Last updated: 2022-11-10
Checks: 6 1
Knit directory:
Genomic-Selection-for-Drought-Tolerance-Using-Genome-Wide-SNPs-in-Casava/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has staged changes. To know which version of the
R Markdown file created these results, you’ll want to first commit it to
the Git repo. If you’re still working on the analysis, you can ignore
this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20221020) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version b78c842. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rproj.user/
Untracked files:
Untracked: data/AllGBSandDArTClones_ReadyForGP_Dos.rds
Untracked: data/DCas22_GBSandDArT_ReadyForGP_Dos.rds
Untracked: data/allchrAR08.txt
Untracked: data/convet_haplo_diplo.txt
Untracked: output/BLUPS.RDS
Untracked: output/BLUPS.csv
Untracked: output/BLUPS_par.Rdata
Untracked: output/herdabilidade.csv
Untracked: output/media_pheno.csv
Untracked: output/resultMM.Rdata
Unstaged changes:
Deleted: BLUPS.RDS
Deleted: BLUPS.csv
Deleted: BLUPs.Rdata
Deleted: RR-BLUP-method.pdf
Modified: analysis/GWS.Rmd
Modified: analysis/phenotype.Rmd
Deleted: cor-methods-RR_BLUP.pdf
Deleted: deregress.Rdata
Deleted: geno.txt
Deleted: geno2.txt
Deleted: herdabilidade.csv
Staged changes:
New: BLUPS.RDS
New: BLUPS.csv
New: BLUPs.Rdata
New: RR-BLUP-method.pdf
New: analysis/GWS.Rmd
Modified: analysis/_site.yml
Modified: analysis/about.Rmd
Modified: analysis/index.Rmd
Modified: analysis/license.Rmd
New: analysis/phenotype.Rmd
New: cor-methods-RR_BLUP.pdf
New: data/Fenótipos Desregressados GBS Todos.RDS
New: data/Genomic Selection for Drought Tolerance Using Genome-Wide SNPs in Maize.pdf
New: data/Phenotyping v2.xlsx
New: data/Phenotyping.xlsx
New: data/SNPs.rds
New: data/deregress.Rdata
New: data/geno.rds
New: data/geno.txt
New: deregress.Rdata
New: geno.txt
New: geno2.txt
New: herdabilidade.csv
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with
wflow_publish() to start tracking its development.
Data and libraries
Load Libraries
library(kableExtra)
library(tidyverse)
require(ComplexHeatmap)
library(data.table)
library(readxl)
library(metan)
library(DataExplorer)
library(doParallel)
theme_set(theme_bw())Data import and manipulation
Vamos importar o conjunto de dados fenotípicos, excluindo as variaveis sem informações e as variaveis Local (redundante com Ano) e Tratamento (só uma observação).
pheno <- read_excel("data/Phenotyping.xlsx",
na = "NA") %>%
select_if( ~ !all(is.na(.))) %>% # Deleting traits without information
select(-c("Local", "Tratamento"))Vamos realizar alguma manipulações para ajustar nosso banco de dados e para facilitar a visualização da análise exploratória.
Primeiro, vamos converter as variaveis que são caracter em fatores. Depois vamos converter as variaveis que são referentes as notas para inteiro e logo em seguida em fatores. Após isso, vamos criar a variável ANo.Bloco para o aninhamento no modelo para obtenção dos BLUPs.
pheno <- pheno %>%
mutate_if(is.character, as.factor) %>%
mutate_at(c("RF", "Ácaro", "Vigor", "Branching_Level"), as.integer) %>%
mutate_if(is.integer, as.factor) %>%
mutate_at(
c(
"Ano",
"Bloco",
"Porte",
"Incidence_Mites",
"Stand_Final",
"Staygreen",
"Flowering"
),
as.factor
) %>% # Convert Ano and Bloco, and traits in factors
mutate(Ano.Bloco = factor(interaction(Ano, Bloco))) # Convert Ano.Bloco interaction in factorsExploratory Data Analysis
Análise introdutória de todo conjunto de dados
introduce(pheno) %>%
kbl(escape = F, align = 'c') %>%
kable_classic(
"hover",
full_width = F,
position = "center",
fixed_thead = T
)| rows | columns | discrete_columns | continuous_columns | all_missing_columns | total_missing_values | complete_rows | total_observations | memory_usage |
|---|---|---|---|---|---|---|---|---|
| 2336 | 28 | 13 | 15 | 0 | 16771 | 440 | 65408 | 449920 |
Não temos nenhuma coluna que tenha todas observações ausentes, mas temos muitos valores ausentes em todo conjunto de dados. Algumas manipulações deverão ser realizadas para melhorar a qualidade dos dados.
Analise de Ano
Vamos produzir um heatmap para verificar a quantidade de clone em cada ano. Vou criar outro conjunto de dados com a contagem de Ano e Clone. Depois vou criar os objetos correspondentes à matriz de clones e de anos. Por fim, criei a matriz que representa a presença e ausencia do clone no ano.
pheno2 <- pheno %>%
count(Ano, Clone)
genmat <- model.matrix(~ -1 + Clone, data = pheno2)
envmat <- model.matrix(~ -1 + Ano, data = pheno2)
genenvmat <- t(envmat) %*% genmat
genenvmat_ch <- ifelse(genenvmat == 1, "Present", "Abscent")
Heatmap(
genenvmat_ch,
col = c("white", "tomato"),
show_column_names = F,
heatmap_legend_param = list(title = ""),
column_title = "Genotypes",
row_title = "Environments"
)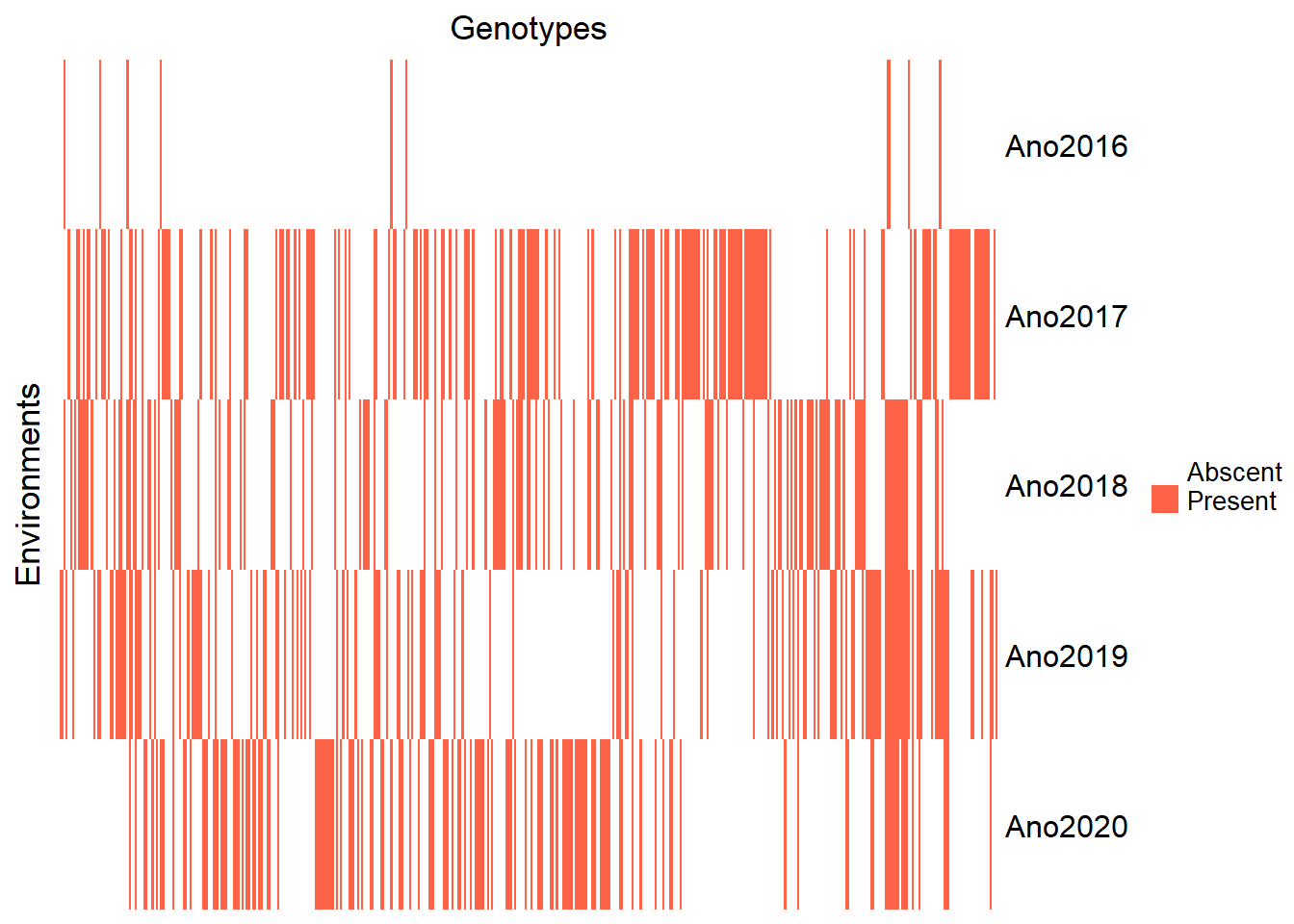
Pelo heatmap, fica claro que o ano 2016 possui muitas poucas observações. Então, devemos eliminá-lo.
pheno <- pheno %>%
filter(Ano != 2016) %>%
droplevels()Apenas para conferência, vamos visualizar novamente o heatmap de clones por ano.
