Syntenic relations and graphs
Emily Baker, Maeva Techer
2025-06-01
Last updated: 2025-06-01
Checks: 6 1
Knit directory:
locust-comparative-genomics/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20221025) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 4e391c3. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: code/.DS_Store
Ignored: code/scripts/.DS_Store
Ignored: code/scripts/pal2nal.v14/.DS_Store
Ignored: data/.DS_Store
Ignored: data/DEG_results/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/americana/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/cancellata/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/cancellata/Thorax/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/cubense/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/davidO/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/gregaria/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/nitens/.DS_Store
Ignored: data/DEG_results/Bulk_RNAseq/piceifrons/.DS_Store
Ignored: data/DEG_results/RNAi/.DS_Store
Ignored: data/DEG_results/RNAi/All/.DS_Store
Ignored: data/DEG_results/RNAi/All_GFP/.DS_Store
Ignored: data/DEG_results/RNAi/All_control/.DS_Store
Ignored: data/DEG_results/RNAi/All_no_rRNA/.DS_Store
Ignored: data/DEG_results/RNAi/Head/.DS_Store
Ignored: data/DEG_results/RNAi/Head_control/.DS_Store
Ignored: data/DEG_results/RNAi/Head_no_rRNA/.DS_Store
Ignored: data/DEG_results/RNAi/Thorax/.DS_Store
Ignored: data/DEG_results/RNAi/Thorax_no_rRNA/.DS_Store
Ignored: data/DEG_results/gregaria/
Ignored: data/DEG_results/single_cell/.DS_Store
Ignored: data/WGCNA/.DS_Store
Ignored: data/WGCNA/input/.DS_Store
Ignored: data/WGCNA/input/Bulk_RNAseq/.DS_Store
Ignored: data/WGCNA/output/.DS_Store
Ignored: data/WGCNA/output/Bulk_RNAseq/.DS_Store
Ignored: data/behavioral_data/.DS_Store
Ignored: data/behavioral_data/Raw_data/.DS_Store
Ignored: data/list/.DS_Store
Ignored: data/list/Bulk_RNAseq/.DS_Store
Ignored: data/list/GO_Annotations/.DS_Store
Ignored: data/list/excluded_loci/.DS_Store
Ignored: data/orthofinder/.DS_Store
Ignored: data/orthofinder/Polyneoptera/.DS_Store
Ignored: data/orthofinder/Polyneoptera/Results_I2_iqtree/.DS_Store
Ignored: data/orthofinder/Polyneoptera/Results_I2_withDaust/.DS_Store
Ignored: data/orthofinder/Polyneoptera/Results_I2_withDaust/Orthogroups/.DS_Store
Ignored: data/orthofinder/Schistocerca/.DS_Store
Ignored: data/orthofinder/Schistocerca/Results_I2/.DS_Store
Ignored: data/orthofinder/Schistocerca/Results_I2/Orthogroups/.DS_Store
Ignored: data/overlap/.DS_Store
Ignored: data/overlap/Bulk_RNAseq/.DS_Store
Ignored: data/overlap/Bulk_RNAseq/cancellata/
Ignored: data/pathway_enrichment/.DS_Store
Ignored: data/pathway_enrichment/custom_sgregaria_orgdb/.DS_Store
Ignored: data/readcounts/.DS_Store
Ignored: data/readcounts/Bulk_RNAseq/.DS_Store
Ignored: data/readcounts/RNAi/.DS_Store
Untracked files:
Untracked: data/RefSeq/
Unstaged changes:
Modified: analysis/2_genome_quality.Rmd
Modified: analysis/2_orthologs-prediction.Rmd
Modified: analysis/2_synteny-graphs.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/2_synteny-graphs.Rmd) and
HTML (docs/2_synteny-graphs.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 4e391c3 | Maeva TECHER | 2025-05-30 | add new analysis orthology, synteny |
| html | 4e391c3 | Maeva TECHER | 2025-05-30 | add new analysis orthology, synteny |
| Rmd | b982319 | Maeva TECHER | 2025-03-03 | update font |
| html | b982319 | Maeva TECHER | 2025-03-03 | update font |
| html | f6a4762 | Maeva TECHER | 2025-02-27 | Build site. |
| Rmd | faf2db3 | Maeva TECHER | 2025-01-13 | update markdown |
| html | faf2db3 | Maeva TECHER | 2025-01-13 | update markdown |
| html | 3fa8e62 | Maeva TECHER | 2024-11-09 | updated analysis |
| html | edb70fe | Maeva TECHER | 2024-11-08 | overlap and deg results created |
| html | ba35b82 | Maeva A. TECHER | 2024-06-20 | Build site. |
| html | 86fd6ad | Maeva A. TECHER | 2024-05-14 | Build site. |
| Rmd | 564d596 | Maeva A. TECHER | 2024-05-14 | wflow_publish("analysis/2_synteny-graphs.Rmd") |
| html | e39d280 | Maeva A. TECHER | 2024-01-30 | Build site. |
| html | f701a01 | Maeva A. TECHER | 2024-01-30 | reupdate |
| html | 50ee4ca | Maeva A. TECHER | 2024-01-24 | Build site. |
| html | 1b09cbe | Maeva A. TECHER | 2024-01-24 | remove |
| html | f77543d | Maeva A. TECHER | 2023-12-18 | Build site. |
| Rmd | 53877fa | Maeva A. TECHER | 2023-12-18 | add pages |
1. Load packages and prepare the R environment
These instructions are for installing GENESPACE within
on the TAMU Grace cluster. While a new version of
OrthoFinder/3.00 has been used for the gene orthology,
GENESPACE works only with OrthoFinder/2.5.5,
thus all downstream modules are loaded in regards to this.
