Post-Alignment QC
Stephen Pederson
06 July, 2022
Last updated: 2022-07-06
Checks: 6 1
Knit directory:
20180328_Atkins_RatFracture/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20220705) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version dd28879. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Untracked files:
Untracked: data/gmt/
Untracked: output/dge.rds
Unstaged changes:
Modified: analysis/alnQC.Rmd
Modified: analysis/dge.Rmd
Modified: analysis/index.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/alnQC.Rmd) and HTML
(docs/alnQC.html) files. If you’ve configured a remote Git
repository (see ?wflow_git_remote), click on the hyperlinks
in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | dd28879 | Steve Pederson | 2022-07-06 | Setup initial DGE after restructure |
| html | dd28879 | Steve Pederson | 2022-07-06 | Setup initial DGE after restructure |
library(ngsReports)
library(tidyverse)
library(yaml)
library(scales)
library(pander)
library(glue)
library(plotly)
library(edgeR)
library(ggfortify)
library(AnnotationHub)
library(ensembldb)
library(magrittr)
library(BSgenome.Rnorvegicus.UCSC.rn6)
library(broom)
library(ggrepel)panderOptions("table.split.table", Inf)
panderOptions("big.mark", ",")
theme_set(theme_bw())
suffix <- "_L001"
pattern <- paste0("_CB2YGANXX_.+fastq.gz")
sp <- "Rnorvegicus"
rn6 <- BSgenome.Rnorvegicus.UCSC.rn6samples <- "data/targets.csv" %>%
here::here() %>%
read_csv() %>%
mutate(
Filename = paste0(File, suffix)
)group_cols <- hcl.colors(
n = length(unique(samples$group)),
palette = "Zissou 1"
) %>%
setNames(unique(samples$group))Annotations
Annotation Setup
ah <- AnnotationHub() %>%
subset(rdataclass == "EnsDb") %>%
subset(species == "Rattus norvegicus") %>%
subset(str_detect(description, "96"))
ensDb <- ah[[1]]
genesGR <- genes(ensDb) %>%
keepStandardChromosomes(species = "Rattus_norvegicus", pruning.mode = "coarse") %>%
sortSeqlevels()
transGR <- transcripts(ensDb) %>%
keepStandardChromosomes(species = "Rattus_norvegicus", pruning.mode = "coarse") %>%
sortSeqlevels()
exonGR <- exonsBy(ensDb, "tx") %>%
reduce() %>%
keepStandardChromosomes(species = "Rattus_norvegicus", pruning.mode = "coarse") %>%
sortSeqlevels()
exonGR_UCSC <- exonGR
seqlevels(exonGR_UCSC) <- paste0("chr", seqlevels(exonGR_UCSC)) %>%
str_replace("chrMT", "chrM")
genome(exonGR_UCSC) <- genome(rn6)
exonSeq <- getSeq(rn6, exonGR_UCSC) %>%
lapply(unlist) %>%
as("DNAStringSet")
transGC <- exonSeq %>%
letterFrequency("GC", as.prob = TRUE) %>%
.[,1] %>%
setNames(names(exonSeq))
transGR$gc_content <- transGC[names(transGR)]
mcols(transGR) <- mcols(transGR) %>%
cbind(
transcriptLengths(ensDb)[rownames(.), c("nexon", "tx_len")]
