Orthology inference: How are genes related across Schistocerca species?
Maeva Techer
2024-05-14
Last updated: 2024-05-14
Checks: 7 0
Knit directory:
locust-comparative-genomics/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20221025) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 3ca5aee. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .RData
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: data/.DS_Store
Ignored: data/.Rhistory
Ignored: data/DEseq2_SAMER_SAMER_HEAD_STARnew_features/.DS_Store
Ignored: data/DEseq2_SAMER_SAMER_THORAX_STARnew_features/.DS_Store
Ignored: data/DEseq2_SAMER_SGREG_HEAD_STARnew_features/.DS_Store
Ignored: data/DEseq2_SAMER_SGREG_THORAX_STARnew_features/.DS_Store
Ignored: data/DEseq2_SCANC_SCANC_HEAD_STARnew_features/.DS_Store
Ignored: data/DEseq2_SCANC_SCANC_THORAX_STARnew_features/.DS_Store
Ignored: data/DEseq2_SCANC_SGREG_HEAD_STARnew_features/.DS_Store
Ignored: data/DEseq2_SCANC_SGREG_THORAX_STARnew_features/.DS_Store
Ignored: data/DEseq2_SCUBE_SCUBE_HEAD_STARnew_features/.DS_Store
Ignored: data/DEseq2_SCUBE_SCUBE_THORAX_STARnew_features/.DS_Store
Ignored: data/DEseq2_SCUBE_SGREG_HEAD_STARnew_features/.DS_Store
Ignored: data/DEseq2_SCUBE_SGREG_THORAX_STARnew_features/.DS_Store
Ignored: data/DEseq2_SGREG_SGREG_HEAD_STARnew_features/.DS_Store
Ignored: data/DEseq2_SGREG_SGREG_THORAX_STARnew_features/.DS_Store
Ignored: data/DEseq2_SNITE_SGREG_HEAD_STARnew_features/.DS_Store
Ignored: data/DEseq2_SNITE_SGREG_THORAX_STARnew_features/.DS_Store
Ignored: data/DEseq2_SNITE_SNITE_HEAD_STARnew_features/.DS_Store
Ignored: data/DEseq2_SNITE_SNITE_THORAX_STARnew_features/.DS_Store
Ignored: data/DEseq2_SPICE_SGREG_HEAD_STARnew_features/.DS_Store
Ignored: data/DEseq2_SPICE_SGREG_THORAX_STARnew_features/.DS_Store
Ignored: data/DEseq2_SPICE_SPICE_HEAD_STARnew_features/.DS_Store
Ignored: data/DEseq2_SPICE_SPICE_THORAX_STARnew_features/.DS_Store
Ignored: data/americana/.DS_Store
Ignored: data/americana/deg_counts/.DS_Store
Ignored: data/americana/deg_counts/STAR/.DS_Store
Ignored: data/americana/deg_counts/STAR/americana/.DS_Store
Ignored: data/americana/deg_counts/STAR/americana/featurecounts/.DS_Store
Ignored: data/americana/deg_counts/STAR/gregaria/.DS_Store
Ignored: data/americana/deg_counts/STAR/gregaria/featurecounts/.DS_Store
Ignored: data/americana/deg_counts/STAR_newparams/.DS_Store
Ignored: data/americana/deg_counts/STAR_newparams/americana/.DS_Store
Ignored: data/americana/deg_counts/STAR_newparams/americana/featurecounts/.DS_Store
Ignored: data/americana/deg_counts/STAR_newparams/gregaria/.DS_Store
Ignored: data/americana/deg_counts/STAR_newparams/gregaria/featurecounts/.DS_Store
Ignored: data/cancellata/.DS_Store
Ignored: data/cancellata/deg_counts/.DS_Store
Ignored: data/cancellata/deg_counts/STAR/.DS_Store
Ignored: data/cancellata/deg_counts/STAR_newparams/.DS_Store
Ignored: data/cubense/.DS_Store
Ignored: data/cubense/deg_counts/.DS_Store
Ignored: data/cubense/deg_counts/STAR/.DS_Store
Ignored: data/cubense/deg_counts/STAR/cubense/.DS_Store
Ignored: data/cubense/deg_counts/STAR/cubense/featurecounts/
Ignored: data/cubense/deg_counts/STAR/gregaria/
Ignored: data/cubense/deg_counts/STAR_newparams/.DS_Store
Ignored: data/cubense/deg_counts/STAR_newparams/cubense/.DS_Store
Ignored: data/cubense/deg_counts/STAR_newparams/gregaria/.DS_Store
Ignored: data/gregaria/.DS_Store
Ignored: data/gregaria/deg_counts/.DS_Store
