Last updated: 2022-11-17
Checks: 6 1
Knit directory:
Genomic-Selection-for-Drought-Tolerance-Using-Genome-Wide-SNPs-in-Casava/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R
Markdown file created these results, you’ll want to first commit it to
the Git repo. If you’re still working on the analysis, you can ignore
this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20221020) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 0f3154e. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rproj.user/
Ignored: data/allchrAR08.txt
Untracked files:
Untracked: analysis/figure/
Untracked: analysis/gws-multi-enviroment.Rmd
Untracked: analysis/phenotype_multi.Rmd
Untracked: data/genomic selection.pptx
Untracked: data/pheno_clean.csv
Untracked: output/BLUPS_Multi.csv
Untracked: output/BLUPS_par_ind.Rdata
Untracked: output/accuracy_multi.png
Untracked: output/media_pheno_Multi.csv
Untracked: output/result_G_BLUP.csv
Untracked: output/varcomp.csv
Unstaged changes:
Modified: analysis/GWS.Rmd
Modified: analysis/_site.yml
Modified: analysis/phenotype.Rmd
Modified: output/BLUPS_par.Rdata
Modified: output/resultMM.Rdata
Modified: output/results_cv.csv
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with
wflow_publish() to start tracking its development.
To perform the analyses, we will need the following packages:
library(readxl)
require(tidyverse)
library(kableExtra)
library(janitor)
library(genomicMateSelectR)
library(AGHmatrix)
require(ComplexHeatmap)
require(rrBLUP)
library(ggpmisc)
library(cvTools)
theme_set(theme_bw())Data
The data set is based in genotypes evalueted in five years (2016 to 2020), each year was considered as environment.
Names marker data
Primeiro vamos buscar os ID dos marcadores para cada clone.
names <-
read_excel("data/Phenotyping.xlsx", sheet = "GBS") %>%
rename(Clone = `Names trials Petrolina`,
ID_Clone = `Nome GBS`) %>%
mutate(ID_Clone = str_replace_all(ID_Clone, ":", ".")) %>%
select(Clone, ID_Clone)
names %>%
head() %>%
kbl(escape = F, align = 'c') %>%
kable_classic(
"hover",
full_width = F,
position = "center",
fixed_thead = T
)| Clone | ID_Clone |
|---|---|
| 4271 | 4271.250494233 |
| 9624-09 | 962409.250370255 |
| Aipim Abacate | ERETA.250437577 |
| Alagoana | Alagoana363.250437472 |
| BGM-0004 | CNPMF4.250370278 |
| BGM-0019 | CNPMF19.250370327 |
Phenotypic data
Agora vamos agrupar os IDs dos marcadores com os nomes dos clones.
pheno <- read.csv("output/BLUPS_Multi.csv") %>%
inner_join(names) %>% # Join Phenotypic data with Genotypics names (ID_Clones) of the Clones
mutate(Clone = as.factor(Clone),
ID_Clone = as.factor(ID_Clone),
Ano = as.factor(Ano))Joining, by = "Clone"pheno %>%
head() %>%
kbl(escape = F, align = 'c') %>%
kable_classic(
"hover",
full_width = F,
position = "center",
fixed_thead = T
)| Clone | Ano | NR.P | PTR | PPA | AP | HI | RF | CR | DR | DCaule | Ácaro | MS | PROD.AMD | AMD | Porte | Nº.Hastes | Stand_Final | Branching_Level | Staygreen | ID_Clone |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Alagoana | 2017 | -0.6795277 | 0.1656796 | 0.3485062 | -0.0284596 | 0.5013653 | -0.0667096 | 0.4854822 | 2.9140433 | 0.0044243 | -0.1333563 | NA | NA | NA | NA | NA | NA | NA | NA | Alagoana363.250437472 |
| BGM-0032 | 2017 | 1.4149888 | 0.3046166 | 0.8963395 | 0.1214955 | 0.4290333 | 0.4001097 | 4.5063002 | -0.6829637 | 0.4800358 | 0.2080576 | NA | NA | NA | NA | NA | NA | NA | NA | CNPMF32.250370305 |
| BGM-0036 | 2017 | -1.3540323 | -0.8137316 | 8.0983401 | 0.2122890 | -11.3135067 | 0.0967386 | 2.1977370 | -0.5302431 | 0.2737945 | -0.4231997 | NA | NA | NA | NA | NA | NA | NA | NA | CNPMF36.250370328 |
| BGM-0046 | 2017 | -0.3322988 | 2.4625915 | 4.9096154 | 0.1085703 | 4.3032975 | 0.0967386 | 3.3122158 | 3.0876838 | 0.1252696 | 0.0502433 | NA | NA | NA | NA | NA | NA | NA | NA | CNPMF46.250370283 |
| BGM-0056 | 2017 | 1.9146996 | 2.3963149 | 0.4202654 | 0.1178382 | 12.1574116 | -0.1420580 | 3.7627115 | 3.3581355 | 0.3203126 | 0.0733478 | NA | NA | NA | NA | NA | NA | NA | NA | CNPMF56.250370248 |
| BGM-0065 | 2017 | 0.7642085 | 0.1967549 | 3.6785055 | 0.0919147 | -3.9200178 | -0.2301578 | 1.5136325 | 3.1620003 | 0.0105003 | -0.2696599 | NA | NA | NA | NA | NA | NA | NA | NA | CNPMF65.250370285 |
Genotypic data
Com o filtro de MAF a 1% restaram 22779 marcadores. Vou carregar a matriz genotipica agora.
geno <- readRDS("data/SNPs.rds")
geno[1:5,1:5] S1_84637 S1_84843 S1_126260 S1_126261 S1_126264
ERETA.250437577 2 2 2 2 2
Alagoana363.250437472 2 2 2 2 2
CNPMF4.250370278 2 2 2 2 2
CNPMF19.250370327 1 2 2 2 2
CNPMF30.250370293 2 1 2 2 2Building the G matrix
Again, we will use the AGHmatrix package [@amadeu_aghmatrix_2016] to build the G matrix:
G_matrix <- Gmatrix(geno,
method = "VanRaden",
ploidy = 2,
missingValue = NA)Initial data:
Number of Individuals: 414
Number of Markers: 22779
Missing data check:
Total SNPs: 22779
0 SNPs dropped due to missing data threshold of 1
Total of: 22779 SNPs
MAF check:
No SNPs with MAF below 0
Monomorphic check:
No monomorphic SNPs
Summary check:
Initial: 22779 SNPs
Final: 22779 SNPs ( 0 SNPs removed)
Completed! Time = 28.46 seconds Now we have the whole G matrix (414 x 414), which we can represent using a heatmap:
Heatmap(
