Mapping reads to Schistocerca RefSeq genome
Maeva Techer
2022-11-07
Last updated: 2022-11-07
Checks: 7 0
Knit directory:
locust-phase-transition-RNAseq/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20221025) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version e6f6c2e. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: analysis/.DS_Store
Ignored: data/.DS_Store
Ignored: data/americana/.DS_Store
Ignored: data/americana/STAR_counts_4thcol/.DS_Store
Ignored: data/cancellata/.DS_Store
Ignored: data/cancellata/STAR_counts_4thcol/.DS_Store
Ignored: data/cubense/.DS_Store
Ignored: data/cubense/STAR_counts_4thcol/.DS_Store
Ignored: data/gregaria/.DS_Store
Ignored: data/gregaria/STAR_counts_4thcol/.DS_Store
Ignored: data/gregaria/list/.DS_Store
Ignored: data/metadata/.DS_Store
Ignored: data/nitens/.DS_Store
Ignored: data/nitens/STAR_counts_4thcol/.DS_Store
Ignored: data/piceifrons/.DS_Store
Ignored: data/piceifrons/.Rhistory
Ignored: data/piceifrons/DEseq2_SPICE_HEAD 2/
Ignored: data/piceifrons/DEseq2_SPICE_HEAD/.DS_Store
Ignored: data/piceifrons/STAR_counts_4thcol/.DS_Store
Ignored: data/piceifrons/edgeR_SPICE_HEAD/.DS_Store
Ignored: data/piceifrons/list/.DS_Store
Unstaged changes:
Modified: data/piceifrons/DEseq2_SPICE_HEAD/SPICE_HEAD_report.html
Modified: data/piceifrons/edgeR_SPICE_HEAD/SPICE_HEAD_report.html
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/map-refseq.Rmd) and HTML
(docs/map-refseq.html) files. If you’ve configured a remote
Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | 61256d8 | MaevaTecher | 2022-11-06 | Build site. |
| Rmd | 4e6386b | MaevaTecher | 2022-11-06 | workflowr::wflow_publish("analysis/map-refseq.RmD") |
| html | 1fa8b99 | MaevaTecher | 2022-11-01 | Build site. |
| html | 40deb86 | MaevaTecher | 2022-11-01 | Build site. |
| html | 83b00b8 | MaevaTecher | 2022-11-01 | Build site. |
| html | 05f27e6 | MaevaTecher | 2022-11-01 | Build site. |
| html | ec83778 | MaevaTecher | 2022-11-01 | Build site. |
| html | 834088c | MaevaTecher | 2022-10-31 | Build site. |
| html | 8481823 | MaevaTecher | 2022-10-31 | Build site. |
| html | ea70380 | MaevaTecher | 2022-10-31 | Build site. |
| Rmd | de64b6b | MaevaTecher | 2022-10-31 | wflow_publish(c("analysis/_site.yml", "analysis/map-refseq.Rmd", |
| html | 8b34f19 | MaevaTecher | 2022-10-30 | Build site. |
| html | bd04bb5 | MaevaTecher | 2022-10-30 | Build site. |
| html | 61f4b75 | MaevaTecher | 2022-10-30 | Build site. |
| Rmd | f65c630 | MaevaTecher | 2022-10-30 | wflow_publish(c("analysis/_site.yml", "analysis/gene-quant.Rmd", |
Load R libraries (install first from CRAN or Bioconductor)
library("knitr")
library("rmdformats")
library("tidyverse")
library("DT") # for making interactive search table
library("plotly") # for interactive plots
library("ggthemes") # for theme_calc
library("reshape2")
## Global options
options(max.print="10000")
knitr::opts_chunk$set(
echo = TRUE,
message = FALSE,
warning = FALSE,
cache = FALSE,
comment = FALSE,
prompt = FALSE,
tidy = TRUE
)
opts_knit$set(width=75)Short Reads mapping and Gene Quantification
STAR only
We used STAR for mapping reads to either 1) their own
species reference genome or 2) an alternate sister reference genome. The
pipeline is the same, except that the code will change index.