)
mcols(genesGR) <- mcols(genesGR) %>%
as.data.frame() %>%
dplyr::select(
gene_id, gene_name, gene_biotype, entrezid
) %>%
left_join(
mcols(transGR) %>%
as.data.frame() %>%
mutate(
tx_support_level = case_when(
is.na(tx_support_level) ~ 1L,
TRUE ~ tx_support_level
)
) %>%
group_by(gene_id) %>%
summarise(
n_tx = n(),
longest_tx = max(tx_len),
ave_tx_len = mean(tx_len),
gc_content = sum(tx_len*gc_content) / sum(tx_len)
) %>%
mutate(
bin_length = cut(
x = ave_tx_len,
labels = seq_len(10),
breaks = quantile(ave_tx_len, probs = seq(0, 1, length.out = 11)),
include.lowest = TRUE
),
bin_gc = cut(
x = gc_content,
labels = seq_len(10),
breaks = quantile(gc_content, probs = seq(0, 1, length.out = 11)),
include.lowest = TRUE
),
bin = paste(bin_gc, bin_length, sep = "_")
),
by = "gene_id"
) %>%
set_rownames(.$gene_id) %>%
as("DataFrame")
trans2Gene <- mcols(transGR) %>%
as.data.frame() %>%
dplyr::select(tx_id, gene_id) %>%
dplyr::filter(!is.na(tx_id), !is.na(gene_id)) %>%
as_tibble()Annotation data was loaded as an EnsDb object, using
Ensembl release 96. Transcript level gene lengths and GC content was
converted to gene level values using:
- GC Content: The total GC content divided by the total length of transcripts
- Gene Length: The mean transcript length
write_rds(genesGR, here::here("output/genesGR.rds"), compress = "gz")counts <- list.files(here::here("data/3_kallisto"), full.names = TRUE) %>%
catchKallisto()
dge <- counts$counts %>%
as.data.frame() %>%
rownames_to_column("tx_id") %>%
as_tibble() %>%
set_colnames(basename(colnames(.))) %>%
set_colnames(str_remove(colnames(.),"_CB2Y.+")) %>%
mutate(tx_id = str_remove(tx_id, "\\.[0-9]+")) %>%
dplyr::filter(tx_id %in% trans2Gene$tx_id) %>%
pivot_longer(cols = all_of(samples$Rat), names_to = "Rat", values_to = "count") %>%
left_join(trans2Gene) %>%
group_by(Rat, gene_id) %>%
summarise(count = sum(count)) %>%
pivot_wider(names_from = "Rat", values_from = "count") %>%
dplyr::filter(grepl("ENSRNOG", gene_id)) %>%
as.data.frame() %>%
column_to_rownames("gene_id") %>%
DGEList()
dge$samples %<>%
mutate(Rat = rownames(.)) %>%
dplyr::select(-group) %>%
left_join(samples, by = "Rat") %>%
set_rownames(.$Rat)
dge$genes <- genesGR[rownames(dge)] %>%
mcols()Read Assignment To Genes
Library Sizes
dge$samples %>%
mutate(lib.size = lib.size / 1e6) %>%
ggplot(aes(Rat, lib.size, fill = group)) +
geom_col() +
facet_wrap(~group, scales = "free_x") +
scale_y_continuous(expand = expansion(c(0, .05))) +
scale_fill_manual(values = group_cols) +
labs(
x = "Sample", y = "Library Size (millions)",
fill = "Group"
)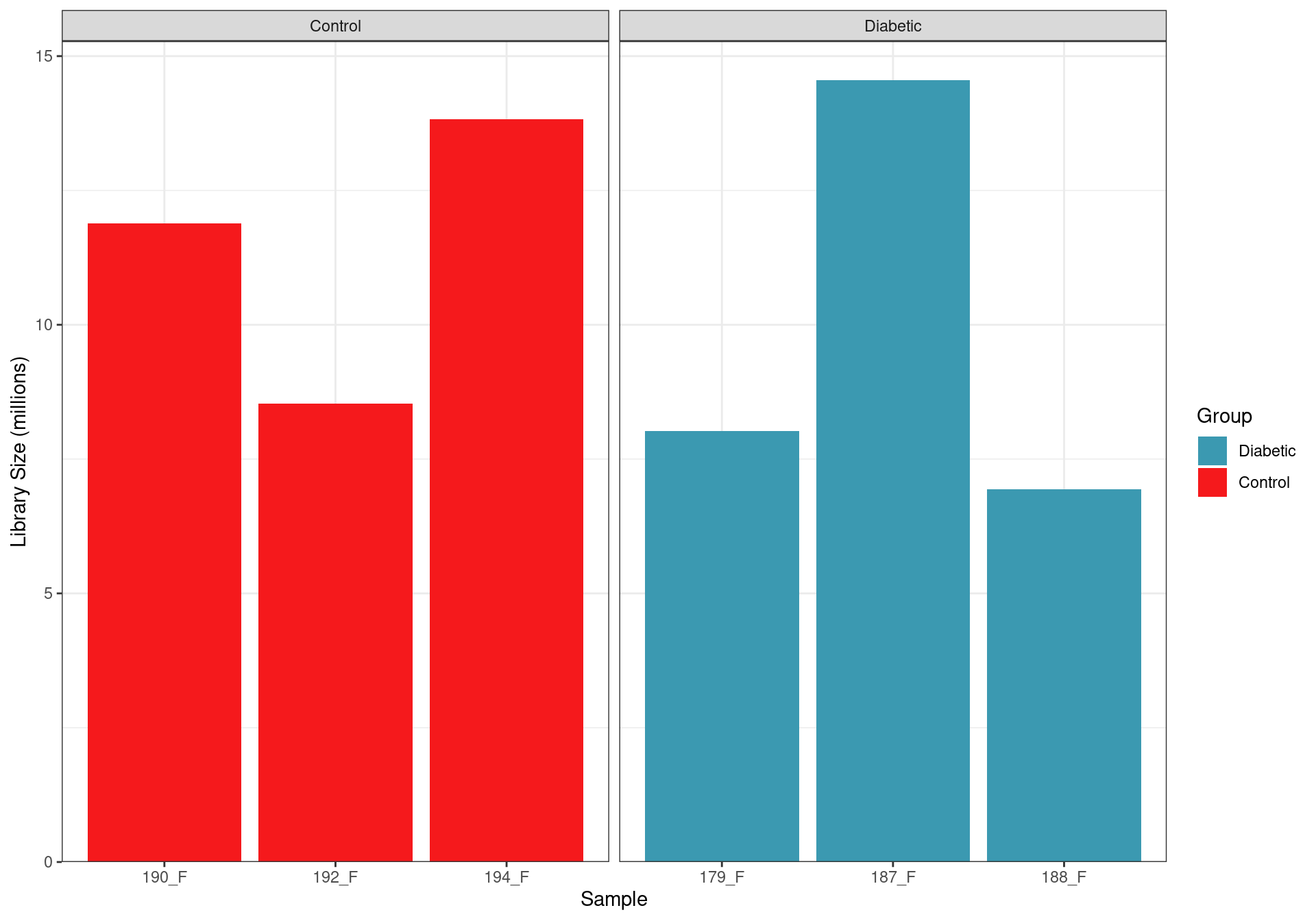
Library sizes after summarising to gene-level counts.