G_matrix,
show_row_names = F,
show_column_names = F,
heatmap_legend_param = list(title = "Res")
)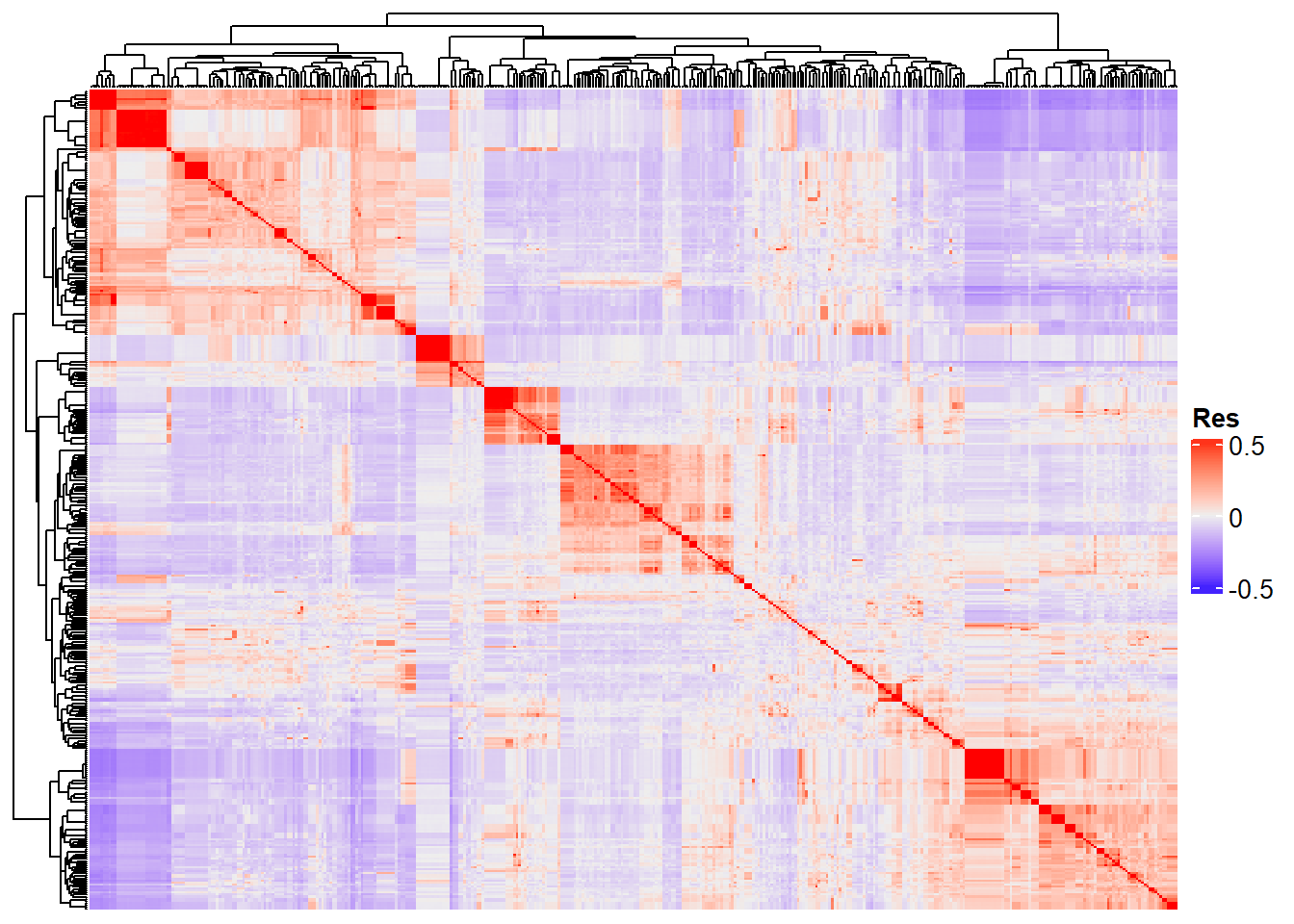
“Res” in the heatmap legend title is for “Resemblance”.
Genomic selection
1. Restraining the genotype means - only adj. means and genotyped:
For this purpose, we will use only individuals with BLUps and SNPs available.
pheno <- pheno %>%
filter(ID_Clone %in% rownames(geno)) %>%
droplevels()2. Building the genotype and environment design matrices:
envmat = model.matrix( ~ -1 + Ano, data = pheno)envmat[1:5, 1:4] %>% kbl(escape = F, align = 'c') %>%
kable_classic(
"hover",
full_width = F,
position = "center",
fixed_thead = T
) %>% footnote("Dimension: 534 $\\times$ 4", general_title = "")| Ano2017 | Ano2018 | Ano2019 | Ano2020 |
|---|---|---|---|
| 1 | 0 | 0 | 0 |
| 1 | 0 | 0 | 0 |
| 1 | 0 | 0 | 0 |
| 1 | 0 | 0 | 0 |
| 1 | 0 | 0 | 0 |
| Dimension: 534 \(\times\) 4 |
genmat = model.matrix( ~ -1 + Clone, data = pheno)genmat[1:5, 1:5] %>% kbl(escape = F, align = 'c') %>%
kable_classic(
"hover",
full_width = F,
position = "center",
fixed_thead = T
) %>% footnote("Dimension: 534 $\\times$ 414", general_title = "")| CloneAipim Abacate | CloneAlagoana | CloneBGM-0004 | CloneBGM-0019 | CloneBGM-0030 |
|---|---|---|---|---|
| 0 | 1 | 0 | 0 | 0 |
| 0 | 0 | 0 | 0 | 0 |
| 0 | 0 | 0 | 0 | 0 |
| 0 | 0 | 0 | 0 | 0 |
| 0 | 0 | 0 | 0 | 0 |
| Dimension: 534 \(\times\) 414 |
3. Building the environmental and genetic covariance matrices:
G = tcrossprod(tcrossprod(genmat, G_matrix), genmat)G[1:5, 1:5] %>% kbl(escape = F, align = 'c') %>%
kable_classic(
"hover",
full_width = F,
position = "center",
fixed_thead = T
) %>% footnote("Dimension: 534 $\\times$ 534", general_title = "")| 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|
| 1.0404654 | -0.0759830 | 0.0667313 | 0.0020280 | 0.0188232 |
| -0.0759830 | 1.2906265 | -0.0615798 | -0.0413384 | -0.0153453 |
| 0.0667313 | -0.0615798 | 0.9818020 | 0.0750449 | 0.1472075 |
| 0.0020280 | -0.0413384 | 0.0750449 | 0.9779189 | 0.0018877 |
| 0.0188232 | -0.0153453 | 0.1472075 | 0.0018877 | 1.0308067 |
| Dimension: 534 \(\times\) 534 |
E = tcrossprod(envmat)E[1:5, 1:5] %>% kbl(escape = F, align = 'c') %>%
kable_classic(
"hover",
full_width = F,
position = "center",
fixed_thead = T
) %>% footnote("Dimension: 534 $\\times$ 534", general_title = "")| 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|
| 1 | 1 | 1 | 1 | 1 |
| 1 | 1 | 1 | 1 | 1 |
| 1 | 1 | 1 | 1 | 1 |
| 1 | 1 | 1 | 1 | 1 |
| 1 | 1 | 1 | 1 | 1 |
| Dimension: 534 \(\times\) 534 |
4. Building the interaction matrix:
GE = G * EGE[1:5, 1:5] %>% kbl(escape = F, align = 'c') %>%
kable_classic(
"hover",
full_width = F,
position = "center",
fixed_thead = T
) %>% footnote("Dimension: 534 $\\times$ 534", general_title = "")| 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|
| 1.0404654 | -0.0759830 | 0.0667313 | 0.0020280 | 0.0188232 |
| -0.0759830 | 1.2906265 | -0.0615798 | -0.0413384 | -0.0153453 |
| 0.0667313 | -0.0615798 | 0.9818020 | 0.0750449 | 0.1472075 |
| 0.0020280 | -0.0413384 | 0.0750449 | 0.9779189 | 0.0018877 |
| 0.0188232 | -0.0153453 | 0.1472075 | 0.0018877 | 1.0308067 |
| Dimension: 534 \(\times\) 534 |
GBLUP
In the GBLUP, we will use the G matrix instead of the SNPs matrix. Thus, we will obtain the GEBV directly. Note that we will need to build the G matrix again, since some genotypes were dropped after our filtering. The rrBLUP package has a function called “A.mat” that build the Additive Genomic Matrix from a SNP matrix with “-1,0,1” codification:
year <- levels(pheno$Ano)
traits <- colnames(pheno[3:20])
result <- data.frame()
result_G_BLUP <- data.frame()
for (i in traits) {
for (j in year) {
data <- pheno %>%
filter(Ano == j) %>%
droplevels()
if (is.na(mean(data[[i]], na.rm = TRUE))) {
next
}
index = rownames(geno) %in% data$ID_Clone
data_gen = geno[index,]
GBLUP <- mixed.solve(data[[i]], K = A.mat(data_gen - 1))
if (dim(result)[1] == 0) {
result <- data.frame(
ID_Clone = rownames(GBLUP$u),
Ano = j,
stringsAsFactors = F
)
result[, i] <- data.frame(GBLUP$u)
} else {
result1 <- data.frame(
ID_Clone = rownames(GBLUP$u),
Ano = j,
stringsAsFactors = F
)
result1[, i] <- data.frame(GBLUP$u)
result <- rbind(result1, result)
}
}
if (i == "NR.P") {
result_G_BLUP <- result
} else{
if (dim(result)[1] == 0) {
next
}
result_G_BLUP1 <- result
result_G_BLUP <- result_G_BLUP %>%
full_join(result_G_BLUP1)
}
result<-data.frame()
}Joining, by = c("ID_Clone", "Ano")
Joining, by = c("ID_Clone", "Ano")
Joining, by = c("ID_Clone", "Ano")
Joining, by = c("ID_Clone", "Ano")
Joining, by = c("ID_Clone", "Ano")
Joining, by = c("ID_Clone", "Ano")
Joining, by = c("ID_Clone", "Ano")
Joining, by = c("ID_Clone", "Ano")
Joining, by = c("ID_Clone", "Ano")
Joining, by = c("ID_Clone", "Ano")
Joining, by = c("ID_Clone", "Ano")
Joining, by = c("ID_Clone", "Ano")
Joining, by = c("ID_Clone", "Ano")
Joining, by = c("ID_Clone", "Ano")
Joining, by = c("ID_Clone", "Ano")
Joining, by = c("ID_Clone", "Ano")
Joining, by = c("ID_Clone", "Ano")write.csv(result_G_BLUP, "output/result_G_BLUP.csv" , row.names = F,
quote = F)Aqui, iremos adicionar novamente os valores médios dos valores fenotípicos aos GEBVs obtidos pelo G-BLUP.