We index each genome using the following bash script:
#!/bin/bash
##NECESSARY JOB SPECIFICATIONS
#SBATCH --job-name=STARindex #Set the job name to "JobExample4"
#SBATCH --time=06:00:00 #Set the wall clock limit to 1hr and 30min
#SBATCH --ntasks=2 #Request 1 task
#SBATCH --cpus-per-task=4 #Request 1 task
#SBATCH --mem=30G #Request 100GB per node
module purge
module load GCC/11.2.0 STAR/2.7.9a
REFDIR="/scratch/user/maeva-techer/refgenomes/locusts_complete/index_rRNA_gregaria/STAR"
GENOME="/scratch/user/maeva-techer/refgenomes/locusts_complete/rRNA_gregaria.fasta"
ANNOTATION="/scratch/user/maeva-techer/refgenomes/locusts_complete/rRNA_gregaria.gtf"
STAR --runMode genomeGenerate --runThreadN 8 --genomeDir $REFDIR --genomeFastaFiles $GENOME --genomeSAindexNbases 10 --sjdbGTFfile $ANNOTATION --sjdbGTFfeatureExon exon --sjdbOverhang 149
The parameters were chosen for the following reasons:
--runMode genomeGenerate indicates we are in the mode to
build genome index
--genomeSAindexNbases 22 should be
min(18,log2[max(GenomeLength/NumberOfReferences,ReadLength)]) according
to the manual. We have a genome of ~9000000000 and with around 1475
contigs so our value is around 22.
--sjdbGTFfile $ANNOTATION --sjdbGTFfeatureExon exon we use
these parameters to indicates that we want the annotation file to be
already accounted for.
--sjdbOverhang 149 this should be ideally (mate_length -1),
and our reads are PE150.
########################################
# Snakefile rule
########################################
#Ahead of the alignment I will build independently the index for STAR, HiSat2 and Segemehl
rule STAR_align:
input:
index = REFdir + "/locusts_complete/index_{genome}/STAR",
annotation = REFdir + "/locusts_complete/{genome}.gtf",
read1 = OUTdir + "/trimming/{locust}_trim1P_1.fastq.gz",
read2 = OUTdir + "/trimming/{locust}_trim2P_2.fastq.gz"
params:
prefix = OUTdir + "/alignment/STAR/{genome}/{locust}_"
output:
OUTdir + "/alignment/STAR/{genome}/{locust}_Aligned.sortedByCoord.out.bam"
shell:
"""
module load GCC/11.2.0 STAR/2.7.9a
STAR --runThreadN 8 --genomeDir {input.index} --readFilesIn {input.read1} {input.read2} --outFileNamePrefix {params.prefix} --readFilesCommand zcat --genomeLoad NoSharedMemory --twopassMode Basic --sjdbGTFfile {input.annotation} --sjdbScore 2 --sjdbOverhang 149 --outFilterMultimapNmax 20 --alignSJoverhangMin 8 --alignSJDBoverhangMin 1 --outFilterMatchNmin 50 --outFilterMismatchNmax 999 --outFilterMismatchNoverReadLmax 0.04 --alignIntronMin 20 --alignIntronMax 1000000 --alignMatesGapMax 1000000 --outSAMunmapped Within --outFilterType BySJout --outSAMattributes NH HI AS NM MD --outSAMtype BAM SortedByCoordinate --quantMode TranscriptomeSAM GeneCounts --quantTranscriptomeBan IndelSoftclipSingleend --limitBAMsortRAM 32000000000 --seedSearchStartLmax 30 --outFilterScoreMinOverLread 0 --outFilterMatchNminOverLread 0
"""
########################################
# Parameters in the cluster.json file
########################################
"STAR_align":
{
"cpus-per-task" : 10,
"partition" : "medium",
"ntasks": 1,
"mem" : "100G",
"time": "0-08:00:00"
}The parameters were chosen as follow:
--runThreadN 8 indicates that we run the mapping process
using 8 threads.
--genomeDir {input.index} indicates where the genome index
is located.
--readFilesIn {input.read1} {input.read2} path to the PE
reads.
--outFileNamePrefix {params.prefix} is the prefix of our
output file.
--readFilesCommand zcat signifies that we our reads are
compressed and need to be read as fastq.gz files.
--genomeLoad NoSharedMemory
--twopassMode Basic indicates that we wish to use a
two-passes mapping mode that will first extract the junctions and insert
them into the genome index and re-map everything during a 2nd pass. The
option basic allows us to perform this on multiple files in
parallel.
--sjdbGTFfile {input.annotation} indicates the path of our
annotation file.
--sjdbScore 2
--sjdbOverhang 149 is the same parameter used for building
our index.
--outFilterMultimapNmax 20
--alignSJoverhangMin 8
--alignSJDBoverhangMin 1
--outFilterMatchNmin 50
--outFilterMismatchNmax 999
--outFilterMismatchNoverReadLmax 0.04
--alignIntronMin 20
--alignIntronMax 1000000
--alignMatesGapMax 1000000
--outSAMunmapped Within
--outFilterType BySJout
--outSAMattributes NH HI AS NM MD indicates that the
temporary .sam alignment file should contain headers.
--outSAMtype BAM SortedByCoordinate indicates that the
output should be in a .bam format and sorted by coordines.
--quantMode TranscriptomeSAM GeneCounts indicates that we
wish to have two outputs, one with the Read Count for each gene and one
with the gene aligned to the transcriptome only.