After assignment to genes, library sizes ranged between 6,934,169 and 14,552,053 reads, with a median library size of 10,208,651 reads.
Count Assignment Rates
trimFqc <- here::here("data/1_trimmedData/FastQC") %>%
list.files(pattern = "zip", full.names = TRUE) %>%
FastqcDataList()
trimFqc %>%
getModule("Basic") %>%
mutate(Filename = str_remove_all(Filename, "_R1.fastq.gz")) %>%
left_join(dge$samples, by = "Filename") %>%
mutate(`% Assigned To Genes` = lib.size / Total_Sequences) %>%
ggplot(aes(Rat, `% Assigned To Genes`, fill = group)) +
geom_col() +
facet_wrap(~group, scales = "free_x") +
scale_fill_manual(values = group_cols) +
scale_y_continuous(labels = percent, expand = expansion(c(0, 0.05))) +
labs(fill = "Group")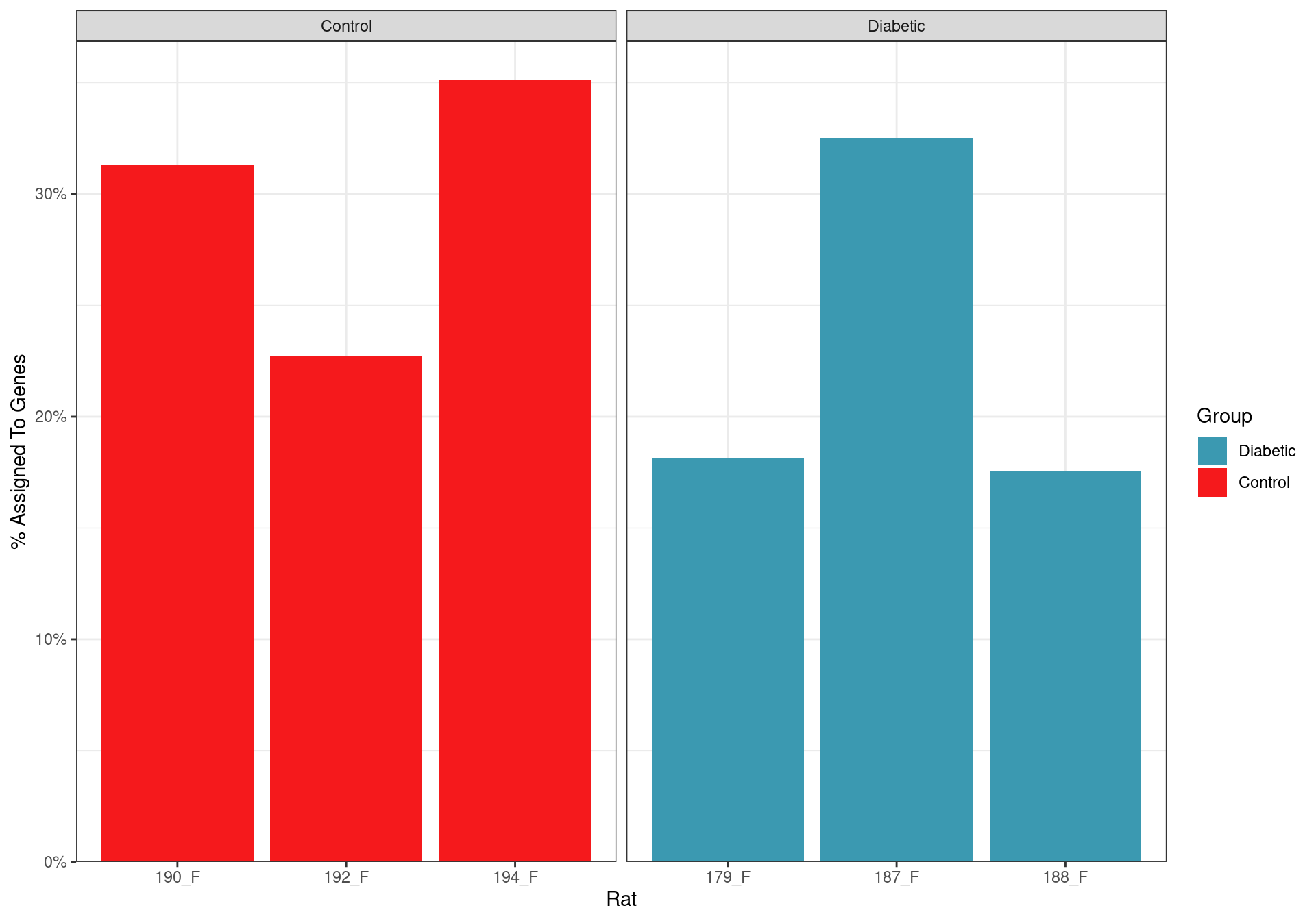
Assignment rate for reads to genes. All samples showed that a large proportion of reads were not derived from mRNA transcripts, and as such, were not informative for the analysis.