media_pheno_Multi <- read.csv("output/media_pheno_Multi.csv") %>%
mutate(Ano = as.factor(Ano))
phen<-
data.frame(pheno %>%
group_by(ID_Clone, Ano) %>%
group_keys() %>%
arrange(ID_Clone, .by_group = TRUE),
stringsAsFactors = F)
media_pheno_Multi <- full_join(phen, media_pheno_Multi)Joining, by = "Ano"traits <- colnames(result_G_BLUP[3:ncol(result_G_BLUP)])
result_G_BLUP <- result_G_BLUP %>%
arrange(ID_Clone, .by_group = TRUE)
for (i in traits) {
phen[,i] <- result_G_BLUP[,i] + media_pheno_Multi[,i]
}GEBVS_G_BLUP <- result_G_BLUP %>%
pivot_longer(NR.P:Staygreen,
names_to = "Variable",
values_to = "GEBVS")
BLUPS_MED <- result_G_BLUP %>%
arrange(ID_Clone, .by_group = TRUE)
for (i in traits) {
BLUPS_MED[,i] <- BLUPS_MED[,i] + media_pheno_Multi[, i]
}
BLUPS_MED <- BLUPS_MED %>%
pivot_longer(NR.P:Staygreen,
names_to = "Variable",
values_to = "BLUPS")
data_gws_G_BLUP <- GEBVS_G_BLUP %>%
full_join(BLUPS_MED)Joining, by = c("ID_Clone", "Ano", "Variable")Agora vamos produzir os gráficos para cada variável comparando a correlação entre os GEBVs e BLUPs.
data_gws_G_BLUP %>%
ggplot(aes(x = BLUPS, y = GEBVS)) +
geom_point(aes(color = GEBVS), show.legend = F) +
geom_smooth(method = lm, se = F) +
stat_poly_eq(formula = y ~ x, label.y = "bottom") +
scale_color_gradient(low = '#c80000', high = '#FFFF00') +
facet_wrap(. ~ Variable, ncol = 6, scales = "free") +
expand_limits(y = 0, x = 0) +
ggtitle("G-BLUP")`geom_smooth()` using formula 'y ~ x'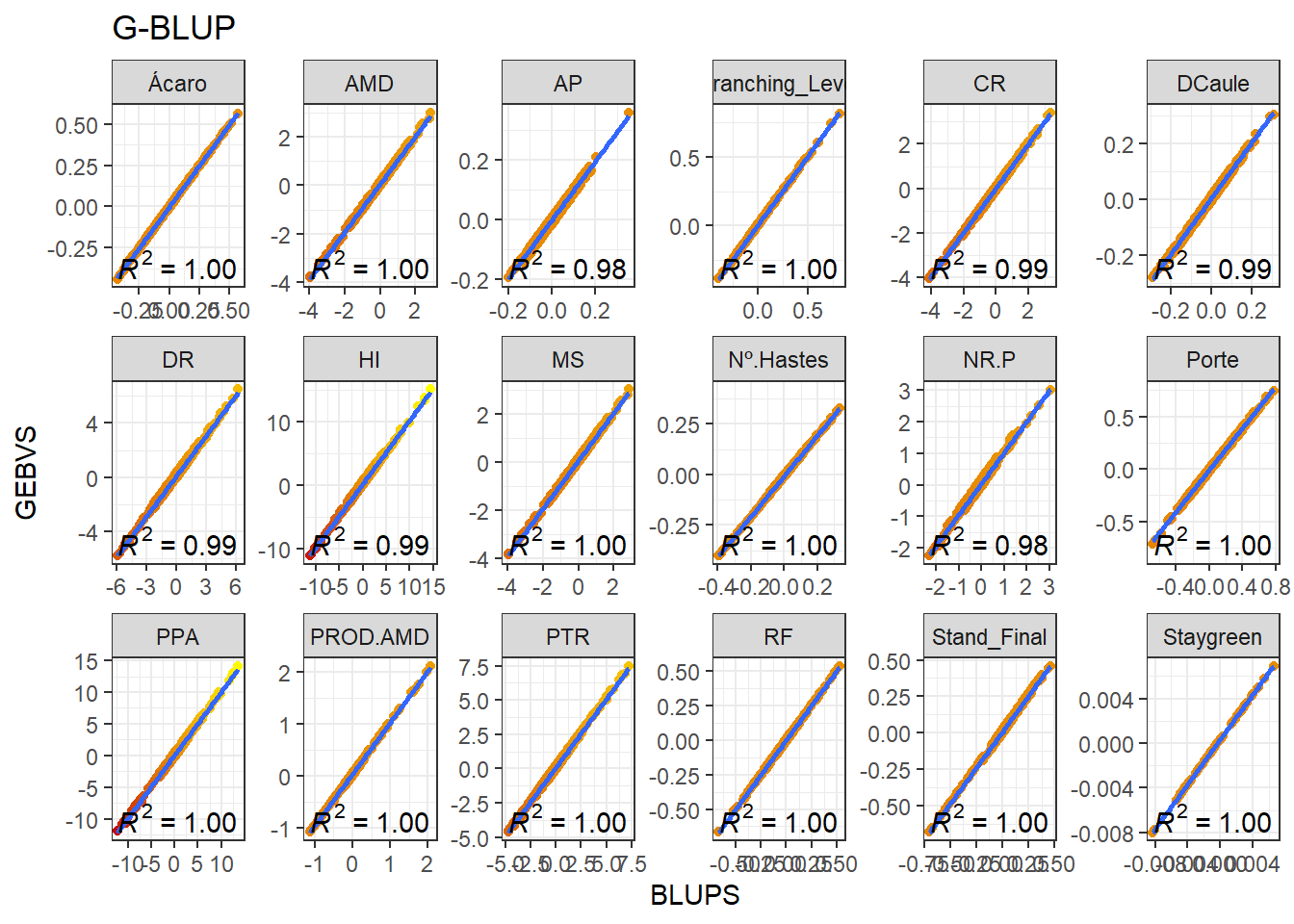
Agora vamos comparar os modelos por meio de um boxplot.