1.To install GENESPACE itself, you first need to load R and specify path for local packages as described here
To run R without bothering other users, we will claim one interactive node to make sure we can proactively update the package if there are some issues:
srun --ntasks 1 --cpus-per-task 16 --mem 50G --time 05:00:00 --pty bashWe need to load the needed modules as follow
ml GCC/12.3.0 OpenMPI/4.1.5 R_tamu/4.3.2 OrthoFinder/2.5.5 MCScanX/2024.12.19
ml WebProxy # if you are on a core and not on the login node
export R_LIBS=$SCRATCH/R_LIBS_USER/4.3.2-foss-2023a:$R_LIBS- Once R is active, run the following command in the R terminal:
R # Launches R terminal
if (!requireNamespace("devtools", quietly = TRUE))
install.packages("devtools")
install.packages("dbscan", type = "source", INSTALL_opts = "--no-test-load") # if some issues with GENESPACE install
devtools::install_github("jtlovell/GENESPACE", force = TRUE)- Verify that GENESPACE have been installed by running
library(GENESPACE)If no error message appears, then GENESPACE has been installed correctly. After this step, you may exit the R terminal with the command
q()2. Running with our data
The code provided below is meant to generate GENESPACE results from a previous OrthoFinder run. Slight modifications can be made to run OrthoFinder within the GENESPACE pipeline.
- Create the genome repository
GENESPACE expects a directory called the genomeRepo with
a specific file structure for each species as follows:
└── genomeRepo
└── species_gene_id
├── species_gene_id_genomic_gff.gz
└── species_gene_id_translated_cds.faa.gzYou must create a folder under the genomeRepo for each
species you want to run GENESPACE on and be sure to name
the folder the same as the gene id you assigned to the species in
OrthoFinder!
- Create a file called
genespace-script.Rand copy and paste the following code into it:
library(GENESPACE)
genomeRepo <- "/scratch/group/songlab/maeva/LocustsGenomeEvolution/Polyneoptera_FINAL/13_GENESPACE/genomeRepo"
wd <- "/scratch/group/songlab/maeva/LocustsGenomeEvolution/Polyneoptera_FINAL/13_GENESPACE/genespace"
path2mcscanx <- "/sw/eb/sw/MCScanX/2024.12.19-foss-2023a/bin/"
########################## CHANGE TO YOUR GENOME NAMES #######################
#genomes2run <- c("Lmigr", "Sgreg", "Scanc", "Samer", "Snite", "Spice", "Sscub", "Asimp", "Gbima", "Glong","Csecu", "Brsri", "Pamer")
#genomes2run <- c( "Sgreg", "Scanc", "Samer", "Snite", "Spice", "Sscub", "Asimp", "Csecu", "Brsri", "Pamer")
genomes2run <- c( "Sgreg", "Scanc", "Samer", "Snite", "Spice", "Sscub", "Lmigr")
################################################################################
parsedPaths <- parse_annotations(
rawGenomeRepo = genomeRepo,
genomeDirs = genomes2run,
genomeIDs = genomes2run,
presets = "ncbi",
genespaceWd = wd)
gpar <- init_genespace(
wd = wd,
path2mcscanx = path2mcscanx,
rawOrthofinderDir = "/scratch/group/songlab/maeva/LocustsGenomeEvolution/Polyneoptera_FINAL/5_OrthoFinder/fasta/Results_May26_iqtree",
genomeIDs = genomes2run
) # Note: If you do not have a previous OrthoFinder run you want to run GENESPACE on,
# just leave this option and only include the wd and path2mcscanx options and
# GENESPACE will automatically run OrthoFinder for you
out <- run_genespace(gpar)
saveRDS(out, file = "genespace_output.rds")- Reload the session to customize the riparian plot
We are making plots below with original sense of the chromosome and reordered ones to see better the synteny. We also made some plots to highlight the inversion blocks. We used S. gregaria as reference for most of the synteny mapping and ordered the species to locusts on top and sedentary on the bottom.