Total Detected Genes
- Of the 23,706 genes defined in this annotation build, 1,621 genes had no reads assigned in any samples.
dge$counts %>%
as_tibble() %>%
mutate(
across(everything(), as.logical)
) %>%
summarise(
across(everything(), sum)
) %>%
pivot_longer(
everything(), names_to = "Rat", values_to = "Detected"
) %>%
left_join(samples)%>%
ggplot(aes(group, Detected, colour = group)) +
geom_point() +
geom_segment(
aes(xend = group, y = 0, yend = Detected),
data = . %>%
group_by(group) %>%
summarise(Detected = min(Detected)),
colour = "black", size = 1/4) +
scale_y_continuous(labels = comma, expand = expansion(c(0, 0.05))) +
scale_colour_manual(values = group_cols) +
labs(
x = "Group",
y = "Genes Detected",
colour = "Group"
)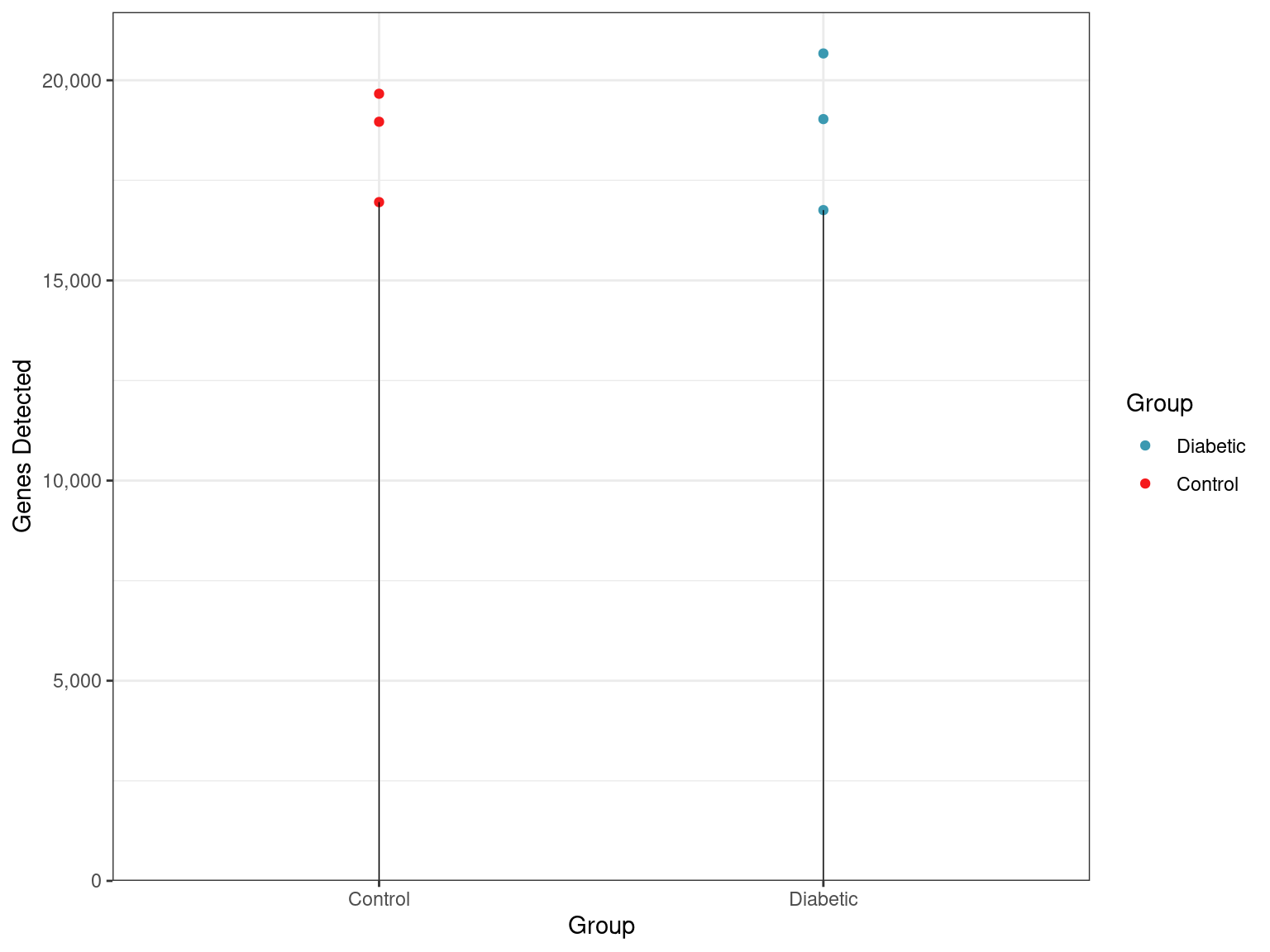
Total numbers of genes detected across all samples and groups.