data_gws_G_BLUP %>%
pivot_longer(4:5, names_to = "Method", values_to = "Values") %>%
ggplot(aes(x = , y = Values, fill = Method)) +
geom_boxplot() +
facet_wrap(. ~ Variable, ncol = 6, scales = "free") +
expand_limits(y = 0, x = 0) +
ggtitle("G-BLUP")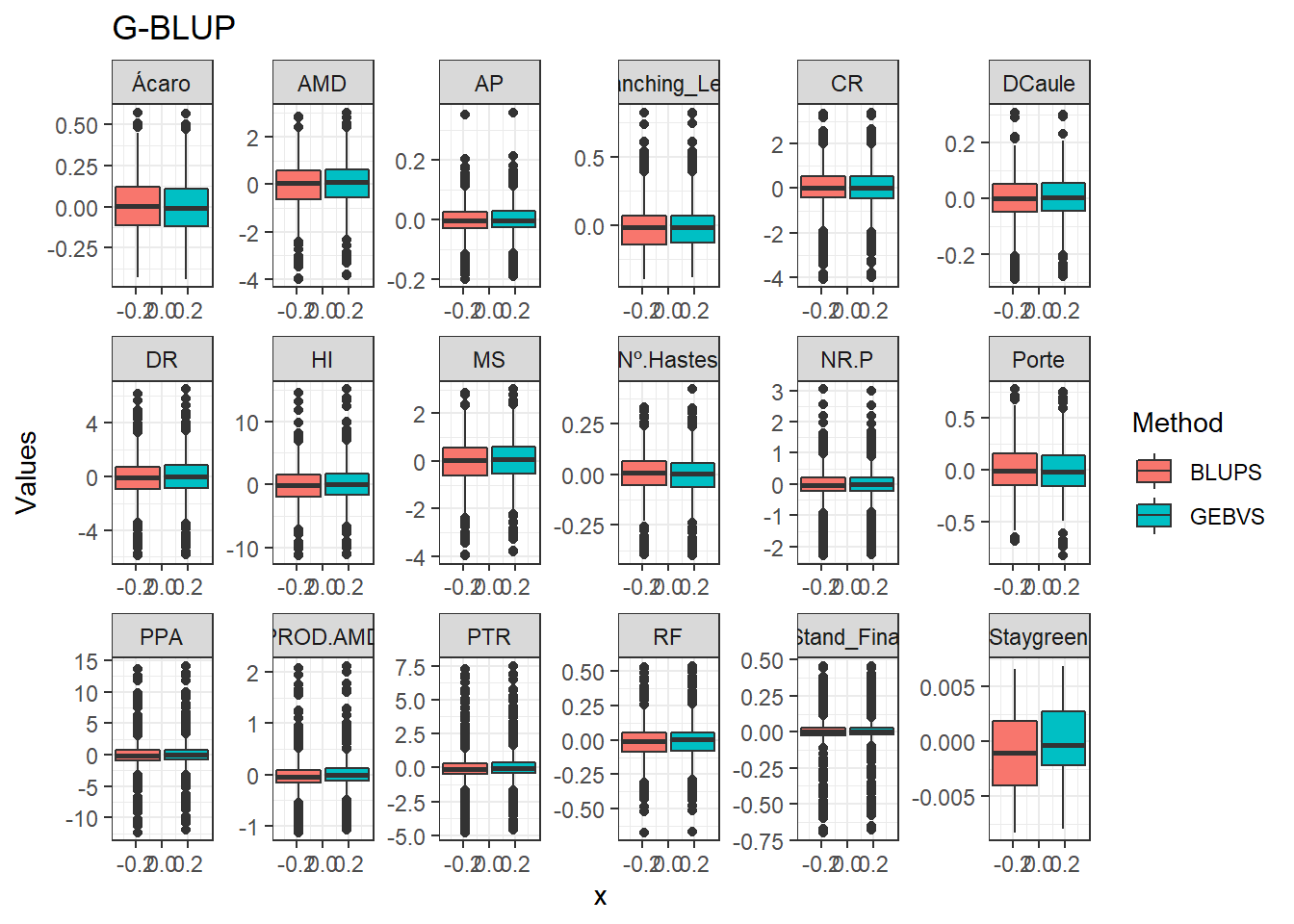
Cross-validation
To prove that the prediction is accurate, we should perform a cross-validation (CV) scheme. For this purpose, we divide the data into a training set and a validation set.
First we separate the data into k folds. Then, we attribute NA for one fold and try to predict the data from this fold based on the others. When selecting the number of folds, one must prioritize the balance between the number of observations in each fold.
In addition, this process should be repeated for further validation. The step-by-step below will guide the CV in the data we are analysing.
1. Determine the number of folds and repetitions
nfolds = 5
nrept = 5Since we defined 5 folds, our data will be divided into 5 parts with 83 observations each.
2. Match the data and the rows of the SNP matrix
all(rownames(geno) %in% pheno$ID_Clone)[1] TRUEall(pheno$ID_Clone %in% rownames(geno))[1] TRUE3. Croos-Validation
set.seed(100)
results <- list()
results_cv <- data.frame()
for (j in traits) {
for (a in year) {
data <- pheno %>%
filter(Ano == a) %>%
droplevels()
if (is.na(mean(data[[j]], na.rm = TRUE))) {
next
}
index = rownames(geno) %in% data$ID_Clone
data_gen = geno[index, ]
data$ID <- factor(1:nrow(data))
sort <- list()
for (r in 1:nrept) {
for (f in 1:nfolds) {
folds <- cvFolds(nlevels(data$ID),
type = "random",
K = 5,
R = 1)
Sample <- cbind(folds$which, folds$subsets)
cv <- split(Sample[, 2], f = Sample[, 1])
}
sort[[r]] <- cv
}
rm(r, folds, f, cv, Sample)
fold.list <- sort
for (z in 1:length(fold.list)) {
for (i in 1:nfolds) {
# Training set
train_data <- data
# Validation set
train_data[train_data$ID %in% fold.list[[z]][[i]], j] <- NA
# Fitting model
GBLUP <-
mixed.solve(train_data[[j]], K = A.mat(data_gen - 1))
# GEBV
Pred <- data.frame(Yhat = GBLUP$u, G = data$ID)
rownames(Pred) <- rownames(data_gen)
# Predicted GEBV
results[[i]] <- Pred[Pred[, "G"] %in% fold.list[[z]][[i]],]
# Remove
rm(GBLUP, Pred, train_data)
}
GEBV <- do.call(rbind, results)
GEBV <- GEBV[order(GEBV$G),]
# Log
log <- all(GEBV$G == data$ID)
# Results
result_cv <- data.frame(
Ano = a,
Trait = j,
Rep = z,
Log = log,
Ac = round(cor(GEBV$Yhat, data[[j]], use = "na.or.complete"), 3),
MSPE = round(mean(((GEBV$Yhat - data[[j]]) ^ 2
), na.rm = T), 3)
)
results_cv <-
rbind(results_cv, result_cv)
}
}
}results_cv %>%
kbl(escape = F, align = 'c') %>%
kable_classic(
"hover",
full_width = F,
position = "center",
fixed_thead = T
)| Ano | Trait | Rep | Log | Ac | MSPE |
|---|---|---|---|---|---|
| 2017 | NR.P | 1 | TRUE | 0.069 | 2.715 |
| 2017 | NR.P | 2 | TRUE | 0.101 | 2.687 |
| 2017 | NR.P | 3 | TRUE | 0.162 | 2.642 |
| 2017 | NR.P | 4 | TRUE | -0.024 | 2.784 |
| 2017 | NR.P | 5 | TRUE | -0.002 | 2.785 |
| 2018 | NR.P | 1 | TRUE | -0.075 | 0.487 |
| 2018 | NR.P | 2 | TRUE | 0.072 | 0.457 |
| 2018 | NR.P | 3 | TRUE | 0.057 | 0.462 |
| 2018 | NR.P | 4 | TRUE | 0.022 | 0.463 |
| 2018 | NR.P | 5 | TRUE | 0.054 | 0.459 |
| 2019 | NR.P | 1 | TRUE | 0.131 | 2.596 |
| 2019 | NR.P | 2 | TRUE | 0.048 | 2.699 |
| 2019 | NR.P | 3 | TRUE | 0.121 | 2.610 |
| 2019 | NR.P | 4 | TRUE | 0.229 | 2.520 |
| 2019 | NR.P | 5 | TRUE | 0.175 | 2.563 |
| 2020 | NR.P | 1 | TRUE | 0.566 | 1.339 |
| 2020 | NR.P | 2 | TRUE | 0.502 | 1.488 |
| 2020 | NR.P | 3 | TRUE | 0.598 | 1.262 |
| 2020 | NR.P | 4 | TRUE | 0.551 | 1.372 |
| 2020 | NR.P | 5 | TRUE | 0.492 | 1.511 |
| 2017 | PTR | 1 | TRUE | 0.147 | 1.810 |
| 2017 | PTR | 2 | TRUE | 0.224 | 1.754 |
| 2017 | PTR | 3 | TRUE | 0.220 | 1.756 |
| 2017 | PTR | 4 | TRUE | 0.108 | 1.856 |
| 2017 | PTR | 5 | TRUE | 0.097 | 1.862 |
| 2018 | PTR | 1 | TRUE | 0.098 | 2.186 |
| 2018 | PTR | 2 | TRUE | 0.055 | 2.243 |
| 2018 | PTR | 3 | TRUE | 0.091 | 2.209 |
| 2018 | PTR | 4 | TRUE | 0.053 | 2.231 |
| 2018 | PTR | 5 | TRUE | 0.108 | 2.179 |
| 2019 | PTR | 1 | TRUE | 0.196 | 5.646 |