--quantTranscriptomeBan IndelSoftclipSingleend
--limitBAMsortRAM 32000000000
--seedSearchStartLmax 30
--outFilterScoreMinOverLread 0
--outFilterMatchNminOverLread 0
After mapping, we obtained alignment statistics from the
*_Log.final.out file and filled out the metadata table with
it.
grep 'Number of input reads' *_Log.final.out
grep 'Average input read length' *_Log.final.out
grep 'Uniquely mapped reads number' *_Log.final.out
grep 'Number of reads mapped to multiple loci' *_Log.final.out
grep 'Number of reads mapped to too many loci' *_Log.final.out
grep 'Number of reads unmapped: too many mismatches' *Log.final.out
grep 'Number of reads unmapped: too short' *Log.final.out
grep 'Number of reads unmapped: other' *Log.final.outSTAR only
Quantification using GeneCount
We can note that the option --quantMode GeneCounts from
STAR produces the same output as the htseq-count tool if we used the
–-sjdbGTFfile option.
In the output file {locust}_ReadsPerGene.out.tab we can
obtain the read counts per gene depending if our data is
unstranded (column 2) or stranded
(columns 3 and 4).
column 1: gene ID
column 2: counts for unstranded RNA-seq.
column 3: counts for the 1st read strand aligned with RNA
column 4: counts for the 2nd read strand aligned with RNA (the most
common protocol nowadays)
For our pilot S. gregaria project, we know we used Illumina stranded kit but to check we can with the following code:
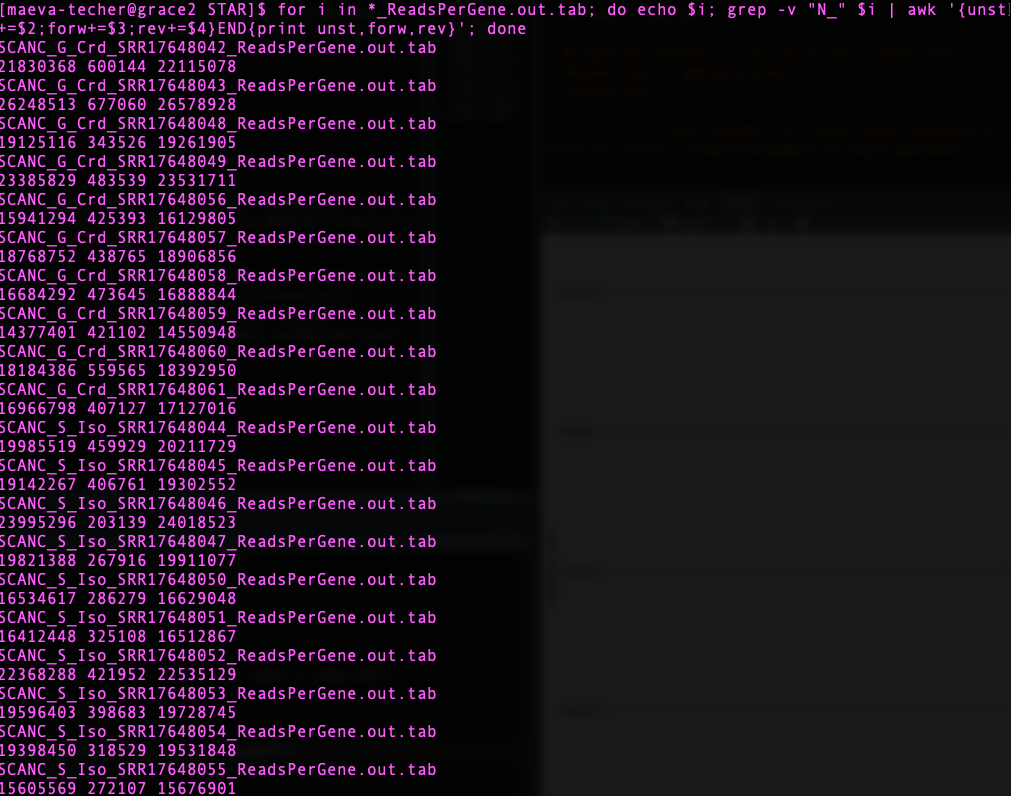
grep -v "N_" {locust}_ReadsPerGene.out.tab | awk '{unst+=$2;forw+=$3;rev+=$4}END{print unst,forw,rev}'
#or as a loop
for i in *_ReadsPerGene.out.tab; do echo $i; grep -v "N_" $i | awk '{unst+=$2;forw+=$3;rev+=$4}END{print unst,forw,rev}'; doneIn a stranded library preparation protocol, there should be a strong imbalance between number of reads mapped to known genes in forward versus reverse strands. This is what we observe for example on S. cancellata libraries here.
PREFERRED OPTION: We need to extract in our case the 1st and 4th columns for each file.