| Version | Author | Date |
|---|---|---|
| dd28879 | Steve Pederson | 2022-07-06 |
plotly::ggplotly(
dge$counts %>%
is_greater_than(0) %>%
rowSums() %>%
table() %>%
enframe(name = "n_samples", value = "n_genes") %>%
mutate(
n_samples = as.integer(n_samples),
n_genes = as.integer(n_genes),
) %>%
arrange(desc(n_samples)) %>%
mutate(
Detectable = cumsum(n_genes),
Undetectable = sum(n_genes) - Detectable
) %>%
pivot_longer(
cols = ends_with("table"),
names_to = "Status",
values_to = "Number of Genes"
) %>%
dplyr::rename(
`Number of Samples` = n_samples,
) %>%
ggplot(aes(`Number of Samples`, `Number of Genes`, colour = Status)) +
geom_line() +
geom_vline(
aes(xintercept = `Mean Sample Number`),
data = . %>%
summarise(`Mean Sample Number` = mean(`Number of Samples`)),
linetype = 2,
colour = "grey50"
) +
scale_x_continuous(expand = expansion(c(0.01, 0.01))) +
scale_y_continuous(labels = comma) +
scale_colour_manual(values = c(rgb(0.1, 0.7, 0.2), rgb(0.7, 0.1, 0.1))) +
labs(
x = "Samples > 0"
)
)Total numbers of genes detected shown against the number of samples with at least one read assigned to each gene.
PCA
Sample Similarity
pca <- dge$counts %>%
.[rowSums(. == 0) < ncol(.)/2,] %>%
cpm(log = TRUE) %>%
t() %>%
