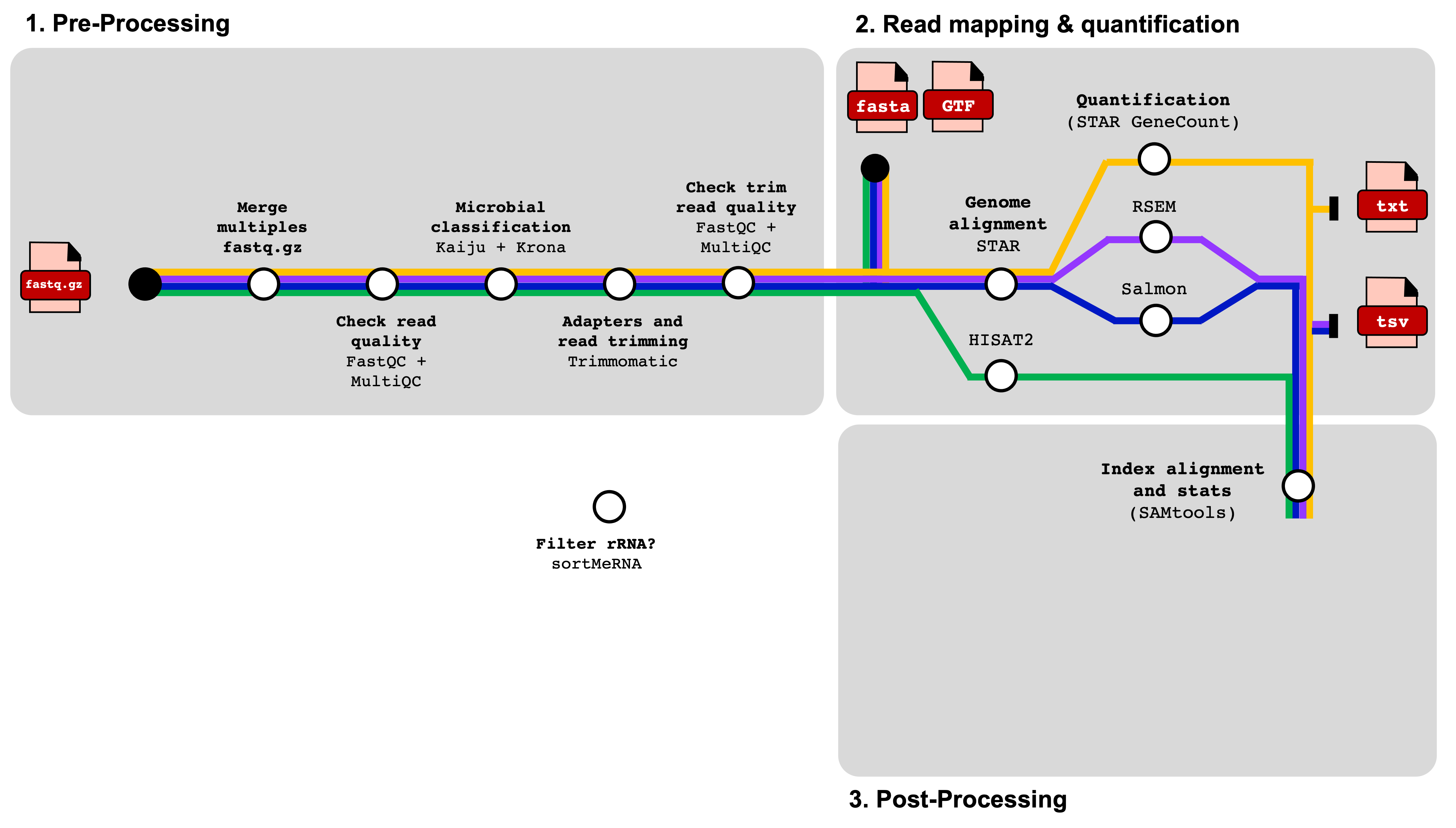
We built our Snakemake pipeline by launching small individual jobs to tailor each cluster parameter for the best memory and time efficiency.
To ease the indexing of our file and folder, we generate some shared
parameters which will be helpful in the future: 1) reference genome
directory path REFdir, 2) output directory path
OUTdir and 3) a list LOCUSTS containing sample
base name referred as locust.
### SET DIRECTORY PATHS FOR REFERENCE AND OUTPUT DATA
REFdir = "/scratch/user/maeva-techer/refgenomes"
OUTdir = "/scratch/user/maeva-techer/locust-rna/data"
### SAMPLES LIST AND OTHER PARAMETERS
LOCUSTS, = glob_wildcards(OUTdir + "/reads/{locust}_1.fastq.gz")
print(LOCUSTS)Softwares and database
Links to each software to add in the future
Use a conda environment
add what to install on the Macpro tower e.g., we install a conda environment called rna-seq
Use modules on Grace cluster
On Grace, each module may requires some dependencies, which is why we
need to ensure they will be loaded together. For this we use the
function module spider [targeted software] w/o the
version.
