Differential Expression
Briana Mittleman
11/11/2019
Last updated: 2019-12-06
Checks: 7 0
Knit directory: Comparative_APA/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.5.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190902) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: code/chimp_log/
Ignored: code/human_log/
Ignored: data/.DS_Store
Ignored: data/DiffSplice_liftedJunc/
Ignored: data/HC_filenames.txt.sb-4426323c-IKIs0S/
Ignored: data/OppositeSpeciesMap.txt.sb-a5794dd2-mayWJf/
Ignored: data/RNASEQ_metadata.txt.sb-4426323c-TE4ns3/
Ignored: data/RNASEQ_metadata.txt.sb-51f67ae1-HXp7Gq/
Ignored: data/RNASEQ_metadata_2Removed.txt.sb-4426323c-a4lBwx/
Ignored: data/RNASEQ_metadata_stranded.txt.sb-a5794dd2-D659m2/
Ignored: data/metadata_HCpanel.txt.sb-a3d92a2d-b9cYoF/
Ignored: data/metadata_HCpanel.txt.sb-f4823d1e-qihGek/
Untracked files:
Untracked: ._.DS_Store
Untracked: Chimp/
Untracked: Human/
Untracked: analysis/assessReadQual.Rmd
Untracked: code/._Config_chimp.yaml
Untracked: code/._Config_chimp_full.yaml
Untracked: code/._Config_human.yaml
Untracked: code/._CountNucleotides.py
Untracked: code/._DiffSplice.sh
Untracked: code/._DiffSplicePlots.sh
Untracked: code/._DiffSplicePlots_gencode.sh
Untracked: code/._DiffSplice_gencode.sh
Untracked: code/._DiffSplice_removebad.sh
Untracked: code/._GetMAPQscore.py
Untracked: code/._GetSecondaryMap.py
Untracked: code/._LiftOrthoPAS2chimp.sh
Untracked: code/._MapBadSamples.sh
Untracked: code/._QuantMergedClusters.sh
Untracked: code/._RunFixLeafCluster.sh
Untracked: code/._Snakefile
Untracked: code/._SnakefilePAS
Untracked: code/._SnakefilePASfilt
Untracked: code/._SortIndexBadSamples.sh
Untracked: code/._bed215upbed.py
Untracked: code/._bed2SAF_gen.py
Untracked: code/._buildIndecpantro5
Untracked: code/._buildIndecpantro5.sh
Untracked: code/._buildStarIndex.sh
Untracked: code/._chimpChromprder.sh
Untracked: code/._cleanbed2saf.py
Untracked: code/._cluster.json
Untracked: code/._cluster2bed.py
Untracked: code/._clusterLiftReverse.sh
Untracked: code/._clusterLiftReverse_removebad.sh
Untracked: code/._clusterLiftprimary.sh
Untracked: code/._clusterLiftprimary_removebad.sh
Untracked: code/._converBam2Junc.sh
Untracked: code/._converBam2Junc_removeBad.sh
Untracked: code/._extraSnakefiltpas
Untracked: code/._filter5percPAS.py
Untracked: code/._filterNumChroms.py
Untracked: code/._filterPASforMP.py
Untracked: code/._filterPostLift.py
Untracked: code/._fixExonFC.py
Untracked: code/._fixLeafCluster.py
Untracked: code/._fixLiftedJunc.py
Untracked: code/._fixUTRexonanno.py
Untracked: code/._formathg38Anno.py
Untracked: code/._formatpantro6Anno.py
Untracked: code/._getRNAseqMapStats.sh
Untracked: code/._hg19MapStats.sh
Untracked: code/._humanChromorder.sh
Untracked: code/._intersectLiftedPAS.sh
Untracked: code/._liftJunctionFiles.sh
Untracked: code/._liftPAS19to38.sh
Untracked: code/._liftedchimpJunc2human.sh
Untracked: code/._makeSamplyGroupsHuman_TvN.py
Untracked: code/._mapRNAseqhg19.sh
Untracked: code/._mapRNAseqhg19_newPipeline.sh
Untracked: code/._maphg19.sh
Untracked: code/._maphg19_subjunc.sh
Untracked: code/._mergedBam2BW.sh
Untracked: code/._nameClusters.py
Untracked: code/._numMultimap.py
Untracked: code/._overlapapaQTLPAS.sh
Untracked: code/._prepareCleanLiftedFC_5perc4LC.py
Untracked: code/._preparePAS4lift.py
Untracked: code/._primaryLift.sh
Untracked: code/._processhg38exons.py
Untracked: code/._quantJunc.sh
Untracked: code/._quantJunc_TEST.sh
Untracked: code/._quantJunc_removeBad.sh
Untracked: code/._quantMerged_seperatly.sh
Untracked: code/._recLiftchim2human.sh
Untracked: code/._revLiftPAShg38to19.sh
Untracked: code/._reverseLift.sh
Untracked: code/._runChimpDiffIso.sh
Untracked: code/._runCountNucleotides.sh
Untracked: code/._runFilterNumChroms.sh
Untracked: code/._runHumanDiffIso.sh
Untracked: code/._runNuclearDifffIso.sh
Untracked: code/._run_chimpverifybam.sh
Untracked: code/._run_verifyBam.sh
Untracked: code/._snakemake.batch
Untracked: code/._snakemakePAS.batch
Untracked: code/._snakemakePASchimp.batch
Untracked: code/._snakemakePAShuman.batch
Untracked: code/._snakemake_chimp.batch
Untracked: code/._snakemake_human.batch
Untracked: code/._snakemakefiltPAS.batch
Untracked: code/._snakemakefiltPAS_chimp
Untracked: code/._snakemakefiltPAS_chimp.sh
Untracked: code/._snakemakefiltPAS_human.sh
Untracked: code/._submit-snakemake-chimp.sh
Untracked: code/._submit-snakemake-human.sh
Untracked: code/._submit-snakemakePAS-chimp.sh
Untracked: code/._submit-snakemakePAS-human.sh
Untracked: code/._submit-snakemakefiltPAS-chimp.sh
Untracked: code/._submit-snakemakefiltPAS-human.sh
Untracked: code/._subset_diffisopheno_Nuclear_HvC.py
Untracked: code/._transcriptDTplotsNuclear.sh
Untracked: code/._verifyBam4973.sh
Untracked: code/._verifyBam4973inHuman.sh
Untracked: code/._wrap_chimpverifybam.sh
Untracked: code/._wrap_verifyBam.sh
Untracked: code/._writeMergecode.py
Untracked: code/.snakemake/
Untracked: code/Config_chimp.yaml
Untracked: code/Config_chimp_full.yaml
Untracked: code/Config_human.yaml
Untracked: code/CountNucleotides.py
Untracked: code/DiffSplice.err
Untracked: code/DiffSplice.out
Untracked: code/DiffSplice.sh
Untracked: code/DiffSplicePlots.err
Untracked: code/DiffSplicePlots.out
Untracked: code/DiffSplicePlots.sh
Untracked: code/DiffSplicePlots_gencode.sh
Untracked: code/DiffSplice_gencode.sh
Untracked: code/DiffSplice_removebad.err
Untracked: code/DiffSplice_removebad.out
Untracked: code/DiffSplice_removebad.sh
Untracked: code/GencodeDiffSplice.err
Untracked: code/GencodeDiffSplice.out
Untracked: code/GetMAPQscore.py
Untracked: code/GetSecondaryMap.py
Untracked: code/HchromOrder.err
Untracked: code/HchromOrder.out
Untracked: code/JunctionLift.err
Untracked: code/JunctionLift.out
Untracked: code/LiftClustersFirst.err
Untracked: code/LiftClustersFirst.out
Untracked: code/LiftClustersFirst_remove.err
Untracked: code/LiftClustersFirst_remove.out
Untracked: code/LiftClustersSecond.err
Untracked: code/LiftClustersSecond.out
Untracked: code/LiftClustersSecond_remove.err
Untracked: code/LiftClustersSecond_remove.out
Untracked: code/LiftOrthoPAS2chimp.sh
Untracked: code/LiftorthoPAS.err
Untracked: code/LiftorthoPASt.out
Untracked: code/Log.out
Untracked: code/MapBadSamples.err
Untracked: code/MapBadSamples.out
Untracked: code/MapBadSamples.sh
Untracked: code/MapStats.err
Untracked: code/MapStats.out
Untracked: code/MergeClusters.sh
Untracked: code/QuantMergeClusters
Untracked: code/QuantMergeClusters.err
Untracked: code/QuantMergeClusters.out
Untracked: code/QuantMergedClusters.sh
Untracked: code/Rev_liftoverPAShg19to38.err
Untracked: code/Rev_liftoverPAShg19to38.out
Untracked: code/RunFixCluster.err
Untracked: code/RunFixCluster.out
Untracked: code/RunFixLeafCluster.sh
Untracked: code/SAF215upbed_gen.py
Untracked: code/Snakefile
Untracked: code/SnakefilePAS
Untracked: code/SnakefilePASfilt
Untracked: code/SortIndexBadSamples.err
Untracked: code/SortIndexBadSamples.out
Untracked: code/SortIndexBadSamples.sh
Untracked: code/TotalTranscriptDTplot.err
Untracked: code/TotalTranscriptDTplot.out
Untracked: code/Upstream10Bases_general.py
Untracked: code/apaQTLsnake.err
Untracked: code/apaQTLsnake.out
Untracked: code/apaQTLsnakePAS.err
Untracked: code/apaQTLsnakePAS.out
Untracked: code/apaQTLsnakePAShuman.err
Untracked: code/bam2junc.err
Untracked: code/bam2junc.out
Untracked: code/bam2junc_remove.err
Untracked: code/bam2junc_remove.out
Untracked: code/bed215upbed.py
Untracked: code/bed2SAF_gen.py
Untracked: code/bed2saf.py
Untracked: code/bg_to_cov.py
Untracked: code/buildIndecpantro5
Untracked: code/buildIndecpantro5.sh
Untracked: code/buildStarIndex.sh
Untracked: code/callPeaksYL.py
Untracked: code/chimpChromprder.sh
Untracked: code/chooseAnno2Bed.py
Untracked: code/chooseAnno2SAF.py
Untracked: code/chromOrder.err
Untracked: code/chromOrder.out
Untracked: code/cleanbed2saf.py
Untracked: code/cluster.json
Untracked: code/cluster2bed.py
Untracked: code/clusterLiftReverse.sh
Untracked: code/clusterLiftReverse_removebad.sh
Untracked: code/clusterLiftprimary.sh
Untracked: code/clusterLiftprimary_removebad.sh
Untracked: code/clusterPAS.json
Untracked: code/clusterfiltPAS.json
Untracked: code/comands2Mege.sh
Untracked: code/converBam2Junc.sh
Untracked: code/converBam2Junc_removeBad.sh
Untracked: code/convertNumeric.py
Untracked: code/environment.yaml
Untracked: code/extraSnakefiltpas
Untracked: code/filter5perc.R
Untracked: code/filter5percPAS.py
Untracked: code/filter5percPheno.py
Untracked: code/filterBamforMP.pysam2_gen.py
Untracked: code/filterJuncChroms.err
Untracked: code/filterJuncChroms.out
Untracked: code/filterMissprimingInNuc10_gen.py
Untracked: code/filterNumChroms.py
Untracked: code/filterPASforMP.py
Untracked: code/filterPostLift.py
Untracked: code/filterSAFforMP_gen.py
Untracked: code/filterSortBedbyCleanedBed_gen.R
Untracked: code/filterpeaks.py
Untracked: code/fixExonFC.py
Untracked: code/fixFChead.py
Untracked: code/fixFChead_bothfrac.py
Untracked: code/fixLeafCluster.py
Untracked: code/fixLiftedJunc.py
Untracked: code/fixUTRexonanno.py
Untracked: code/formathg38Anno.py
Untracked: code/generateStarIndex.err
Untracked: code/generateStarIndex.out
Untracked: code/generateStarIndexHuman.err
Untracked: code/generateStarIndexHuman.out
Untracked: code/getRNAseqMapStats.sh
Untracked: code/hg19MapStats.err
Untracked: code/hg19MapStats.out
Untracked: code/hg19MapStats.sh
Untracked: code/humanChromorder.sh
Untracked: code/humanFiles
Untracked: code/humanJunc_pooled
Untracked: code/intersectAnno.err
Untracked: code/intersectAnno.out
Untracked: code/intersectLiftedPAS.sh
Untracked: code/leafcutter_merge_regtools_redo.py
Untracked: code/liftJunctionFiles.sh
Untracked: code/liftPAS19to38.sh
Untracked: code/liftoverPAShg19to38.err
Untracked: code/liftoverPAShg19to38.out
Untracked: code/log/
Untracked: code/make5percPeakbed.py
Untracked: code/makeFileID.py
Untracked: code/makePheno.py
Untracked: code/makeSamplyGroupsChimp_TvN.py
Untracked: code/makeSamplyGroupsHuman_TvN.py
Untracked: code/mapRNAseqhg19.sh
Untracked: code/mapRNAseqhg19_newPipeline.sh
Untracked: code/maphg19.err
Untracked: code/maphg19.out
Untracked: code/maphg19.sh
Untracked: code/maphg19_new.err
Untracked: code/maphg19_new.out
Untracked: code/maphg19_sub.err
Untracked: code/maphg19_sub.out
Untracked: code/maphg19_subjunc.sh
Untracked: code/merge.err
Untracked: code/merge_leafcutter_clusters_redo.py
Untracked: code/mergedBam2BW.sh
Untracked: code/mergedbam2bw.err
Untracked: code/mergedbam2bw.out
Untracked: code/nameClusters.py
Untracked: code/namePeaks.py
Untracked: code/nuclearTranscriptDTplot.err
Untracked: code/nuclearTranscriptDTplot.out
Untracked: code/numMultimap.py
Untracked: code/overlapPAS.err
Untracked: code/overlapPAS.out
Untracked: code/overlapapaQTLPAS.sh
Untracked: code/peak2PAS.py
Untracked: code/pheno2countonly.R
Untracked: code/prepareCleanLiftedFC_5perc4LC.py
Untracked: code/preparePAS4lift.py
Untracked: code/prepare_phenotype_table.py
Untracked: code/primaryLift.err
Untracked: code/primaryLift.out
Untracked: code/primaryLift.sh
Untracked: code/processhg38exons.py
Untracked: code/quantJunc.sh
Untracked: code/quantJunc_TEST.sh
Untracked: code/quantJunc_removeBad.sh
Untracked: code/quantLiftedPAS.err
Untracked: code/quantLiftedPAS.out
Untracked: code/quantLiftedPAS.sh
Untracked: code/quatJunc.err
Untracked: code/quatJunc.out
Untracked: code/recChimpback2Human.err
Untracked: code/recChimpback2Human.out
Untracked: code/recLiftchim2human.sh
Untracked: code/revLift.err
Untracked: code/revLift.out
Untracked: code/revLiftPAShg38to19.sh
Untracked: code/reverseLift.sh
Untracked: code/runChimpDiffIso.sh
Untracked: code/runCountNucleotides.err
Untracked: code/runCountNucleotides.out
Untracked: code/runCountNucleotides.sh
Untracked: code/runFilterNumChroms.sh
Untracked: code/runHumanDiffIso.sh
Untracked: code/runNuclearDifffIso.sh
Untracked: code/run_Chimpleafcutter_ds.err
Untracked: code/run_Chimpleafcutter_ds.out
Untracked: code/run_Chimpverifybam.err
Untracked: code/run_Chimpverifybam.out
Untracked: code/run_Humanleafcutter_ds.err
Untracked: code/run_Humanleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_ds.err
Untracked: code/run_Nuclearleafcutter_ds.out
Untracked: code/run_chimpverifybam.sh
Untracked: code/run_verifyBam.sh
Untracked: code/run_verifybam.err
Untracked: code/run_verifybam.out
Untracked: code/slurm-62824013.out
Untracked: code/slurm-62825841.out
Untracked: code/slurm-62826116.out
Untracked: code/slurm-64108209.out
Untracked: code/slurm-64108521.out
Untracked: code/slurm-64108557.out
Untracked: code/snakePASChimp.err
Untracked: code/snakePASChimp.out
Untracked: code/snakePAShuman.out
Untracked: code/snakemake.batch
Untracked: code/snakemakeChimp.err
Untracked: code/snakemakeChimp.out
Untracked: code/snakemakeHuman.err
Untracked: code/snakemakeHuman.out
Untracked: code/snakemakePAS.batch
Untracked: code/snakemakePASFiltChimp.err
Untracked: code/snakemakePASFiltChimp.out
Untracked: code/snakemakePASFiltHuman.err
Untracked: code/snakemakePASFiltHuman.out
Untracked: code/snakemakePASchimp.batch
Untracked: code/snakemakePAShuman.batch
Untracked: code/snakemake_chimp.batch
Untracked: code/snakemake_human.batch
Untracked: code/snakemakefiltPAS.batch
Untracked: code/snakemakefiltPAS_chimp.sh
Untracked: code/snakemakefiltPAS_human.sh
Untracked: code/submit-snakemake-chimp.sh
Untracked: code/submit-snakemake-human.sh
Untracked: code/submit-snakemakePAS-chimp.sh
Untracked: code/submit-snakemakePAS-human.sh
Untracked: code/submit-snakemakefiltPAS-chimp.sh
Untracked: code/submit-snakemakefiltPAS-human.sh
Untracked: code/subset_diffisopheno.py
Untracked: code/subset_diffisopheno_Chimp_tvN.py
Untracked: code/subset_diffisopheno_Huma_tvN.py
Untracked: code/subset_diffisopheno_Nuclear_HvC.py
Untracked: code/test
Untracked: code/transcriptDTplotsNuclear.sh
Untracked: code/transcriptDTplotsTotal.sh
Untracked: code/verifyBam4973.sh
Untracked: code/verifyBam4973inHuman.sh
Untracked: code/verifybam4973.err
Untracked: code/verifybam4973.out
Untracked: code/verifybam4973HumanMap.err
Untracked: code/verifybam4973HumanMap.out
Untracked: code/wrap_Chimpverifybam.err
Untracked: code/wrap_Chimpverifybam.out
Untracked: code/wrap_chimpverifybam.sh
Untracked: code/wrap_verifyBam.sh
Untracked: code/wrap_verifybam.err
Untracked: code/wrap_verifybam.out
Untracked: code/writeMergecode.py
Untracked: data/._.DS_Store
Untracked: data/._HC_filenames.txt
Untracked: data/._HC_filenames.txt.sb-4426323c-IKIs0S
Untracked: data/._HC_filenames.xlsx
Untracked: data/._OppositeSpeciesMap.txt
Untracked: data/._OppositeSpeciesMap.txt.sb-a5794dd2-mayWJf
Untracked: data/._OppositeSpeciesMap.xlsx
Untracked: data/._RNASEQ_metadata.txt
Untracked: data/._RNASEQ_metadata.txt.sb-4426323c-TE4ns3
Untracked: data/._RNASEQ_metadata.txt.sb-51f67ae1-HXp7Gq
Untracked: data/._RNASEQ_metadata.xlsx
Untracked: data/._RNASEQ_metadata_2Removed.txt
Untracked: data/._RNASEQ_metadata_2Removed.txt.sb-4426323c-a4lBwx
Untracked: data/._RNASEQ_metadata_2Removed.xlsx
Untracked: data/._RNASEQ_metadata_stranded.txt
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-a5794dd2-D659m2
Untracked: data/._RNASEQ_metadata_stranded.xlsx
Untracked: data/._metadata_HCpanel.txt
Untracked: data/._metadata_HCpanel.txt.sb-a3d92a2d-b9cYoF
Untracked: data/._metadata_HCpanel.txt.sb-f4823d1e-qihGek
Untracked: data/._metadata_HCpanel.xlsx
Untracked: data/._~$RNASEQ_metadata.xlsx
Untracked: data/._~$metadata_HCpanel.xlsx
Untracked: data/CompapaQTLpas/
Untracked: data/DTmatrix/
Untracked: data/DiffExpression/
Untracked: data/DiffIso_Nuclear/
Untracked: data/DiffSplice/
Untracked: data/DiffSplice_removeBad/
Untracked: data/EvalPantro5/
Untracked: data/HC_filenames.txt
Untracked: data/HC_filenames.xlsx
Untracked: data/MapStats/
Untracked: data/NuclearHvC/
Untracked: data/OppositeSpeciesMap.txt
Untracked: data/OppositeSpeciesMap.xlsx
Untracked: data/Peaks_5perc/
Untracked: data/Pheno_5perc/
Untracked: data/Pheno_5perc_nuclear/
Untracked: data/Pheno_5perc_total/
Untracked: data/RNASEQ_metadata.txt
Untracked: data/RNASEQ_metadata.xlsx
Untracked: data/RNASEQ_metadata_2Removed.txt
Untracked: data/RNASEQ_metadata_2Removed.xlsx
Untracked: data/RNASEQ_metadata_stranded.txt
Untracked: data/RNASEQ_metadata_stranded.xlsx
Untracked: data/TwoBadSampleAnalysis/
Untracked: data/chainFiles/
Untracked: data/cleanPeaks_anno/
Untracked: data/cleanPeaks_byspecies/
Untracked: data/cleanPeaks_lifted/
Untracked: data/liftover_files/
Untracked: data/metadata_HCpanel.txt
Untracked: data/metadata_HCpanel.xlsx
Untracked: data/primaryLift/
Untracked: data/reverseLift/
Untracked: data/~$RNASEQ_metadata.xlsx
Untracked: data/~$metadata_HCpanel.xlsx
Untracked: output/dtPlots/
Untracked: projectNotes.Rmd
Unstaged changes:
Modified: analysis/CorrbetweenInd.Rmd
Modified: analysis/PASnumperSpecies.Rmd
Modified: analysis/annotatePAS.Rmd
Modified: analysis/annotationInfo.Rmd
Modified: analysis/diffSplicing.Rmd
Modified: analysis/liftoverPAS.Rmd
Modified: analysis/multiMap.Rmd
Modified: analysis/verifyBAM.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 6d93e4d | brimittleman | 2019-12-06 | update stranded |
| html | a288a29 | brimittleman | 2019-12-04 | Build site. |
| Rmd | 558b39f | brimittleman | 2019-12-04 | add current error for splice and write out DE genes |
| html | 8fca47f | brimittleman | 2019-11-22 | Build site. |
| Rmd | f13781e | brimittleman | 2019-11-22 | fixed mapping and indivs |
| html | 25971ed | brimittleman | 2019-11-21 | Build site. |
| Rmd | db0484c | brimittleman | 2019-11-21 | add PC corr |
| html | 712106e | brimittleman | 2019-11-19 | Build site. |
| Rmd | 8dc9ea0 | brimittleman | 2019-11-19 | first pipeline for de |
| html | 586c9ec | brimittleman | 2019-11-13 | Build site. |
| Rmd | bedfa41 | brimittleman | 2019-11-13 | question PCA methods |
| html | a22bae9 | brimittleman | 2019-11-13 | Build site. |
| Rmd | a52c26d | brimittleman | 2019-11-13 | look at pca and tech factors |
| html | da4bab0 | brimittleman | 2019-11-12 | Build site. |
| Rmd | 98d7f9b | brimittleman | 2019-11-12 | add cpm pca |
| html | 32b435b | brimittleman | 2019-11-12 | Build site. |
| Rmd | 1ce8433 | brimittleman | 2019-11-12 | start normalization |
| html | 2c02d70 | brimittleman | 2019-11-12 | Build site. |
| Rmd | 53642f7 | brimittleman | 2019-11-12 | add mapp stats |
| html | dc91b0a | brimittleman | 2019-11-11 | Build site. |
| Rmd | b5ba82e | brimittleman | 2019-11-11 | add diff expression and diff splicing |
library(workflowr)This is workflowr version 1.5.0
Run ?workflowr for help getting startedlibrary(tidyverse)── Attaching packages ──────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.1.1 ✔ purrr 0.3.2
✔ tibble 2.1.1 ✔ dplyr 0.8.0.1
✔ tidyr 0.8.3 ✔ stringr 1.3.1
✔ readr 1.3.1 ✔ forcats 0.3.0 ── Conflicts ─────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library("scales")
Attaching package: 'scales'The following object is masked from 'package:purrr':
discardThe following object is masked from 'package:readr':
col_factorlibrary("gplots")
Attaching package: 'gplots'The following object is masked from 'package:stats':
lowesslibrary("edgeR")Loading required package: limmalibrary("R.utils")Loading required package: R.ooLoading required package: R.methodsS3R.methodsS3 v1.7.1 (2016-02-15) successfully loaded. See ?R.methodsS3 for help.R.oo v1.22.0 (2018-04-21) successfully loaded. See ?R.oo for help.