pheno2<- pheno %>%
count(Ano, Clone)
genmat = model.matrix( ~ -1 + Clone, data = pheno2)
envmat = model.matrix( ~ -1 + Ano, data = pheno2)
genenvmat = t(envmat) %*% genmat
genenvmat_ch = ifelse(genenvmat == 1, "Present", "Abscent")
Heatmap(
genenvmat_ch,
col = c("white", "tomato"),
show_column_names = F,
heatmap_legend_param = list(title = ""),
column_title = "Genotypes",
row_title = "Environments"
)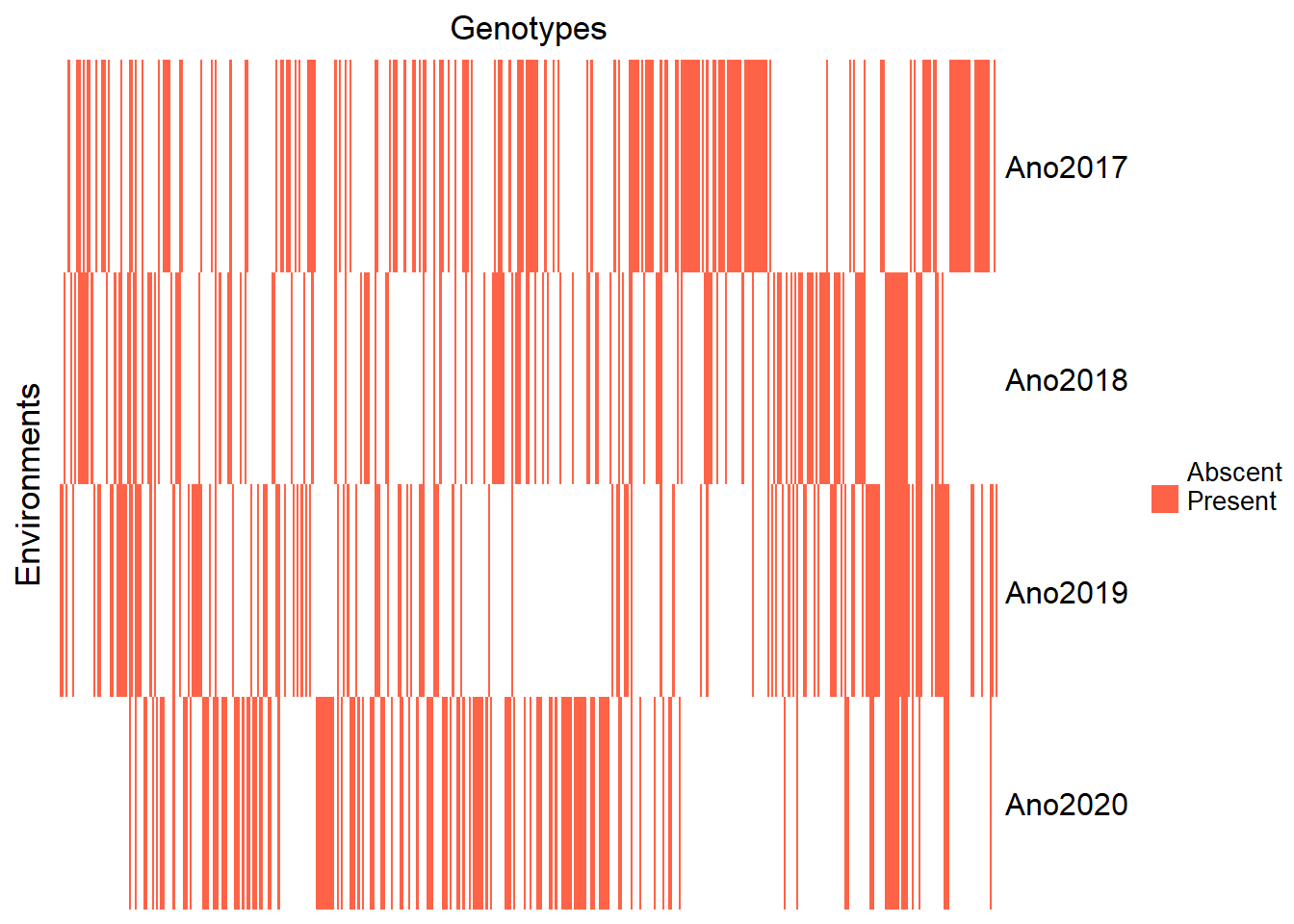
Aqui, é possível obervar que nosso conjunto de dados possui clones que foram avaliados em apenas um ano. Vamos visualizar isso, para verificarmos quantos clones foram avaliados de acordo com o número de anos.
pheno2 %>%
count(Clone) %>%
count(n) %>%
kbl(
escape = F,
align = 'c',
col.names = c("N of Environments", "Number of genotypes")
) %>%
kable_classic(
"hover",
full_width = F,
position = "center",
fixed_thead = T
)Storing counts in `nn`, as `n` already present in input
i Use `name = "new_name"` to pick a new name.| N of Environments | Number of genotypes |
|---|---|
| 1 | 350 |
| 2 | 72 |
| 3 | 20 |
| 4 | 5 |
Apenas 5 clones foram avaliados em todos os anos, isso possivelmente diminuirá nossa acurácia do modelos.
Além disso, observe que os anos diferem quanto ao número de clones avaliados:
pheno2 %>%
group_by(Ano) %>%
summarise(length(Clone)) %>%
kbl(
escape = F,
align = 'c',
col.names = c("Environments", "Number of genotypes")) %>%
kable_classic(
"hover",
full_width = F,
position = "center",
