Leucegene Validation
Last updated: 2020-05-06
Checks: 7 0
Knit directory: MINTIE-paper-analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200415) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 44d8c37. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: analysis/cache/
Ignored: data/.DS_Store
Ignored: data/leucegene/.DS_Store
Ignored: data/leucegene/KMT2A-PTD_results/.DS_Store
Ignored: data/leucegene/normals_controls_test_results/.DS_Store
Ignored: data/leucegene/salmon_out/
Ignored: data/leucegene/sample_info/.KMT2A-PTD_samples.txt.swp
Ignored: data/leucegene/sample_info/.variant_validation_table.tsv.swp
Ignored: data/leucegene/sample_info/KMT2A-PTD_8-2.fa.xls
Ignored: data/simu/.DS_Store
Ignored: output/Leucegene_gene_counts.tsv
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/Leucegene_Validation.Rmd) and HTML (docs/Leucegene_Validation.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 44d8c37 | Marek Cmero | 2020-05-06 | Build leucegene validation notebook. |
| Rmd | ff4b1dc | Marek Cmero | 2020-05-06 | Leucegene results |
# util
library(data.table)
library(dplyr)
library(here)
library(stringr)
# plotting
library(ggplot2)
# bioinformatics/stats helpers
library(edgeR)options(stringsAsFactors = FALSE)source(here("code/leucegene_helper.R"))Leucegene Validation
# load SRX to patient ID lookup table
kmt2a_patient_lookup <- read.delim(here("data/leucegene/sample_info/KMT2A-PTD_samples.txt"),
header = FALSE,
col.names = c("sample", "patient"))
kmt2a_results_dir <- here("data/leucegene/KMT2A-PTD_results")
# load comparisons against all other controls
kmt2a_results <- NULL
control_sets <- list.files(kmt2a_results_dir)
for (controls in control_sets) {
tmp <- str_c(kmt2a_results_dir, controls, sep = "/") %>%
list.files(., full.names = TRUE) %>%
lapply(., read.delim) %>%
rbindlist() %>%
filter(logFC > 5) %>%
mutate(controls = controls)
kmt2a_results <- rbind(kmt2a_results, tmp)
}
kmt2a_results <- inner_join(kmt2a_results, kmt2a_patient_lookup, by = "sample")KMT2A-PTD controls comparison