Attaching package: 'R.oo'The following objects are masked from 'package:methods':
getClasses, getMethodsThe following objects are masked from 'package:base':
attach, detach, gc, load, saveR.utils v2.7.0 successfully loaded. See ?R.utils for help.
Attaching package: 'R.utils'The following object is masked from 'package:tidyr':
extractThe following object is masked from 'package:utils':
timestampThe following objects are masked from 'package:base':
cat, commandArgs, getOption, inherits, isOpen, parse, warningslibrary("limma")
library("VennDiagram")Loading required package: gridLoading required package: futile.loggerlibrary("RColorBrewer")
library(reshape2)
Attaching package: 'reshape2'The following object is masked from 'package:tidyr':
smithsFor this analysis I do preprocessing with the Snakemake pipeline. The snakemake will map the RNA seq and quantify orthologous exons.
From FastQC:
Does not look like there is adapter contamination
No reads tagged as bad quality
Assess mapping:
metaData=read.table("../data/RNASEQ_metadata_stranded.txt", header = T, stringsAsFactors = F)
metaData$Species=as.factor(metaData$Species)
metaData$Collection=as.factor(metaData$Collection)readInfo=metaData %>% mutate(AAUnMapped= Reads-Mapped, ABNotOrtho= Mapped-AssignedOrtho) %>% select(Line, Species, AAUnMapped, ABNotOrtho, AssignedOrtho) %>% gather(key="Category", value="Number", -Line, -Species)
ggplot(readInfo, aes(x=Line,y=Number, fill=Category)) + geom_bar(stat="identity") + scale_fill_brewer(palette = "Dark2",name = "Type", labels = c("Unmapped", "Mapped not ortho", "Assigned Ortho Exon"))+theme(axis.text.x = element_text( hjust = 0,vjust = 1, size = 6, angle = 90)) + labs(y="Reads", title="Human and chimp read statistics") 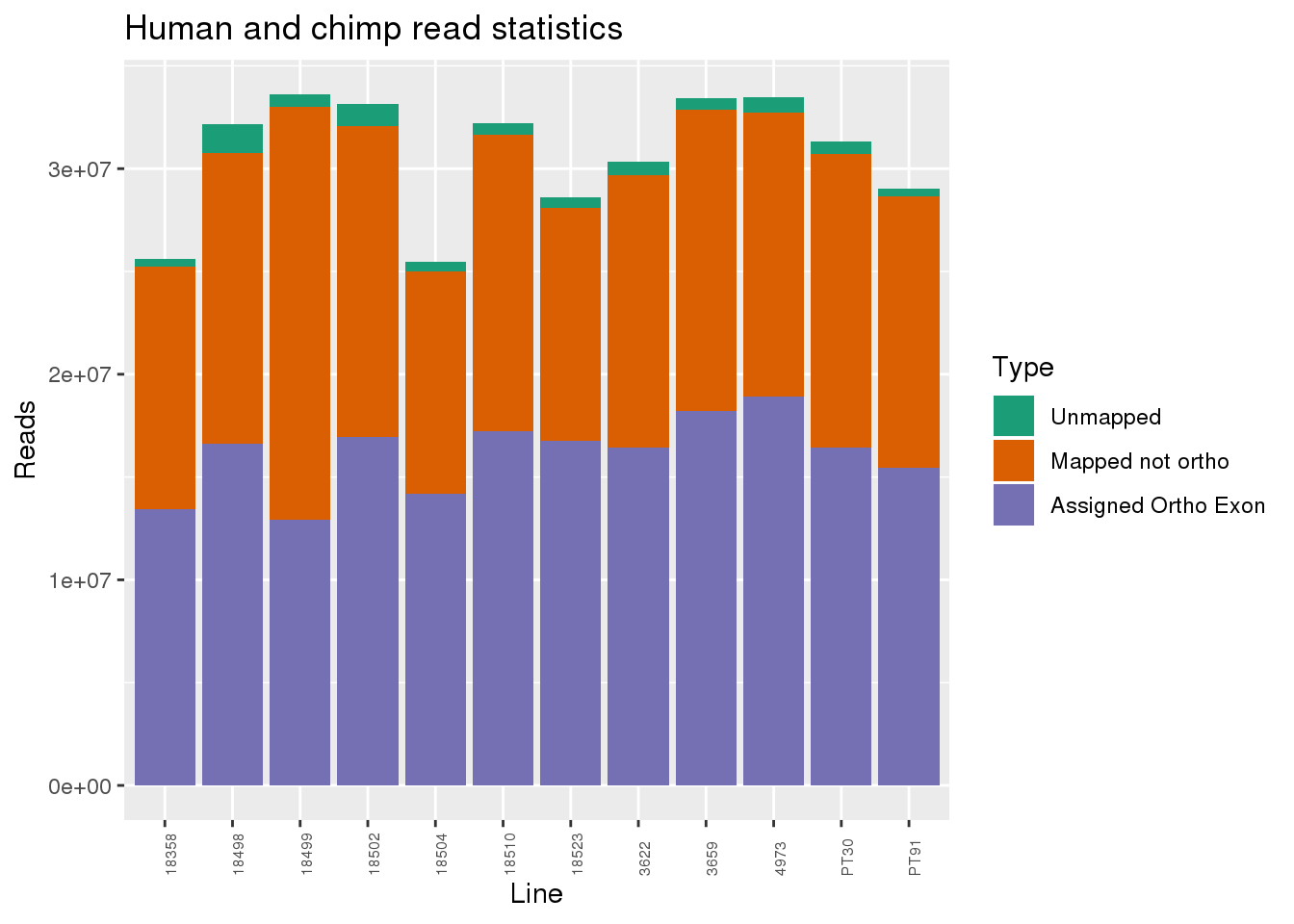
Proportion of reads.
readProp=metaData %>% mutate(Aunmapped=1-percentMapped, MappednotOrtho=percentMapped-percentOrtho) %>% select(Line,Species, percentOrtho, MappednotOrtho, Aunmapped) %>% gather(key="Category", value="Proportion", -Line, -Species)
ggplot(readProp, aes(x=Line,y=Proportion, fill=Category)) + geom_bar(stat="identity") + scale_fill_brewer(palette = "Dark2", name="", labels = c("Unmapped", "Mapped not ortho", "Assigned Ortho Exon"))+theme(axis.text.x = element_text( hjust = 0,vjust = 1, size = 6, angle = 90)) + labs(y="Reads", title="Human and chimp read proportions") 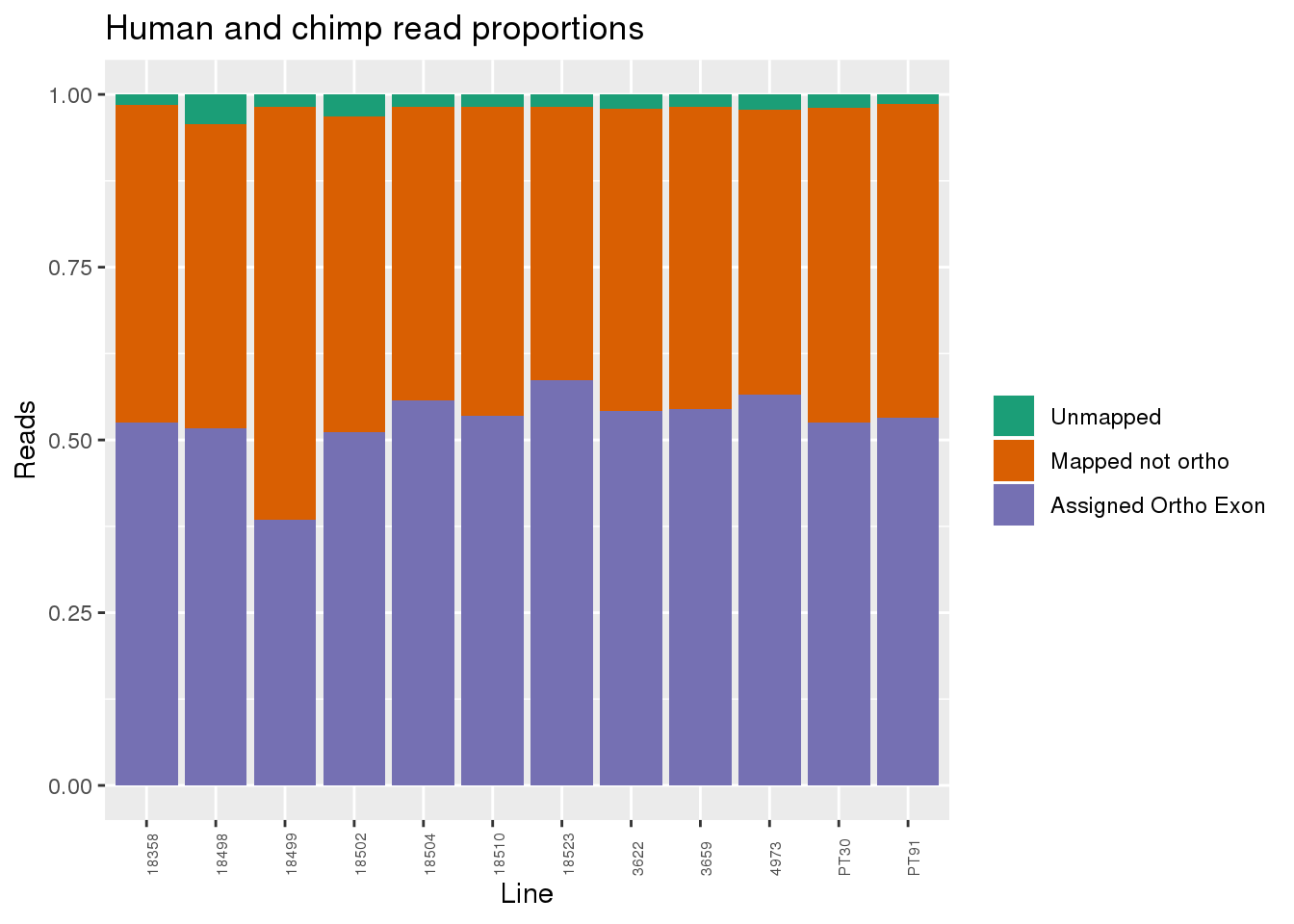
By species:
ggplot(readInfo,aes(x=Category, y=Number, by=Species, fill=Species)) + geom_boxplot() +scale_x_discrete( breaks=c("AAUnMapped","ABNotOrtho","AssignedOrtho"),labels=c("Unmapped", "Not in OrthoExon", "Assigned to OrthoExon")) + scale_fill_brewer(palette = "Dark2") + labs(title="Mapped reads by Species", y="Reads", x="")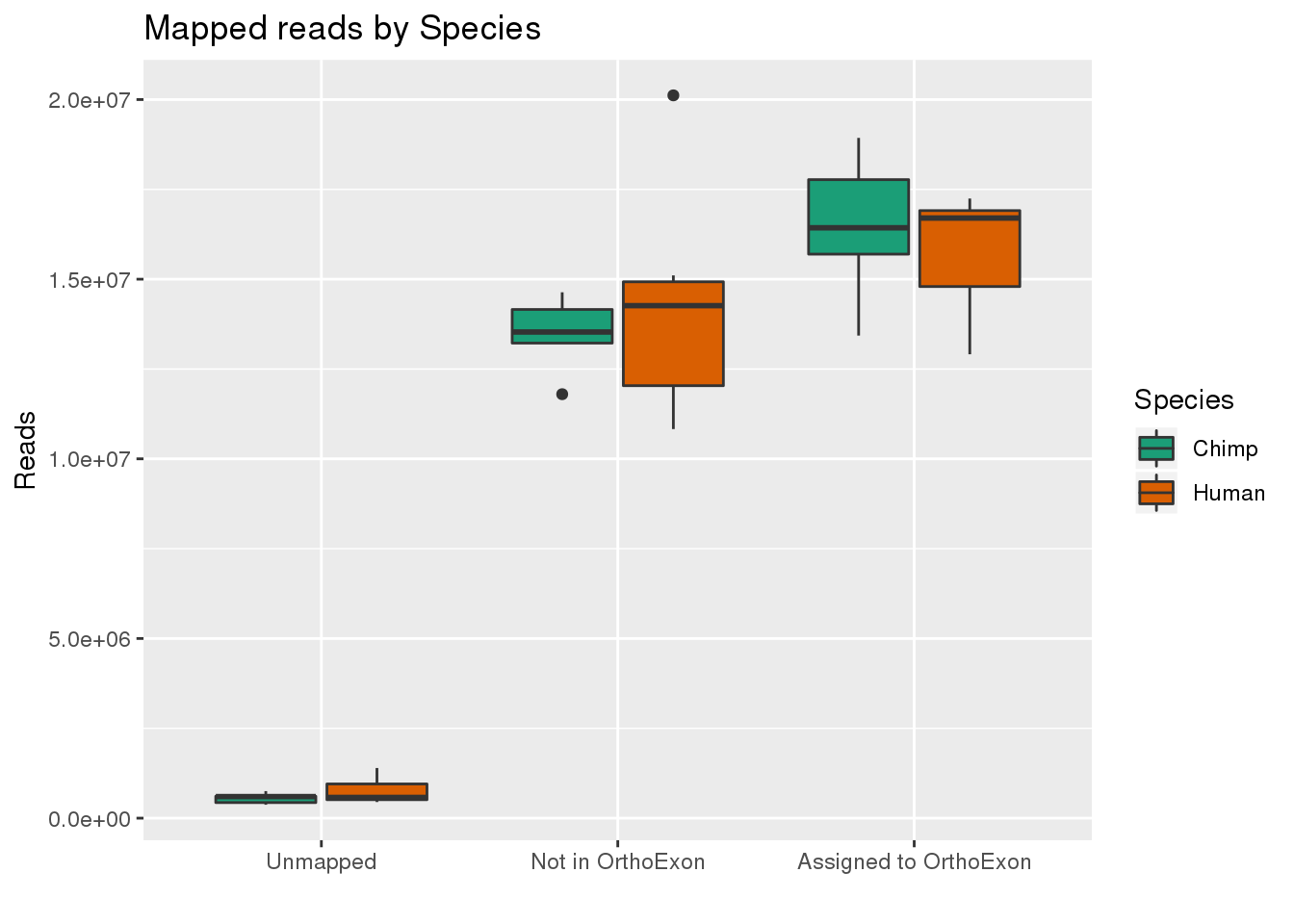
ggplot(readProp,aes(x=Category, y=Proportion, by=Species, fill=Species)) + geom_boxplot() + scale_fill_brewer(palette = "Dark2") + labs(title="Map Proportion by Species", y="Proportion", x="") + scale_x_discrete( breaks=c("Aunmapped","MappedNotOrtho","percentOrtho"),labels=c("Unmapped", "Not in OrthoExon", "Assigned to OrthoExon"))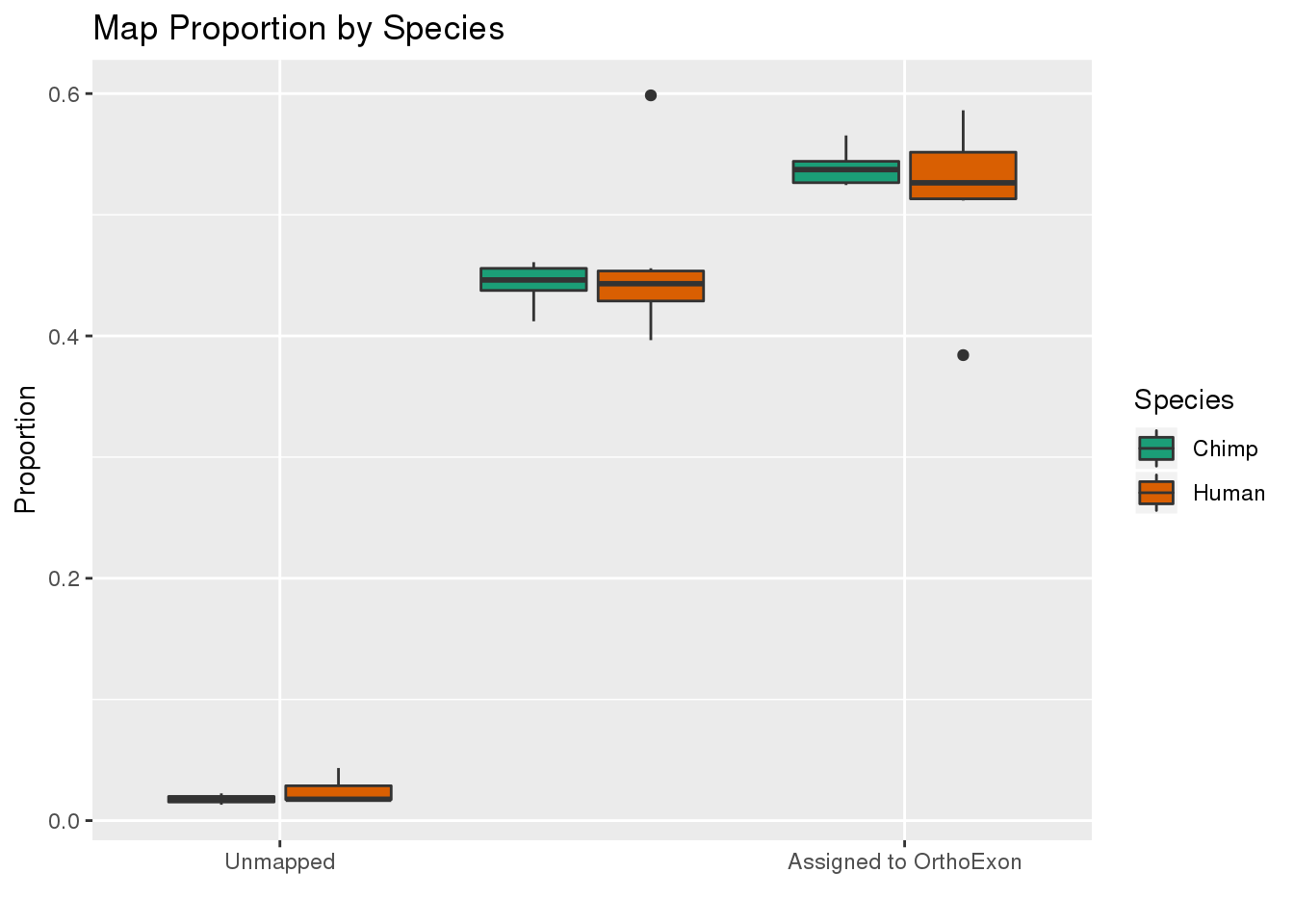
Diffferential Expression
Code originally from Lauren Blake (http://lauren-blake.github.io/Reg_Evo_Primates/analysis/Normalization_plots.html)
Raw Counts
Fix header for fc files:
python fixExonFC.py /project2/gilad/briana/Comparative_APA/Human/data/RNAseq/ExonCounts/RNAseqOrthoExon.fc /project2/gilad/briana/Comparative_APA/Human/data/RNAseq/ExonCounts/RNAseqOrthoExon.fixed.fc