fixed_thead = T
)| Environments | Number of genotypes |
|---|---|
| 2017 | 165 |
| 2018 | 138 |
| 2019 | 133 |
| 2020 | 138 |
Outro fator que diminui a acurácia, e por isso adotar modelos mistos na análise é o mais indicado para a obtenção dos BLUPS.
Podemos verificar quantos clones temos em comum entre os anos:
genenvmat %*% t(genenvmat) %>%
kbl(escape = F, align = 'c') %>%
kable_classic(
"hover",
full_width = F,
position = "center",
fixed_thead = T
)| Ano2017 | Ano2018 | Ano2019 | Ano2020 | |
|---|---|---|---|---|
| Ano2017 | 165 | 42 | 22 | 14 |
| Ano2018 | 42 | 138 | 39 | 16 |
| Ano2019 | 22 | 39 | 133 | 29 |
| Ano2020 | 14 | 16 | 29 | 138 |
O ano 2020 apresenta menor número de clones em comum, no entanto, vamos mantê-lo para realizar as análises.
Análise das variáveis
Agora, iremos analisar a frequência para cada característica discreta.
plot_bar(pheno)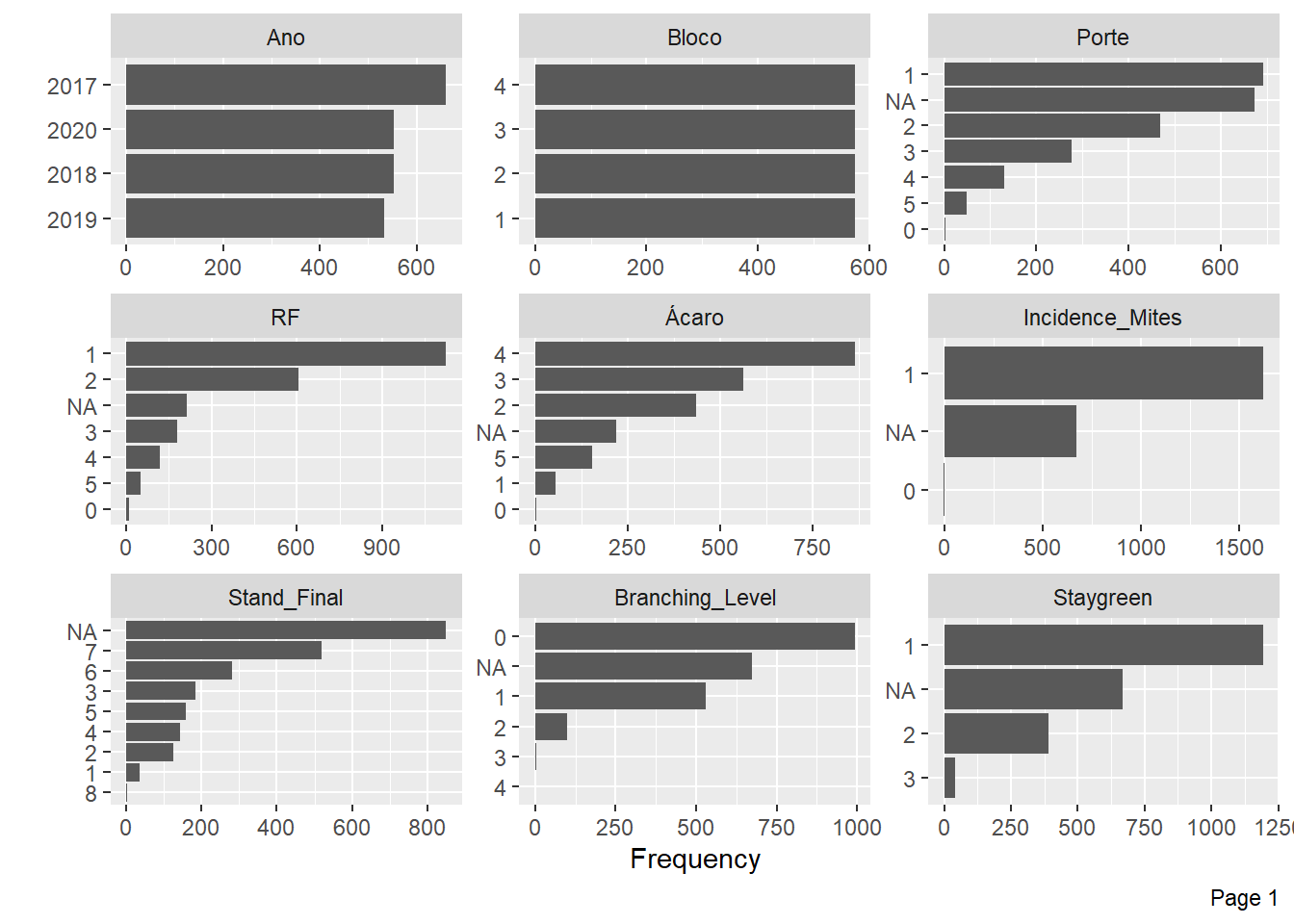
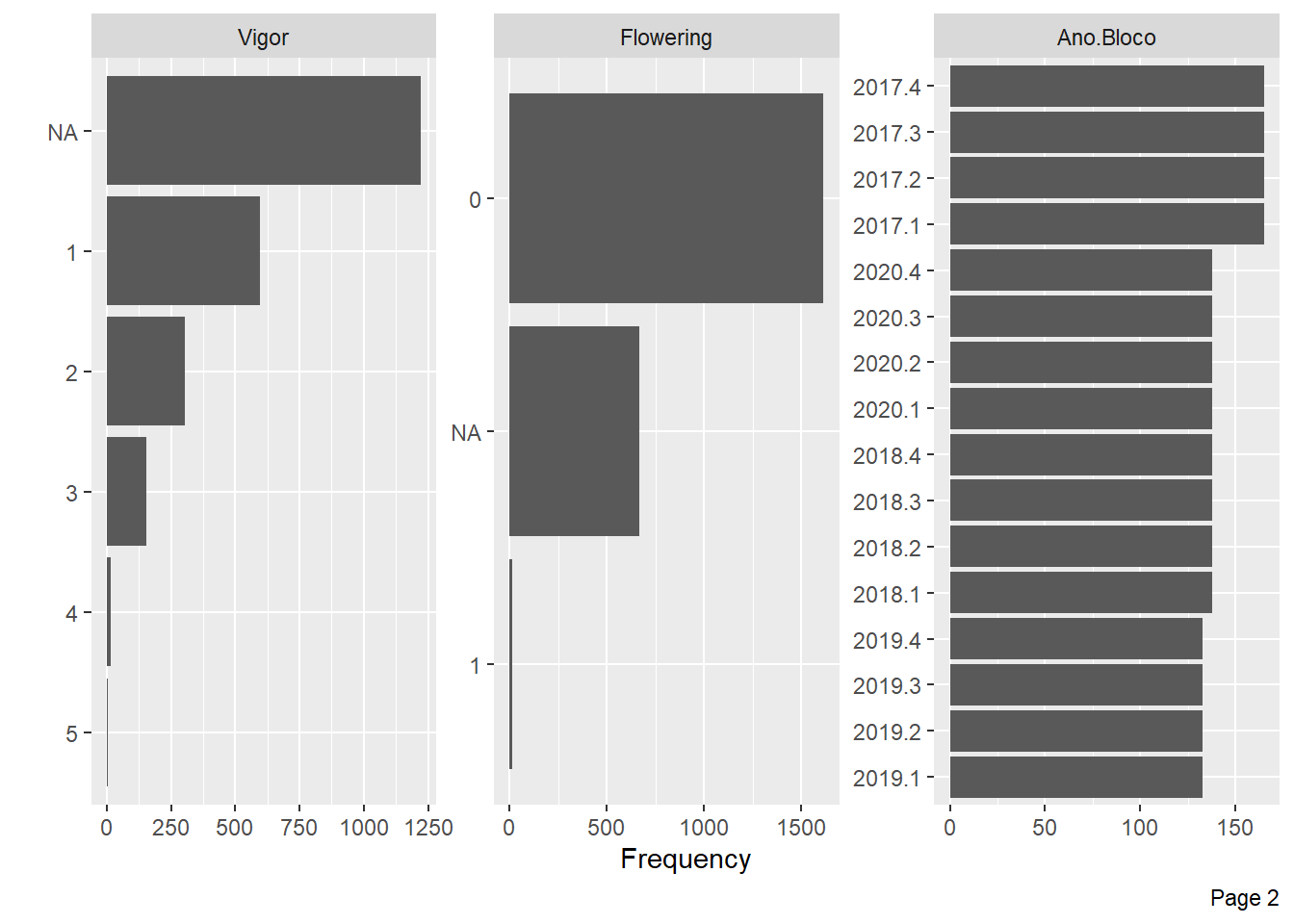
A Incidencia de ácaros e Florescimento possuem poucas informações para alguns níveis e muitos NA’s, também vamos excluir essas variáveis.
pheno <- pheno %>%
select(-c(Incidence_Mites, Flowering))
plot_bar(pheno)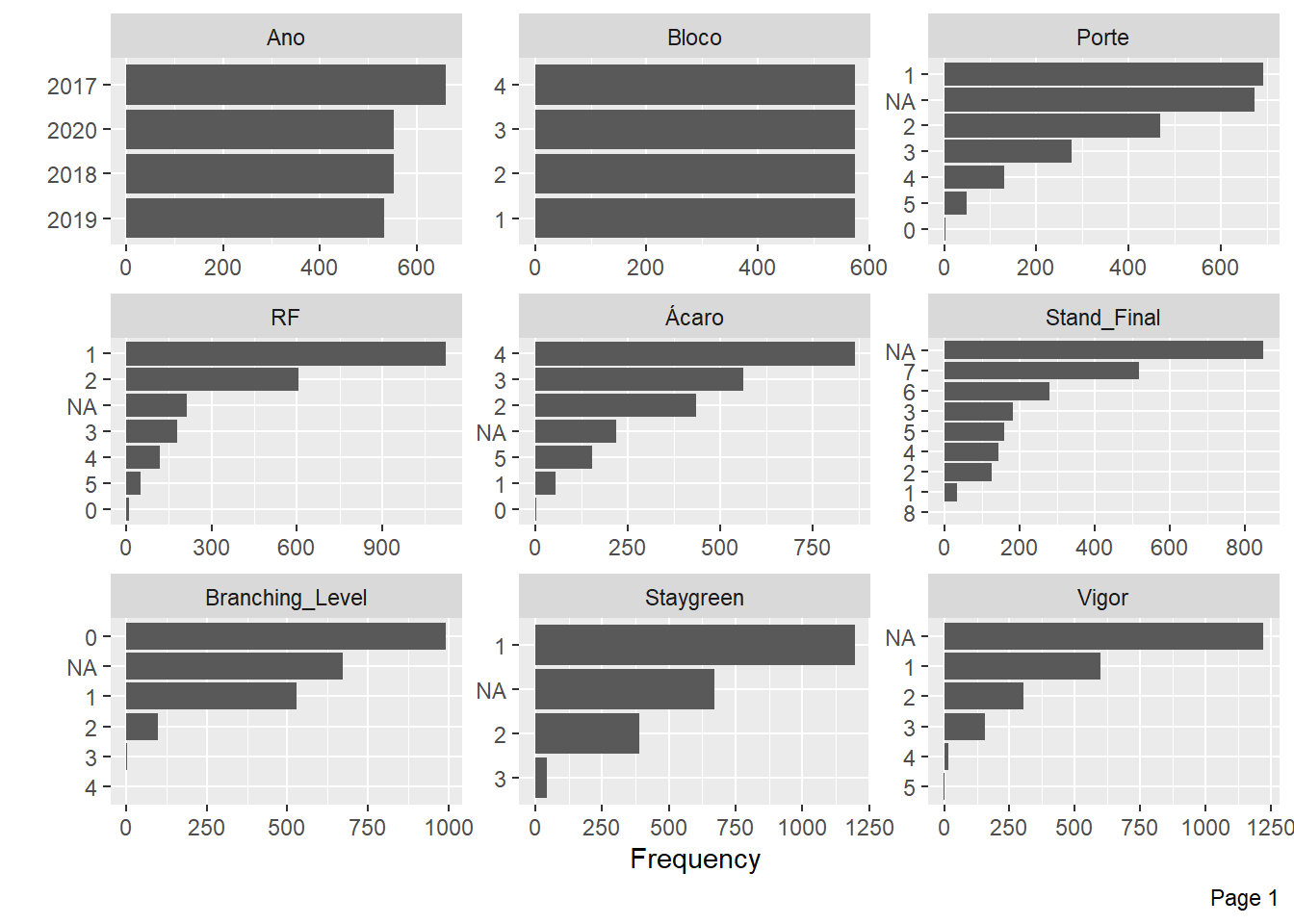
Vamos observar apenas os valores ausentes agora, para verificar as proporções.
plot_missing(pheno)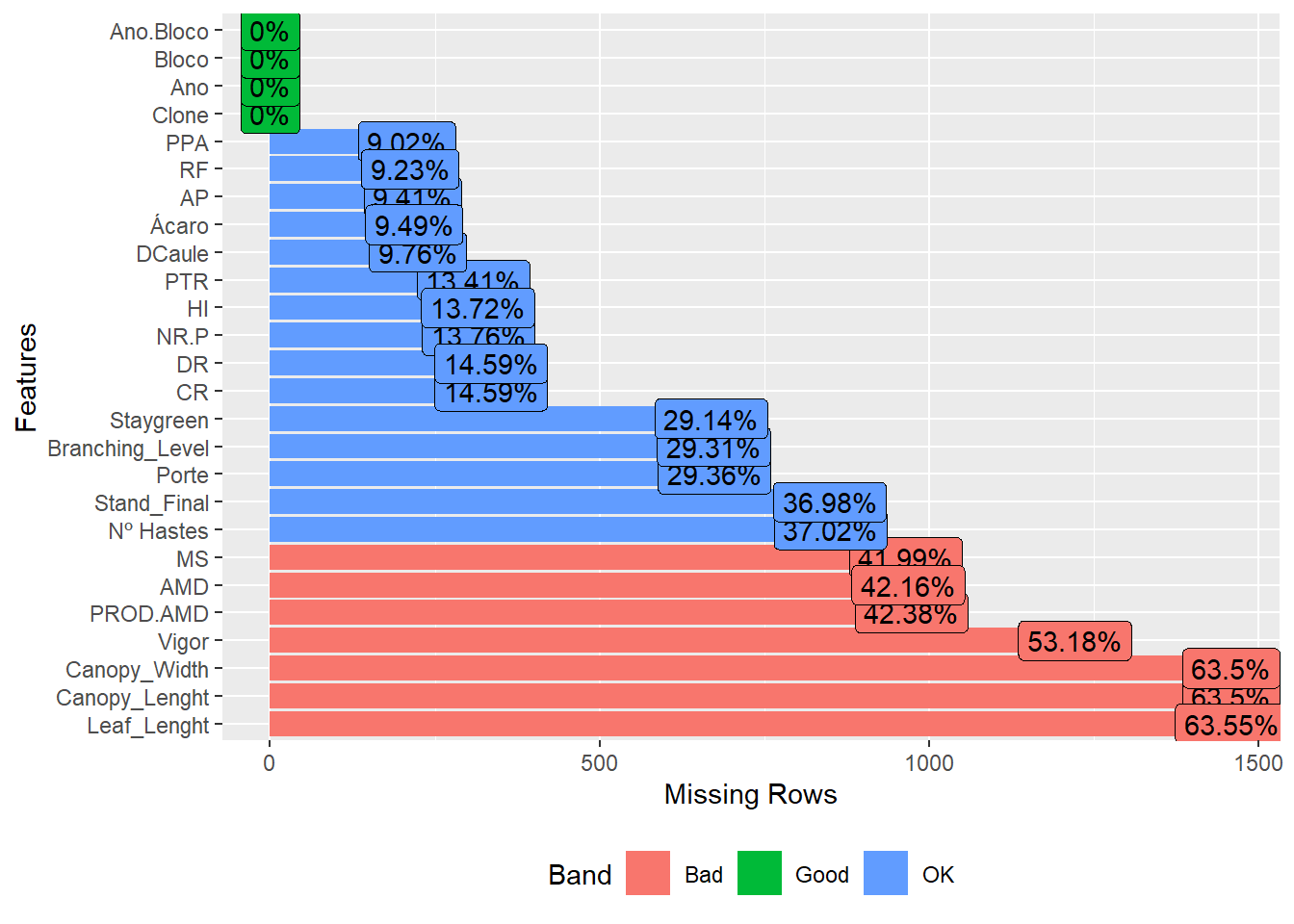
Temos uma proporção alta de valor ausente para Vigor, Leaf_Lenght, Canopy_Width e Canopy_Lenght, também vou excluir-lás.