Ignored: data/gregaria/deg_counts/STAR/.DS_Store
Ignored: data/gregaria/deg_counts/STAR_newparams/.DS_Store
Ignored: data/gregaria/deg_counts/STAR_newparams/gregaria/.DS_Store
Ignored: data/gregaria/deg_counts/STAR_newparams/gregaria/featurecounts/.DS_Store
Ignored: data/list/.DS_Store
Ignored: data/metadata/.DS_Store
Ignored: data/nitens/.DS_Store
Ignored: data/nitens/deg_counts/.DS_Store
Ignored: data/nitens/deg_counts/STAR_newparams/.DS_Store
Ignored: data/nitens/deg_counts/STAR_newparams/gregaria/.DS_Store
Ignored: data/nitens/deg_counts/STAR_newparams/gregaria/featurecounts/.DS_Store
Ignored: data/nitens/deg_counts/STAR_newparams/nitens/.DS_Store
Ignored: data/nitens/deg_counts/STAR_newparams/nitens/featurecounts/.DS_Store
Ignored: data/piceifrons/.DS_Store
Ignored: data/piceifrons/deg_counts/.DS_Store
Ignored: data/piceifrons/deg_counts/STAR/.DS_Store
Ignored: data/piceifrons/deg_counts/STAR/gregaria/.DS_Store
Ignored: data/piceifrons/deg_counts/STAR/gregaria/featurecounts/.DS_Store
Ignored: data/piceifrons/deg_counts/STAR/piceifrons/.DS_Store
Ignored: data/piceifrons/deg_counts/STAR/piceifrons/featurecounts/.DS_Store
Ignored: data/piceifrons/deg_counts/STAR_newparams/.DS_Store
Ignored: data/piceifrons/deg_counts/STAR_newparams/gregaria/.DS_Store
Ignored: data/piceifrons/deg_counts/STAR_newparams/piceifrons/.DS_Store
Ignored: figures/
Ignored: tables/
Untracked files:
Untracked: analysis/2_repeat-landscape.Rmd
Unstaged changes:
Modified: analysis/2_synteny-graphs.Rmd
Modified: analysis/_site.yml
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown
(analysis/2_orthologs-prediction.Rmd) and HTML
(docs/2_orthologs-prediction.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 3ca5aee | Maeva A. TECHER | 2024-05-14 | wflow_publish("analysis/2_orthologs-prediction.Rmd") |
| html | ac06f45 | Maeva A. TECHER | 2024-05-13 | Build site. |
| Rmd | be09a11 | Maeva A. TECHER | 2024-05-13 | update markdown |
| html | be09a11 | Maeva A. TECHER | 2024-05-13 | update markdown |
| html | 0837617 | Maeva A. TECHER | 2024-01-29 | Build site. |
| html | f701a01 | Maeva A. TECHER | 2024-01-29 | reupdate |
| html | 2ca8696 | Maeva A. TECHER | 2024-01-29 | Build site. |
| html | 5c5487e | Maeva A. TECHER | 2024-01-29 | Build site. |
| html | 0135a6e | Maeva A. TECHER | 2024-01-29 | Build site. |
| Rmd | 505a8dc | Maeva A. TECHER | 2024-01-29 | wflow_publish("analysis/2_orthologs-prediction.Rmd") |
| html | 579056a | Maeva A. TECHER | 2024-01-29 | Build site. |
| Rmd | f50a3dd | Maeva A. TECHER | 2024-01-29 | wflow_publish("analysis/2_orthologs-prediction.Rmd") |
| html | f831779 | Maeva A. TECHER | 2024-01-24 | Build site. |
| html | 1b09cbe | Maeva A. TECHER | 2024-01-24 | remove |
| html | 79006db | Maeva A. TECHER | 2024-01-24 | Build site. |
| Rmd | b195f57 | Maeva A. TECHER | 2024-01-23 | refresh |
| html | b195f57 | Maeva A. TECHER | 2024-01-23 | refresh |
| html | dfc68c7 | Maeva A. TECHER | 2023-12-18 | Build site. |
| Rmd | 53877fa | Maeva A. TECHER | 2023-12-18 | add pages |
Comparative genomics and ortholog genes with OrthoFinder
We wanted to compare the six genomes of Schistocerca to get insights on gene evolution and relationships regarding their numbers, content, function and location. In order to achieve this, we need to identify groups of orthologous genes among our species of interest, considering at least one outgroup.