python fixExonFC.py /project2/gilad/briana/Comparative_APA/Chimp/data/RNAseq/ExonCounts/RNAseqOrthoExon.fc /project2/gilad/briana/Comparative_APA/Chimp/data/RNAseq/ExonCounts/RNAseqOrthoExon.fixed.fcHumanCounts=read.table("../Human/data/RNAseq/ExonCounts/RNAseqOrthoExon.fixed.fc", header = T, stringsAsFactors = F) %>% select(-Chr,-Start,-End,-Strand, -Length)
ChimpCounts=read.table("../Chimp/data/RNAseq/ExonCounts/RNAseqOrthoExon.fixed.fc", header = T, stringsAsFactors = F) %>% select(-Chr,-Start,-End,-Strand, -Length)
counts_genes=HumanCounts %>% inner_join(ChimpCounts,by="Geneid") %>% column_to_rownames(var="Geneid")
head(counts_genes) NA18498 NA18504 NA18510 NA18523 NA18499 NA18502 NA4973
ENSG00000188976 48 24 50 31 34 58 2
ENSG00000188157 148 106 59 106 128 39 6
ENSG00000273443 69 40 43 54 48 11 24
ENSG00000217801 77 60 36 164 61 19 62
ENSG00000237330 1 0 1 0 1 1 0
ENSG00000223823 0 0 0 0 0 0 0
NAPT30 NAPT91 NA3622 NA3659 NA18358
ENSG00000188976 2 1 1 1 0
ENSG00000188157 5 7 7 8 6
ENSG00000273443 21 2 3 78 16
ENSG00000217801 34 8 19 139 31
ENSG00000237330 0 0 0 2 0
ENSG00000223823 0 0 0 0 0# Load colors
colors <- colorRampPalette(c(brewer.pal(9, "Blues")[1],brewer.pal(9, "Blues")[9]))(100)
pal <- c(brewer.pal(9, "Set1"), brewer.pal(8, "Set2"), brewer.pal(12, "Set3"))
labels <- paste(metaData$Species,metaData$Line, sep=" ")#PCA function (original code from Julien Roux)
#Load in the plot_scores function
plot_scores <- function(pca, scores, n, m, cols, points=F, pchs =20, legend=F){
xmin <- min(scores[,n]) - (max(scores[,n]) - min(scores[,n]))*0.05
if (legend == T){ ## let some room (35%) for a legend
xmax <- max(scores[,n]) + (max(scores[,n]) - min(scores[,n]))*0.50
}
else {
xmax <- max(scores[,n]) + (max(scores[,n]) - min(scores[,n]))*0.05
}
ymin <- min(scores[,m]) - (max(scores[,m]) - min(scores[,m]))*0.05
ymax <- max(scores[,m]) + (max(scores[,m]) - min(scores[,m]))*0.05
plot(scores[,n], scores[,m], xlab=paste("PC", n, ": ", round(summary(pca)$importance[2,n],3)*100, "% variance explained", sep=""), ylab=paste("PC", m, ": ", round(summary(pca)$importance[2,m],3)*100, "% variance explained", sep=""), xlim=c(xmin, xmax), ylim=c(ymin, ymax), type="n")
if (points == F){
text(scores[,n],scores[,m], rownames(scores), col=cols, cex=1)
}
else {
points(scores[,n],scores[,m], col=cols, pch=pchs, cex=1.3)
}
}# Clustering (original code from Julien Roux)
cors <- cor(counts_genes, method="spearman", use="pairwise.complete.obs")
heatmap.2( cors, scale="none", col = colors, margins = c(12, 12), trace='none', denscol="white", labCol=labels, ColSideColors=pal[as.integer(as.factor(metaData$Species))], RowSideColors=pal[as.integer(as.factor(metaData$Collection))+9], cexCol = 0.2 + 1/log10(15), cexRow = 0.2 + 1/log10(15))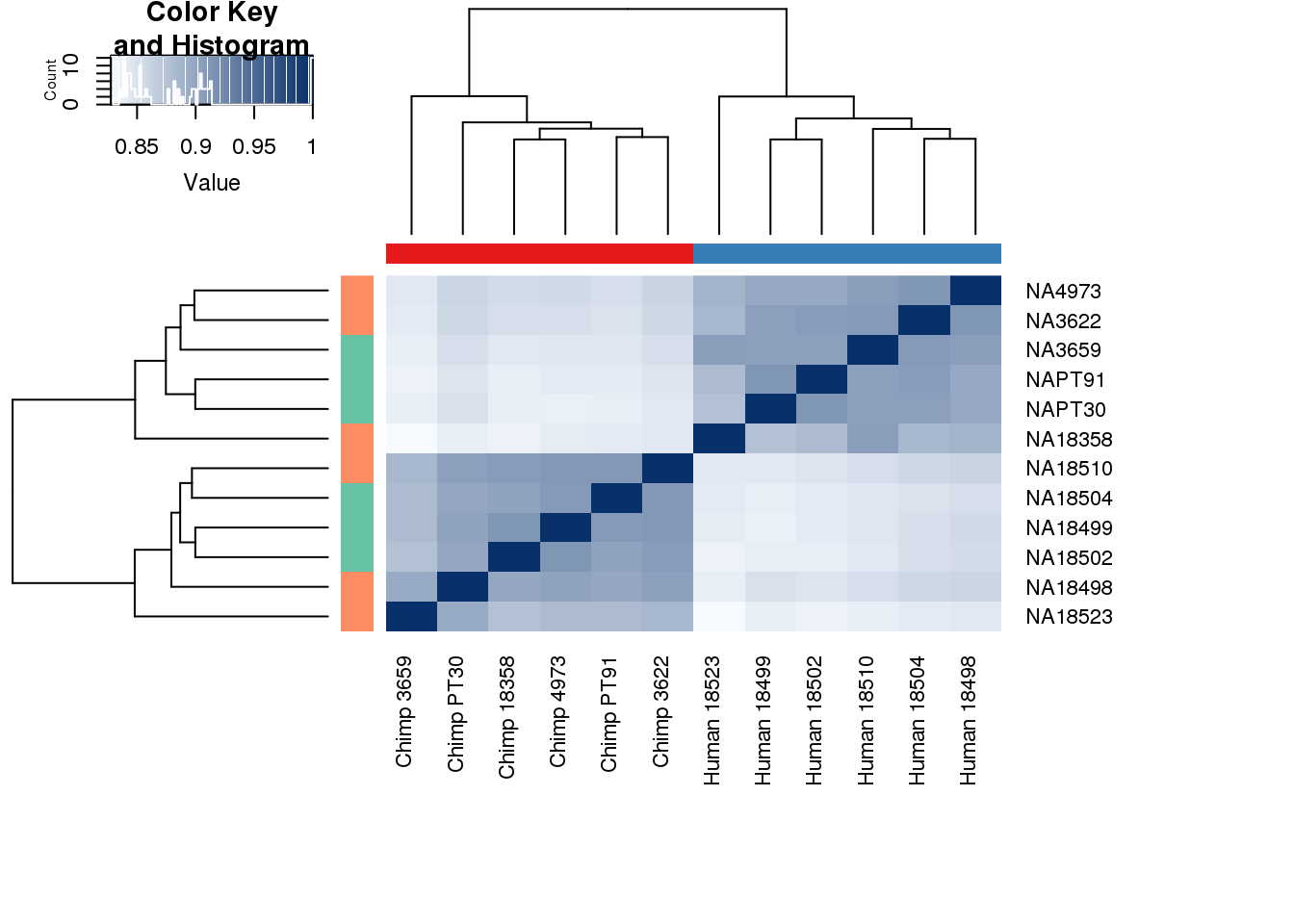
select <- counts_genes
summary(apply(select, 1, var) == 0) Mode FALSE TRUE
logical 32592 11533 # Perform PCA
pca_genes <- prcomp(t(counts_genes), scale = F)
scores <- pca_genes$x
#Make PCA plots with the factors colored by species
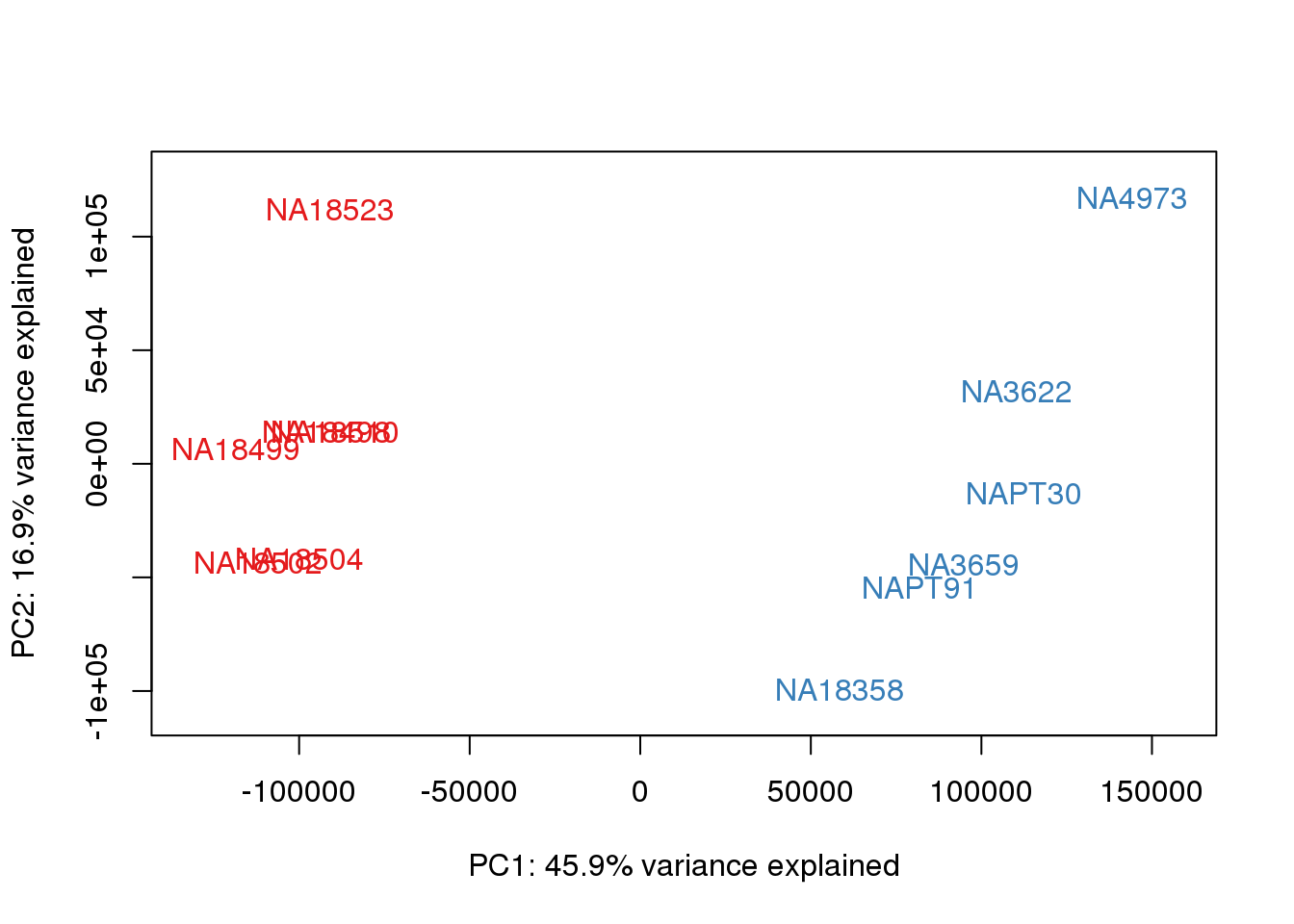
### PCs 1 and 2 Raw Data
for (n in 1:1){
col.v <- pal[as.integer(metaData$Species)]
plot_scores(pca_genes, scores, n, n+1, col.v)
}
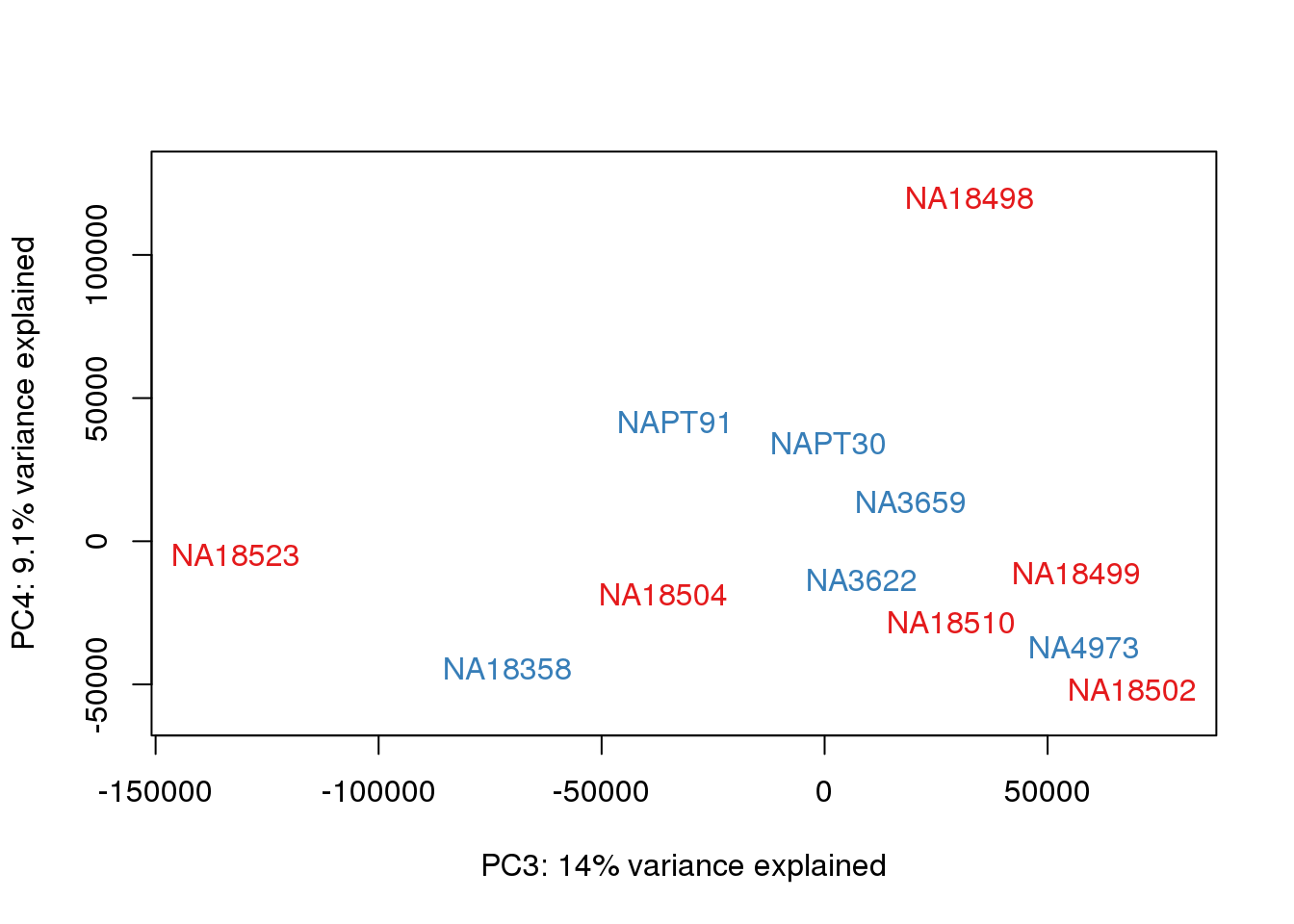
### PCs 3 and 4 Raw Data
for (n in 3:3){
col.v <- pal[as.integer(metaData$Species)]
plot_scores(pca_genes, scores, n, n+1, col.v)
}
Plot density for raw data:
density_plot_18504 <- ggplot(counts_genes, aes(x = NA18504)) + geom_density() + labs(title = "Density plot of raw gene counts of NA18504") + labs(x = "Raw counts for each gene")
density_plot_18504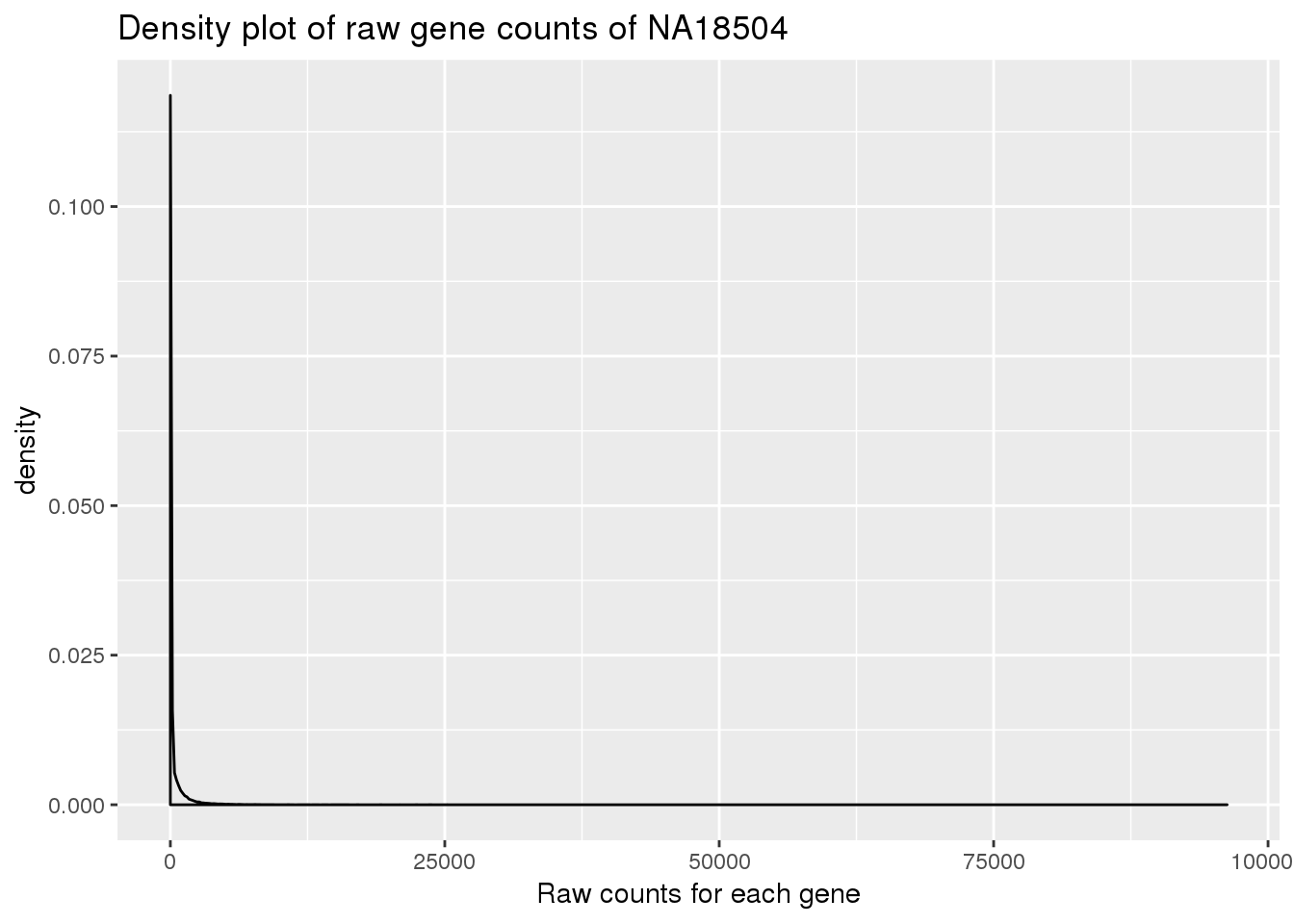