out <- readRDS("genespace_output.rds")
# View available genome IDs and paths
out$genomes
# Visualize dotplot for two species
plot_dotplot(out, refGenome = "Sgreg", qryGenome = "Spice")
# Plot summary synteny blocks
plot_genomespace(out)
library(ggplot2)
ggthemes <- ggplot2::theme(
panel.background = ggplot2::element_rect(fill = "white"))
customPal <- colorRampPalette(
c("darkorange", "skyblue", "darkblue", "purple", "darkred", "salmon"))
customPal2 <- colorRampPalette(
c("blue2", "darkblue"))
# Set output PDF file
pdf("riparian_plots.pdf", width = 10, height = 7) # adjust width/height as needed
# Original chromosome sense from NCBI and order from GENESPACE
ripDat1 <- plot_riparian(
gsParam = out,
palette = customPal,
braidAlpha = .75,
chrFill = "lightgrey",
addThemes = ggthemes,
refGenome = "Sgreg",
chrLabFontSize = 10)
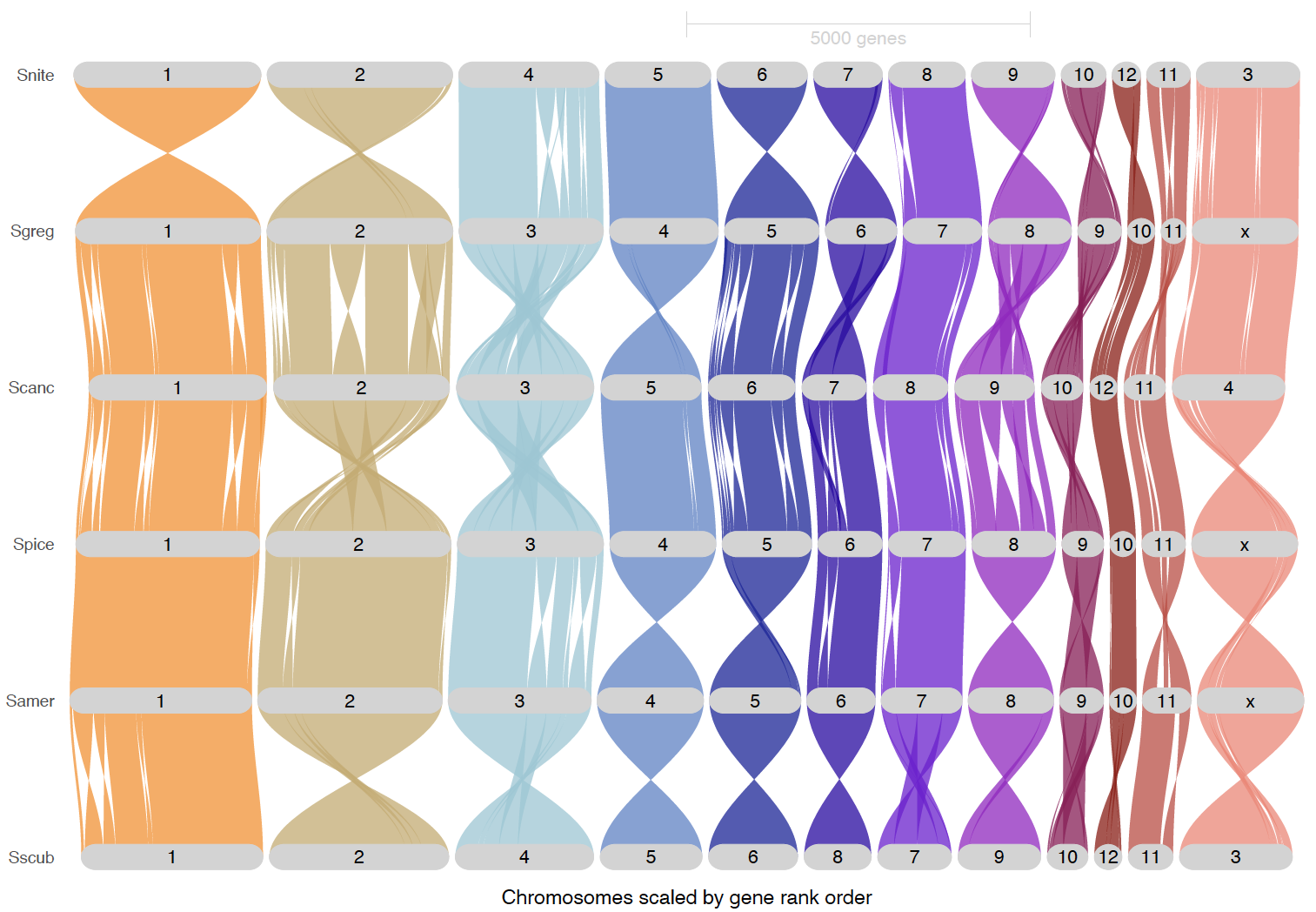
# Original chromosome sense from NCBI
ripDat2 <- plot_riparian(
gsParam = out,
palette = customPal,
invertTheseChrs = invchr,
genomeIDs = c("Snite", "Sscub", "Samer","Scanc", "Spice", "Sgreg"),
braidAlpha = .75,
chrFill = "lightgrey",
addThemes = ggthemes,
refGenome = "Sgreg",
chrLabFontSize = 10)
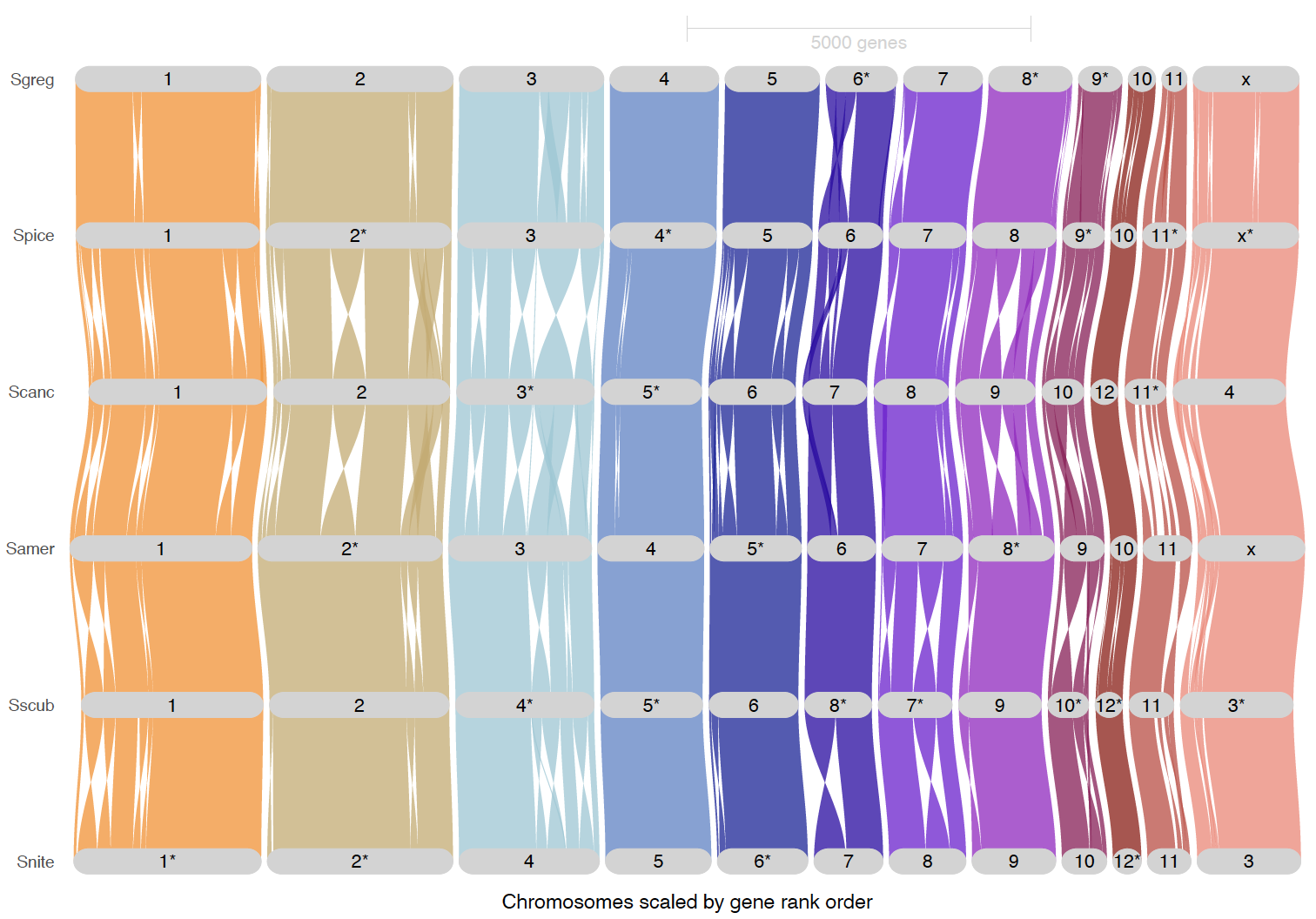
# Chromosome rearranged
invchr <- data.frame(
genome = c( "Snite", "Snite", "Spice", "Samer", "Scanc", "Sscub", "Scanc","Spice", "Sscub", "Snite", "Samer", "Sgreg", "Sscub", "Sscub", "Sgreg", "Samer", "Sgreg", "Spice", "Sscub", "Snite","Sscub", "Scanc", "Spice", "Spice", "Sscub"),
chr = c(1, 2, 2, 2, 3, 4, 5, 4, 5, 6, 5, 6, 8, 7, 8, 8, 9, 9, 10, 12, 12, 11, 11, "X", 3))
ripDat3 <- plot_riparian(
gsParam = out,
minChrLen2plot = 10,
invertTheseChrs = invchr,
refGenome = "Sgreg",
genomeIDs = c("Snite", "Sscub", "Samer","Scanc", "Spice", "Sgreg"),
chrLabFontSize = 7)
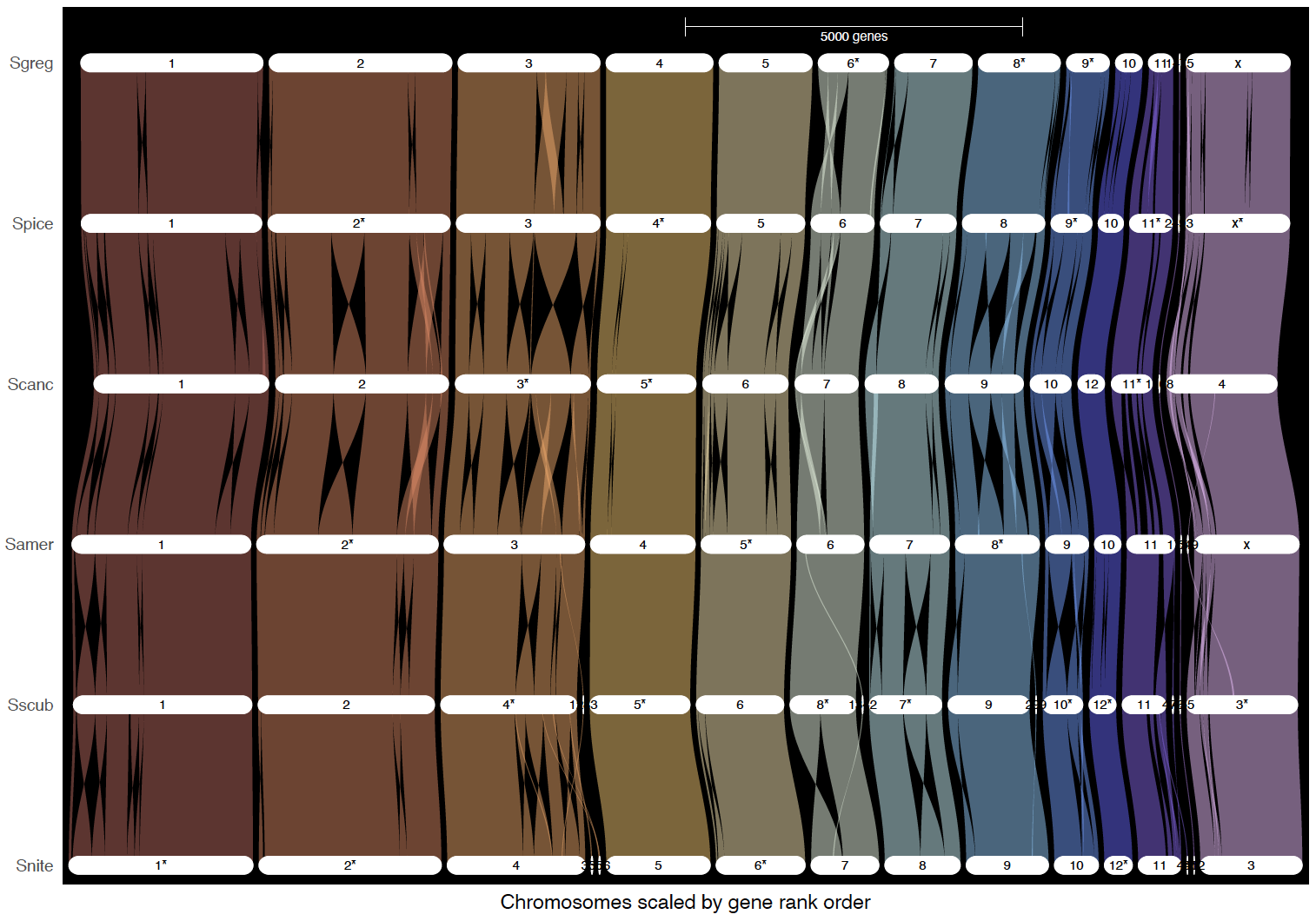
ripDat4 <- plot_riparian(
gsParam = out,
palette = customPal,
invertTheseChrs = invchr,
genomeIDs = c("Snite", "Sscub", "Samer","Scanc", "Spice", "Sgreg"),
braidAlpha = .75,
chrFill = "lightgrey",
addThemes = ggthemes,
refGenome = "Sgreg",