| 2019 | PTR | 2 | TRUE | 0.078 | 6.027 |
| 2019 | PTR | 3 | TRUE | 0.069 | 6.034 |
| 2019 | PTR | 4 | TRUE | 0.134 | 5.806 |
| 2019 | PTR | 5 | TRUE | 0.071 | 6.055 |
| 2020 | PTR | 1 | TRUE | 0.439 | 10.569 |
| 2020 | PTR | 2 | TRUE | 0.465 | 10.164 |
| 2020 | PTR | 3 | TRUE | 0.466 | 10.105 |
| 2020 | PTR | 4 | TRUE | 0.483 | 9.895 |
| 2020 | PTR | 5 | TRUE | 0.456 | 10.285 |
| 2017 | PPA | 1 | TRUE | 0.240 | 9.100 |
| 2017 | PPA | 2 | TRUE | 0.308 | 8.717 |
| 2017 | PPA | 3 | TRUE | 0.343 | 8.463 |
| 2017 | PPA | 4 | TRUE | 0.270 | 8.992 |
| 2017 | PPA | 5 | TRUE | 0.423 | 8.015 |
| 2018 | PPA | 1 | TRUE | 0.228 | 2.781 |
| 2018 | PPA | 2 | TRUE | 0.191 | 2.848 |
| 2018 | PPA | 3 | TRUE | 0.227 | 2.786 |
| 2018 | PPA | 4 | TRUE | 0.249 | 2.746 |
| 2018 | PPA | 5 | TRUE | 0.223 | 2.794 |
| 2019 | PPA | 1 | TRUE | 0.073 | 8.448 |
| 2019 | PPA | 2 | TRUE | 0.178 | 8.042 |
| 2019 | PPA | 3 | TRUE | 0.128 | 8.215 |
| 2019 | PPA | 4 | TRUE | 0.064 | 8.461 |
| 2019 | PPA | 5 | TRUE | 0.179 | 8.032 |
| 2020 | PPA | 1 | TRUE | 0.368 | 64.976 |
| 2020 | PPA | 2 | TRUE | 0.325 | 68.539 |
| 2020 | PPA | 3 | TRUE | 0.286 | 71.632 |
| 2020 | PPA | 4 | TRUE | 0.408 | 62.612 |
| 2020 | PPA | 5 | TRUE | 0.384 | 64.212 |
| 2017 | AP | 1 | TRUE | 0.388 | 0.022 |
| 2017 | AP | 2 | TRUE | 0.381 | 0.023 |
| 2017 | AP | 3 | TRUE | 0.381 | 0.022 |
| 2017 | AP | 4 | TRUE | 0.389 | 0.022 |
| 2017 | AP | 5 | TRUE | 0.396 | 0.022 |
| 2018 | AP | 1 | TRUE | 0.043 | 0.009 |
| 2018 | AP | 2 | TRUE | 0.037 | 0.009 |
| 2018 | AP | 3 | TRUE | -0.083 | 0.010 |
| 2018 | AP | 4 | TRUE | -0.069 | 0.010 |
| 2018 | AP | 5 | TRUE | -0.131 | 0.010 |
| 2019 | AP | 1 | TRUE | 0.153 | 0.014 |
| 2019 | AP | 2 | TRUE | 0.076 | 0.014 |
| 2019 | AP | 3 | TRUE | 0.047 | 0.015 |
| 2019 | AP | 4 | TRUE | 0.093 | 0.014 |
| 2019 | AP | 5 | TRUE | 0.152 | 0.014 |
| 2020 | AP | 1 | TRUE | 0.315 | 0.025 |
| 2020 | AP | 2 | TRUE | 0.260 | 0.026 |
| 2020 | AP | 3 | TRUE | 0.254 | 0.026 |
| 2020 | AP | 4 | TRUE | 0.210 | 0.027 |
| 2020 | AP | 5 | TRUE | 0.365 | 0.024 |
| 2017 | HI | 1 | TRUE | 0.133 | 60.564 |
| 2017 | HI | 2 | TRUE | 0.101 | 61.948 |
| 2017 | HI | 3 | TRUE | 0.153 | 60.296 |
| 2017 | HI | 4 | TRUE | 0.153 | 60.382 |
| 2017 | HI | 5 | TRUE | 0.171 | 59.205 |
| 2018 | HI | 1 | TRUE | 0.150 | 38.174 |
| 2018 | HI | 2 | TRUE | 0.029 | 39.828 |
| 2018 | HI | 3 | TRUE | 0.204 | 37.183 |
| 2018 | HI | 4 | TRUE | 0.097 | 38.903 |
| 2018 | HI | 5 | TRUE | 0.058 | 40.630 |
| 2019 | HI | 1 | TRUE | -0.014 | 73.880 |
| 2019 | HI | 2 | TRUE | -0.134 | 76.365 |
| 2019 | HI | 3 | TRUE | -0.115 | 77.022 |
| 2019 | HI | 4 | TRUE | -0.131 | 77.614 |
| 2019 | HI | 5 | TRUE | -0.127 | 77.601 |
| 2020 | HI | 1 | TRUE | 0.408 | 39.800 |
| 2020 | HI | 2 | TRUE | 0.427 | 38.740 |
| 2020 | HI | 3 | TRUE | 0.454 | 37.223 |
| 2020 | HI | 4 | TRUE | 0.369 | 41.692 |
| 2020 | HI | 5 | TRUE | 0.472 | 36.383 |
| 2017 | RF | 1 | TRUE | 0.217 | 0.079 |
| 2017 | RF | 2 | TRUE | 0.206 | 0.080 |
| 2017 | RF | 3 | TRUE | 0.174 | 0.081 |
| 2017 | RF | 4 | TRUE | 0.259 | 0.078 |
| 2017 | RF | 5 | TRUE | 0.177 | 0.081 |
| 2018 | RF | 1 | TRUE | 0.401 | 0.143 |
| 2018 | RF | 2 | TRUE | 0.367 | 0.149 |
| 2018 | RF | 3 | TRUE | 0.396 | 0.144 |
| 2018 | RF | 4 | TRUE | 0.335 | 0.155 |
| 2018 | RF | 5 | TRUE | 0.409 | 0.143 |
| 2020 | RF | 1 | TRUE | 0.342 | 0.012 |
| 2020 | RF | 2 | TRUE | 0.389 | 0.012 |
| 2020 | RF | 3 | TRUE | 0.424 | 0.012 |
| 2020 | RF | 4 | TRUE | 0.310 | 0.013 |
| 2020 | RF | 5 | TRUE | 0.330 | 0.013 |
| 2017 | CR | 1 | TRUE | 0.200 | 5.347 |
| 2017 | CR | 2 | TRUE | 0.219 | 5.281 |
| 2017 | CR | 3 | TRUE | 0.289 | 5.097 |
| 2017 | CR | 4 | TRUE | 0.143 | 5.505 |
| 2017 | CR | 5 | TRUE | 0.177 | 5.428 |
| 2018 | CR | 1 | TRUE | 0.119 | 1.033 |
| 2018 | CR | 2 | TRUE | 0.109 | 1.037 |
| 2018 | CR | 3 | TRUE | 0.045 | 1.052 |
| 2018 | CR | 4 | TRUE | 0.016 | 1.072 |
| 2018 | CR | 5 | TRUE | 0.073 | 1.044 |
| 2019 | CR | 1 | TRUE | 0.276 | 3.833 |
| 2019 | CR | 2 | TRUE | 0.236 | 3.974 |
| 2019 | CR | 3 | TRUE | 0.326 | 3.691 |
| 2019 | CR | 4 | TRUE | 0.188 | 4.077 |
| 2019 | CR | 5 | TRUE | 0.254 | 3.883 |