MINTIE paper Supplementary Figure 2. Shows the number of variant genes found in the Leucegene cohort containing KMT2A PTDs.
# extract variant genes and make summary
var_genes <- kmt2a_results$overlapping_genes %>%
str_split("\\||:")
repeat_rows <- rep(1:nrow(kmt2a_results), sapply(var_genes, length))
results_by_gene <- data.table(kmt2a_results[repeat_rows,])
results_by_gene$gene <- unlist(var_genes)
results_summary <- results_by_gene[, length(unique(gene)), by = c("patient", "controls")]
results_summary <- results_summary %>% arrange(controls, V1) %>% data.table()
results_summary$patient <- factor(results_summary$patient,
levels = unique(results_summary$patient))
# reorder by totals across different controls
results_totals <- results_summary[, sum(V1), by = c("controls")] %>%
arrange(V1)
results_summary$controls <- factor(results_summary$controls,
levels = results_totals$controls)
print("Total variant genes called using different controls:")[1] "Total variant genes called using different controls:"results_summary %>%
group_by(controls) %>%
summarise(min = min(V1), median = median(V1), max = max(V1)) %>%
data.frame() %>%
print() controls min median max
1 AML_controls 129 213.5 2093
2 normal_controls 266 508.0 2365
3 normal_controls_reduced 501 794.0 2562ggplot(results_summary, aes(patient, V1, fill=controls)) +
geom_bar(position = position_dodge2(width =0.9, preserve = "single"), stat = "identity") +
theme_bw() +
xlab("") +
ylab("Genes with variants") +
coord_flip() +
theme(legend.position = "bottom") +
scale_fill_brewer(palette="Dark2",
labels = c("AML_controls" = "13 AMLs",
"normal_controls" = "13 normals",
"normal_controls_reduced" = "3 normals"))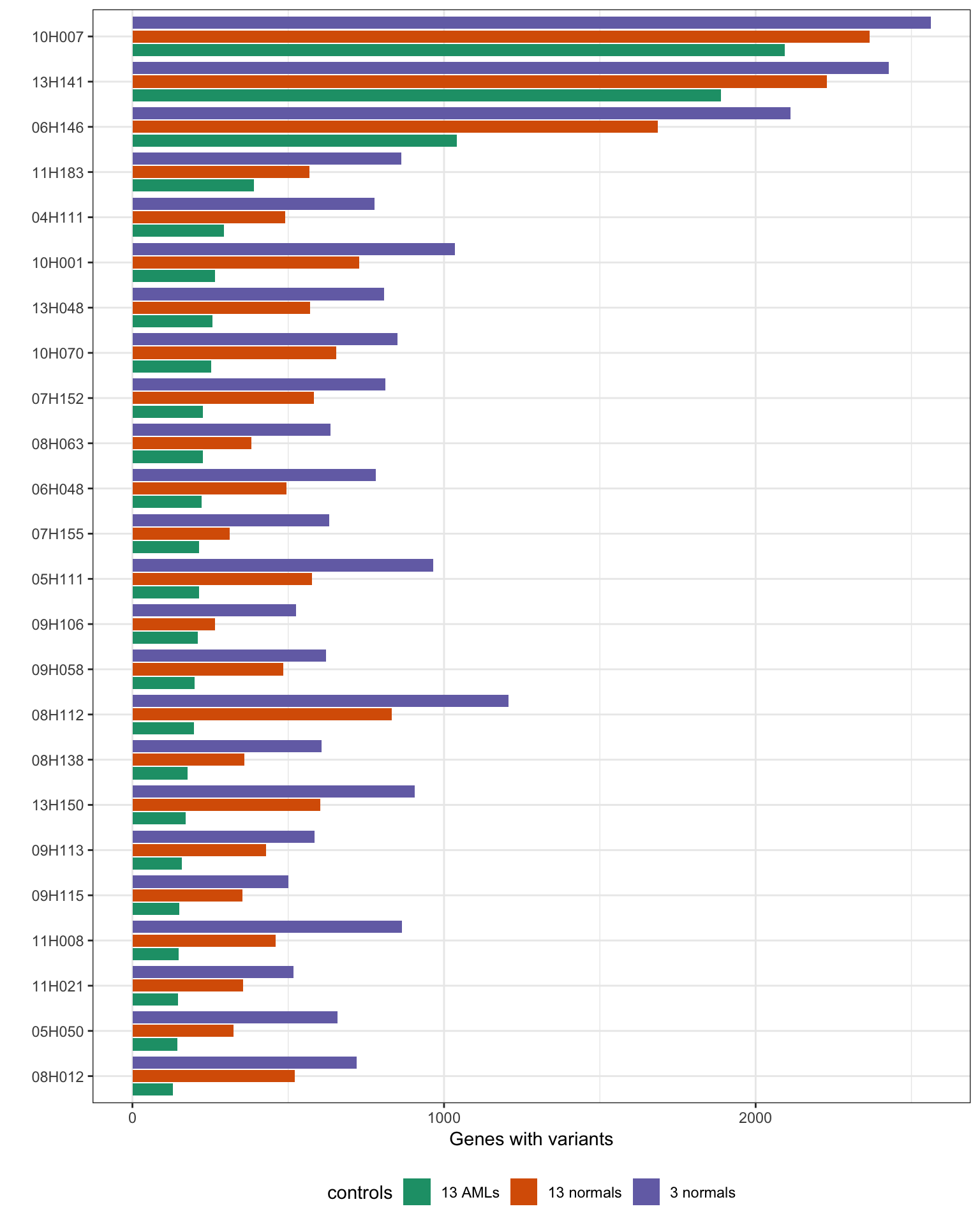
KMT2A variants found in cohort
MINTIE paper Supplementary Table 1. Shows whether MINITE found a KMT2A SV in each sample for the given control group. Coverage obtained from Audemard et al. spreadsheet containing the Leucegene results must be manually added to data/leucegene/sample_info to run the code.