chrLabFontSize = 10)
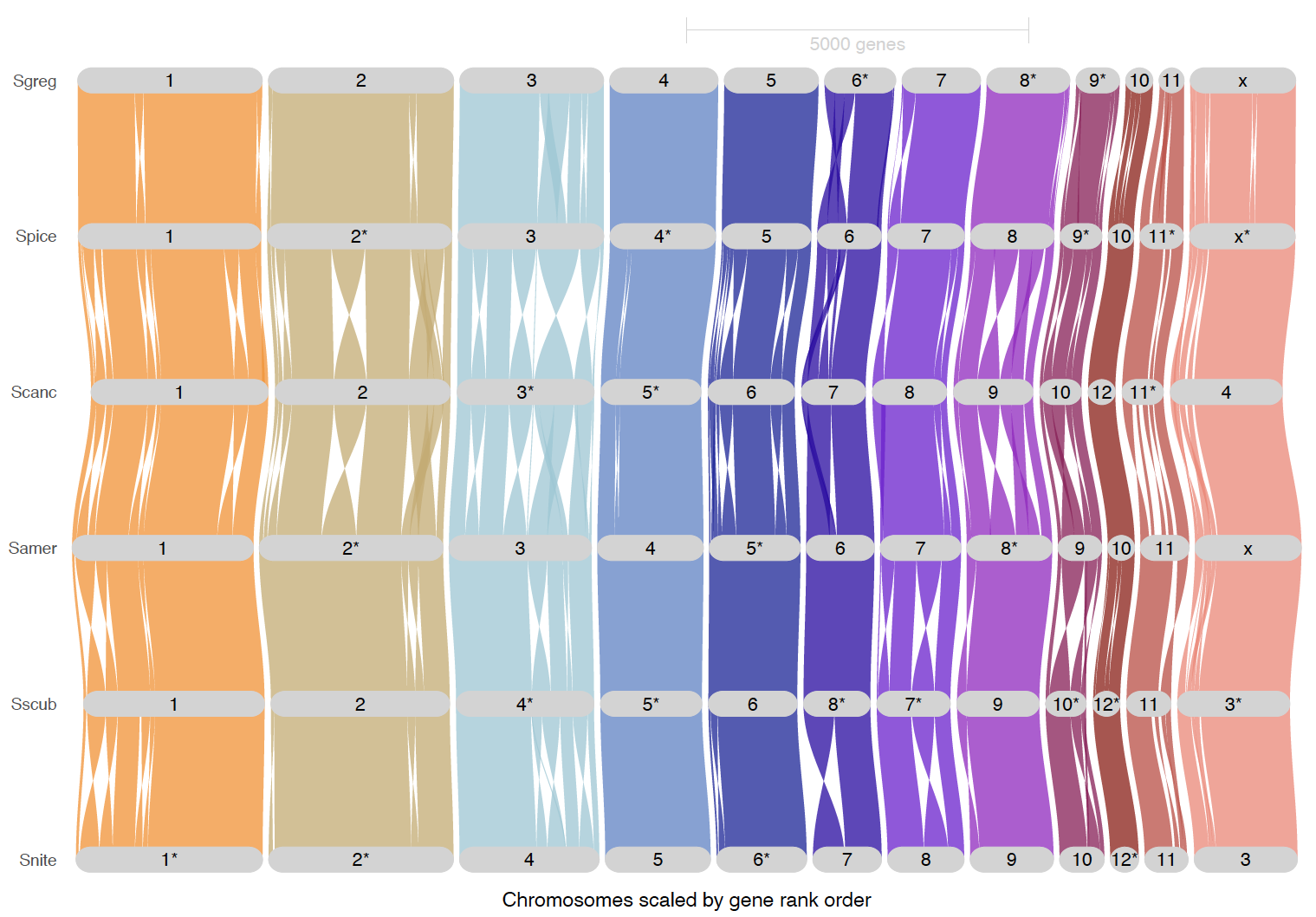
# Inversions highlighted
ripDat5 <- plot_riparian(
gsParam = out,
palette = customPal,
braidAlpha = .75,
chrFill = "lightgrey",
addThemes = ggthemes,
refGenome = "Sgreg",
chrLabFontSize = 10,
inversionColor = "black")
ripDat6 <- plot_riparian(
gsParam = out,
palette = customPal,
invertTheseChrs = invchr,
genomeIDs = c("Snite", "Sscub", "Samer","Scanc", "Spice", "Sgreg"),
braidAlpha = .75,
chrFill = "lightgrey",
addThemes = ggthemes,
refGenome = "Sgreg",
chrLabFontSize = 10,
inversionColor = "black")
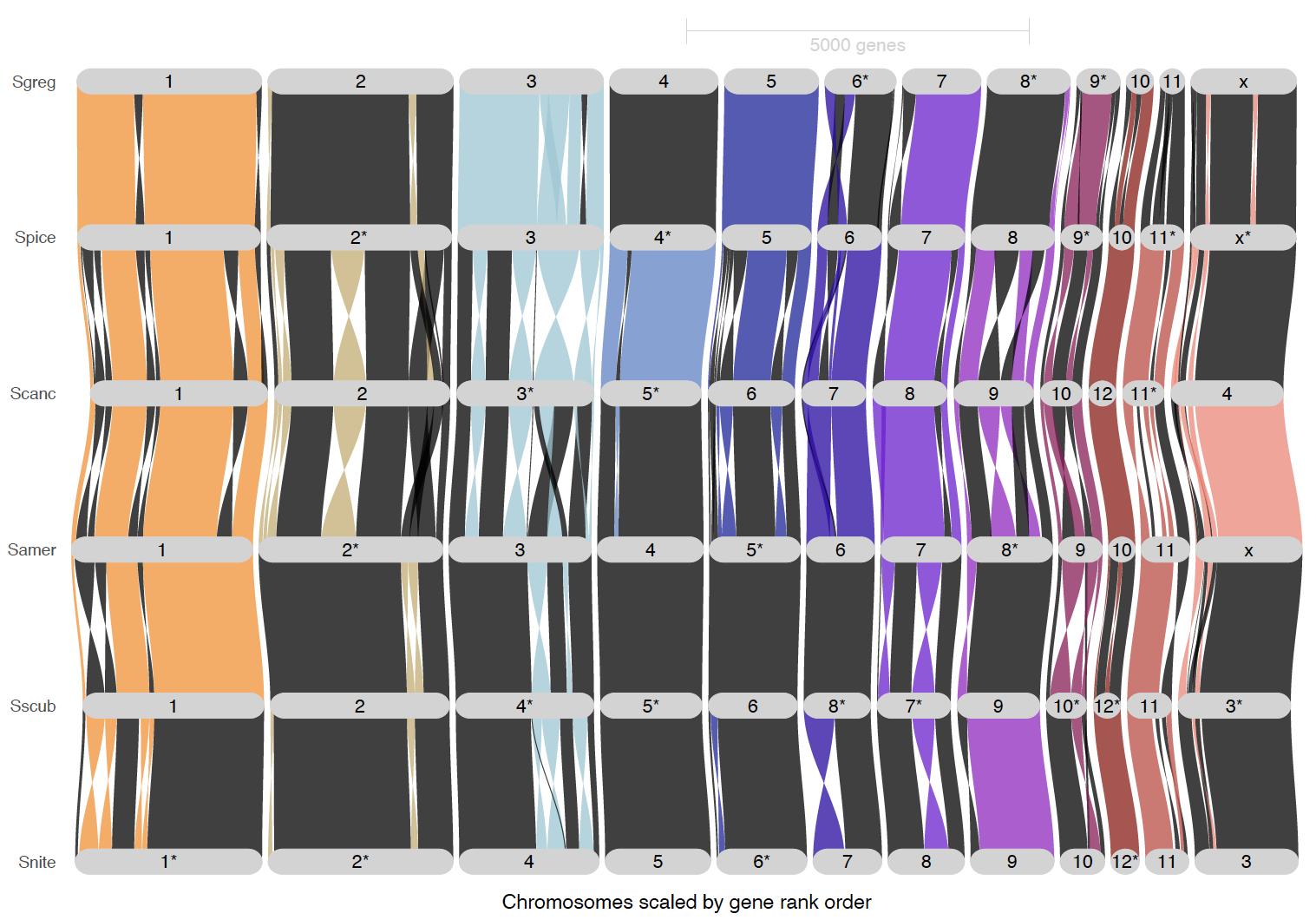
ripDat7 <- plot_riparian(
gsParam = out,