prcomp()A PCA was performed using logCPM values from the subset of 18,017 genes with at least one read in more than half of the samples.
showLabel <- nrow(samples) <= 20
pca %>%
tidy() %>%
dplyr::rename(Rat = row) %>%
left_join(dge$samples, by = "Rat") %>%
dplyr::filter(PC %in% 1:2) %>%
pivot_wider(names_from = "PC", names_prefix = "PC", values_from = "value") %>%
ggplot(
aes(PC1, PC2, colour = group, size = lib.size/1e6)
) +
geom_point() +
geom_text_repel(aes(label = Rat), show.legend = FALSE) +
scale_colour_manual(values = group_cols) +
scale_size_continuous(limits = c(5, 15), breaks = seq(5, 15, by = 5)) +
labs(
x = glue("PC1 ({percent(pca$sdev[[1]]^2 / sum(pca$sdev^2), 0.1)})"),
y = glue("PC2 ({percent(pca$sdev[[2]]^2 / sum(pca$sdev^2), 0.1)})"),
colour = "Group",
size = "Library Size\n(millions)"
)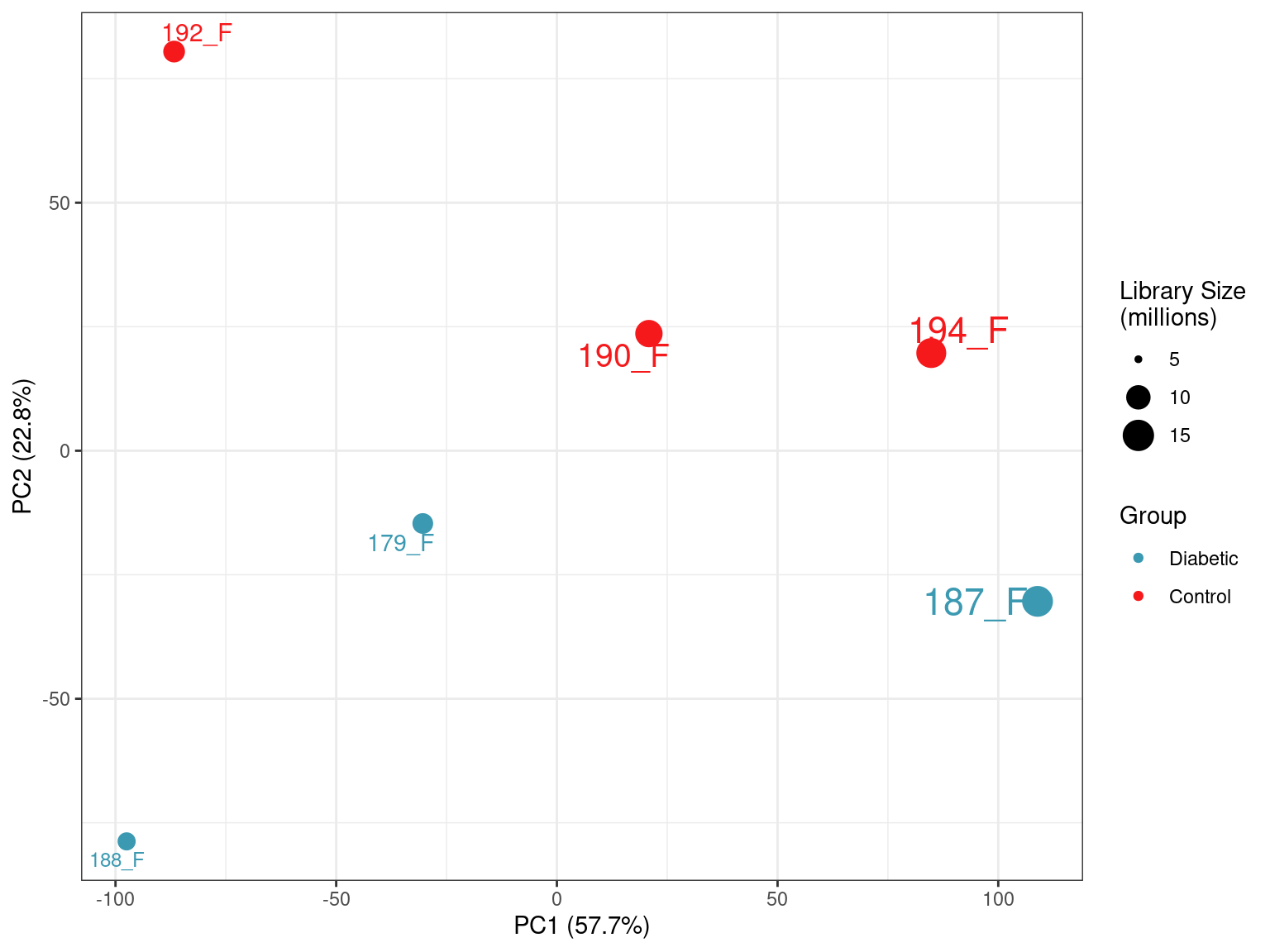
PCA plot of all samples. PC1 is most strongly correlated with library size, whilst PC2 appears to capture the majority of the biological variability
| Version | Author | Date |
|---|---|---|
| dd28879 | Steve Pederson | 2022-07-06 |
GC and Length Biases
mcols(genesGR) %>%
as.data.frame() %>%
dplyr::filter(gene_id %in% rownames(pca$rotation)) %>%
as_tibble() %>%
mutate(
bin_length = cut(
x = ave_tx_len,
labels = seq_len(10),