########################################
# Snakefile rule
########################################
#either ran the following rule
rule reads_count:
input:
readtable = OUTdir + "/alignment/STAR2/{locust}_ReadsPerGene.out.tab",
output:
OUTdir + "/DESeq2/counts_4thcol/{locust}_counts.txt"
shell:
"""
cut -f1,4 {input.readtable} | grep -v "_" > {output}
"""
#or simply this loop for less core usage
# for i in $SCRATCH/locust_phase/data/alignment/STAR/*ReadsPerGene.out.tab; do echo $i; cut -f1,4 $i | grep -v "_" > $SCRATCH/locust_phase/data/DESeq2/counts_4thcol/`basename $i ReadsPerGene.out.tab`counts.txt; doneALTERNATIVE OPTION: We can also build a single matrix of expression with all individuals targeted. Below is the example for S. piceifrons:
paste SPICE_*_ReadsPerGene.out.tab | grep -v "_" | awk '{printf "%s\t", $1}{for (i=4;i<=NF;i+=4) printf "%s\t", $i; printf "\n" }' > tmp
sed -e "1igene_name\t$(ls SPICE_*ReadsPerGene.out.tab | tr '\n' '\t' | sed 's/_ReadsPerGene.out.tab//g')" tmp > raw_counts_piceifrons_matrix.txtSTAR and RSEM
[NEED TO DO]
STAR and Salmon
[NEED TO DO]
HISAT2
[NEED TO DO]
sessionInfo()FALSE R version 4.2.1 (2022-06-23)
FALSE Platform: x86_64-apple-darwin17.0 (64-bit)
FALSE Running under: macOS Big Sur ... 10.16
FALSE
FALSE Matrix products: default
FALSE BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
FALSE LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
FALSE
FALSE locale:
FALSE [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
FALSE
FALSE attached base packages:
FALSE [1] stats graphics grDevices utils datasets methods base
FALSE
FALSE other attached packages:
FALSE [1] reshape2_1.4.4 ggthemes_4.2.4 plotly_4.10.0 DT_0.26
FALSE [5] forcats_0.5.2 stringr_1.4.1 dplyr_1.0.10 purrr_0.3.5
FALSE [9] readr_2.1.3 tidyr_1.2.1 tibble_3.1.8 ggplot2_3.3.6
FALSE [13] tidyverse_1.3.2 rmdformats_1.0.4 knitr_1.40
FALSE
FALSE loaded via a namespace (and not attached):
FALSE [1] httr_1.4.4 sass_0.4.2 jsonlite_1.8.3
FALSE [4] viridisLite_0.4.1 modelr_0.1.9 bslib_0.4.0
FALSE [7] assertthat_0.2.1 googlesheets4_1.0.1 cellranger_1.1.0
FALSE [10] yaml_2.3.6 pillar_1.8.1 backports_1.4.1
FALSE [13] glue_1.6.2 digest_0.6.30 promises_1.2.0.1
FALSE [16] rvest_1.0.3 colorspace_2.0-3 htmltools_0.5.3
FALSE [19] httpuv_1.6.6 plyr_1.8.7 pkgconfig_2.0.3
FALSE [22] broom_1.0.1 haven_2.5.1 bookdown_0.29
FALSE [25] scales_1.2.1 whisker_0.4 later_1.3.0
FALSE [28] tzdb_0.3.0 git2r_0.30.1 googledrive_2.0.0
FALSE [31] generics_0.1.3 ellipsis_0.3.2 cachem_1.0.6
FALSE [34] withr_2.5.0 lazyeval_0.2.2 cli_3.4.1
FALSE [37] magrittr_2.0.3 crayon_1.5.2 readxl_1.4.1
FALSE [40] evaluate_0.17 fs_1.5.2 fansi_1.0.3
FALSE [43] xml2_1.3.3 tools_4.2.1 data.table_1.14.4
FALSE [46] hms_1.1.2 formatR_1.12 gargle_1.2.1
FALSE [49] lifecycle_1.0.3 munsell_0.5.0 reprex_2.0.2
FALSE [52] compiler_4.2.1 jquerylib_0.1.4 rlang_1.0.6
FALSE [55] grid_4.2.1 rstudioapi_0.14 htmlwidgets_1.5.4
FALSE [58] rmarkdown_2.17 gtable_0.3.1 DBI_1.1.3
FALSE [61] R6_2.5.1 lubridate_1.8.0 fastmap_1.1.0
FALSE [64] utf8_1.2.2 workflowr_1.7.0 rprojroot_2.0.3
FALSE [67] stringi_1.7.8 Rcpp_1.0.9 vctrs_0.5.0
FALSE [70] dbplyr_2.2.1 tidyselect_1.2.0 xfun_0.34