Orthologs are genes from different species that originated from a single ancestral gene and evolved through speciation events. However since genes can be lost or duplicated during evolution, some genes may not have exactly one orthologue in the genome of another species. Here we will separate the 1:1 orthologs to the the concept of orthogroups. Orthogroups can contain 1:1 orthologs but also several several orthologs from different species, including paralogs and one-to-many orthologs. Paralogs are genes within the same species that have originated from a shared ancestral genes but have diverged over time following gene duplication events.
IMPORTANT
We used OrthoFinder to identify the orthogroups
using amino acid sequences from the longest isoform of each gene. For
this part, refers to the AMAZINGLY WELL CURATED
pipeline FormicidaeMolecularEvolution
by Megan Barkdull
(PhD Student at Cornell University). We describe below the modifications
made and mostly copied the workflow from her Github.
1. Downloading data
We created the file “input-XXX.txt” as described to automatically download the coding sequence, protein sequence, GFF annotation data for each of our six Schistocerca species and annotated outgroups. The outgroups were choosen as close phylogenetic species with a RefSeq genome showing 1) a chromosome length, 2) associated with an annotation, 3) large size and 4) hybrid techniques used for assembly (last NCBI search: 28 April 2024).
We choose the following outgroups:
- Phasmatodea: European stick insect Bacillus
rossius (1.6 Gb), 19,298 annotated genes and 96.8% BUSCO
completeness.
- Blattodea: drywood termite Cryptotermes
secundus (1Gb when Zootermopsis nevadensis was only
485 Mb), 15,413 annotated genes and 96.9% BUSCO completeness.
- Hymenoptera: Western honey bee Apis
mellifera (225.2 Mb), 12,398 annotated genes and 98.6% BUSCO
completeness.
- Hymenoptera: red fire ant Solenopsis
invicta (378.1 Mb), 16,996 annotated genes and 98.3% BUSCO
completeness.
- Hemiptera: pea aphid Acyrthosiphon
pisum (533.6 Mb) and 20,307 annotated genes.
- Diptera: fruit fly Drosophila
melanogaster (143.7 Mb) and 17,872 annotated genes.
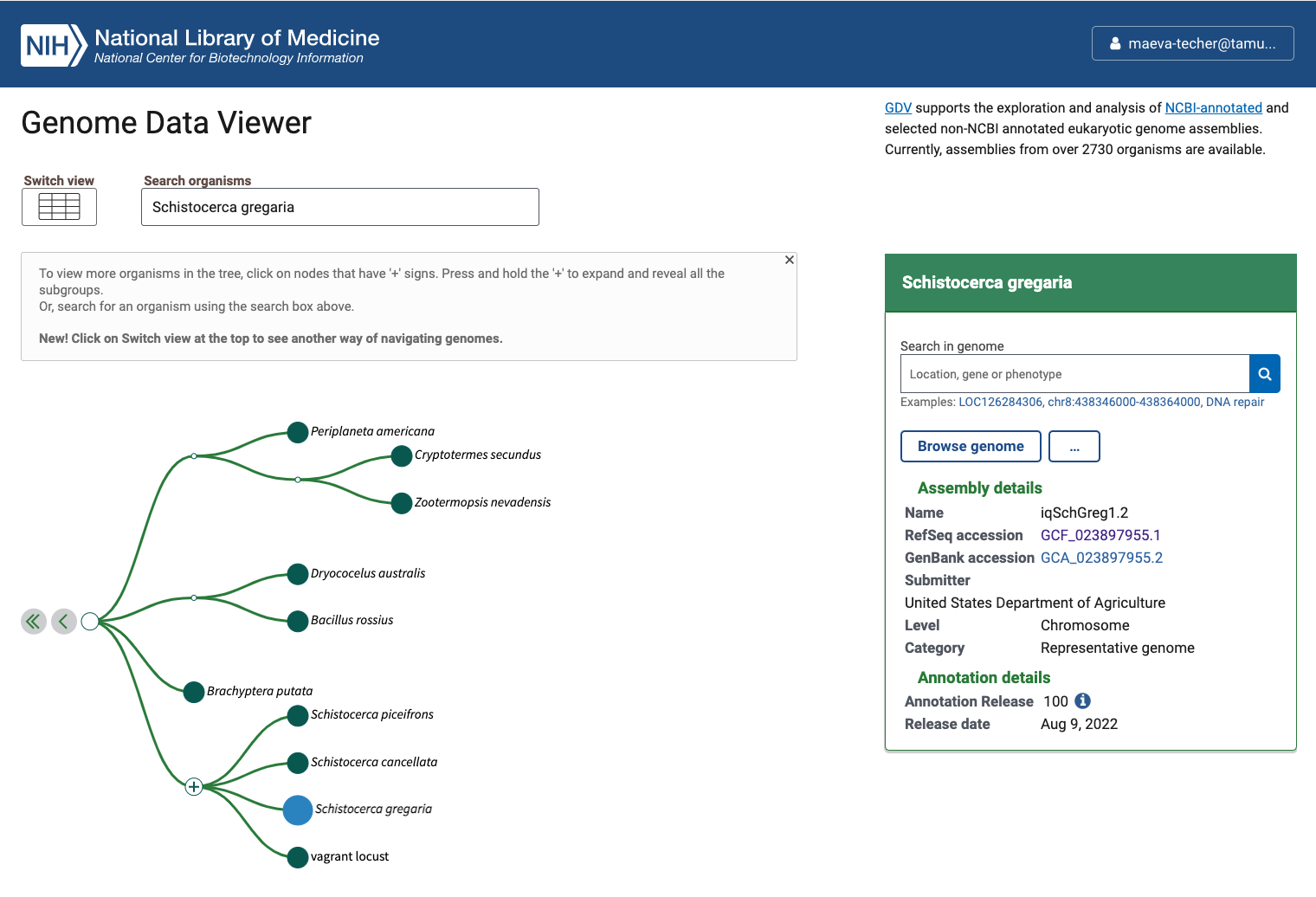
Status of Genome Data Viewer on NCBI for selecting our
outgroups
During our search, we identified very valuable genomes but for which no annotation was available (e.g., CAU_Lmig_1.0 for Locusta migratoria , iqMecThal1.2 Meconema thalassinum). However upon request to NCBI team, we were informed that the Locusta genome has not been annotated because of the presence of large bacterial contamination and Meconema does not possess RNA SRAs to help with the annotation.
./scripts/DataDownload ./scripts/inputurls_May2024.txtOn the TAMU Grace cluster, when you download do not run it on a node
as it does not work. Run it from the login node or transfer the files
already preloaded. This will creates a folder ./1_RawData
with 3 files for each species.
2. Selecting longest isoforms:
Here again, we will follow the pipeline except that to run it on Grace cluster we will make small modifications. The idea is that we will use only the single longest isoform of each gene to ease the orthology analysis. While this might not always be the principal isoform of said gene, we will apply the same bias to all genes the same way.
First, to run R without bothering other users, we will claim one interactive node to make sure we can proactively update the package if there are some issues:
srun --ntasks 1 --cpus-per-task 4 --mem 10G --time 01:00:00 --pty bashThen we will use any package preloaded on the cluster before needed to install our own on our user library if needed. We will need Pandoc for loading orthologr
ml GCC/12.2.0 OpenMPI/4.1.4 R_tamu/4.3.1
export R_LIBS=$SCRATCH/R_LIBS_USER/
ml Pandoc/2.13We now simply run the script as indicated on the pipeline page:
./scripts/GeneRetrieval.R ./scripts/inputurls_May2024.txt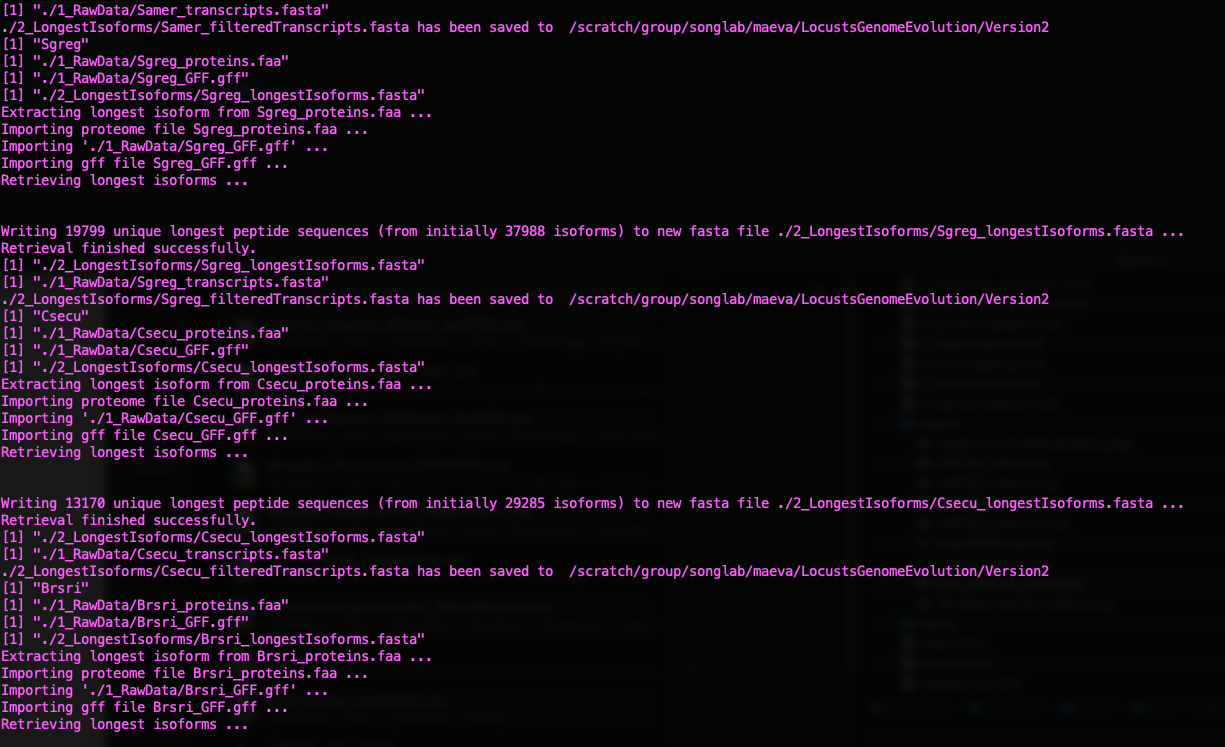
Status of Gene Retrieval script when successful
Initially we wanted to also include the genome of Dryococelus
australis (3.4Gb). However this will not be possible as the
user submitted annotation does not match the need for
orthologr here. There are some missing columns in the
transcripts regarding “gene”.
3. Cleaning the raw data:
For the cleaning step of the mbarkdull’s pipeline we simply followed the command line with no modifications.
./scripts/DataCleaning ./scripts/inputurls_May2024.txt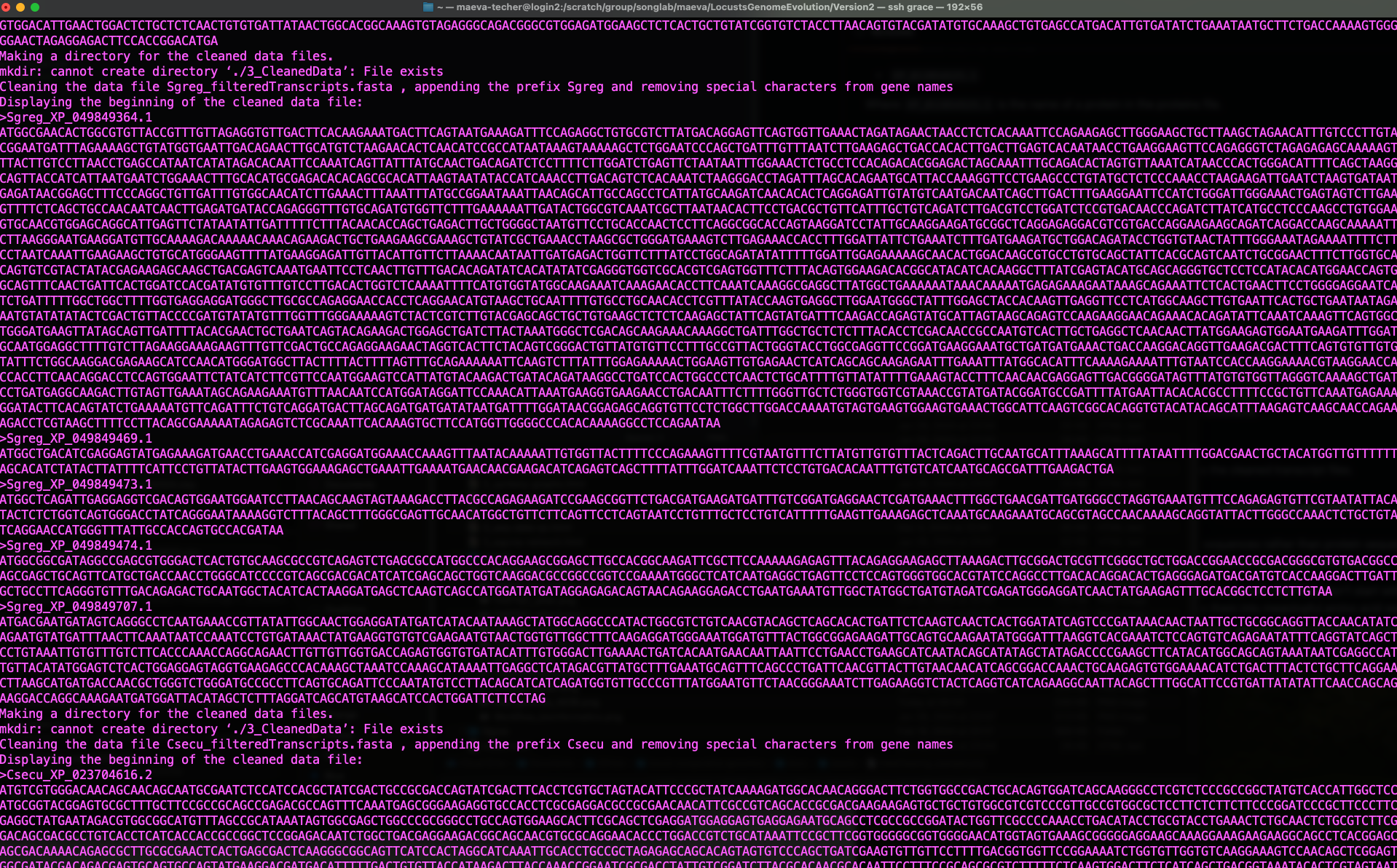
Status of Data Cleaning script when successful