breaks = quantile(ave_tx_len, probs = seq(0, 1, length.out = 11)),
include.lowest = TRUE
),
bin_gc = cut(
x = gc_content,
labels = seq_len(10),
breaks = quantile(gc_content, probs = seq(0, 1, length.out = 11)),
include.lowest = TRUE
),
bin = paste(bin_gc, bin_length, sep = "_")
) %>%
dplyr::select(gene_id, contains("bin")) %>%
mutate(
PC1 = pca$rotation[gene_id, "PC1"],
PC2 = pca$rotation[gene_id, "PC2"]
) %>%
pivot_longer(
cols = c("PC1", "PC2"),
names_to = "PC",
values_to = "value"
) %>%
group_by(PC, bin_gc, bin_length, bin) %>%
summarise(
Size = n(),
mean = mean(value),
sd = sd(value),
t = t.test(value)$statistic,
p = t.test(value)$p.value,
adjP = p.adjust(p, method = "bonf")
) %>%
ggplot(
aes(bin_length, bin_gc, colour = t, alpha = -log10(adjP), size = Size)
) +
geom_point() +
facet_wrap(~PC) +
scale_colour_gradient2() +
scale_size_continuous(range = c(1, 10)) +
labs(
x = "Average Transcript Length",
y = "GC Content",
alpha = expression(paste(-log[10], p[adj]))) +
theme(
panel.grid = element_blank(),
legend.position = "bottom"
) 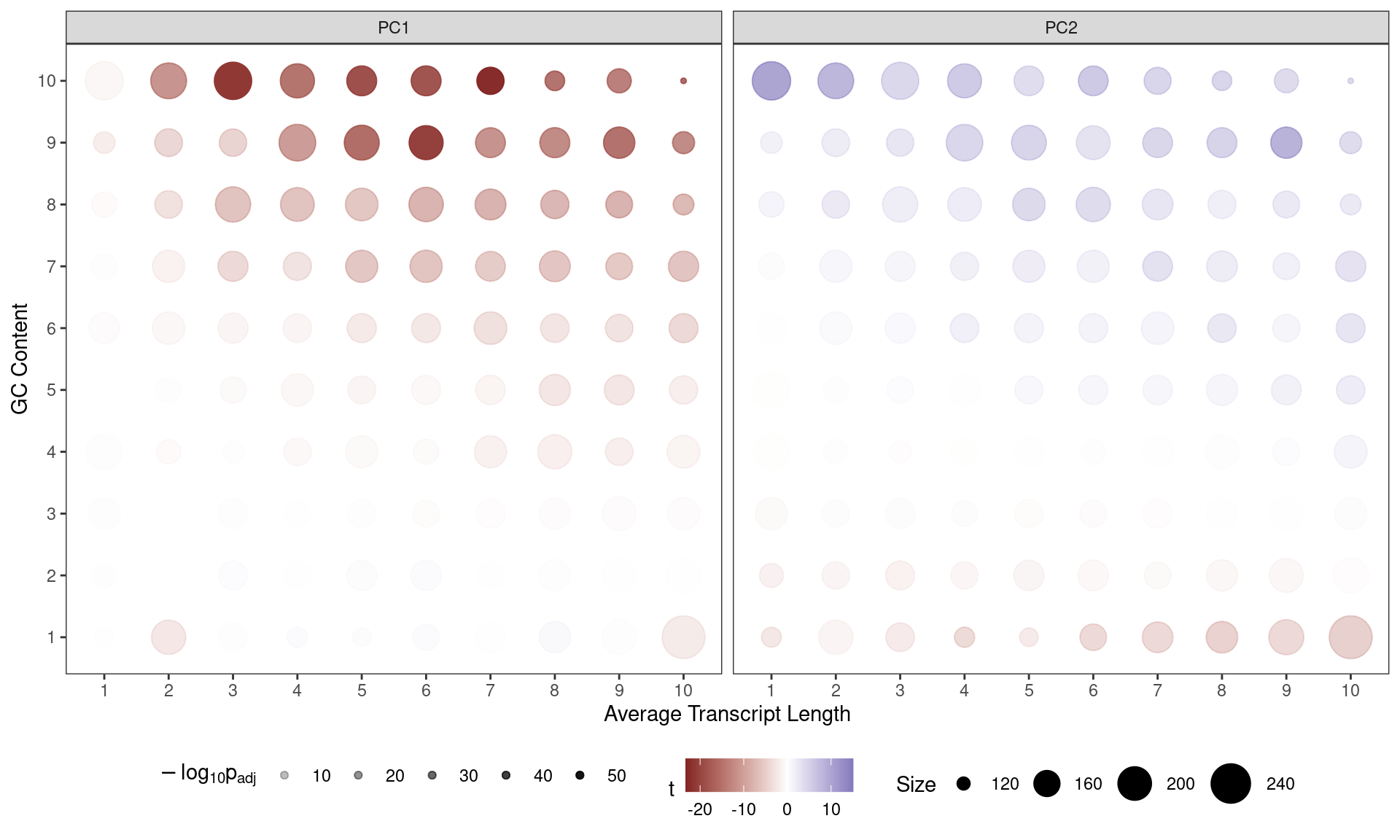
Contribution of each GC/Length Bin to PC1 and PC2. Fill colours indicate the t-statistic, with tranparency denoting significance as -log10(p), using Bonferroni-adjusted p-values.