| 2020 | CR | 1 | TRUE | 0.356 | 7.249 |
| 2020 | CR | 2 | TRUE | 0.357 | 7.245 |
| 2020 | CR | 3 | TRUE | 0.363 | 7.186 |
| 2020 | CR | 4 | TRUE | 0.364 | 7.205 |
| 2020 | CR | 5 | TRUE | 0.348 | 7.353 |
| 2017 | DR | 1 | TRUE | 0.203 | 8.381 |
| 2017 | DR | 2 | TRUE | 0.137 | 8.616 |
| 2017 | DR | 3 | TRUE | 0.168 | 8.513 |
| 2017 | DR | 4 | TRUE | 0.200 | 8.389 |
| 2017 | DR | 5 | TRUE | 0.173 | 8.499 |
| 2018 | DR | 1 | TRUE | 0.366 | 6.054 |
| 2018 | DR | 2 | TRUE | 0.203 | 6.834 |
| 2018 | DR | 3 | TRUE | 0.341 | 6.152 |
| 2018 | DR | 4 | TRUE | 0.304 | 6.307 |
| 2018 | DR | 5 | TRUE | 0.276 | 6.443 |
| 2019 | DR | 1 | TRUE | 0.129 | 14.179 |
| 2019 | DR | 2 | TRUE | 0.114 | 14.038 |
| 2019 | DR | 3 | TRUE | 0.229 | 13.356 |
| 2019 | DR | 4 | TRUE | 0.039 | 14.774 |
| 2019 | DR | 5 | TRUE | 0.213 | 13.399 |
| 2020 | DR | 1 | TRUE | 0.480 | 11.290 |
| 2020 | DR | 2 | TRUE | 0.436 | 11.921 |
| 2020 | DR | 3 | TRUE | 0.435 | 11.961 |
| 2020 | DR | 4 | TRUE | 0.469 | 11.446 |
| 2020 | DR | 5 | TRUE | 0.385 | 12.679 |
| 2017 | DCaule | 1 | TRUE | 0.259 | 0.048 |
| 2017 | DCaule | 2 | TRUE | 0.246 | 0.048 |
| 2017 | DCaule | 3 | TRUE | 0.235 | 0.048 |
| 2017 | DCaule | 4 | TRUE | 0.237 | 0.048 |
| 2017 | DCaule | 5 | TRUE | 0.212 | 0.049 |
| 2018 | DCaule | 1 | TRUE | 0.168 | 0.024 |
| 2018 | DCaule | 2 | TRUE | 0.023 | 0.025 |
| 2018 | DCaule | 3 | TRUE | 0.134 | 0.024 |
| 2018 | DCaule | 4 | TRUE | 0.094 | 0.024 |
| 2018 | DCaule | 5 | TRUE | 0.140 | 0.024 |
| 2019 | DCaule | 1 | TRUE | 0.282 | 0.019 |
| 2019 | DCaule | 2 | TRUE | 0.176 | 0.020 |
| 2019 | DCaule | 3 | TRUE | 0.128 | 0.020 |
| 2019 | DCaule | 4 | TRUE | 0.238 | 0.019 |
| 2019 | DCaule | 5 | TRUE | 0.292 | 0.019 |
| 2020 | DCaule | 1 | TRUE | 0.341 | 0.031 |
| 2020 | DCaule | 2 | TRUE | 0.178 | 0.035 |
| 2020 | DCaule | 3 | TRUE | 0.302 | 0.033 |
| 2020 | DCaule | 4 | TRUE | 0.417 | 0.029 |
| 2020 | DCaule | 5 | TRUE | 0.388 | 0.030 |
| 2017 | Ácaro | 1 | TRUE | 0.233 | 0.074 |
| 2017 | Ácaro | 2 | TRUE | 0.325 | 0.069 |
| 2017 | Ácaro | 3 | TRUE | 0.188 | 0.075 |
| 2017 | Ácaro | 4 | TRUE | 0.319 | 0.070 |
| 2017 | Ácaro | 5 | TRUE | 0.236 | 0.073 |
| 2018 | Ácaro | 1 | TRUE | 0.279 | 0.106 |
| 2018 | Ácaro | 2 | TRUE | 0.330 | 0.101 |
| 2018 | Ácaro | 3 | TRUE | 0.236 | 0.108 |
| 2018 | Ácaro | 4 | TRUE | 0.325 | 0.102 |
| 2018 | Ácaro | 5 | TRUE | 0.295 | 0.104 |
| 2020 | Ácaro | 1 | TRUE | 0.608 | 0.070 |
| 2020 | Ácaro | 2 | TRUE | 0.555 | 0.076 |
| 2020 | Ácaro | 3 | TRUE | 0.570 | 0.074 |
| 2020 | Ácaro | 4 | TRUE | 0.532 | 0.079 |
| 2020 | Ácaro | 5 | TRUE | 0.582 | 0.073 |
| 2018 | MS | 1 | TRUE | 0.256 | 3.738 |
| 2018 | MS | 2 | TRUE | 0.069 | 3.951 |
| 2018 | MS | 3 | TRUE | 0.064 | 3.962 |
| 2018 | MS | 4 | TRUE | 0.080 | 3.957 |
| 2018 | MS | 5 | TRUE | 0.120 | 3.889 |
| 2019 | MS | 1 | TRUE | 0.317 | 3.378 |
| 2019 | MS | 2 | TRUE | 0.298 | 3.430 |
| 2019 | MS | 3 | TRUE | 0.230 | 3.618 |
| 2019 | MS | 4 | TRUE | 0.305 | 3.395 |
| 2019 | MS | 5 | TRUE | 0.340 | 3.308 |
| 2020 | MS | 1 | TRUE | 0.156 | 19.956 |
| 2020 | MS | 2 | TRUE | 0.188 | 19.558 |
| 2020 | MS | 3 | TRUE | 0.144 | 20.024 |
| 2020 | MS | 4 | TRUE | 0.193 | 19.513 |
| 2020 | MS | 5 | TRUE | 0.180 | 19.591 |
| 2018 | PROD.AMD | 1 | TRUE | 0.045 | 0.175 |
| 2018 | PROD.AMD | 2 | TRUE | 0.121 | 0.167 |
| 2018 | PROD.AMD | 3 | TRUE | 0.183 | 0.162 |
| 2018 | PROD.AMD | 4 | TRUE | 0.134 | 0.166 |
| 2018 | PROD.AMD | 5 | TRUE | 0.077 | 0.176 |
| 2019 | PROD.AMD | 1 | TRUE | 0.105 | 0.246 |
| 2019 | PROD.AMD | 2 | TRUE | 0.129 | 0.244 |
| 2019 | PROD.AMD | 3 | TRUE | 0.063 | 0.251 |
| 2019 | PROD.AMD | 4 | TRUE | 0.126 | 0.245 |
| 2019 | PROD.AMD | 5 | TRUE | 0.174 | 0.239 |
| 2020 | PROD.AMD | 1 | TRUE | 0.376 | 0.760 |
| 2020 | PROD.AMD | 2 | TRUE | 0.380 | 0.753 |
| 2020 | PROD.AMD | 3 | TRUE | 0.351 | 0.785 |
| 2020 | PROD.AMD | 4 | TRUE | 0.352 | 0.780 |
| 2020 | PROD.AMD | 5 | TRUE | 0.411 | 0.727 |
| 2018 | AMD | 1 | TRUE | 0.106 | 3.859 |
| 2018 | AMD | 2 | TRUE | 0.126 | 3.821 |
| 2018 | AMD | 3 | TRUE | 0.179 | 3.751 |
| 2018 | AMD | 4 | TRUE | 0.039 | 4.016 |
| 2018 | AMD | 5 | TRUE | 0.138 | 3.828 |