Convert to log2
log_counts_genes <- as.data.frame(log2(counts_genes))
head(log_counts_genes) NA18498 NA18504 NA18510 NA18523 NA18499 NA18502
ENSG00000188976 5.584963 4.584963 5.643856 4.954196 5.087463 5.857981
ENSG00000188157 7.209453 6.727920 5.882643 6.727920 7.000000 5.285402
ENSG00000273443 6.108524 5.321928 5.426265 5.754888 5.584963 3.459432
ENSG00000217801 6.266787 5.906891 5.169925 7.357552 5.930737 4.247928
ENSG00000237330 0.000000 -Inf 0.000000 -Inf 0.000000 0.000000
ENSG00000223823 -Inf -Inf -Inf -Inf -Inf -Inf
NA4973 NAPT30 NAPT91 NA3622 NA3659 NA18358
ENSG00000188976 1.000000 1.000000 0.000000 0.000000 0.000000 -Inf
ENSG00000188157 2.584963 2.321928 2.807355 2.807355 3.000000 2.584963
ENSG00000273443 4.584963 4.392317 1.000000 1.584963 6.285402 4.000000
ENSG00000217801 5.954196 5.087463 3.000000 4.247928 7.118941 4.954196
ENSG00000237330 -Inf -Inf -Inf -Inf 1.000000 -Inf
ENSG00000223823 -Inf -Inf -Inf -Inf -Inf -Infdensity_plot_18504 <- ggplot(log_counts_genes, aes(x = 18504)) + geom_density()
density_plot_18504 + labs(title = "Density plot of log2 counts of 18504") + labs(x = "Log2 counts for each gene") + geom_vline(xintercept = 1) 
| Version | Author | Date |
|---|---|---|
| 32b435b | brimittleman | 2019-11-12 |
plotDensities(log_counts_genes, col=pal[as.numeric(metaData$Species)], legend="topright")
Convert to CPM
cpm <- cpm(counts_genes, log=TRUE)
head(cpm) NA18498 NA18504 NA18510 NA18523 NA18499
ENSG00000188976 1.588820 0.9875278 1.619299 1.208070 1.067263
ENSG00000188157 3.172567 3.0589636 1.849178 2.925953 2.915718
ENSG00000273443 2.094172 1.6879541 1.411153 1.975405 1.539652
ENSG00000217801 2.248076 2.2542940 1.167830 3.547237 1.872256
ENSG00000237330 -2.441803 -3.0119508 -2.450600 -3.011951 -2.458357
ENSG00000223823 -3.011951 -3.0119508 -3.011951 -3.011951 -3.011951
NA18502 NA4973 NAPT30 NAPT91 NA3622
ENSG00000188976 1.8417218 -2.1227985 -2.0245732 -2.4061117 -2.4360702
ENSG00000188157 1.2932493 -1.1815889 -1.2226619 -0.7937756 -0.8633065
ENSG00000273443 -0.3579863 0.4766993 0.4884775 -1.9807482 -1.7064115
ENSG00000217801 0.3306162 1.7649277 1.1340383 -0.6404174 0.3555815
ENSG00000237330 -2.4451184 -3.0119508 -3.0119508 -3.0119508 -3.0119508
ENSG00000223823 -3.0119508 -3.0119508 -3.0119508 -3.0119508 -3.0119508
NA3659 NA18358
ENSG00000188976 -2.483011 -3.0119508
ENSG00000188157 -0.828322 -0.8091641
ENSG00000273443 2.139573 0.3953756
ENSG00000217801 2.955188 1.2822287
ENSG00000237330 -2.096822 -3.0119508
ENSG00000223823 -3.011951 -3.0119508plotDensities(cpm, col=pal[as.numeric(metaData$Species)], legend="topright")
Log2 CPM
TMM/log2(CPM)
## Create edgeR object (dge) to calculate TMM normalization
dge_original <- DGEList(counts=as.matrix(counts_genes), genes=rownames(counts_genes), group = as.character(t(labels)))
dge_original <- calcNormFactors(dge_original)
tmm_cpm <- cpm(dge_original, normalized.lib.sizes=TRUE, log=TRUE, prior.count = 0.25)
head(cpm) NA18498 NA18504 NA18510 NA18523 NA18499
ENSG00000188976 1.588820 0.9875278 1.619299 1.208070 1.067263
ENSG00000188157 3.172567 3.0589636 1.849178 2.925953 2.915718
ENSG00000273443 2.094172 1.6879541 1.411153 1.975405 1.539652
ENSG00000217801 2.248076 2.2542940 1.167830 3.547237 1.872256
ENSG00000237330 -2.441803 -3.0119508 -2.450600 -3.011951 -2.458357
ENSG00000223823 -3.011951 -3.0119508 -3.011951 -3.011951 -3.011951
NA18502 NA4973 NAPT30 NAPT91 NA3622
ENSG00000188976 1.8417218 -2.1227985 -2.0245732 -2.4061117 -2.4360702
ENSG00000188157 1.2932493 -1.1815889 -1.2226619 -0.7937756 -0.8633065
ENSG00000273443 -0.3579863 0.4766993 0.4884775 -1.9807482 -1.7064115