pheno <- pheno %>%
select(-c(Vigor, Leaf_Lenght, Canopy_Width, Canopy_Lenght))Vamos verificar a distribuição das características por ano agora.
plot_bar(pheno, by = "Ano")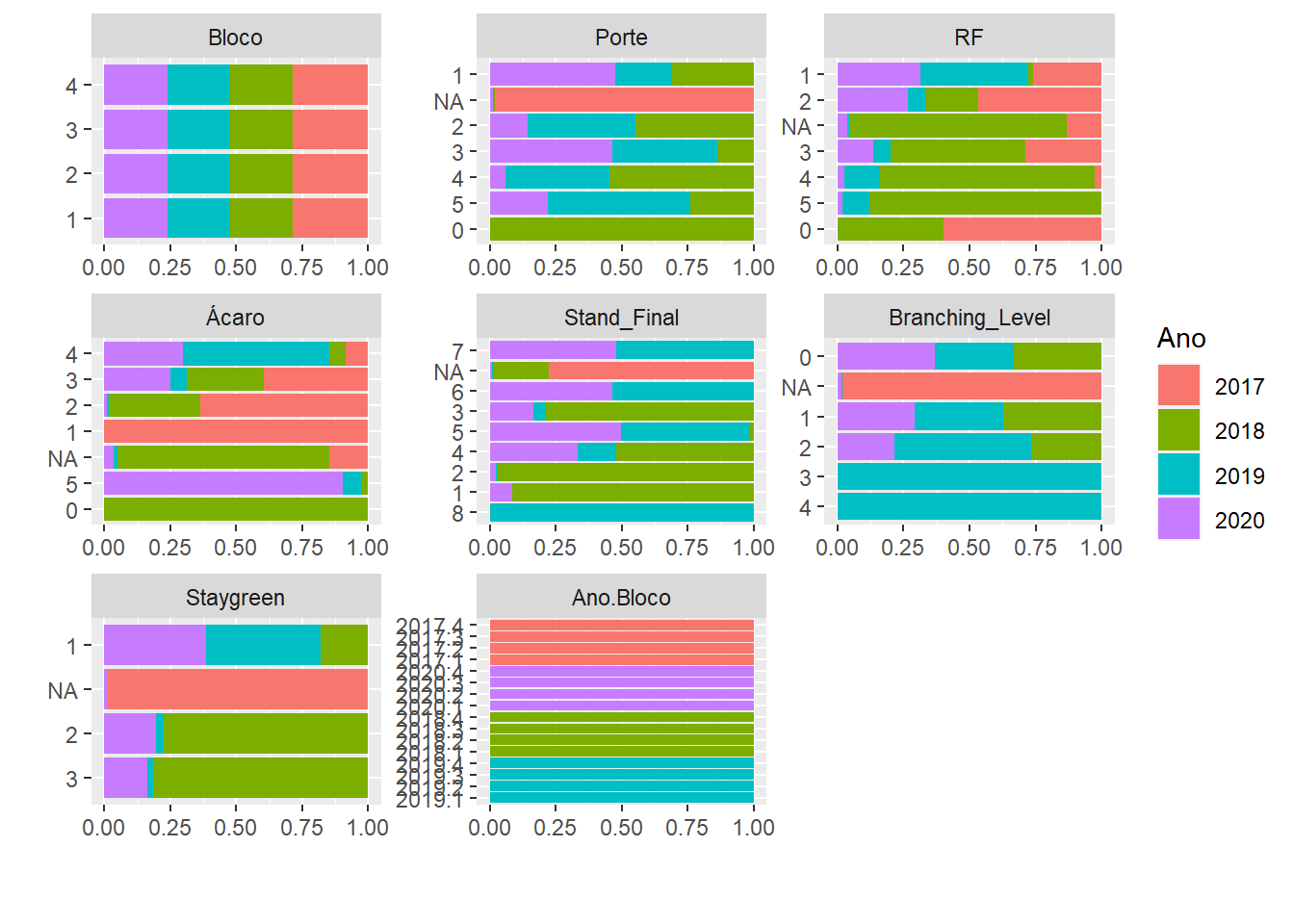
Para Porte, Branching_Level e Staygreen temos muitos valores ausentes para o ano de 2017, possivelmente não houve avaliação nesse ano para essas características. Para obter os BLUPs, teremos que remover esse Ano do banco de dados.
Agora vamos observar os histogramas das varaiveis quantitativas:
plot_histogram(pheno)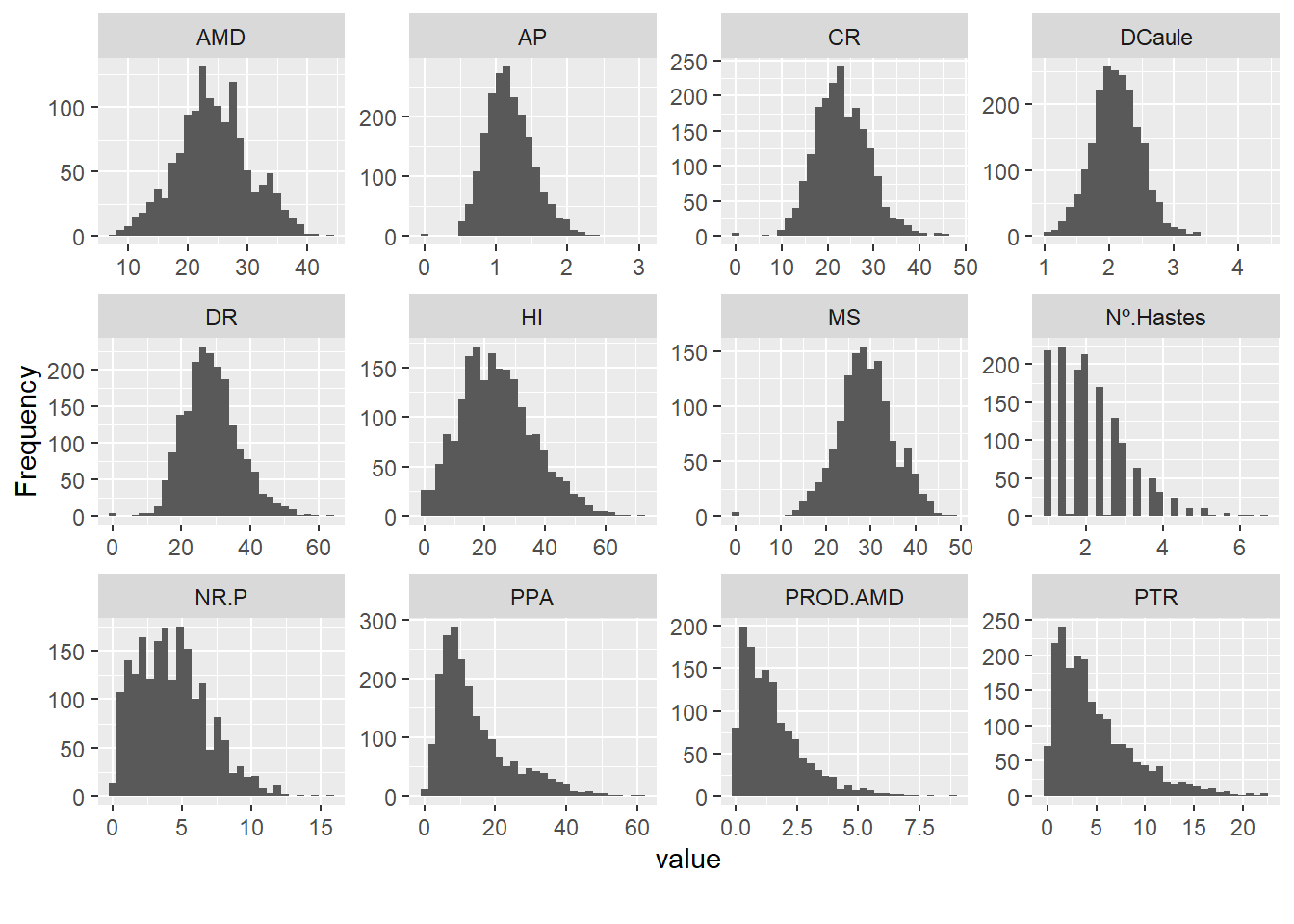
Vimos aqui que as variáveis quantitativas apresentam correlações entre si, principalmente entre PROD.AMD com PTR e AMD com MS
Vamos avaliar as estatisticas descritivas da combinação entre clone e ano para as variaveis.