4. Translating nucleotide sequences to amino acid sequences:
As mentioned in the pipeline page, we will mostly need to use amino
acid sequences rather than protein and we need to translate the data we
downloaded. We will be using the Python script from mbarkdull
./scripts/TranscriptFilesTranslateScript.py but before that
we will modify our input file to only include only the 12 essential
species.
We changed the script in head lines and lines 25-27, to call the module directly from the cluster as follow:
#!/bin/bash
##NECESSARY JOB SPECIFICATIONS
#SBATCH --job-name=Transdecoder #Set the job name to "JobExample4"
#SBATCH --time=02:00:00 #Set the wall clock limit to 1hr and 30min
#SBATCH --ntasks=2 #Request 2 task
#SBATCH --cpus-per-task=4 #Request 8 task
#SBATCH --mem=20G #Request 50GB per node
ml GCC/10.2.0 OpenMPI/4.0.5 TransDecoder/5.5.0 Biopython/1.78 # Now we can run Transdecoder on the cleaned file:
echo "First, attempting TransDecoder run on $cleanName"
ml GCC/10.2.0 OpenMPI/4.0.5 TransDecoder/5.5.0
TransDecoder.LongOrfs -t $cleanName
TransDecoder.Predict -t $cleanName --single_best_onlyand then we launched the script by changing the top lines to put in sbatch:
sbatch ./scripts/DataTranslating_modif ./scripts/inputurls_May2024.txt5. Running Orthofinder
Now we will finally run Orthofinder to identify groups of orthologous genones in our translated amino acid sequences. We will also use MAFFT to produce multiple sequence alignment across our six species of Schistocerca and the outgroups.