ENSG00000217801 0.3306162 1.7649277 1.1340383 -0.6404174 0.3555815
ENSG00000237330 -2.4451184 -3.0119508 -3.0119508 -3.0119508 -3.0119508
ENSG00000223823 -3.0119508 -3.0119508 -3.0119508 -3.0119508 -3.0119508
NA3659 NA18358
ENSG00000188976 -2.483011 -3.0119508
ENSG00000188157 -0.828322 -0.8091641
ENSG00000273443 2.139573 0.3953756
ENSG00000217801 2.955188 1.2822287
ENSG00000237330 -2.096822 -3.0119508
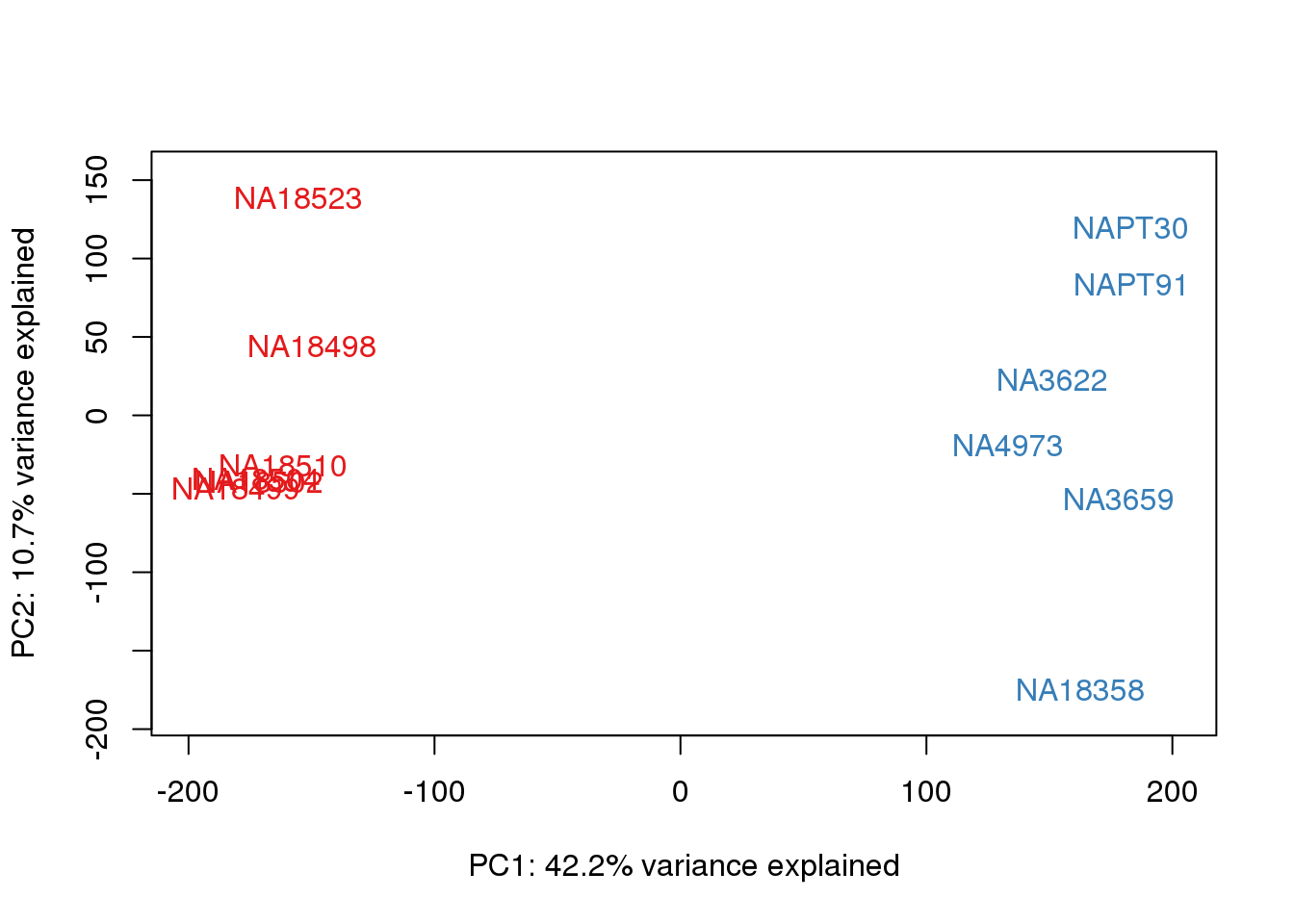
ENSG00000223823 -3.011951 -3.0119508pca_genes <- prcomp(t(tmm_cpm), scale = F)
scores <- pca_genes$x
for (n in 1:2){
col.v <- pal[as.integer(metaData$Species)]
plot_scores(pca_genes, scores, n, n+1, col.v)
}
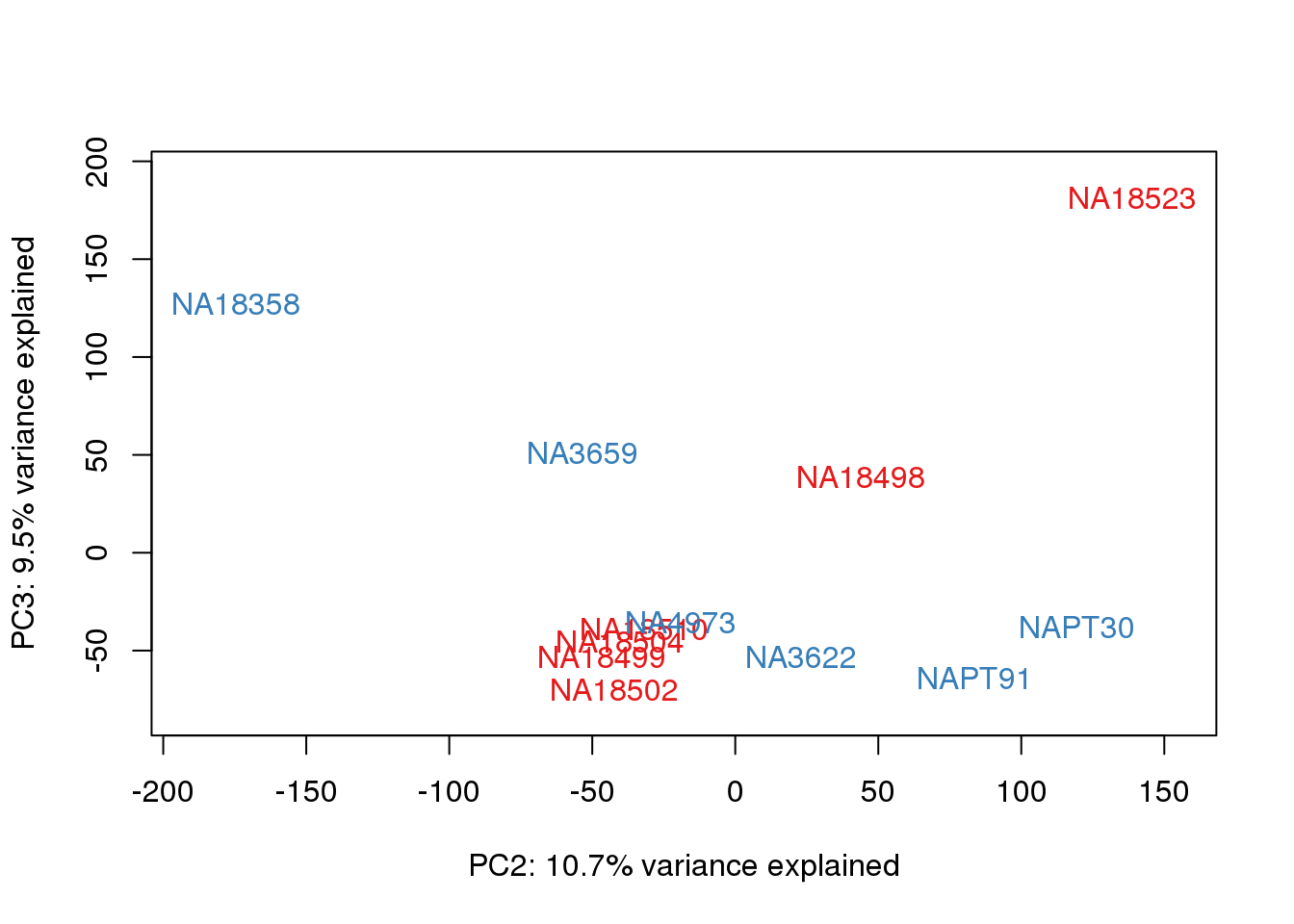
# Plot library size
boxplot_library_size <- ggplot(dge_original$samples, aes(x=metaData$Species, y = dge_original$samples$lib.size, fill = metaData$Species)) + geom_boxplot()
boxplot_library_size + labs(title = "Library size by Species") + labs(y = "Library size") + labs(x = "Species") + guides(fill=guide_legend(title="Species"))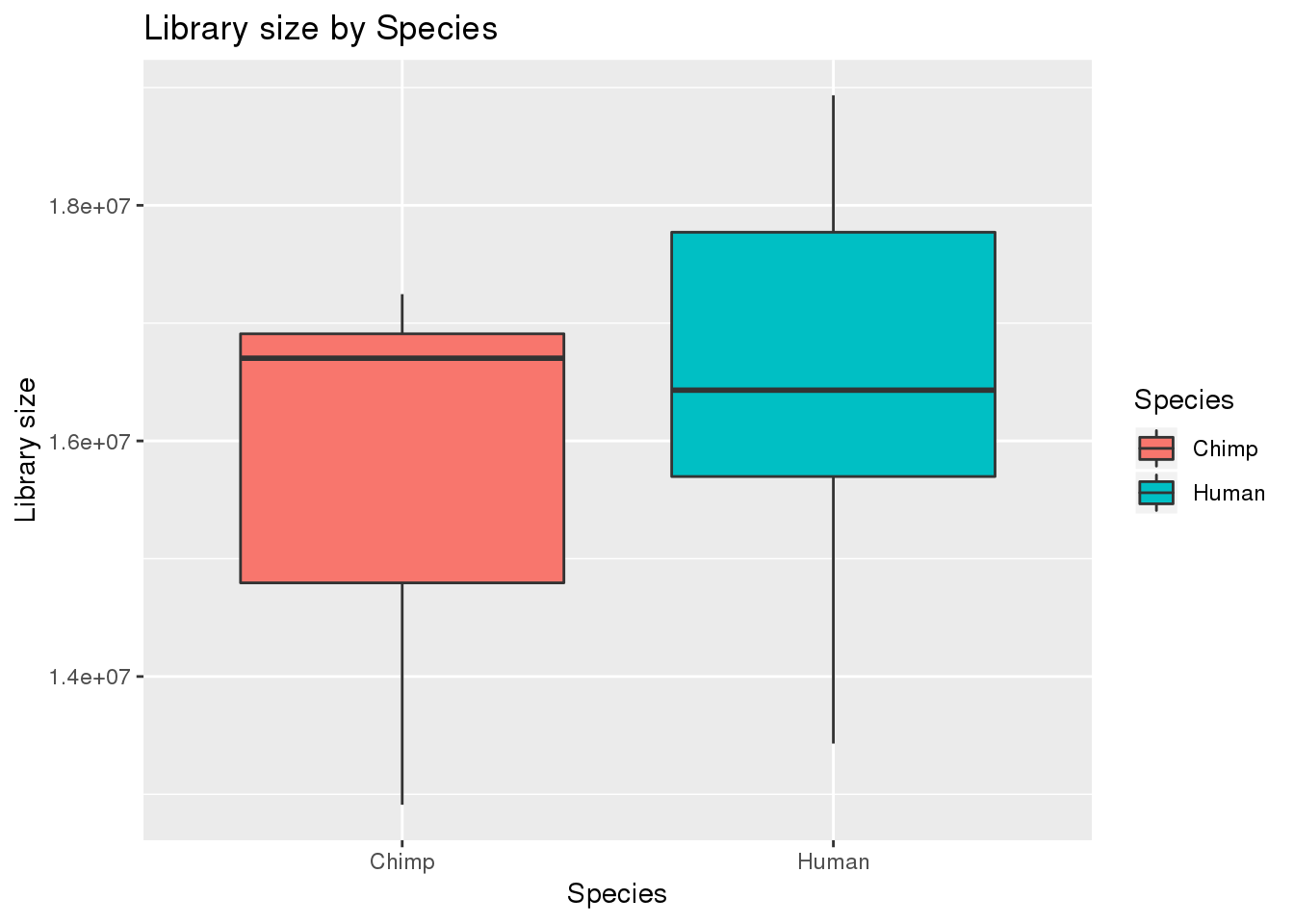
plotDensities(tmm_cpm, col=pal[as.numeric(metaData$Species)], legend="topright")
Filter low expressed gene
Filter based on log2 cpm
filter log2(cpm >1) in at least 10 of the samples (2/3)
#filter counts
keep.exprs=rowSums(tmm_cpm>1) >8
counts_filtered= counts_genes[keep.exprs,]
plotDensities(counts_filtered, col=pal[as.numeric(metaData$Species)], legend="topright")
labels <- paste(metaData$Species, metaData$Line, sep=" ")
dge_in_cutoff <- DGEList(counts=as.matrix(counts_filtered), genes=rownames(counts_filtered), group = as.character(t(labels)))
dge_in_cutoff <- calcNormFactors(dge_in_cutoff)
cpm_in_cutoff <- cpm(dge_in_cutoff, normalized.lib.sizes=TRUE, log=TRUE, prior.count = 0.25)
head(cpm_in_cutoff) NA18498 NA18504 NA18510 NA18523 NA18499 NA18502
ENSG00000217801 2.256506 2.234115 1.0781627 3.660723 1.851408 0.2044122
ENSG00000186891 6.449667 5.009505 5.0323421 4.887082 5.704206 3.3563261
ENSG00000186827 5.245211 3.093190 4.6496753 1.700807 3.965561 1.8966473
ENSG00000078808 7.201153 6.859702 7.0260235 7.463228 6.617502 6.7324642
ENSG00000176022 4.796263 4.753597 4.6761916 4.790014 4.605178 4.6953254
ENSG00000184163 1.692492 1.418453 -0.1695685 1.630775 1.054722 0.3468831
NA4973 NAPT30 NAPT91 NA3622 NA3659 NA18358
ENSG00000217801 1.778862 1.1607241 -0.791047 0.2757475 2.998563 1.300872
ENSG00000186891 6.669085 4.6336365 3.159521 4.7270793 6.447786 6.482146
ENSG00000186827 1.778862 0.2523816 2.006912 3.1506928 0.646502 2.175264
ENSG00000078808 7.024471 6.6878664 6.765572 6.7760951 6.783817 7.061063
ENSG00000176022 5.263508 4.8384442 5.550276 4.8453842 5.276059 5.561721
ENSG00000184163 1.381411 2.8988501 2.195679 1.4096039 1.766196 3.222359hist(cpm_in_cutoff, xlab = "Log2(CPM)", main = "Log2(CPM) values for genes meeting the filtering criteria", breaks = 100 )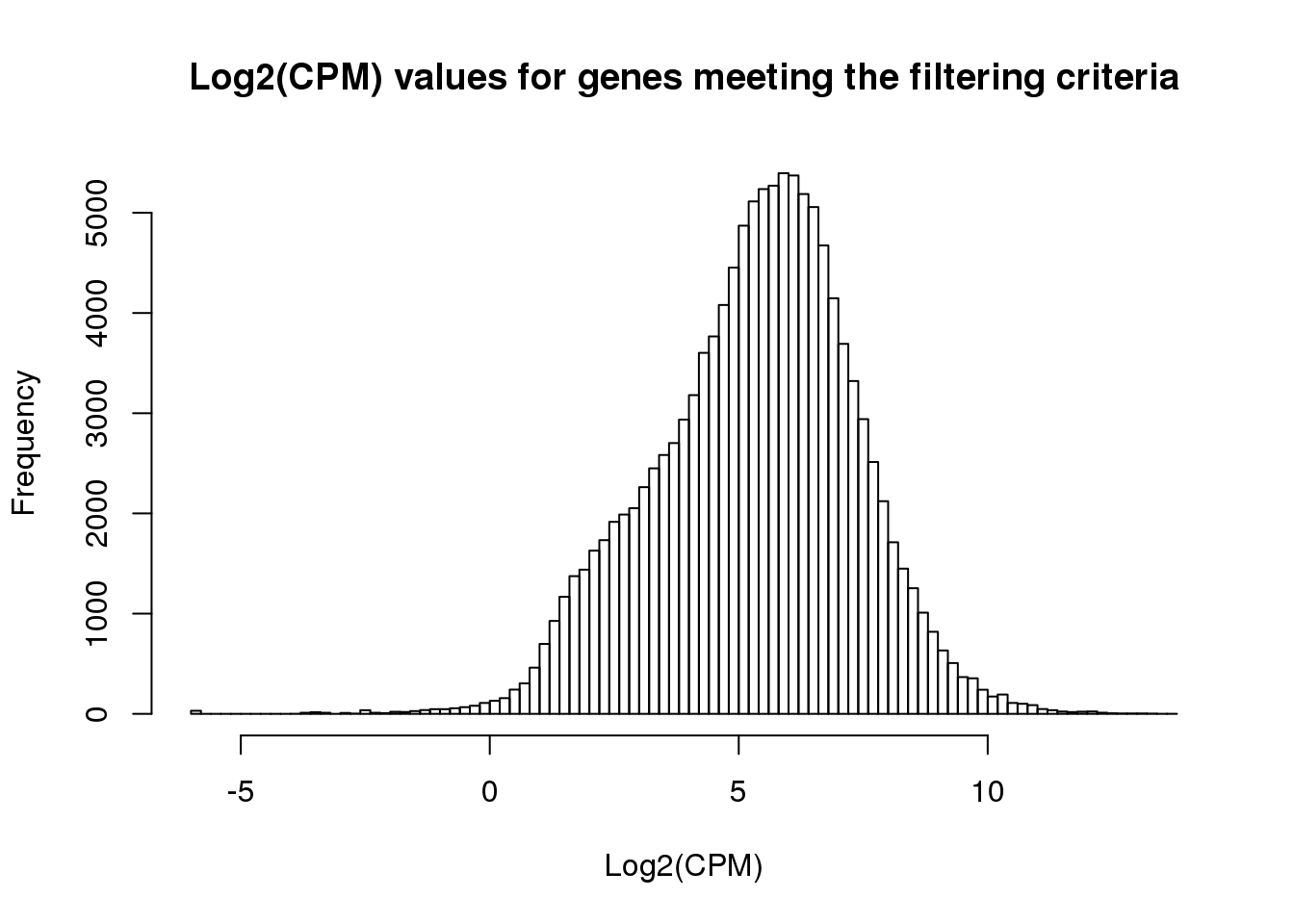
Voom transformation:
Species <- factor(metaData$Species)
design <- model.matrix(~ 0 + Species)
head(design) SpeciesChimp SpeciesHuman
1 1 0
2 1 0
3 1 0
4 1 0
5 1 0
6 1 0colnames(design) <- gsub("Species", "", dput(colnames(design)))c("SpeciesChimp", "SpeciesHuman")Voom creates a random effect.
# Voom with individual as a random variable
cpm.voom<- voom(counts_filtered, design, normalize.method="quantile", plot=T)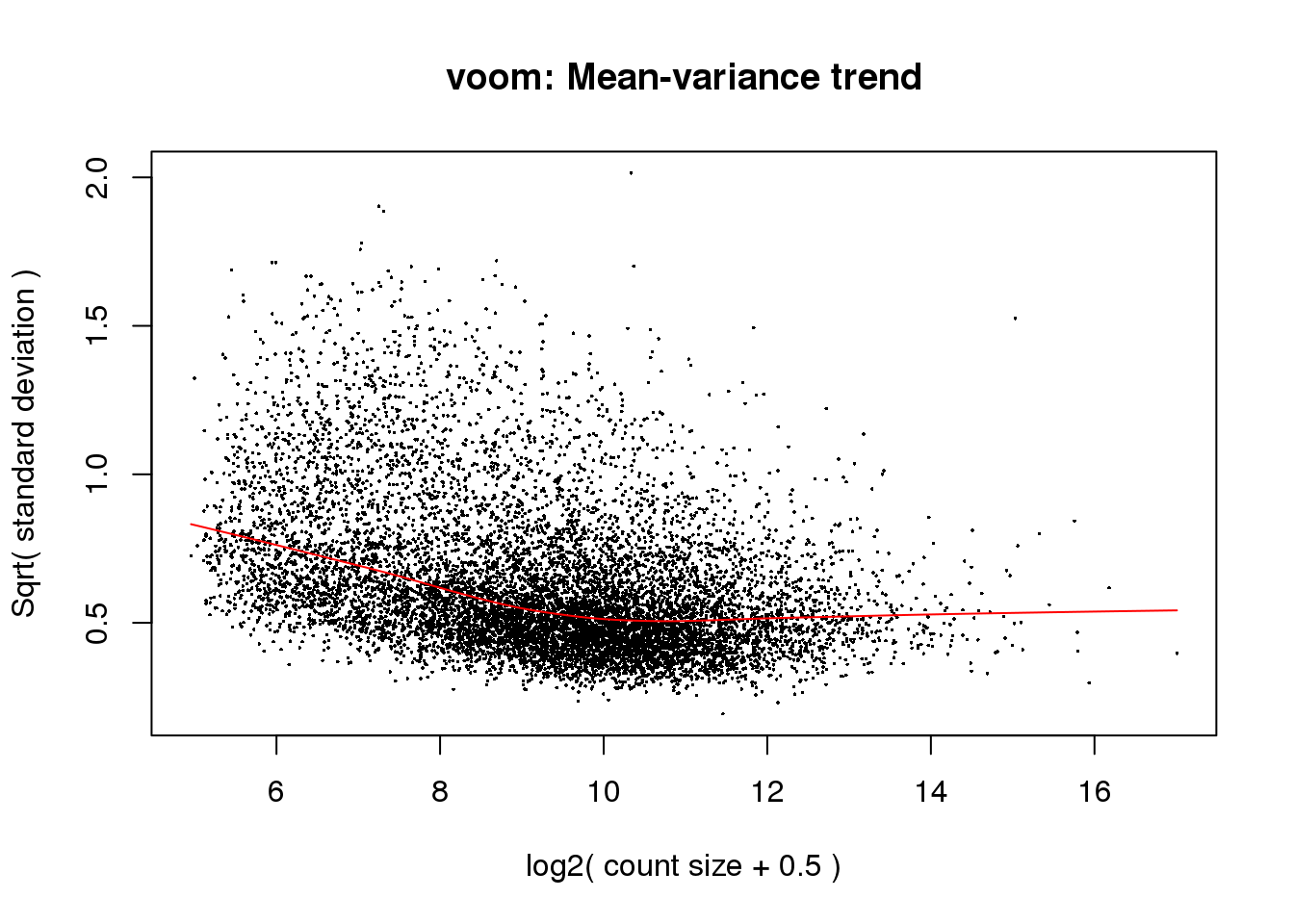
boxplot(cpm.voom$E, col = pal[as.numeric(metaData$Species)],las=2)
plotDensities(cpm.voom, col = pal[as.numeric(metaData$Species)], legend = "topleft") 
Looks like i still have a skew on the lower side of the distribution.