ge_details(pheno, Ano, Clone, resp = everything()) %>% kbl(escape = F, align = 'c') %>%
kable_classic(
"hover",
full_width = F,
position = "center",
fixed_thead = T
)| Parameters | NR.P | PTR | PPA | MS | PROD.AMD | AP | HI | AMD | CR | DR | DCaule | Nº Hastes |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | 4.28 | 4.88 | 14.2 | 28.97 | 1.51 | 1.19 | 24.23 | 24.42 | 23.17 | 28.82 | 2.11 | 2.13 |
| SE | 0.06 | 0.09 | 0.22 | 0.17 | 0.04 | 0.01 | 0.27 | 0.17 | 0.13 | 0.18 | 0.01 | 0.03 |
| SD | 2.52 | 4.06 | 10.17 | 6.29 | 1.27 | 0.33 | 12.13 | 6.11 | 5.89 | 7.99 | 0.38 | 0.95 |
| CV | 58.9 | 83.19 | 71.66 | 21.73 | 84.4 | 27.81 | 50.08 | 25.05 | 25.42 | 27.75 | 17.9 | 44.53 |
| Min | 0 (BGM-0019 in 2019) | 0 (BGM-0019 in 2019) | 0 (BGM-0044 in 2018) | 0 (BGM-0044 in 2018) | 0 (BGM-0044 in 2018) | 0 (BGM-0044 in 2018) | 0 (BGM-0019 in 2019) | 7.33 (BGM-0626 in 2020) | 0 (BGM-0044 in 2018) | 0 (BGM-0044 in 2018) | 1.01 (BGM-0592 in 2018) | 1 (BGM-0036 in 2018) |
| Max | 15.67 (2012-107-002 in 2019) | 22.2 (BGM-1267 in 2018) | 61.17 (BGM-2124 in 2020) | 48.34 (BGM-1015 in 2020) | 8.87 (BGM-0396 in 2018) | 3.03 (BR-11-24-156 in 2020) | 71.97 (BGM-1315 in 2018) | 43.69 (BGM-1015 in 2020) | 47.33 (BGM-0396 in 2018) | 63.3 (BRS Mulatinha in 2018) | 4.37 (BRS Tapioqueira in 2020) | 6.67 (BGM-0714 in 2019) |
| MinENV | 2018 (1.6) | 2017 (2.75) | 2017 (8.47) | 2020 (26.21) | 2019 (1.32) | 2017 (1) | 2020 (18.61) | 2020 (21.56) | 2017 (19.72) | 2017 (24.49) | 2018 (2.02) | 2018 (1.44) |
| MaxENV | 2019 (5.74) | 2020 (6.52) | 2020 (25.87) | 2018 (34.91) | 2018 (1.81) | 2019 (1.48) | 2018 (31.84) | 2018 (30.78) | 2019 (27.14) | 2018 (34.28) | 2017 (2.16) | 2019 (2.71) |
| MinGEN | BGM-0044 (0) | BGM-0044 (0) | BGM-0044 (0) | BGM-0044 (0) | BGM-0044 (0) | BGM-0044 (0) | BGM-0044 (0) | BGM-0626 (10.33) | BGM-0044 (0) | BGM-0044 (0) | BGM-0048 (1.25) | BGM-0066 (1) |
| MaxGEN | 2012-107-002 (11.33) | IAC-14 (14.07) | BGM-2124 (54.33) | BGM-1015 (45.02) | BGM-1023 (4.76) | BGM-1200 (1.91) | Mata_Fome_Branca (52.78) | BGM-1015 (40.37) | BGM-1956 (35.5) | BGM-1956 (48.91) | BGM-1523 (2.93) | BGM-0451 (4.44) |
O genótipo BGM-0044 apresentou valores nulos para a maioria das características, como foi avaliado apenas no ano de 2018, é melhor excluí-lo.
pheno<- pheno %>%
filter(Clone != "BGM-0044")%>%
droplevels()Aparentemente não temos mais um genótipo que possa prejudicar nossa análise. Agora devemos avaliar as estatisticas descritivas apenas de clone para as variaveis.
desc_stat(pheno, by = Ano) %>%
kbl(escape = F, align = 'c') %>%
kable_classic(
"hover",
full_width = F,
position = "center",
fixed_thead = T
)| Ano | variable | cv | max | mean | median | min | sd.amo | se | ci.t | n.valid |
|---|---|---|---|---|---|---|---|---|---|---|
| 2017 | AMD | NA | -Inf | NaN | NA | Inf | 0.0000 | NA | NaN | 0 |
| 2017 | AP | 21.0398 | 1.6767 | 1.0023 | 1.0000 | 0.4500 | 0.2109 | 0.0084 | 0.0165 | 632 |
| 2017 | CR | 21.5823 | 31.6667 | 19.7236 | 19.6667 | 7.0000 | 4.2568 | 0.1719 | 0.3376 | 613 |
| 2017 | DCaule | 16.7774 | 3.3333 | 2.1581 | 2.1667 | 1.2000 | 0.3621 | 0.0144 | 0.0284 | 629 |
| 2017 | DR | 21.0039 | 39.6900 | 24.4889 | 24.4733 | 8.9867 | 5.1436 | 0.2077 | 0.4080 | 613 |
| 2017 | HI | 48.2942 | 66.6827 | 23.5691 | 22.7255 | 1.5744 | 11.3825 | 0.4612 | 0.9058 | 609 |
| 2017 | MS | NA | -Inf | NaN | NA | Inf | 0.0000 | NA | NaN | 0 |
| 2017 | Nº Hastes | NA | -Inf | NaN | NA | Inf | 0.0000 | NA | NaN | 0 |
| 2017 | NR.P | 48.8708 | 12.0000 | 4.3530 | 4.3330 | 0.1250 | 2.1273 | 0.0860 | 0.1689 | 612 |
| 2017 | PPA | 45.4973 | 22.2220 | 8.4677 | 8.0750 | 0.6940 | 3.8526 | 0.1542 | 0.3029 | 624 |
| 2017 | PROD.AMD | NA | -Inf | NaN | NA | Inf | 0.0000 | NA | NaN | 0 |
| 2017 | PTR | 66.8379 | 9.6700 | 2.7517 | 2.4310 | 0.1160 | 1.8392 | 0.0743 | 0.1459 | 613 |
| 2018 | AMD | 15.6937 | 41.4284 | 30.7820 | 31.3500 | 13.5318 | 4.8309 | 0.2794 | 0.5498 | 299 |
| 2018 | AP | 28.0409 | 1.9967 | 1.0953 | 1.0600 | 0.4867 | 0.3071 | 0.0159 | 0.0313 | 373 |
| 2018 | CR | 27.1625 | 47.3333 | 23.6102 | 23.0000 | 9.5000 | 6.4131 | 0.3715 | 0.7311 | 298 |
| 2018 | DCaule | 22.3814 | 3.7317 | 2.0189 | 1.9800 | 1.0133 | 0.4519 | 0.0235 | 0.0463 | 369 |
| 2018 | DR | 23.3943 | 63.3033 | 34.7356 | 34.4400 | 14.1300 | 8.1262 | 0.4707 | 0.9264 | 298 |
| 2018 | HI | 42.0024 | 71.9673 | 32.2503 | 32.6091 | 0.0000 | 13.5459 | 0.7719 | 1.5188 | 308 |
| 2018 | MS | 13.6086 | 46.0784 | 35.3819 | 36.0000 | 18.1818 | 4.8150 | 0.2785 | 0.5480 | 299 |
| 2018 | Nº Hastes | 33.4885 | 3.3333 | 1.4379 | 1.3333 | 1.0000 | 0.4815 | 0.0249 | 0.0490 | 373 |
| 2018 | NR.P | 63.2913 | 6.2500 | 1.6249 | 1.4085 | 0.1900 | 1.0284 | 0.0588 | 0.1157 | 306 |
| 2018 | PPA | 69.0625 | 31.6000 | 8.7610 | 7.2000 | 1.0000 | 6.0506 | 0.3022 | 0.5940 | 401 |
| 2018 | PROD.AMD | 91.8742 | 8.8669 | 1.8356 | 1.3539 | 0.0839 | 1.6864 | 0.0984 | 0.1936 | 294 |
| 2018 | PTR | 86.6619 | 22.2000 | 5.6274 | 4.2500 | 0.0000 | 4.8768 | 0.2770 | 0.5450 | 310 |
| 2019 | AMD | 16.5407 | 35.9441 | 23.6326 | 23.6328 | 10.7346 | 3.9090 | 0.1745 | 0.3428 | 502 |
| 2019 | AP | 19.1125 | 2.4233 | 1.4843 | 1.4633 | 0.7400 | 0.2837 | 0.0123 | 0.0243 | 528 |
| 2019 | CR | 21.1089 | 45.6667 | 27.1361 | 27.0000 | 11.0000 | 5.7281 | 0.2529 | 0.4969 | 513 |
| 2019 | DCaule | 15.7037 | 3.3203 | 2.1061 | 2.0963 | 1.1197 | 0.3307 | 0.0144 | 0.0283 | 528 |
| 2019 | DR | 24.3155 | 58.9267 | 33.4940 | 33.6500 | 6.1200 | 8.1442 | 0.3596 | 0.7064 | 513 |
| 2019 | HI | 47.3227 | 60.1626 | 26.1706 | 26.6117 | 0.0000 | 12.3846 | 0.5390 | 1.0588 | 528 |
| 2019 | MS | 13.8212 | 40.5941 | 28.2826 | 28.2828 | 15.3846 | 3.9090 | 0.1745 | 0.3428 | 502 |
| 2019 | Nº Hastes | 37.3740 | 6.6667 | 2.7113 | 2.6667 | 1.0000 | 1.0133 | 0.0441 | 0.0867 | 527 |
| 2019 | NR.P | 47.7261 | 15.6670 | 5.7402 | 5.6670 | 0.0000 | 2.7396 | 0.1192 | 0.2342 | 528 |
| 2019 | PPA | 47.4901 | 47.5710 | 13.4607 | 12.7735 | 1.2860 | 6.3925 | 0.2782 | 0.5465 | 528 |
| 2019 | PROD.AMD | 72.5347 | 5.4021 | 1.3211 | 1.1082 | 0.0000 | 0.9583 | 0.0428 | 0.0841 | 501 |
| 2019 | PTR | 74.3122 | 22.2000 | 5.2850 | 4.5000 | 0.0000 | 3.9274 | 0.1709 | 0.3358 | 528 |
| 2020 | AMD | 27.3600 | 43.6860 | 21.5594 | 21.3900 | 7.3260 | 5.8986 | 0.2569 | 0.5048 | 527 |
| 2020 | AP | 24.0927 | 3.0333 | 1.1945 | 1.1600 | 0.3600 | 0.2878 | 0.0124 | 0.0243 | 543 |
| 2020 | CR | 18.9730 | 36.6667 | 23.2345 | 23.3333 | 9.0000 | 4.4083 | 0.1909 | 0.3751 | 533 |
| 2020 | DCaule | 17.5042 | 4.3733 | 2.1321 | 2.1207 | 1.0547 | 0.3732 | 0.0160 | 0.0315 | 543 |
| 2020 | DR | 20.5858 | 43.2733 | 26.2101 | 26.1800 | 11.8533 | 5.3956 | 0.2337 | 0.4591 | 533 |
| 2020 | HI | 43.3204 | 45.9340 | 18.6129 | 18.0486 | 1.9608 | 8.0632 | 0.3496 | 0.6867 | 532 |
| 2020 | MS | 22.5059 | 48.3360 | 26.2094 | 26.0400 | 11.9760 | 5.8986 | 0.2569 | 0.5048 | 527 |
| 2020 | Nº Hastes | 37.1507 | 4.6667 | 2.0463 | 2.0000 | 1.0000 | 0.7602 | 0.0326 | 0.0641 | 543 |
| 2020 | NR.P | 47.0839 | 10.3330 | 4.3060 | 4.3330 | 0.3330 | 2.0274 | 0.0881 | 0.1730 | 530 |
| 2020 | PPA | 42.7475 | 61.1670 | 25.8653 | 25.9165 | 3.3330 | 11.0568 | 0.4794 | 0.9417 | 532 |
| 2020 | PROD.AMD | 81.2220 | 6.0724 | 1.5211 | 1.2326 | 0.0483 | 1.2354 | 0.0540 | 0.1060 | 524 |
| 2020 | PTR | 68.4455 | 20.5670 | 6.5246 | 5.9000 | 0.4000 | 4.4658 | 0.1934 | 0.3800 | 533 |
Novamente, algumas variáveis não foram computadas para o ano de 2017 então temos que eliminar esse ano na hora de realizar a análise para essas variaveis.