| Version | Author | Date |
|---|---|---|
| dd28879 | Steve Pederson | 2022-07-06 |
write_rds(dge, here::here("output/dge.rds"))
sessionInfo()R version 4.2.0 (2022-04-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 20.04.4 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.9.0
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.9.0
locale:
[1] LC_CTYPE=en_AU.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_AU.UTF-8 LC_COLLATE=en_AU.UTF-8
[5] LC_MONETARY=en_AU.UTF-8 LC_MESSAGES=en_AU.UTF-8
[7] LC_PAPER=en_AU.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_AU.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] ggrepel_0.9.1 broom_0.8.0
[3] BSgenome.Rnorvegicus.UCSC.rn6_1.4.1 BSgenome_1.64.0
[5] rtracklayer_1.56.0 Biostrings_2.64.0
[7] XVector_0.36.0 magrittr_2.0.3
[9] ensembldb_2.20.1 AnnotationFilter_1.20.0
[11] GenomicFeatures_1.48.0 AnnotationDbi_1.58.0
[13] Biobase_2.56.0 GenomicRanges_1.48.0
[15] GenomeInfoDb_1.32.1 IRanges_2.30.0
[17] S4Vectors_0.34.0 AnnotationHub_3.4.0
[19] BiocFileCache_2.4.0 dbplyr_2.1.1
[21] ggfortify_0.4.14 edgeR_3.38.0
[23] limma_3.52.0 plotly_4.10.0
[25] glue_1.6.2 pander_0.6.5
[27] scales_1.2.0 yaml_2.3.5
[29] forcats_0.5.1 stringr_1.4.0
[31] dplyr_1.0.9 purrr_0.3.4
[33] readr_2.1.2 tidyr_1.2.0
[35] tidyverse_1.3.1 ngsReports_1.13.0
[37] tibble_3.1.7 ggplot2_3.3.6
[39] BiocGenerics_0.42.0 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] readxl_1.4.0 backports_1.4.1
[3] lazyeval_0.2.2 crosstalk_1.2.0
[5] BiocParallel_1.30.0 digest_0.6.29
[7] htmltools_0.5.2 fansi_1.0.3
[9] memoise_2.0.1 tzdb_0.3.0
[11] modelr_0.1.8 matrixStats_0.62.0
[13] vroom_1.5.7 prettyunits_1.1.1
[15] colorspace_2.0-3 blob_1.2.3
[17] rvest_1.0.2 rappdirs_0.3.3
[19] haven_2.5.0 xfun_0.30
[21] callr_3.7.0 crayon_1.5.1
[23] RCurl_1.98-1.6 jsonlite_1.8.0
[25] zoo_1.8-10 gtable_0.3.0
[27] zlibbioc_1.42.0 DelayedArray_0.22.0
[29] Rhdf5lib_1.18.0 DBI_1.1.2
[31] Rcpp_1.0.8.3 viridisLite_0.4.0
[33] xtable_1.8-4 progress_1.2.2
[35] bit_4.0.4 DT_0.22
[37] htmlwidgets_1.5.4 httr_1.4.3
[39] ellipsis_0.3.2 farver_2.1.0
[41] pkgconfig_2.0.3 XML_3.99-0.9
[43] sass_0.4.1 here_1.0.1
[45] locfit_1.5-9.5 utf8_1.2.2
[47] labeling_0.4.2 tidyselect_1.1.2
[49] rlang_1.0.2 later_1.3.0
[51] munsell_0.5.0 BiocVersion_3.15.2
[53] cellranger_1.1.0 tools_4.2.0
[55] cachem_1.0.6 cli_3.3.0
[57] generics_0.1.2 RSQLite_2.2.13
[59] evaluate_0.15 fastmap_1.1.0
[61] ggdendro_0.1.23 processx_3.5.3
[63] knitr_1.39 bit64_4.0.5
[65] fs_1.5.2 KEGGREST_1.36.0
[67] whisker_0.4 mime_0.12
[69] xml2_1.3.3 biomaRt_2.52.0
[71] compiler_4.2.0 rstudioapi_0.13
[73] filelock_1.0.2 curl_4.3.2
[75] png_0.1-7 interactiveDisplayBase_1.34.0
[77] reprex_2.0.1 bslib_0.3.1
[79] stringi_1.7.6 highr_0.9
[81] ps_1.7.0 lattice_0.20-45
[83] ProtGenerics_1.28.0 Matrix_1.4-1
[85] vctrs_0.4.1 rhdf5filters_1.8.0
[87] pillar_1.7.0 lifecycle_1.0.1
[89] BiocManager_1.30.17 jquerylib_0.1.4
[91] data.table_1.14.2 bitops_1.0-7
[93] httpuv_1.6.5 R6_2.5.1
[95] BiocIO_1.6.0 promises_1.2.0.1
[97] gridExtra_2.3 MASS_7.3-57
[99] assertthat_0.2.1 rhdf5_2.40.0
[101] SummarizedExperiment_1.26.1 rprojroot_2.0.3
[103] rjson_0.2.21 withr_2.5.0
[105] GenomicAlignments_1.32.0 Rsamtools_2.12.0
[107] GenomeInfoDbData_1.2.8 parallel_4.2.0
[109] hms_1.1.1 grid_4.2.0
[111] rmarkdown_2.14 MatrixGenerics_1.8.0
[113] git2r_0.30.1 getPass_0.2-2
[115] shiny_1.7.1 lubridate_1.8.0
[117] restfulr_0.0.13