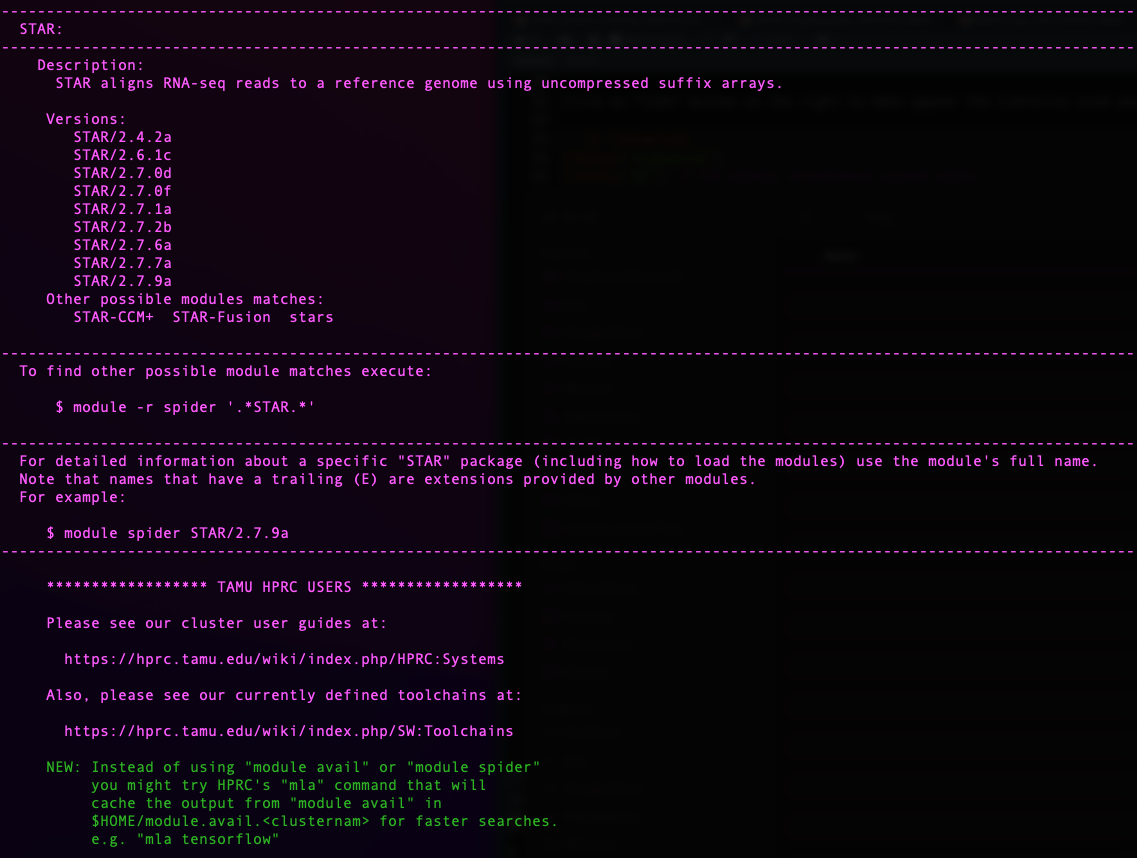 Today we will need the
following software:
Today we will need the
following software:
- to launch the Snakemake pipeline
module load GCC/11.2.0 OpenMPI/4.1.1 snakemake/6.10.0 Biopython/1.79- to perform adapter trimming and QC
module load Trimmomatic/0.39-Java-11
module load FastQC/0.11.9-Java-11- to perform short read mapping
module load GCC/11.2.0 STAR/2.7.9aR packages neccessary
List of R packages used
CRAN
data.table: https://cran.r-project.org/package=data.table
dplyr: https://cran.r-project.org/package=dplyr
reshape2: https://cran.r-project.org/package=reshape2
ggplot2: https://cran.r-project.org/package=ggplot2
ggrepel: https://cran.r-project.org/package=ggrepel
Bioconductor
DESeq2: https://bioconductor.org/packages/DESeq2/
apeglm: https://bioconductor.org/packages/apeglm/
EnhancedVolcano: https://bioconductor.org/packages/EnhancedVolcano/
edgeR: https://www.bioconductor.org/packages/edgeR/
sessionInfo()R version 4.3.1 (2023-06-16)
Platform: x86_64-apple-darwin20 (64-bit)
Running under: macOS Sonoma 14.1.2
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-x86_64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-x86_64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Asia/Tokyo
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] workflowr_1.7.1
loaded via a namespace (and not attached):
[1] vctrs_0.6.4 httr_1.4.7 cli_3.6.1 knitr_1.43
[5] rlang_1.1.1 xfun_0.40 stringi_1.7.12 processx_3.8.2
[9] promises_1.2.1 jsonlite_1.8.7 glue_1.6.2 rprojroot_2.0.3
[13] git2r_0.32.0 htmltools_0.5.6 httpuv_1.6.11 ps_1.7.5
[17] sass_0.4.7 fansi_1.0.5 rmarkdown_2.24 jquerylib_0.1.4
[21] tibble_3.2.1 evaluate_0.21 fastmap_1.1.1 yaml_2.3.7
[25] lifecycle_1.0.3 whisker_0.4.1 stringr_1.5.0 compiler_4.3.1
[29] fs_1.6.3 pkgconfig_2.0.3 Rcpp_1.0.11 rstudioapi_0.15.0
[33] later_1.3.1 digest_0.6.33 R6_2.5.1 utf8_1.2.3
[37] pillar_1.9.0 callr_3.7.3 magrittr_2.0.3 bslib_0.5.1
[41] tools_4.3.1 cachem_1.0.8 getPass_0.2-2