O que chama atenção nessa tabela são os cv altos para algumas característica, especialmente: HI, Nº de Hastes, NR.P, PPA, PROD.AMD e PTR.
Isso pode se dá devido a presença de outliers, vamos fazer uma inspeção em todo conjunto de dados para avaliar se há outliers:
inspect(pheno %>%
select(-c(Clone)), verbose = FALSE) %>% kbl(escape = F, align = 'c') %>%
kable_classic(
"hover",
full_width = F,
position = "center",
fixed_thead = T
)| Variable | Class | Missing | Levels | Valid_n | Min | Median | Max | Outlier | Text |
|---|---|---|---|---|---|---|---|---|---|
| Ano | factor | No | 4 | 2292 | NA | NA | NA | NA | NA |
| Bloco | factor | No | 4 | 2292 | NA | NA | NA | NA | NA |
| NR.P | numeric | Yes |
|
1976 | 0.00 | 4.00 | 15.67 | 16 | NA |
| PTR | numeric | Yes |
|
1984 | 0.00 | 3.70 | 22.20 | 76 | NA |
| PPA | numeric | Yes |
|
2085 | 0.69 | 11.17 | 61.17 | 89 | NA |
| MS | numeric | Yes |
|
1328 | 11.98 | 28.80 | 48.34 | 6 | NA |
| PROD.AMD | numeric | Yes |
|
1319 | 0.00 | 1.21 | 8.87 | 49 | NA |
| AP | numeric | Yes |
|
2076 | 0.36 | 1.16 | 3.03 | 23 | NA |
| HI | numeric | Yes |
|
1977 | 0.00 | 23.21 | 71.97 | 14 | NA |
| AMD | numeric | Yes |
|
1328 | 7.33 | 24.15 | 43.69 | 6 | NA |
| Porte | factor | Yes | 5 | 1618 | NA | NA | NA | NA | NA |
| RF | factor | Yes | 6 | 2080 | NA | NA | NA | NA | NA |
| CR | numeric | Yes |
|
1957 | 7.00 | 23.00 | 47.33 | 19 | NA |
| DR | numeric | Yes |
|
1957 | 6.12 | 28.17 | 63.30 | 32 | NA |
| DCaule | numeric | Yes |
|
2069 | 1.01 | 2.10 | 4.37 | 22 | NA |
| Ácaro | factor | Yes | 5 | 2074 | NA | NA | NA | NA | NA |
| Nº Hastes | numeric | Yes |
|
1443 | 1.00 | 2.00 | 6.67 | 30 | NA |
| Stand_Final | factor | Yes | 8 | 1444 | NA | NA | NA | NA | NA |
| Branching_Level | factor | Yes | 5 | 1619 | NA | NA | NA | NA | NA |
| Staygreen | factor | Yes | 3 | 1623 | NA | NA | NA | NA | NA |
| Ano.Bloco | factor | No | 16 | 2292 | NA | NA | NA | NA | NA |
Confirmando o que foi descrito antes, a maioria das variáveis com alto cv apresenta muitos outliers e por isso iremos excluir elas no loop para obtenção dos blups.
Geral Inspection
Agora vamos realizar apenas uma inspeção geral dos dados para finalizar as manipulações.
corr_plot(pheno, col.by = Ano)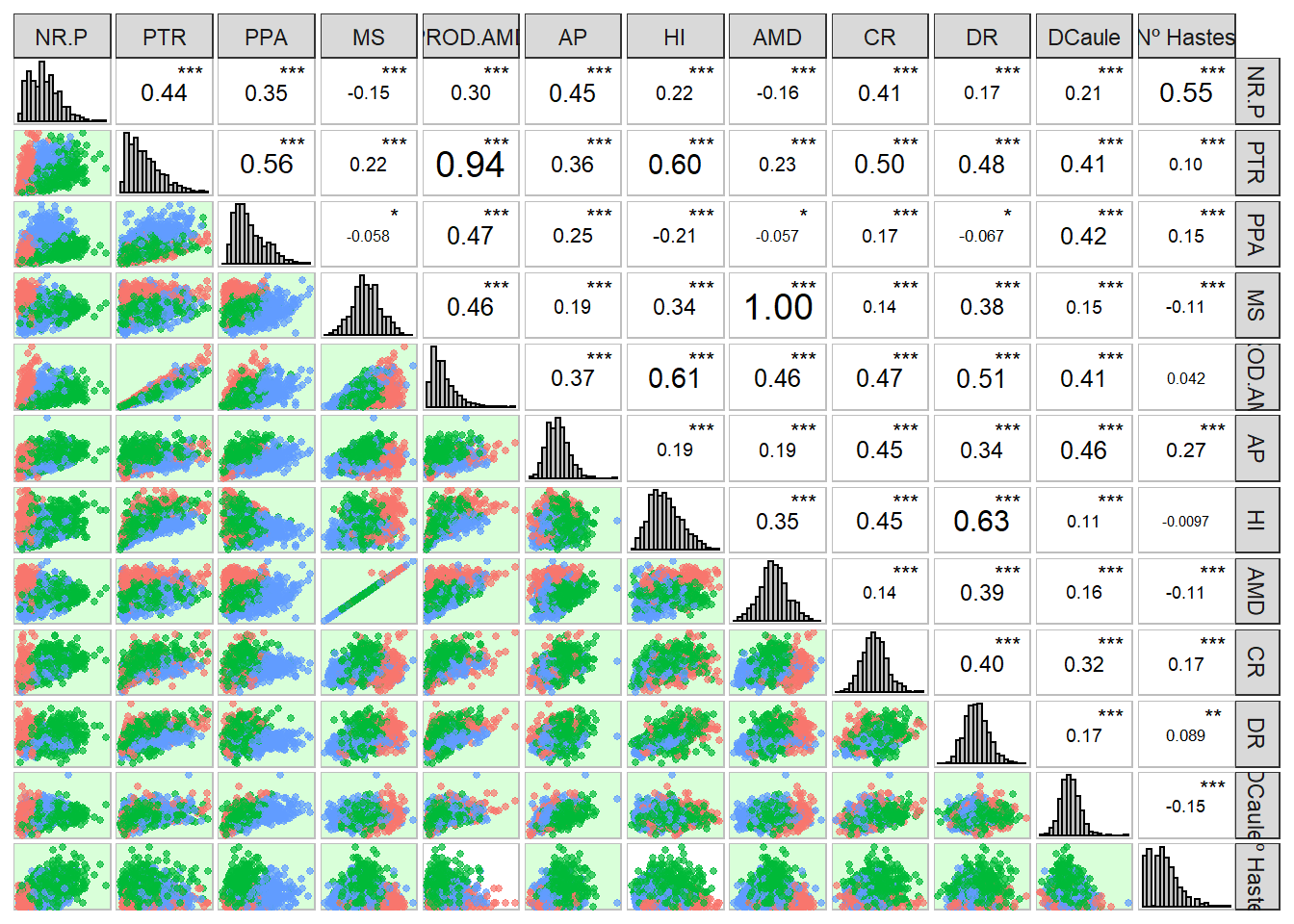
Amido com MS e PROD.AMD com PTR apresentam alta correlação.