| 2019 | AMD | 1 | TRUE | 0.256 | 3.556 |
| 2019 | AMD | 2 | TRUE | 0.393 | 3.188 |
| 2019 | AMD | 3 | TRUE | 0.293 | 3.432 |
| 2019 | AMD | 4 | TRUE | 0.315 | 3.382 |
| 2019 | AMD | 5 | TRUE | 0.253 | 3.533 |
| 2020 | AMD | 1 | TRUE | 0.166 | 19.855 |
| 2020 | AMD | 2 | TRUE | 0.279 | 18.558 |
| 2020 | AMD | 3 | TRUE | 0.182 | 19.655 |
| 2020 | AMD | 4 | TRUE | 0.203 | 19.435 |
| 2020 | AMD | 5 | TRUE | 0.213 | 19.264 |
| 2018 | Porte | 1 | TRUE | 0.264 | 0.057 |
| 2018 | Porte | 2 | TRUE | 0.344 | 0.053 |
| 2018 | Porte | 3 | TRUE | 0.283 | 0.055 |
| 2018 | Porte | 4 | TRUE | 0.357 | 0.052 |
| 2018 | Porte | 5 | TRUE | 0.383 | 0.050 |
| 2019 | Porte | 1 | TRUE | 0.396 | 0.432 |
| 2019 | Porte | 2 | TRUE | 0.346 | 0.449 |
| 2019 | Porte | 3 | TRUE | 0.383 | 0.437 |
| 2019 | Porte | 4 | TRUE | 0.301 | 0.463 |
| 2019 | Porte | 5 | TRUE | 0.367 | 0.442 |
| 2020 | Porte | 1 | TRUE | 0.263 | 0.182 |
| 2020 | Porte | 2 | TRUE | 0.284 | 0.179 |
| 2020 | Porte | 3 | TRUE | 0.258 | 0.184 |
| 2020 | Porte | 4 | TRUE | 0.267 | 0.182 |
| 2020 | Porte | 5 | TRUE | 0.265 | 0.183 |
| 2018 | Nº.Hastes | 1 | TRUE | 0.179 | 0.039 |
| 2018 | Nº.Hastes | 2 | TRUE | 0.182 | 0.039 |
| 2018 | Nº.Hastes | 3 | TRUE | 0.134 | 0.040 |
| 2018 | Nº.Hastes | 4 | TRUE | 0.014 | 0.042 |
| 2018 | Nº.Hastes | 5 | TRUE | 0.104 | 0.040 |
| 2019 | Nº.Hastes | 1 | TRUE | -0.028 | 0.349 |
| 2019 | Nº.Hastes | 2 | TRUE | 0.024 | 0.346 |
| 2019 | Nº.Hastes | 3 | TRUE | 0.007 | 0.345 |
| 2019 | Nº.Hastes | 4 | TRUE | 0.096 | 0.336 |
| 2019 | Nº.Hastes | 5 | TRUE | 0.127 | 0.334 |
| 2020 | Nº.Hastes | 1 | TRUE | 0.314 | 0.148 |
| 2020 | Nº.Hastes | 2 | TRUE | 0.335 | 0.145 |
| 2020 | Nº.Hastes | 3 | TRUE | 0.318 | 0.147 |
| 2020 | Nº.Hastes | 4 | TRUE | 0.382 | 0.140 |
| 2020 | Nº.Hastes | 5 | TRUE | 0.326 | 0.146 |
| 2018 | Stand_Final | 1 | TRUE | 0.073 | 0.004 |
| 2018 | Stand_Final | 2 | TRUE | 0.203 | 0.003 |
| 2018 | Stand_Final | 3 | TRUE | 0.153 | 0.003 |
| 2018 | Stand_Final | 4 | TRUE | 0.091 | 0.004 |
| 2018 | Stand_Final | 5 | TRUE | 0.199 | 0.003 |
| 2019 | Stand_Final | 1 | TRUE | 0.014 | 0.126 |
| 2019 | Stand_Final | 2 | TRUE | -0.010 | 0.125 |
| 2019 | Stand_Final | 3 | TRUE | -0.082 | 0.128 |
| 2019 | Stand_Final | 4 | TRUE | -0.093 | 0.128 |
| 2019 | Stand_Final | 5 | TRUE | -0.063 | 0.125 |
| 2020 | Stand_Final | 1 | TRUE | 0.249 | 0.534 |
| 2020 | Stand_Final | 2 | TRUE | 0.193 | 0.549 |
| 2020 | Stand_Final | 3 | TRUE | 0.295 | 0.527 |
| 2020 | Stand_Final | 4 | TRUE | 0.206 | 0.545 |
| 2020 | Stand_Final | 5 | TRUE | 0.136 | 0.563 |
| 2018 | Branching_Level | 1 | TRUE | 0.033 | 0.034 |
| 2018 | Branching_Level | 2 | TRUE | 0.092 | 0.033 |
| 2018 | Branching_Level | 3 | TRUE | 0.096 | 0.033 |
| 2018 | Branching_Level | 4 | TRUE | 0.118 | 0.033 |
| 2018 | Branching_Level | 5 | TRUE | 0.159 | 0.033 |
| 2019 | Branching_Level | 1 | TRUE | 0.182 | 0.281 |
| 2019 | Branching_Level | 2 | TRUE | 0.143 | 0.286 |
| 2019 | Branching_Level | 3 | TRUE | 0.183 | 0.281 |
| 2019 | Branching_Level | 4 | TRUE | 0.193 | 0.280 |
| 2019 | Branching_Level | 5 | TRUE | 0.202 | 0.277 |
| 2020 | Branching_Level | 1 | TRUE | 0.436 | 0.150 |
| 2020 | Branching_Level | 2 | TRUE | 0.465 | 0.144 |
| 2020 | Branching_Level | 3 | TRUE | 0.468 | 0.143 |
| 2020 | Branching_Level | 4 | TRUE | 0.452 | 0.146 |
| 2020 | Branching_Level | 5 | TRUE | 0.529 | 0.132 |
| 2018 | Staygreen | 1 | TRUE | -0.029 | 0.001 |
| 2018 | Staygreen | 2 | TRUE | -0.130 | 0.001 |
| 2018 | Staygreen | 3 | TRUE | -0.145 | 0.001 |
| 2018 | Staygreen | 4 | TRUE | -0.003 | 0.001 |
| 2018 | Staygreen | 5 | TRUE | 0.032 | 0.001 |
write.csv(results_cv, "output/results_cv.csv", row.names = F,
quote = F)The object “result_cv” is divided by repetition. In the “result_cv” objects for each repetition, “Rep” is the number of the repetition, “Log” is a diagnostic indicating if the order of the predicted breeding values matches the order of the adjusted means, “Ac” is the prediction accuracy (correlation between the GEBV and adjusted means), and “MSPE” is the mean square prediction error (the lower, the better).