genomeIDs = c("Snite", "Sscub", "Samer","Scanc", "Spice", "Sgreg"),
braidAlpha = .75,
chrFill = "lightgrey",
refGenome = "Sgreg",
chrLabFontSize = 10,
addThemes = ggthemes,
inversionColor = "darkred")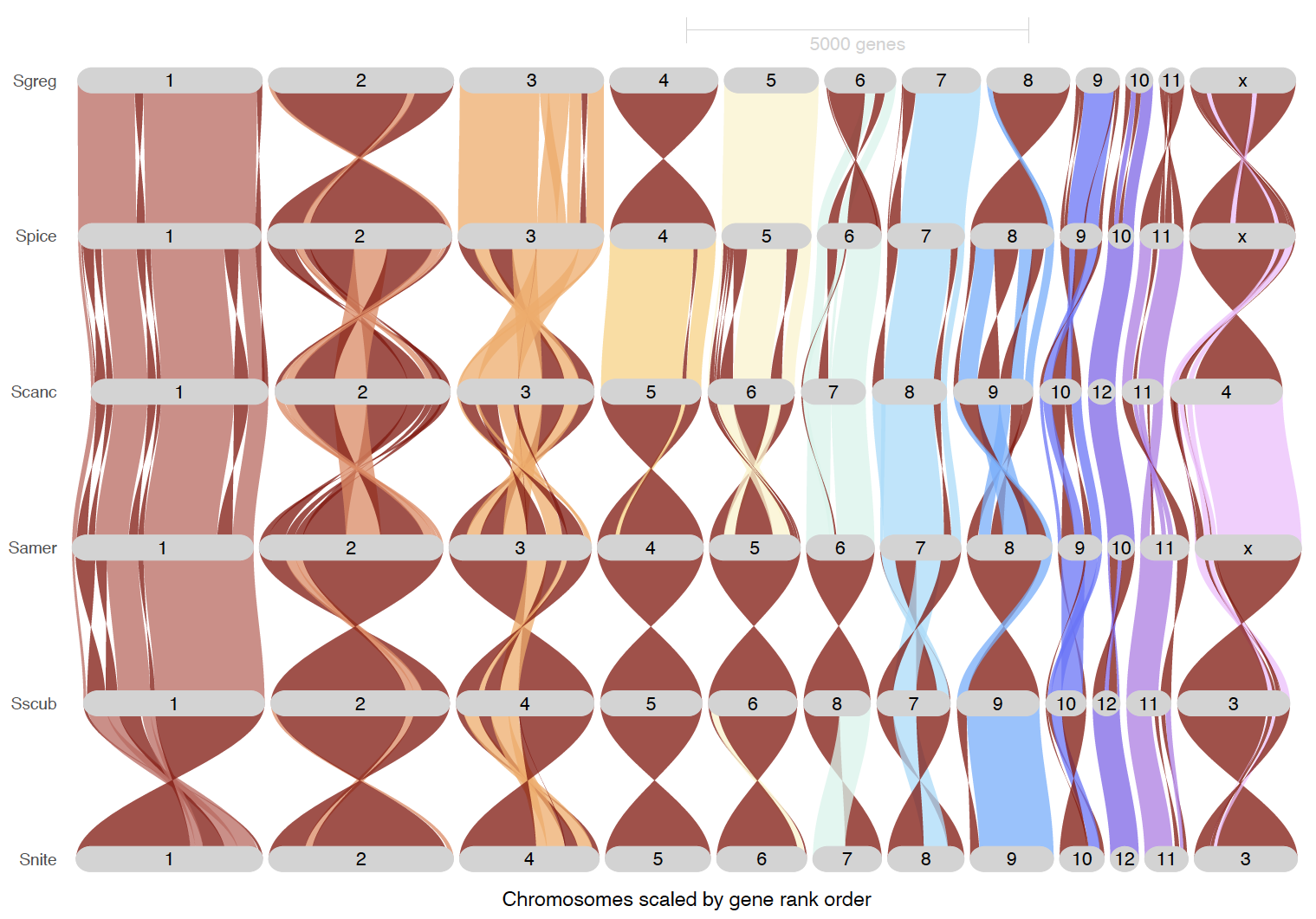
ripDat8 <- plot_riparian(
gsParam = out,
invertTheseChrs = invchr,
palette = customPal2,
genomeIDs = c("Snite", "Sscub", "Samer","Scanc", "Spice", "Sgreg"),
braidAlpha = .75,
chrFill = "lightgrey",
refGenome = "Sgreg",
chrLabFontSize = 10,
addThemes = ggthemes,
inversionColor = "darkred")
dev.off()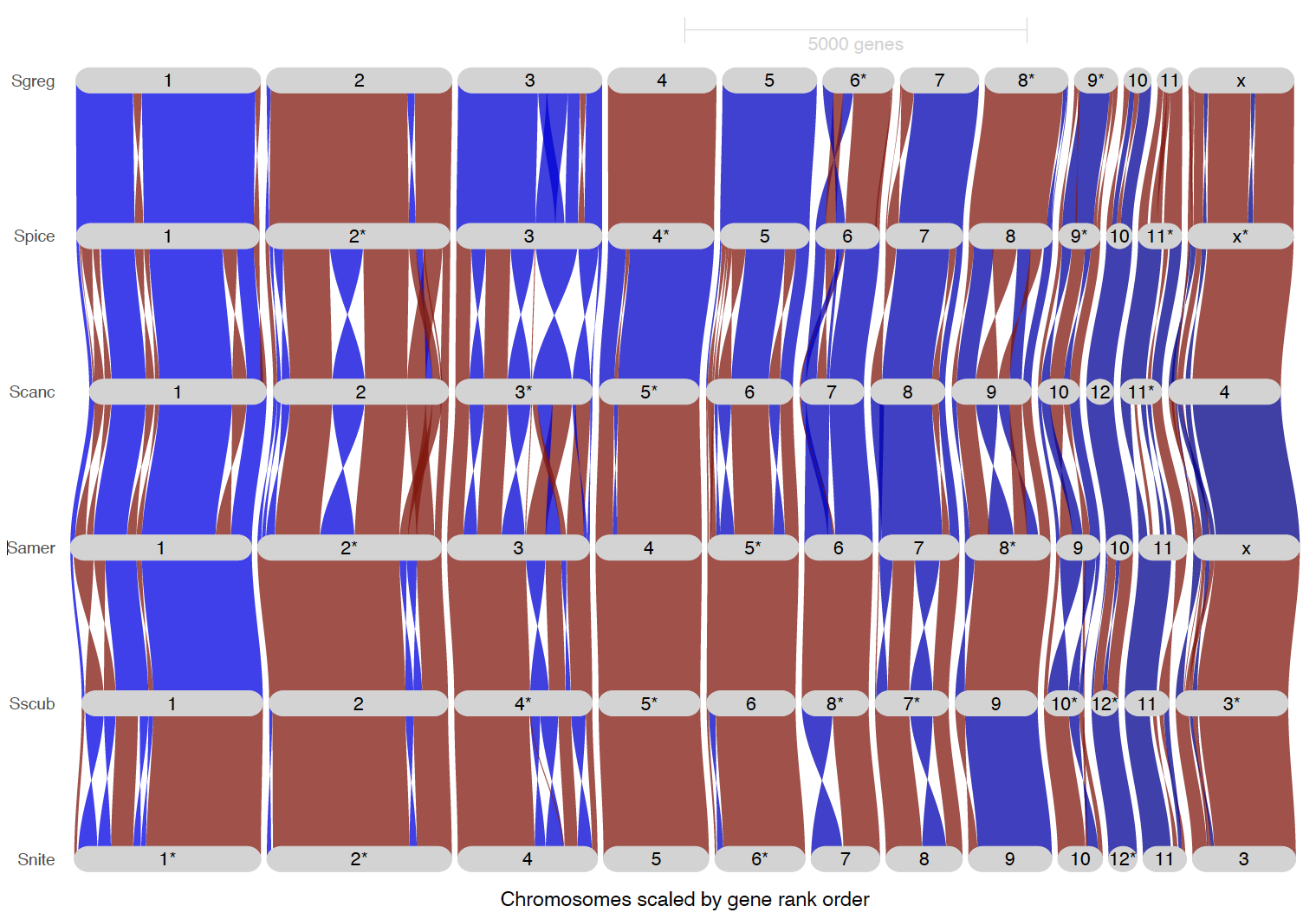
sessionInfo()R version 4.4.2 (2024-10-31)
Platform: aarch64-apple-darwin20
Running under: macOS Sequoia 15.5
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Asia/Tokyo
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
loaded via a namespace (and not attached):
[1] vctrs_0.6.5 cli_3.6.5 knitr_1.49 rlang_1.1.6
[5] xfun_0.51 stringi_1.8.4 promises_1.3.2 jsonlite_1.9.1
[9] workflowr_1.7.1 glue_1.8.0 rprojroot_2.0.4 git2r_0.35.0
[13] htmltools_0.5.8.1 httpuv_1.6.15 sass_0.4.9 rmarkdown_2.29
[17] evaluate_1.0.3 jquerylib_0.1.4 tibble_3.2.1 fastmap_1.2.0
[21] yaml_2.3.10 lifecycle_1.0.4 whisker_0.4.1 stringr_1.5.1
[25] compiler_4.4.2 fs_1.6.5 Rcpp_1.0.14 pkgconfig_2.0.3
[29] rstudioapi_0.17.1 later_1.4.1 digest_0.6.37 R6_2.6.1
[33] pillar_1.10.2 magrittr_2.0.3 bslib_0.9.0 tools_4.4.2
[37] cachem_1.1.0