Além disso a maioria das variáveis aparentemente apresentação distribuição normal dos dados fenotípicos. Dessa forma, vamos prosseguir para a obtenção dos blups.
Genotype-environment analysis by mixed-effect models
Primeiro, vou criar uma função para obter os blups e alguns parâmetros do nosso modelo.
BLUPS_par <- function(model, trait) {
BLUP <- ranef(model, condVar = TRUE)$Clone
PEV <-
c(attr(BLUP, "postVar")) # PEV is a vector of error variances associated with each individual BLUP... # it tells you about how confident you should be in the estimate of an individual CLONE's BLUP value.
Clone.var <-
c(VarCorr(model)$Clone) # Extract the variance component for CLONE
ResidVar <-
(attr(VarCorr(model), "sc")) ^ 2 # Extract the residual variance component
Ano <-
c(VarCorr(model)$Ano) # Extract the variance component for Ano.Bloco
Ano.Bloco <-
c(VarCorr(model)$Ano.Bloco) # Extract the variance component for Ano.Bloco
# You will need a line like the one above for every random effect (not for fixed effects)
out <-
BLUP / (1 - (PEV / Clone.var)) # This is the actual de-regress part (the BLUP for CLONE is divided by (1 - PEV/CLONE.var))
r2 <-
1 - (PEV / Clone.var) # Reliability: a confidence value for a BLUP (0 to 1 scale)
H2 = Clone.var / (Clone.var + Ano.Bloco + ResidVar) # An estimate of the broad-sense heritability, must change this formula when you change the model analysis
wt = (1 - H2) / ((0.1 + (1 - r2) / r2) * H2) # Weights for each de-regressed BLUP
# There is a paper the determined this crazy formula, Garrick et al. 2009. I wouldn't pay much attn. to it.
# These weights will be used in the second-step (e.g. cross-validation) to account for what we've done in this step
# The weights will be fit as error variances associated with each residual value
VarComps <- as.data.frame(VarCorr(model))
return(
list(
Trait = trait,
drgBLUP = out,
BLUP = BLUP,
weights = wt,
varcomps = VarComps,
H2 = H2,
Reliability = r2,
model = model
)
)
}
save(BLUPS_par, file = "output/BLUPS_par.Rdata")The BLUP model
Aqui temos que lembrar que possuímos outliers para algumas características e também que devemos excluir o ano 2017 para algumas.
Vou criar um loop onde informo quais características onde esse ano deverá ser excluído e também utilizar a função de remover outliers.
As características que devemos excluir o ano 2017 são Porte, Branching_Level, Staygreen, AMD, MS, Nº Hastes e PROD.AMD.
excluir_2017 <- c("Porte", "Branching_Level", "Staygreen", "AMD", "MS", "Nº Hastes" , "PROD.AMD")Usaremos esse vetor dentro do loop para exclusão do ano 2017 para essas variáveis.
Vamos converter todas as variáveis para numéricas agora.
traits <- colnames(pheno)[4:21]
pheno<- pheno %>%
mutate_at(traits, as.numeric)Agora vamos realizar a análise de modelos mistos para obter os blups.
load("output/BLUPS_par.Rdata")
registerDoParallel(cores = 6) # Specify the number of cores (my lab computer has 8; I will use 6 of them)
resultMM <- foreach(a = traits, i = icount(), .inorder = TRUE) %dopar% {
require(lme4)
require(dplyr)
library(purrr)
# Loop para exclusão do ano 2017 de acordo com o vetor com os nomes da variáveis descritos anteriormente.
if (a %in% excluir_2017) {
data <- pheno %>%
filter(Ano != 2017) %>%
droplevels()
} else{
data <- pheno
}
# Exclusão dos primeiros outliers encontrados
outliers <- boxplot(data[i+3], plot = FALSE)$out
if(!is_empty(outliers)){
data <- filter(data,data[i+3] != outliers)
}
model <- lmer(data = data,
formula = get(traits[i]) ~ (1 |Clone) + Ano + (1|Ano.Bloco)) # Clone and Ano.Bloco are random and Ano is fixed
result <- BLUPS_par(model, traits[i])
}
save(resultMM, file = "output/resultMM.Rdata")BLUPS for Clone
Como usei “foreach” para executar cada análise do estágio 1 em paralelo, cada característica está em um elemento separado de uma lista Precisamos processar o objeto resultMM em um data.frame ou matriz para análise posterior.
load("output/resultMM.Rdata")
BLUPS <-
data.frame(Clone = unique(pheno$Clone), stringsAsFactors = F)
H2 <- data.frame(H2 = "H2",
stringsAsFactors = F)
varcomp <-
data.frame(
grp = c("Clone", "Ano", "Ano.Bloco", "Residual"),
stringsAsFactors = F
)
# Aqui vamos obter os BLUPS para cada clone
for (i in 1:length(resultMM)) {
data <-
data.frame(Clone = rownames(resultMM[[i]]$BLUP),
stringsAsFactors = F)
data[, resultMM[[i]]$Trait] <- resultMM[[i]]$BLUP
BLUPS <- merge(BLUPS, data, by = "Clone", all.x = T)
H2[, resultMM[[i]]$Trait] <- resultMM[[i]]$H2
colnames(resultMM[[i]]$varcomps) <-
c(
"grp",
"var1",
"var2",
paste("vcov", resultMM[[i]]$Trait, sep = "."),
paste("sdcor", resultMM[[i]]$Trait, sep = ".")
)
varcomp <- varcomp %>%
right_join(resultMM[[i]]$varcomps)
}Joining, by = "grp"
Joining, by = c("grp", "var1", "var2")
Joining, by = c("grp", "var1", "var2")
Joining, by = c("grp", "var1", "var2")
Joining, by = c("grp", "var1", "var2")
Joining, by = c("grp", "var1", "var2")
Joining, by = c("grp", "var1", "var2")
Joining, by = c("grp", "var1", "var2")
Joining, by = c("grp", "var1", "var2")
Joining, by = c("grp", "var1", "var2")
Joining, by = c("grp", "var1", "var2")
Joining, by = c("grp", "var1", "var2")
Joining, by = c("grp", "var1", "var2")
Joining, by = c("grp", "var1", "var2")
Joining, by = c("grp", "var1", "var2")
Joining, by = c("grp", "var1", "var2")
Joining, by = c("grp", "var1", "var2")
Joining, by = c("grp", "var1", "var2")rownames(BLUPS) <- BLUPS$CloneSalvando os resultados dos BLUPs e dos parâmetros
saveRDS(BLUPS, file = "output/BLUPS.RDS")
write.csv(BLUPS,
"output/BLUPS.csv",
row.names = F,
quote = F)
write.csv(H2,
"output/herdabilidade.csv",
row.names = F,
quote = F)Ploting BLUPS for all traits
Primeiros, vou somar a média das variáveis com os BLUPS para melhor interpretação.
BLUPS<-readRDS("output/BLUPS.RDS")
media_pheno <- as.data.frame(pheno %>%
summarise_if(is.numeric, mean, na.rm = TRUE))
write.table(media_pheno, "output/media_pheno.csv")
phen<-
data.frame(Clone = unique(pheno$Clone), stringsAsFactors = F)
for (i in traits) {
phen[i] <- BLUPS[i] + media_pheno[, i]
}Vamos plotar os boxplot das variaveis.
phen %>%
pivot_longer(2:19, names_to = "Variable", values_to = "Values") %>%
ggplot() +
geom_boxplot(aes(y = Values, fill = Variable), show.legend = FALSE) +
facet_wrap(. ~ Variable, ncol = 6, scales = "free") +
expand_limits(y = 0) +
theme_bw()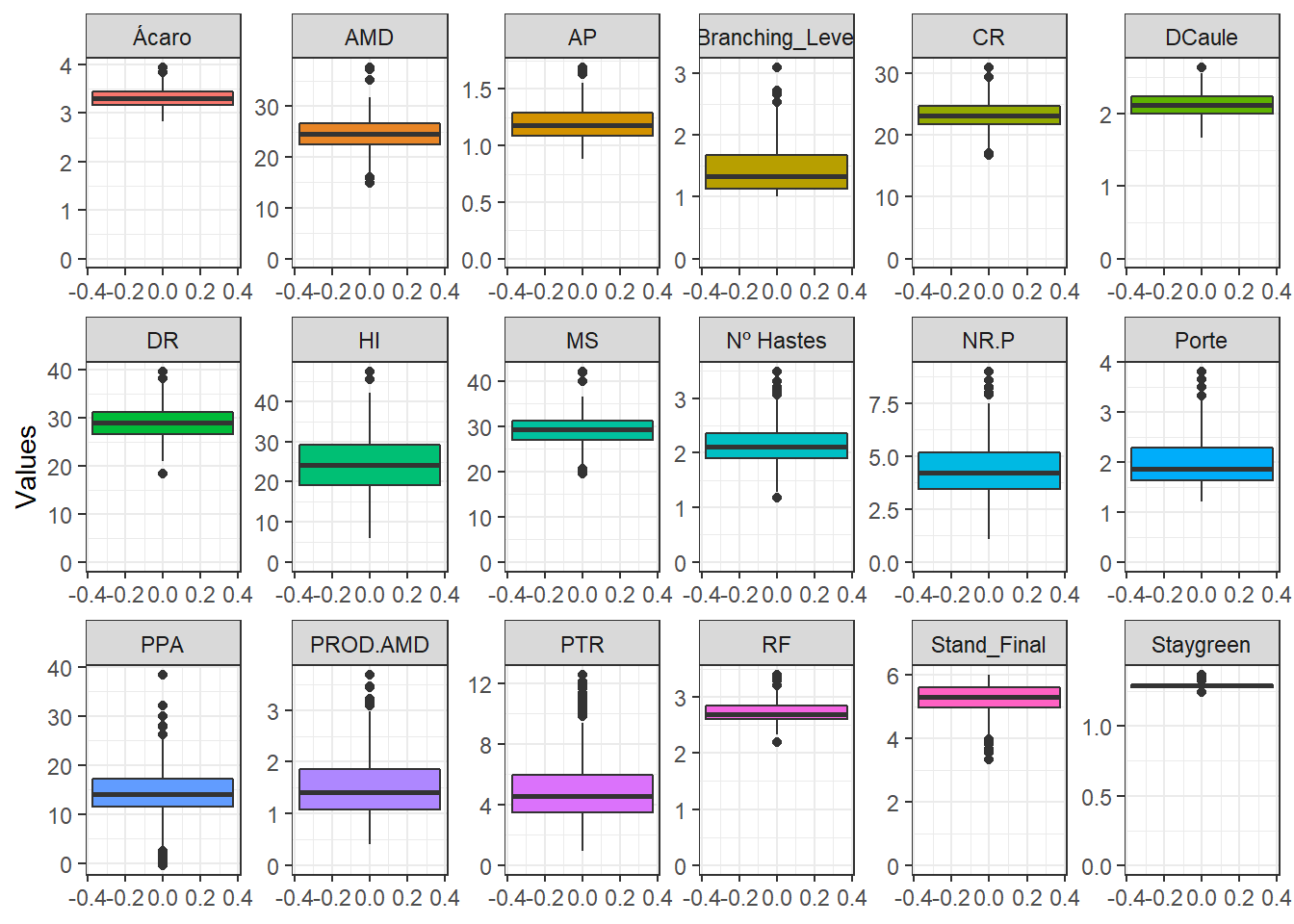
Aqui vamos avaliar apenas a distribuição dos BLUPs sem a média.