Agora vamos plotar os resultados de acurácias para cada característica
results_cv %>%
group_by(Ano, Trait) %>%
summarise(
mean.Ac = mean(Ac),
sd.Ac = sd(Ac),
mean.MSPE = mean(MSPE),
sd.MSPE = sd(MSPE)
) %>%
ggplot(aes(x = Ano, y = mean.Ac , fill = Trait)) +
geom_col(alpha = 0.8,
width = 0.5,
show.legend = F) +
geom_errorbar(
aes(ymin = mean.Ac - sd.Ac, ymax = mean.Ac + sd.Ac),
width = .2,
position = position_dodge(.9)
) +
facet_wrap(. ~ Trait, ncol = 6) +
expand_limits(y = c(0,1)) +
theme(axis.text.x = element_text(angle = 60, vjust = 1, hjust=1),
text = element_text(size = 15)) +
labs(y = "Accuracy")`summarise()` has grouped output by 'Ano'. You can override using the `.groups`
argument.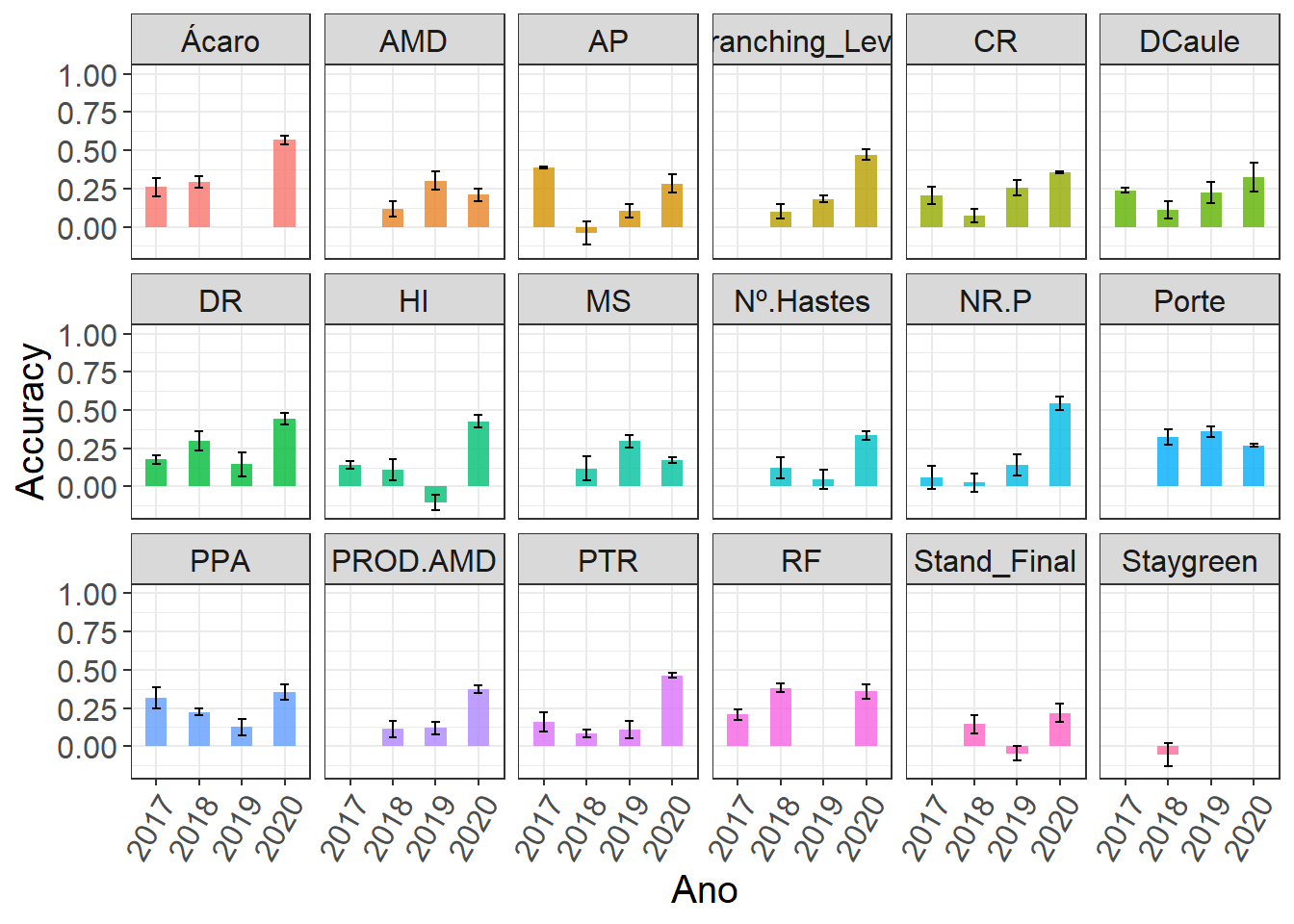
ggsave("output/accuracy_multi.png", width = 16, height = 8, dpi =300)
sessionInfo()R version 4.1.3 (2022-03-10)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 19042)
Matrix products: default
locale:
[1] LC_COLLATE=Portuguese_Brazil.1252 LC_CTYPE=Portuguese_Brazil.1252
[3] LC_MONETARY=Portuguese_Brazil.1252 LC_NUMERIC=C
[5] LC_TIME=Portuguese_Brazil.1252
attached base packages:
[1] grid stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] cvTools_0.3.2 robustbase_0.95-0 lattice_0.20-45
[4] ggpmisc_0.5.1 ggpp_0.4.5 rrBLUP_4.6.1
[7] ComplexHeatmap_2.10.0 AGHmatrix_2.0.4 genomicMateSelectR_0.2.0
[10] janitor_2.1.0 kableExtra_1.3.4 forcats_0.5.2
[13] stringr_1.4.1 dplyr_1.0.10 purrr_0.3.4
[16] readr_2.1.2 tidyr_1.2.1 tibble_3.1.8
[19] ggplot2_3.3.6 tidyverse_1.3.2 readxl_1.4.1
loaded via a namespace (and not attached):
[1] googledrive_2.0.0 colorspace_2.0-3 rjson_0.2.21
[4] ellipsis_0.3.2 rprojroot_2.0.3 circlize_0.4.15
[7] snakecase_0.11.0 GlobalOptions_0.1.2 fs_1.5.2
[10] clue_0.3-61 rstudioapi_0.14 farver_2.1.1
[13] MatrixModels_0.5-1 fansi_1.0.3 lubridate_1.8.0
[16] xml2_1.3.3 splines_4.1.3 codetools_0.2-18
[19] doParallel_1.0.17 cachem_1.0.6 confintr_0.2.0
[22] knitr_1.40 polynom_1.4-1 jsonlite_1.8.0
[25] workflowr_1.7.0 broom_1.0.1 cluster_2.1.2
[28] dbplyr_2.2.1 png_0.1-7 compiler_4.1.3
[31] httr_1.4.4 backports_1.4.1 assertthat_0.2.1
[34] Matrix_1.5-1 fastmap_1.1.0 gargle_1.2.1
[37] cli_3.3.0 later_1.3.0 htmltools_0.5.3
[40] quantreg_5.94 tools_4.1.3 gtable_0.3.1
[43] glue_1.6.2 Rcpp_1.0.9 cellranger_1.1.0
[46] jquerylib_0.1.4 vctrs_0.4.1 nlme_3.1-159
[49] svglite_2.1.0 iterators_1.0.14 xfun_0.32
[52] rvest_1.0.3 lifecycle_1.0.3 googlesheets4_1.0.1
[55] DEoptimR_1.0-11 MASS_7.3-58.1 zoo_1.8-11
[58] scales_1.2.1 ragg_1.2.3 hms_1.1.2
[61] promises_1.2.0.1 parallel_4.1.3 SparseM_1.81
[64] RColorBrewer_1.1-3 yaml_2.3.5 sass_0.4.2
[67] stringi_1.7.6 highr_0.9 S4Vectors_0.32.4
[70] foreach_1.5.2 BiocGenerics_0.40.0 boot_1.3-28
[73] shape_1.4.6 rlang_1.0.6 pkgconfig_2.0.3
[76] systemfonts_1.0.4 matrixStats_0.62.0 evaluate_0.17
[79] labeling_0.4.2 tidyselect_1.2.0 magrittr_2.0.3
[82] R6_2.5.1 magick_2.7.3 IRanges_2.28.0
[85] generics_0.1.3 DBI_1.1.3 mgcv_1.8-40
[88] pillar_1.8.1 haven_2.5.1 withr_2.5.0
[91] survival_3.4-0 modelr_0.1.9 crayon_1.5.2
[94] utf8_1.2.2 tzdb_0.3.0 rmarkdown_2.17
[97] GetoptLong_1.0.5 git2r_0.30.1 reprex_2.0.2
[100] digest_0.6.29 webshot_0.5.4 httpuv_1.6.5
[103] textshaping_0.3.6 stats4_4.1.3 munsell_0.5.0
[106] viridisLite_0.4.1 bslib_0.4.0 Weverton Gomes da Costa, Pós-Doutorando, Embrapa Mandioca e Fruticultura, wevertonufv@gmail.com↩︎