# load results from km paper for coverage of KMT2A PTDs
kmt2a_lgene_km_results <- read.csv(here("data/leucegene/sample_info/KMT2A-PTD_8-2.fa.xls"), sep="\t") %>%
mutate(patient = Sample) %>%
group_by(patient) %>%
summarise(coverage = max(Min.coverage))
# check whether MINTIE found a KMT2A SV in each control set
found_using_cancon <- get_samples_with_kmt2a_sv(kmt2a_results, "AML_controls")
found_using_norcon <- get_samples_with_kmt2a_sv(kmt2a_results, "normal_controls")
found_using_redcon <- get_samples_with_kmt2a_sv(kmt2a_results, "normal_controls_reduced")
# make the table
kmt2a_control_comp <- inner_join(kmt2a_patient_lookup, kmt2a_lgene_km_results, by = "patient") %>%
arrange(desc(coverage))
kmt2a_control_comp$found_using_cancon <- kmt2a_control_comp$sample %in% found_using_cancon
kmt2a_control_comp$found_using_norcon <- kmt2a_control_comp$sample %in% found_using_norcon
kmt2a_control_comp$found_using_redcon <- kmt2a_control_comp$sample %in% found_using_redcon
print(kmt2a_control_comp) sample patient coverage found_using_cancon found_using_norcon
1 SRX958906 07H152 158 TRUE TRUE
2 SRX332646 09H115 125 TRUE TRUE
3 SRX957230 06H146 87 TRUE TRUE
4 SRX957223 05H111 79 TRUE TRUE
5 SRX332659 11H021 63 TRUE TRUE
6 SRX332633 05H050 58 TRUE TRUE
7 SRX959061 13H150 58 TRUE TRUE
8 SRX959044 13H048 57 TRUE TRUE
9 SRX958974 10H070 53 TRUE TRUE
10 SRX958963 10H007 50 TRUE TRUE
11 SRX958959 09H106 49 TRUE TRUE
12 SRX959060 13H141 45 TRUE TRUE
13 SRX958945 09H058 29 TRUE TRUE
14 SRX958907 07H155 23 TRUE TRUE
15 SRX381854 08H112 22 TRUE TRUE
16 SRX332645 09H113 17 TRUE TRUE
17 SRX959001 11H183 16 FALSE FALSE
18 SRX381852 08H012 15 FALSE FALSE
19 SRX958932 08H138 15 FALSE FALSE
20 SRX381865 11H008 13 FALSE FALSE
21 SRX958873 06H048 10 FALSE FALSE
22 SRX958922 08H063 6 FALSE FALSE
23 SRX958961 10H001 6 FALSE FALSE
24 SRX958844 04H111 3 FALSE FALSE
found_using_redcon
1 TRUE
2 TRUE
3 TRUE
4 TRUE
5 TRUE
6 TRUE
7 TRUE
8 TRUE
9 TRUE
10 TRUE
11 TRUE
12 TRUE
13 TRUE
14 TRUE
15 TRUE
16 TRUE
17 FALSE
18 FALSE
19 FALSE
20 FALSE
21 FALSE
22 FALSE
23 FALSE
24 FALSESelected Leucegene variants found by MINTIE
truth <- read.delim(here("data/leucegene/sample_info/variant_validation_table.tsv"), sep = "\t")
leucegene_results_dir <- here("data/leucegene/validation_results")
validation <- list.files(leucegene_results_dir, full.names = TRUE) %>%
lapply(., read.delim) %>%
rbindlist() %>%
filter(logFC > 5)
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS 10.14.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_AU.UTF-8/en_AU.UTF-8/en_AU.UTF-8/C/en_AU.UTF-8/en_AU.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] edgeR_3.24.3 limma_3.38.3 ggplot2_3.1.0 stringr_1.4.0
[5] here_0.1 dplyr_0.8.1 data.table_1.12.0
loaded via a namespace (and not attached):
[1] Rcpp_1.0.1 RColorBrewer_1.1-2 plyr_1.8.4
[4] compiler_3.5.1 pillar_1.3.1 later_1.0.0
[7] git2r_0.26.1 workflowr_1.6.1 tools_3.5.1
[10] digest_0.6.18 lattice_0.20-38 evaluate_0.13
[13] tibble_2.1.1 gtable_0.3.0 pkgconfig_2.0.2
[16] rlang_0.4.2 yaml_2.2.0 xfun_0.5
[19] withr_2.1.2 knitr_1.22 fs_1.2.7
[22] locfit_1.5-9.1 rprojroot_1.3-2 grid_3.5.1
[25] tidyselect_0.2.5 glue_1.3.1 R6_2.4.0
[28] rmarkdown_1.12 purrr_0.3.2 magrittr_1.5
[31] whisker_0.3-2 backports_1.1.3 scales_1.0.0
[34] promises_1.1.0 htmltools_0.3.6 assertthat_0.2.1
[37] colorspace_1.4-1 httpuv_1.5.2 labeling_0.3
[40] stringi_1.4.3 lazyeval_0.2.2 munsell_0.5.0
[43] crayon_1.3.4