# PCA
pca_genes <- prcomp(t(cpm.voom$E), scale = T)
scores <- pca_genes$x
eigsGene <- pca_genes$sdev^2
proportionG = eigsGene/sum(eigsGene)
plot(proportionG)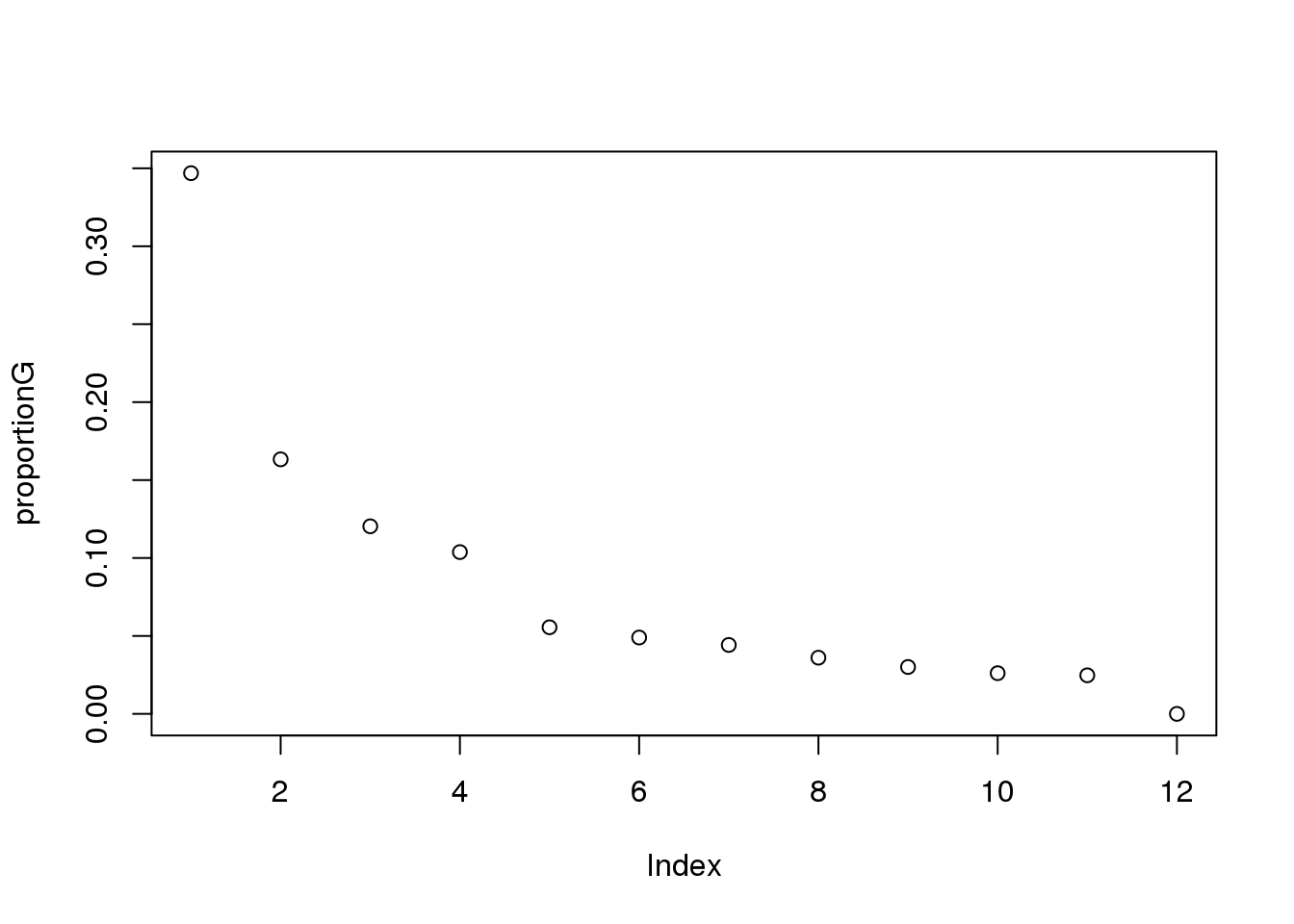
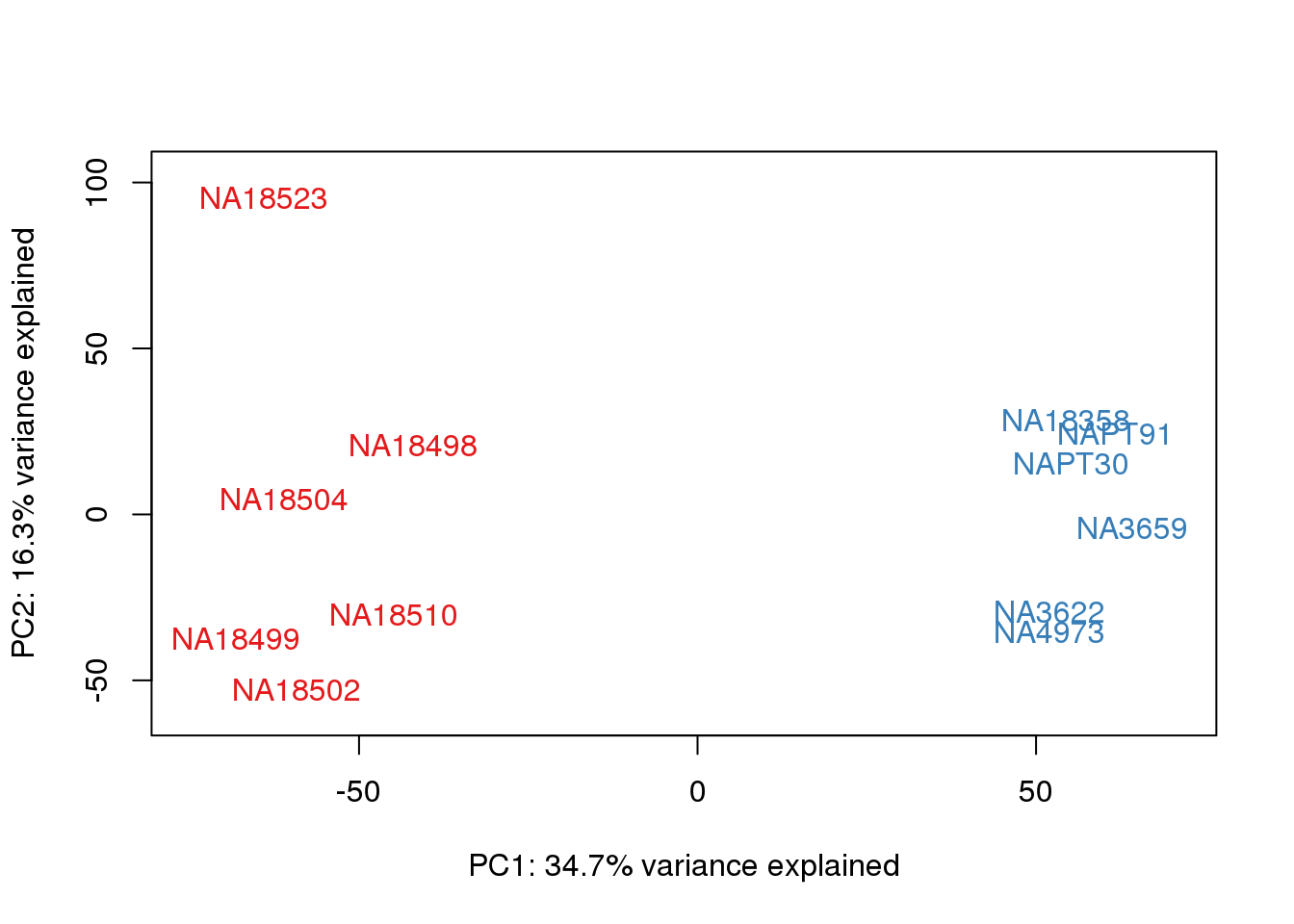
for (n in 1:2){
col.v <- pal[as.integer(metaData$Species)]
plot_scores(pca_genes, scores, n, n+1, col.v)
}
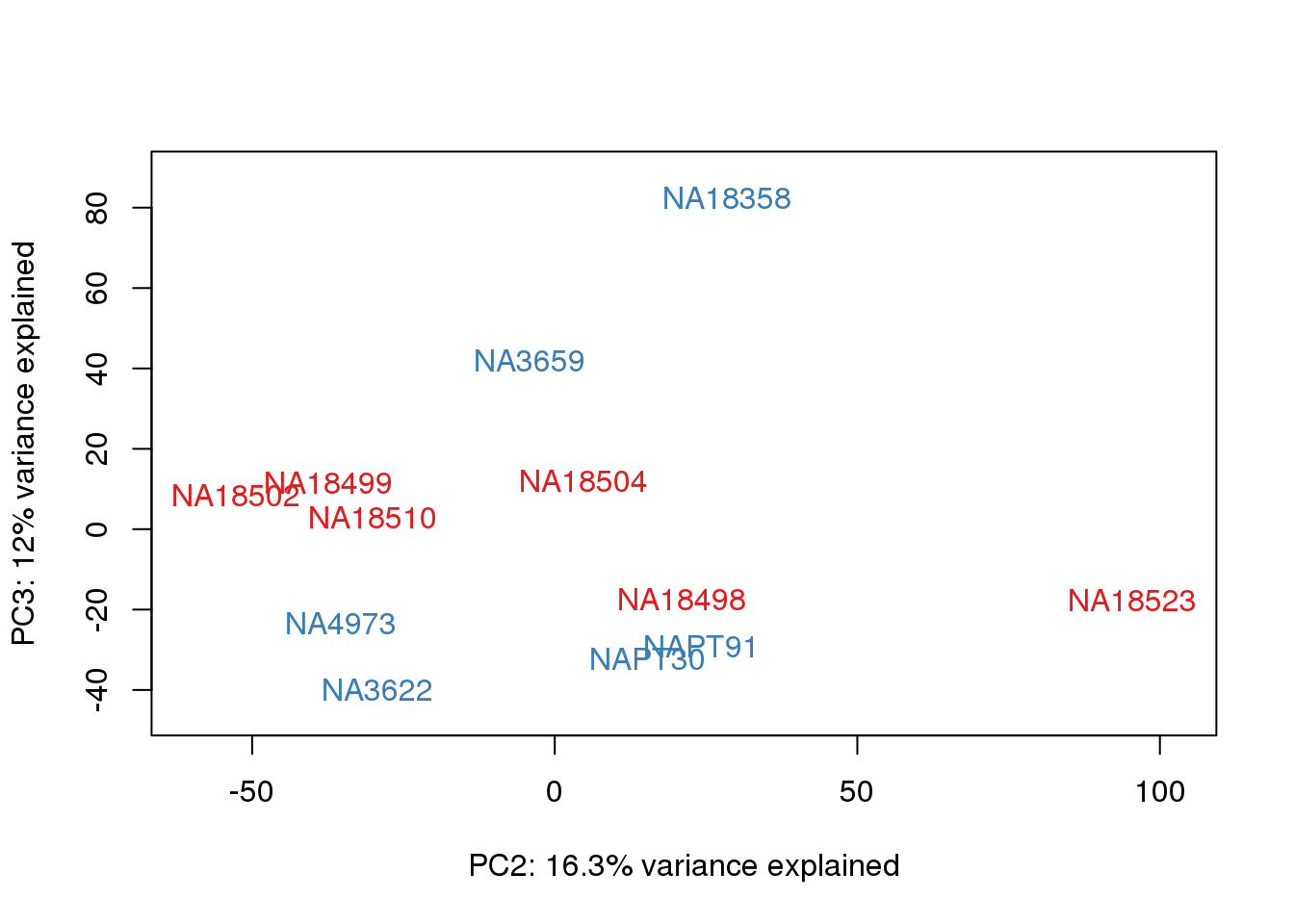
#Clustering (original code from Julien Roux)
cors <- cor(cpm.voom$E, method="spearman", use="pairwise.complete.obs")
heatmap.2( cors, scale="none", col = colors, margins = c(12, 12), trace='none', denscol="white", labCol=labels, ColSideColors=pal[as.integer(as.factor(metaData$Species))], RowSideColors=pal[as.integer(as.factor(metaData$Species))+9], cexCol = 0.2 + 1/log10(15), cexRow = 0.2 + 1/log10(15))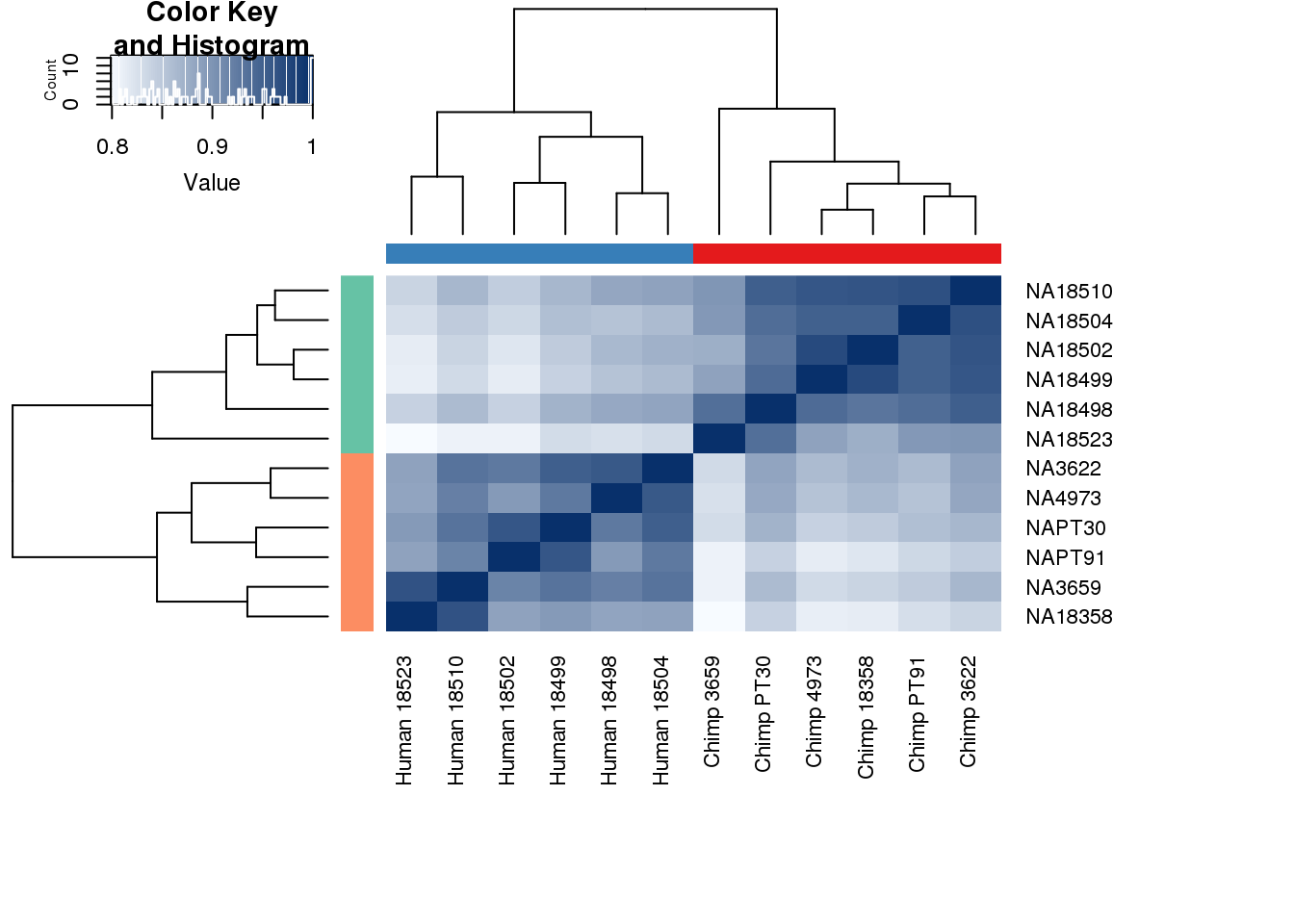
One thing I can do is look at the correlation between the PCs and other factors in the data.
# PCA
pca_genes <- prcomp(t(cpm.voom$E), scale = F)