BLUPS %>%
pivot_longer(2:19, names_to = "Variable", values_to = "Values") %>%
ggplot() +
geom_density(aes(x = Values), show.legend = FALSE) +
facet_wrap(. ~ Variable, ncol = 6, scales = "free") +
expand_limits(y = 0) +
theme_bw()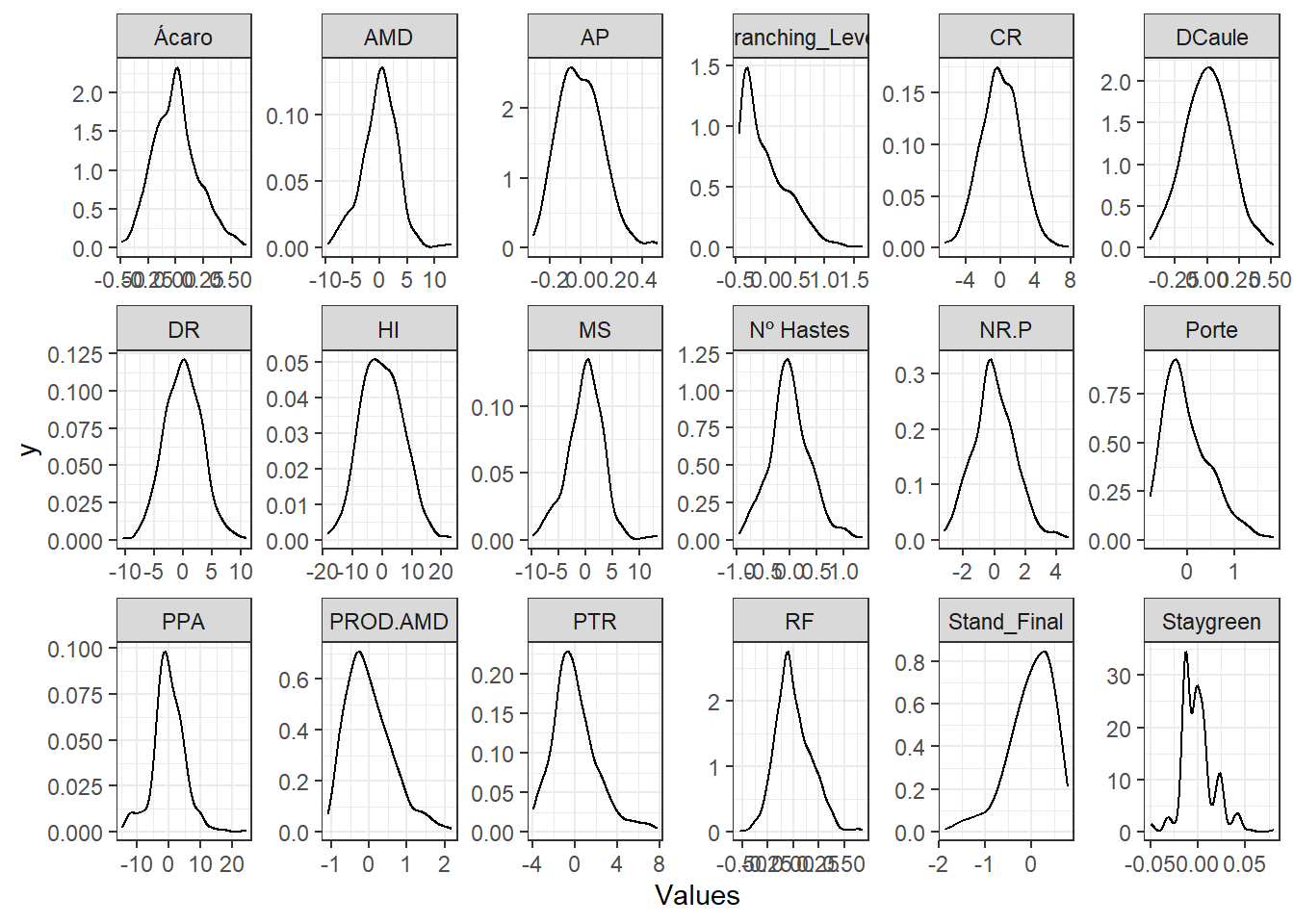
Aparentemente a maioria dos BLUPs para as variáveis seguem distribuição normal e pode ser aplicada a GWS pelos métodos convencionais.
sessionInfo()R version 4.1.3 (2022-03-10)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 19042)
Matrix products: default
locale:
[1] LC_COLLATE=Portuguese_Brazil.1252 LC_CTYPE=Portuguese_Brazil.1252
[3] LC_MONETARY=Portuguese_Brazil.1252 LC_NUMERIC=C
[5] LC_TIME=Portuguese_Brazil.1252
attached base packages:
[1] parallel grid stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] doParallel_1.0.17 iterators_1.0.14 foreach_1.5.2
[4] DataExplorer_0.8.2 metan_1.17.0 readxl_1.4.1
[7] data.table_1.14.2 ComplexHeatmap_2.10.0 forcats_0.5.2
[10] stringr_1.4.1 dplyr_1.0.10 purrr_0.3.4
[13] readr_2.1.2 tidyr_1.2.1 tibble_3.1.8
[16] ggplot2_3.3.6 tidyverse_1.3.2 kableExtra_1.3.4
loaded via a namespace (and not attached):
[1] minqa_1.2.4 googledrive_2.0.0 colorspace_2.0-3
[4] rjson_0.2.21 ellipsis_0.3.2 rprojroot_2.0.3
[7] circlize_0.4.15 GlobalOptions_0.1.2 fs_1.5.2
[10] clue_0.3-61 rstudioapi_0.14 farver_2.1.1
[13] ggrepel_0.9.1 fansi_1.0.3 lubridate_1.8.0
[16] mathjaxr_1.6-0 xml2_1.3.3 codetools_0.2-18
[19] splines_4.1.3 cachem_1.0.6 knitr_1.40
[22] polyclip_1.10-0 jsonlite_1.8.0 nloptr_2.0.3
[25] workflowr_1.7.0 broom_1.0.1 cluster_2.1.2
[28] dbplyr_2.2.1 png_0.1-7 ggforce_0.4.1
[31] compiler_4.1.3 httr_1.4.4 backports_1.4.1
[34] Matrix_1.5-1 assertthat_0.2.1 fastmap_1.1.0
[37] gargle_1.2.1 cli_3.3.0 later_1.3.0
[40] tweenr_2.0.2 htmltools_0.5.3 tools_4.1.3
[43] igraph_1.3.5 lmerTest_3.1-3 gtable_0.3.1
[46] glue_1.6.2 Rcpp_1.0.9 cellranger_1.1.0
[49] jquerylib_0.1.4 vctrs_0.4.1 nlme_3.1-159
[52] svglite_2.1.0 xfun_0.32 networkD3_0.4
[55] lme4_1.1-30 rvest_1.0.3 lifecycle_1.0.3
[58] googlesheets4_1.0.1 MASS_7.3-58.1 scales_1.2.1
[61] hms_1.1.2 promises_1.2.0.1 RColorBrewer_1.1-3
[64] yaml_2.3.5 gridExtra_2.3 sass_0.4.2
[67] reshape_0.8.9 stringi_1.7.6 highr_0.9
[70] S4Vectors_0.32.4 BiocGenerics_0.40.0 boot_1.3-28
[73] shape_1.4.6 rlang_1.0.6 pkgconfig_2.0.3
[76] systemfonts_1.0.4 matrixStats_0.62.0 evaluate_0.17
[79] lattice_0.20-45 labeling_0.4.2 htmlwidgets_1.5.4
[82] patchwork_1.1.2 tidyselect_1.2.0 GGally_2.1.2
[85] plyr_1.8.7 magrittr_2.0.3 R6_2.5.1
[88] magick_2.7.3 IRanges_2.28.0 generics_0.1.3
[91] DBI_1.1.3 pillar_1.8.1 haven_2.5.1
[94] withr_2.5.0 modelr_0.1.9 crayon_1.5.2
[97] utf8_1.2.2 tzdb_0.3.0 rmarkdown_2.17
[100] GetoptLong_1.0.5 git2r_0.30.1 reprex_2.0.2
[103] digest_0.6.29 webshot_0.5.4 numDeriv_2016.8-1.1
[106] httpuv_1.6.5 stats4_4.1.3 munsell_0.5.0
[109] viridisLite_0.4.1 bslib_0.4.0 Weverton Gomes da Costa, Pós-Doutorando, Embrapa Mandioca e Fruticultura, wevertonufv@gmail.com↩︎