Instead of using the ./scripts/DataOrthofinder from the
pipeline, we will be running our own command line here. Before doing so,
we did manually the creating of folders and moving of fasta files as
follow:
mkdir /LocustsGenomeEvolution/tmp
mkdir ./5_OrthoFinder/fasta
cp ./4_1_TranslatedData/OutputFiles/translated* ./5_OrthoFinder/fasta
cd ./5_OrthoFinder/fasta
rename translated '' translated*
cd ../To run orthofinder onto our Grace cluster, I checked the
compatibility of the modules required and these are the versions
acceptable to run in sbatchorthofinder.sh:
#!/bin/bash
##NECESSARY JOB SPECIFICATIONS
#SBATCH --job-name=orthofinder1 #Set the job name to "JobExample4"
#SBATCH --time=06:00:00 #Set the wall clock limit to 1hr and 30min
#SBATCH --ntasks=2 #Request 1 task
#SBATCH --cpus-per-task=4 #Request 1 task
#SBATCH --mem=30G #Request 100GB per node
ml iccifort/2019.5.281 impi/2018.5.288 OrthoFinder/2.3.11-Python-3.7.4
ml IQ-TREE/1.6.12 FastTree/2.1.11
ml MAFFT/7.453-with-extensions
orthofinder -S diamond -T iqtree -A mafft -I 5 -t 32 -a 4 -M msa -f /scratch/group/songlab/maeva/LocustsGenomeEvolution/Version2/5_OrthoFinder/fasta -p /scratch/group/songlab/maeva/LocustsGenomeEvolution/tmpHere we use:
-S diamond DIAMOND as a sequence search program
-T iqtree IQTREE as a tree inference program
-A mafft MAFFT as the multiple sequence alignment (MSA)
program
-I 5 MCL inflation parameter (default from the
pipeline)
-t 32 number of threads
We want to run also orthofinder using BLAST as it can give -2% accuracy increase over DIAMOND but will be computationally more heavy. For this we will only change:
ml BLAST+/2.9.0
orthofinder -S blastand we will run the script with the blast, iqtree and mafft options:
sbatch scripts/orthofinder_blast.shNow we are all done, we can explore the results and go on for the next steps.
6. Gene orthology results
References
If you use this script, and orthologr please cite:
Drost et al. 2015. Evidence for Active Maintenance of Phylotranscriptomic Hourglass Patterns in Animal and Plant Embryogenesis. Mol. Biol. Evol. 32 (5): 1221-1231. doi:10.1093/molbev/msv012
If you use Transdecoder, please cite it as:
Haas, B., and A. Papanicolaou. “TransDecoder.” (2017).
If you use Orthofinder, please cite it:
OrthoFinder’s orthogroup and ortholog inference are described here:
Emms, D.M., Kelly, S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol 16, 157 (2015).
Emms, D.M., Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol 20, 238 (2019).
If you use the OrthoFinder species tree then also cite:
Emms D.M. & Kelly S. STRIDE: Species Tree Root Inference from Gene Duplication Events (2017), Mol Biol Evol 34(12): 3267-3278.
Emms D.M. & Kelly S. STAG: Species Tree Inference from All Genes (2018), bioRxiv https://doi.org/10.1101/267914.
Please also cite MAFFT:
K. Katoh, K. Misawa, K. Kuma, and T. Miyata. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30(14): 3059-3066.
sessionInfo()R version 4.3.1 (2023-06-16)
Platform: x86_64-apple-darwin20 (64-bit)
Running under: macOS Sonoma 14.4.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-x86_64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-x86_64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: America/Chicago
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] workflowr_1.7.1
loaded via a namespace (and not attached):
[1] vctrs_0.6.5 httr_1.4.7 cli_3.6.2 knitr_1.45
[5] rlang_1.1.3 xfun_0.43 stringi_1.8.4 processx_3.8.4
[9] promises_1.3.0 jsonlite_1.8.8 glue_1.7.0 rprojroot_2.0.4
[13] git2r_0.33.0 htmltools_0.5.8.1 httpuv_1.6.15 ps_1.7.6
[17] sass_0.4.9 fansi_1.0.6 rmarkdown_2.26 jquerylib_0.1.4
[21] tibble_3.2.1 evaluate_0.23 fastmap_1.1.1 yaml_2.3.8
[25] lifecycle_1.0.4 whisker_0.4.1 stringr_1.5.1 compiler_4.3.1
[29] fs_1.6.4 pkgconfig_2.0.3 Rcpp_1.0.12 rstudioapi_0.16.0
[33] later_1.3.2 digest_0.6.35 R6_2.5.1 utf8_1.2.4
[37] pillar_1.9.0 callr_3.7.6 magrittr_2.0.3 bslib_0.7.0
[41] tools_4.3.1 cachem_1.0.8 getPass_0.2-4