scores <- pca_genes$x
for (n in 1:2){
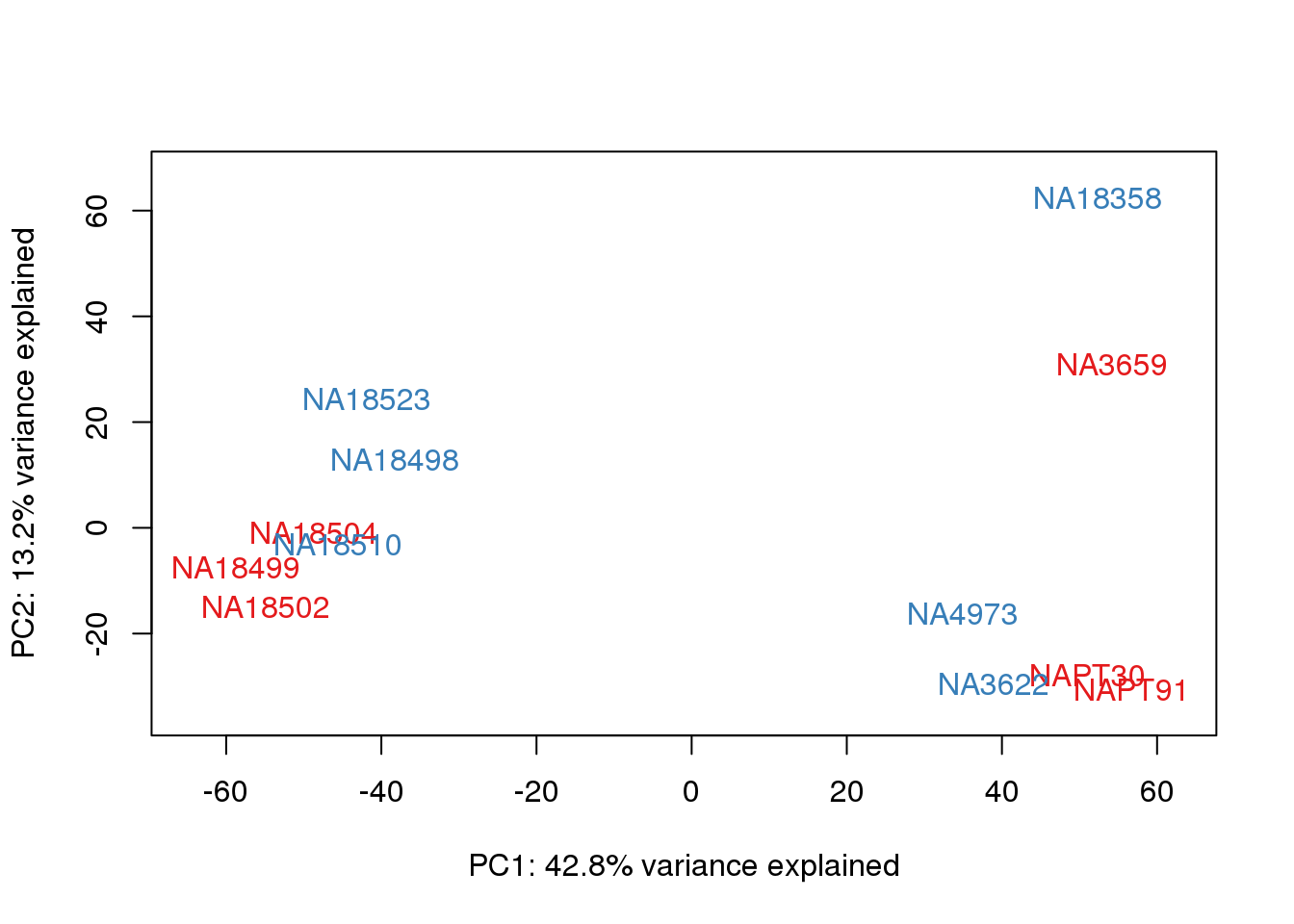
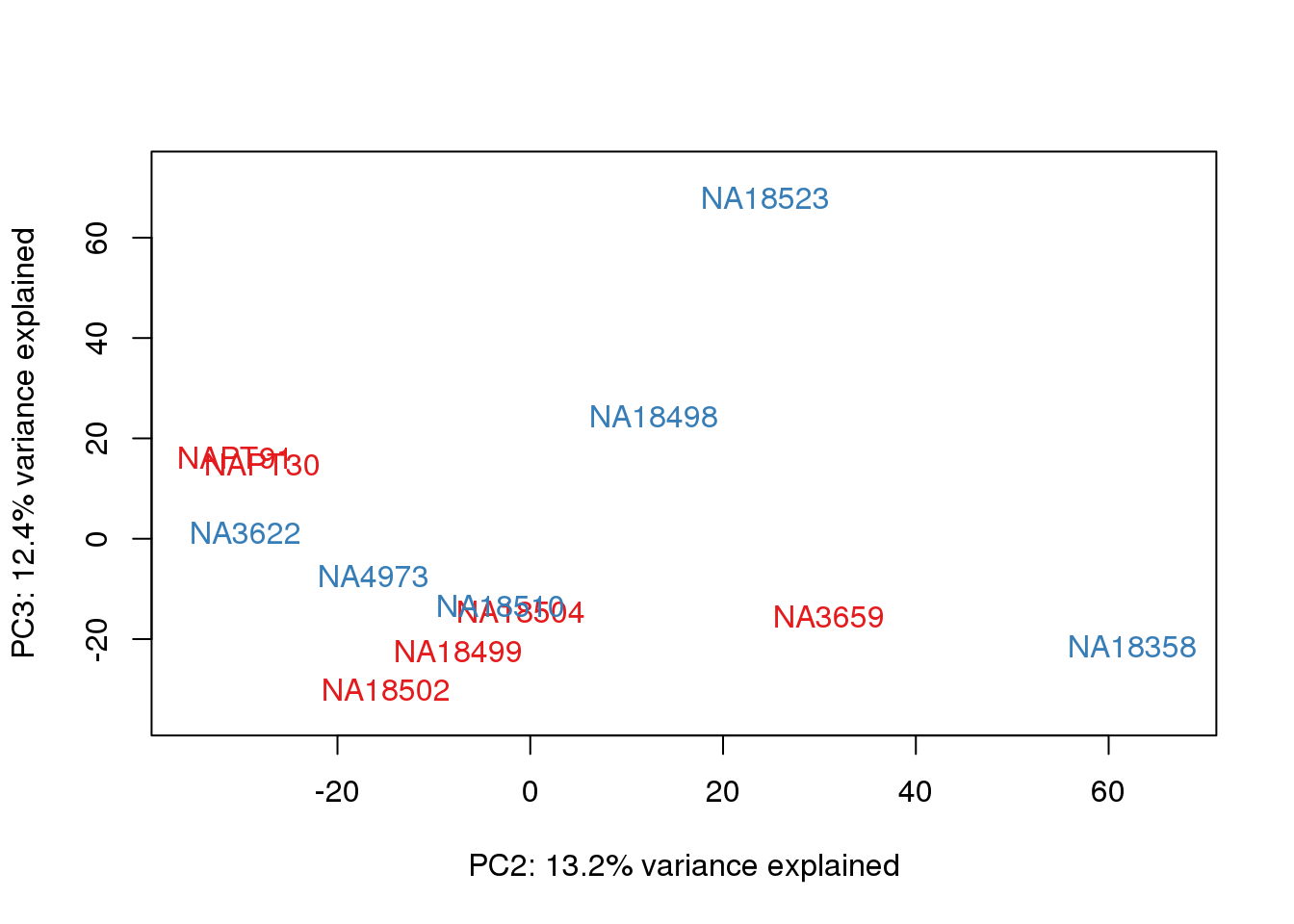
col.v <- pal[as.integer(metaData$Collection)]
plot_scores(pca_genes, scores, n, n+1, col.v)
}
metaData$Extraction=as.factor(metaData$Extraction)
for (n in 1:2){
col.v <- pal[as.integer(metaData$Extraction)]
plot_scores(pca_genes, scores, n, n+1, col.v)
}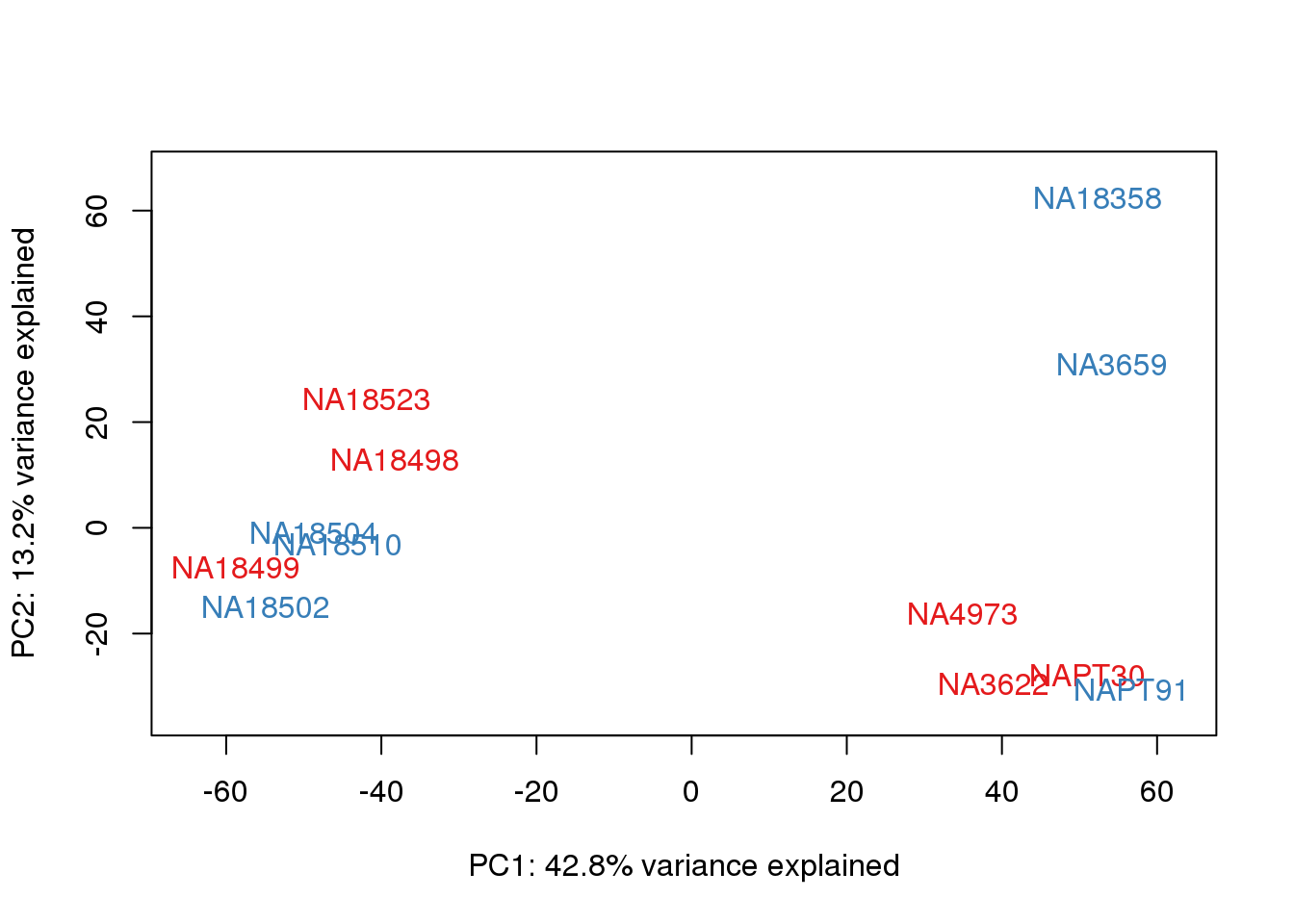
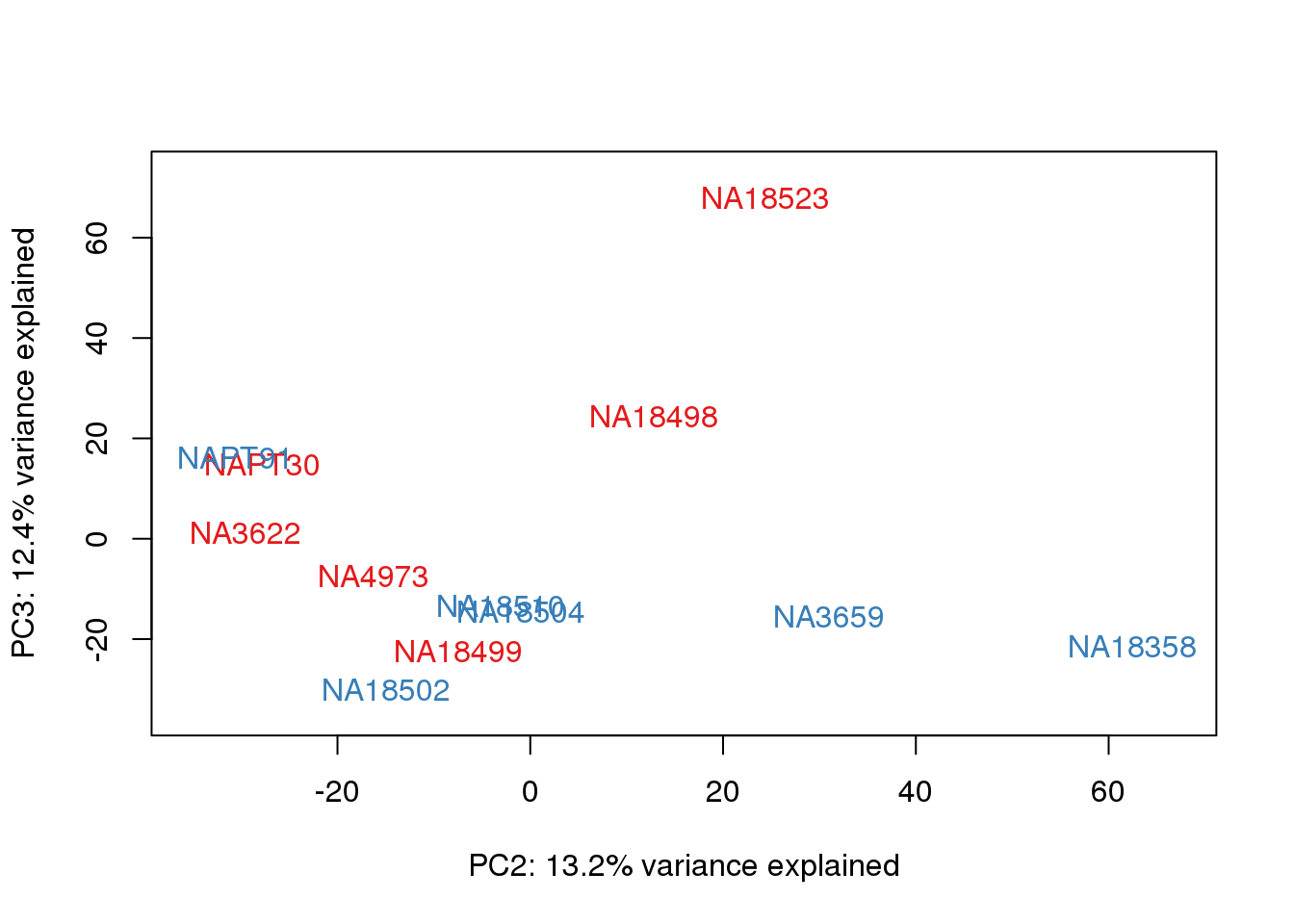
It does not look like batch (who collected or extraction date batch)
cols = brewer.pal(9, "Blues")
palC = colorRampPalette(cols)
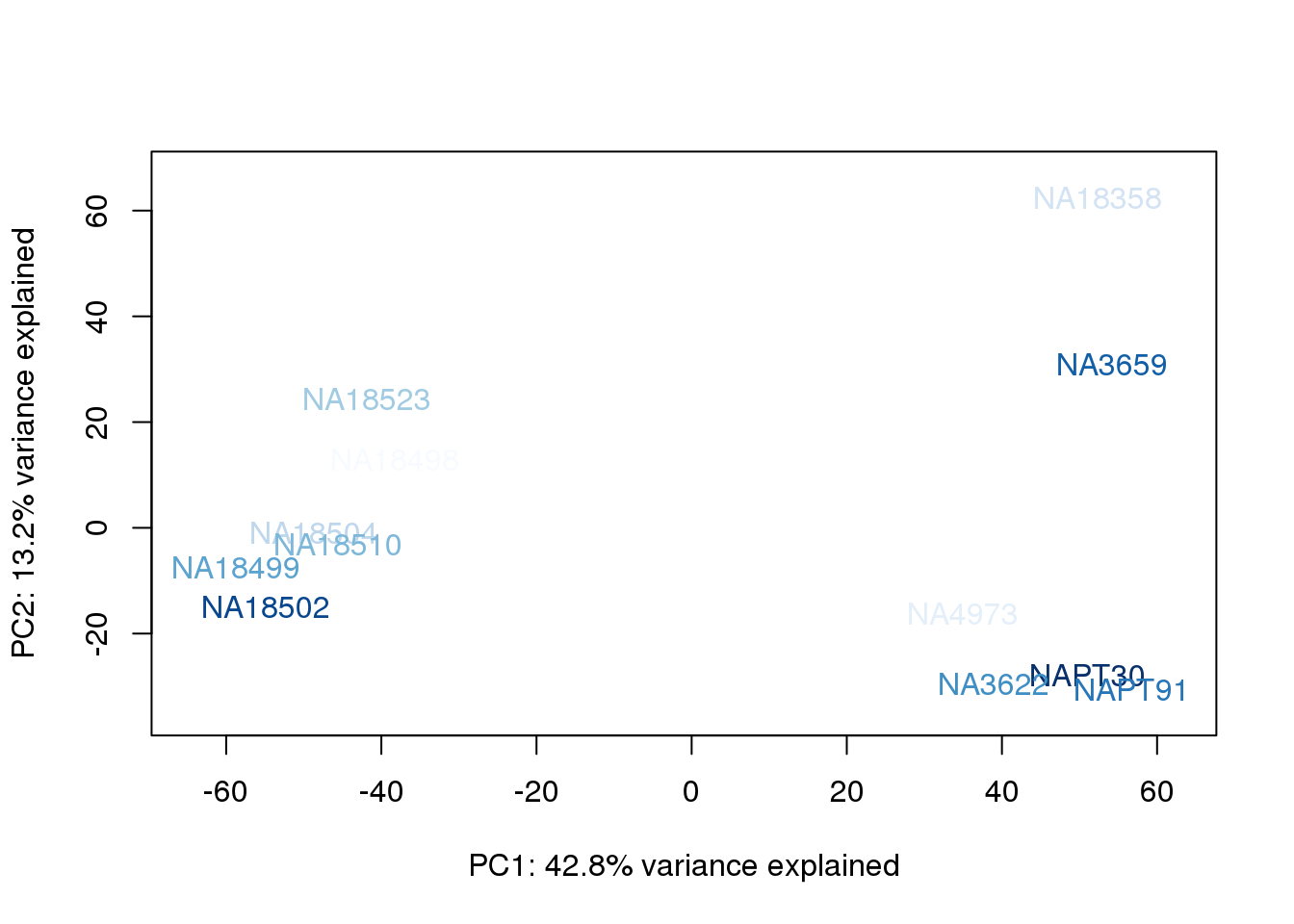
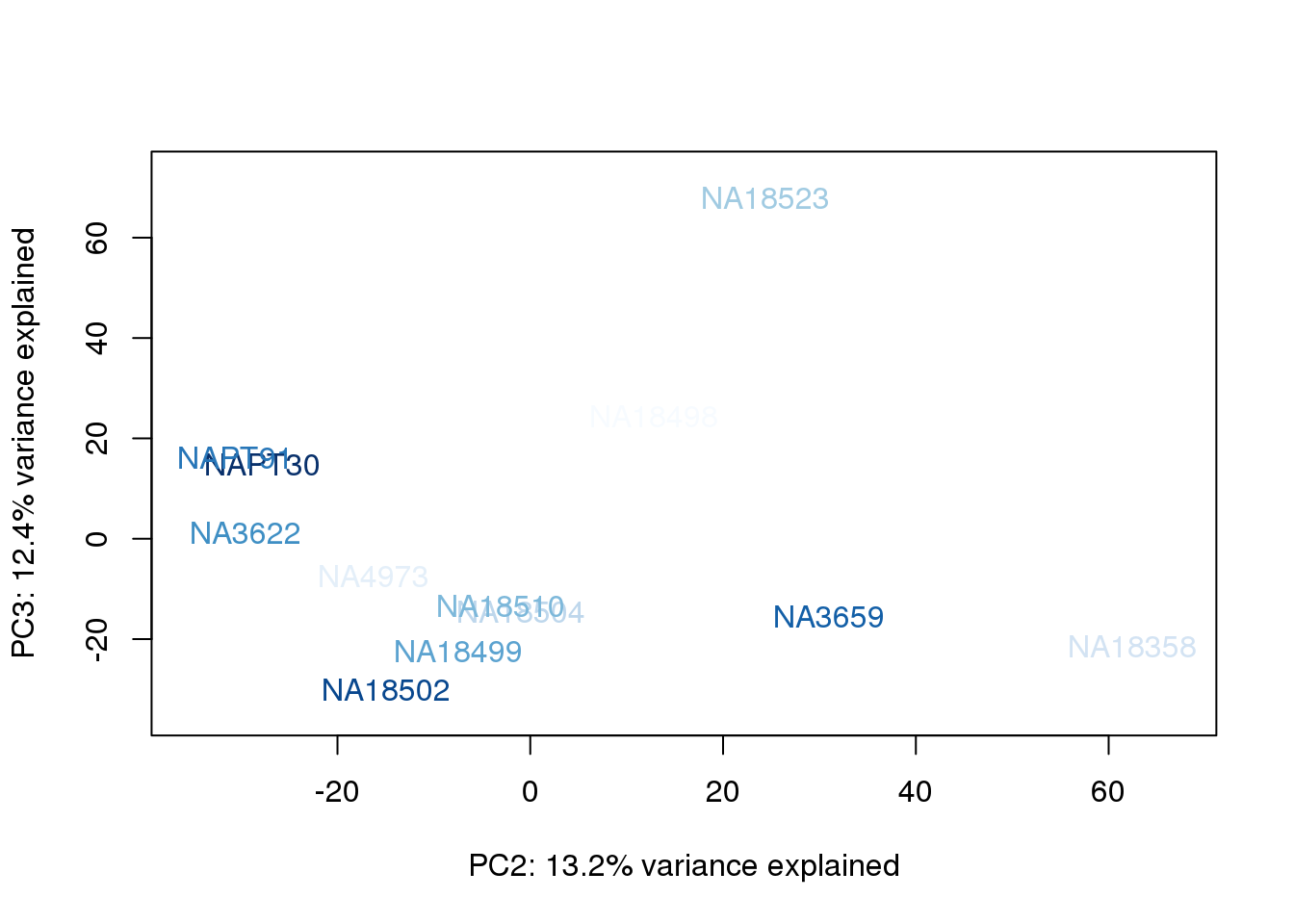
metaData$UndilutedAverageorder = findInterval(metaData$UndilutedAverage, sort(metaData$UndilutedAverage))
for (n in 1:2){
col.v <- palC(nrow(metaData))[metaData$UndilutedAverageorder]
plot_scores(pca_genes, scores, n, n+1, col.v)
}
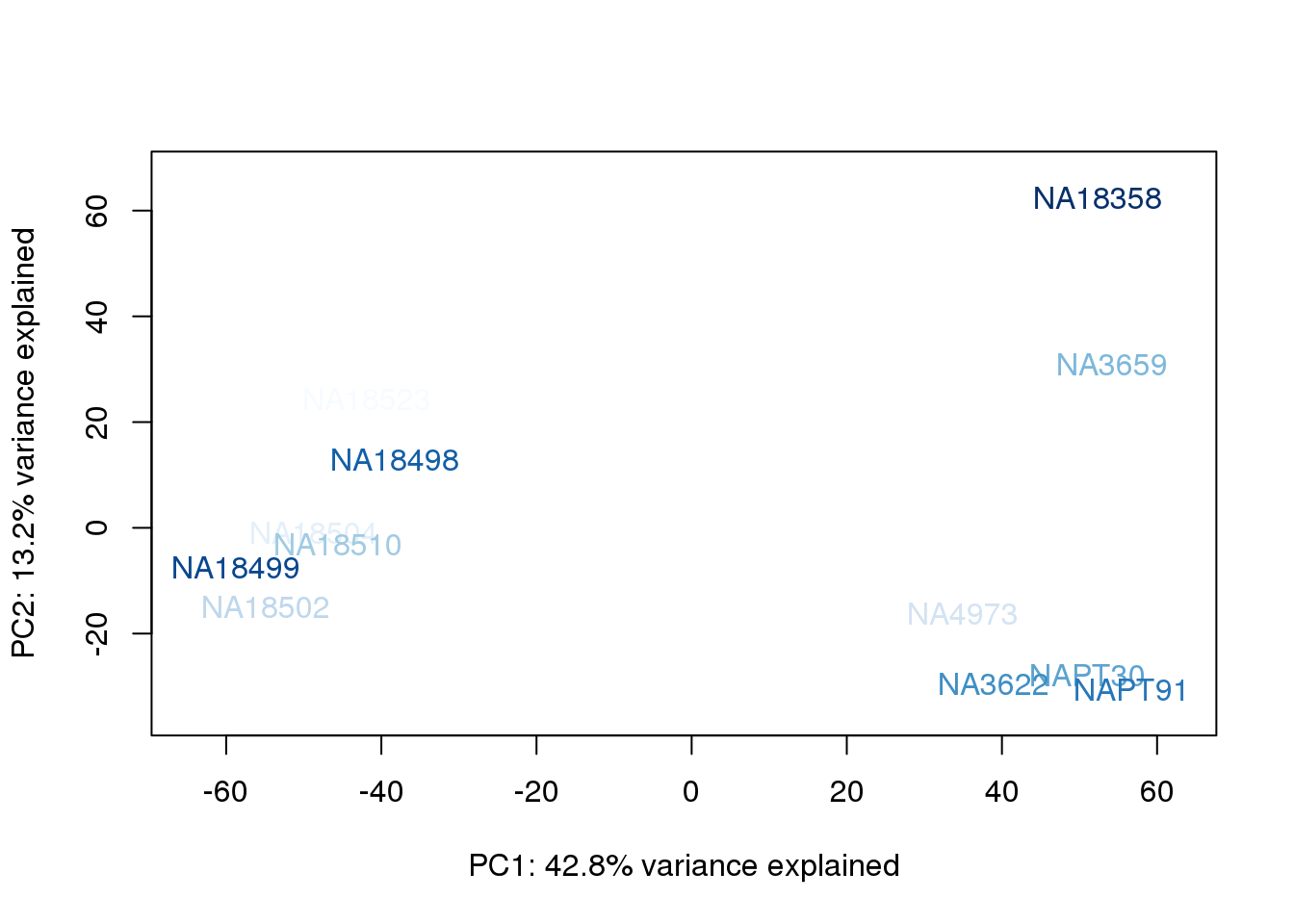
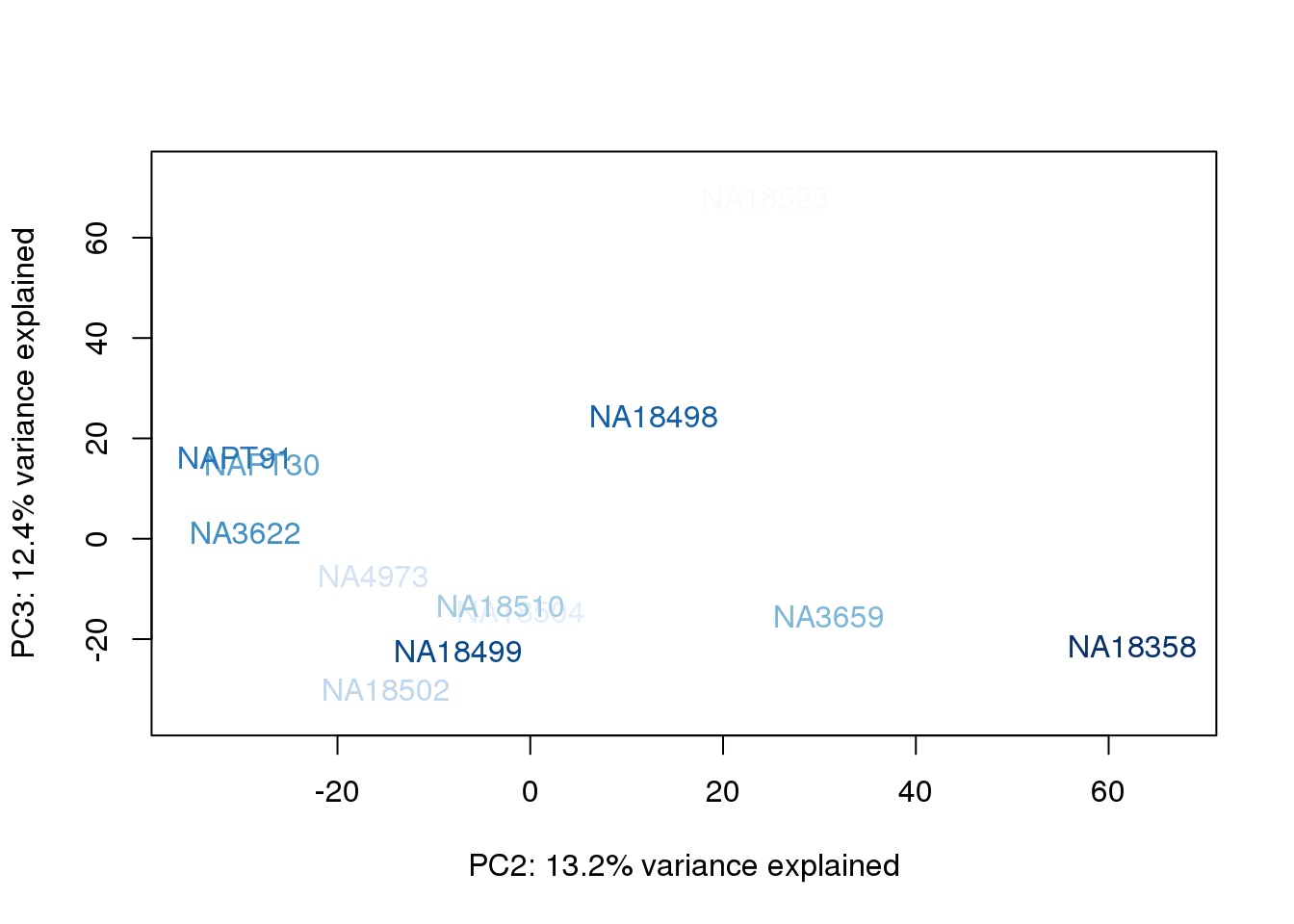
metaData$BioAConcorder = findInterval(metaData$BioAConc, sort(metaData$BioAConc))
for (n in 1:2){
col.v <- palC(nrow(metaData))[metaData$BioAConcorder]
plot_scores(pca_genes, scores, n, n+1, col.v)
}
metaData$RinConcorder = findInterval(metaData$Rin, sort(metaData$Rin))
for (n in 1:2){
col.v <- palC(nrow(metaData))[metaData$RinConcorder]
plot_scores(pca_genes, scores, n, n+1, col.v)
}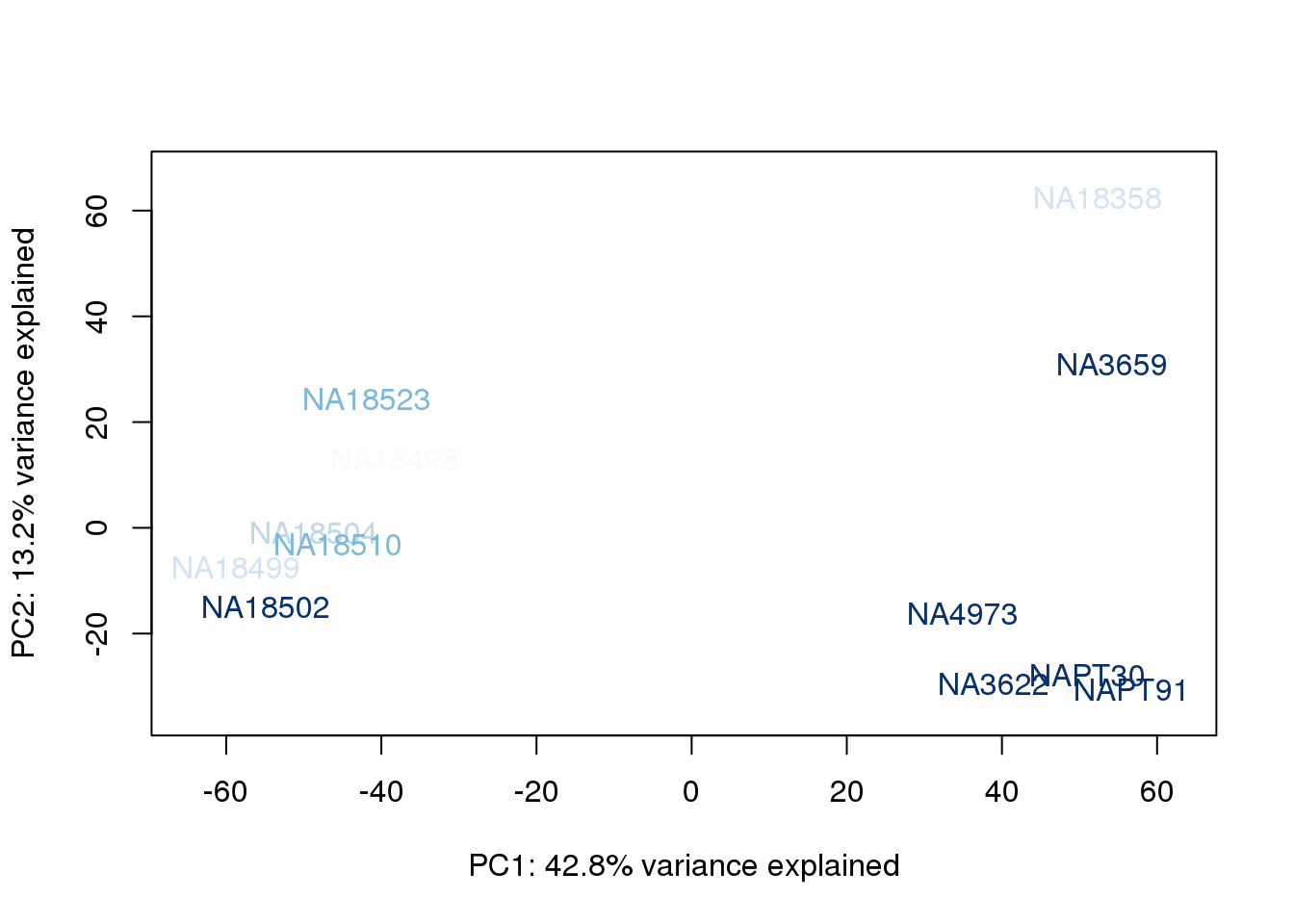
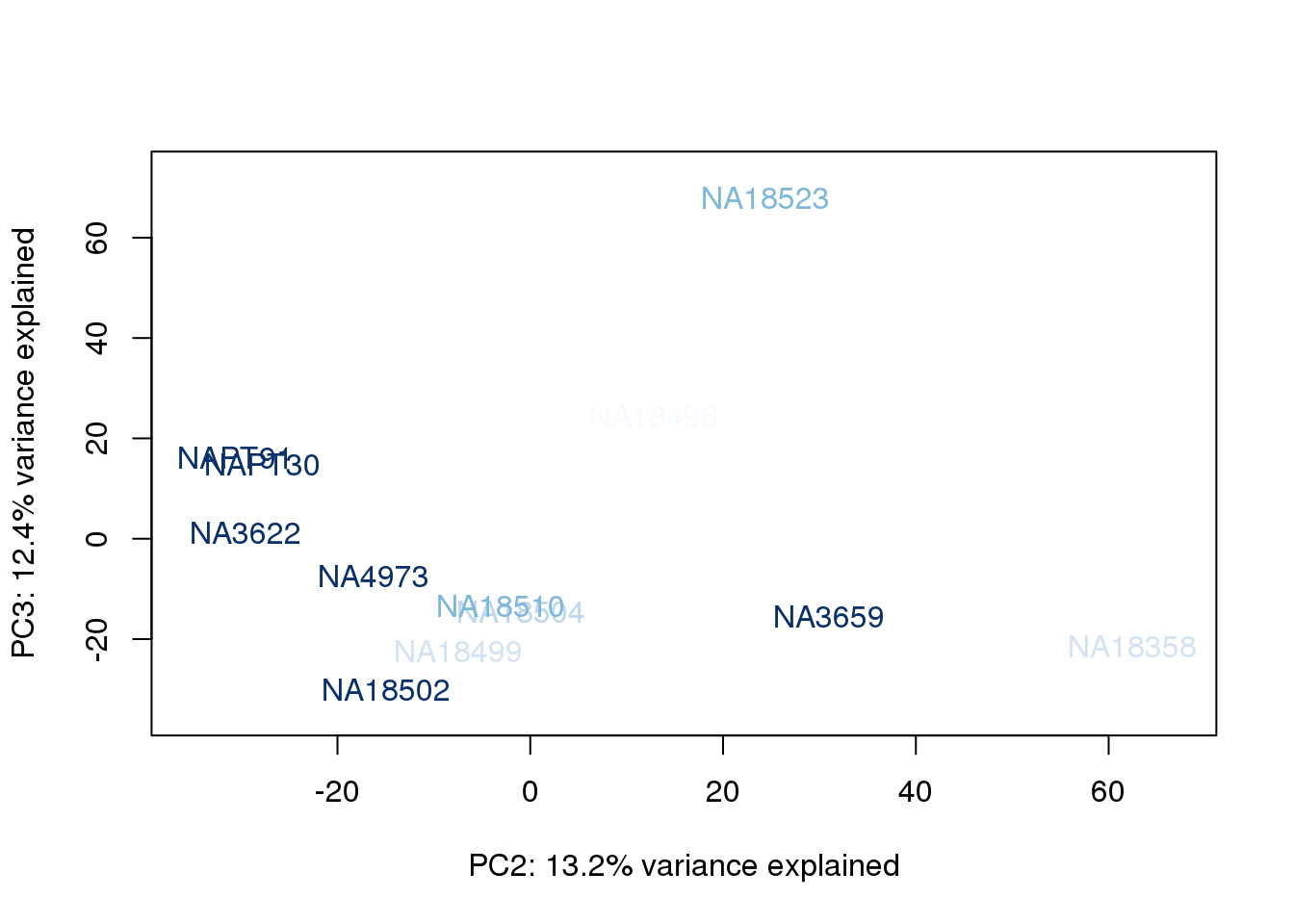
The samples do not cluster by collection concentration, RNA rin score or RNA concentration.
metaData$AssignedOrthoorder = findInterval(metaData$AssignedOrtho, sort(metaData$AssignedOrtho))
for (n in 1:2){
col.v <- palC(nrow(metaData))[metaData$AssignedOrthoorder]
plot_scores(pca_genes, scores, n, n+1, col.v)
}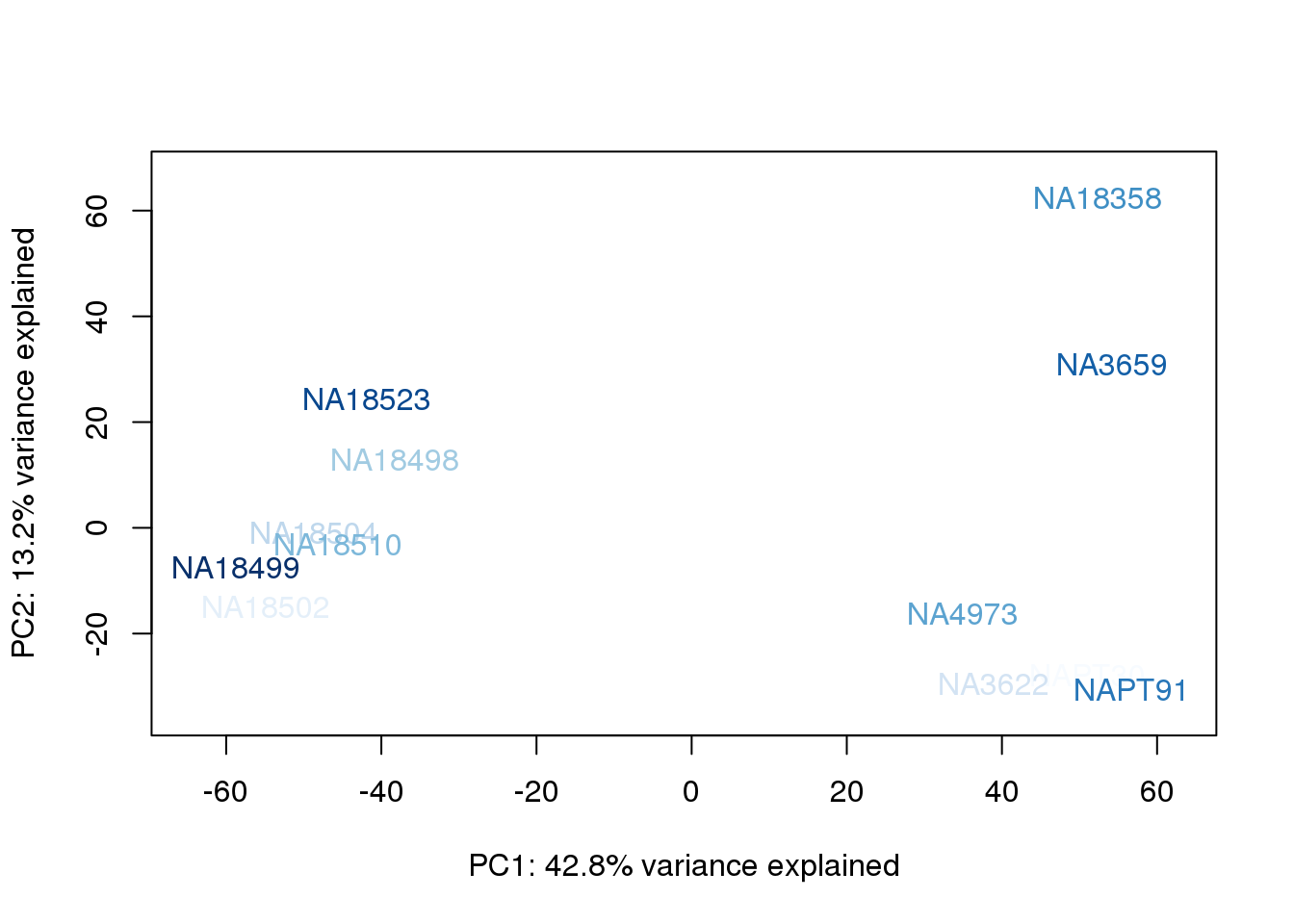
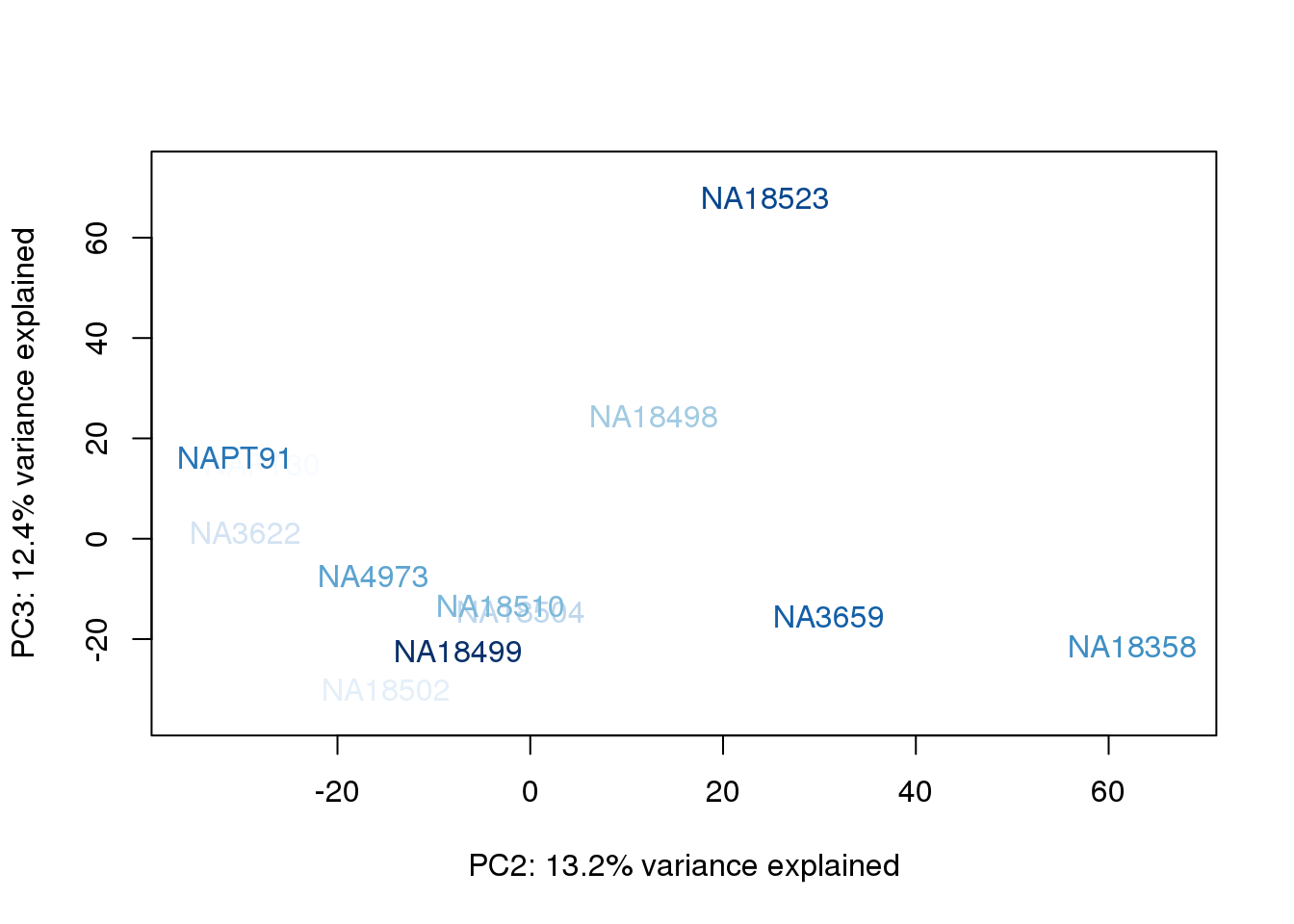
They also do not cluster by number of reads mapping to ortho exons.
PCA heatmap: Code from Michelle Ward:
x.pca <- pca_genes
tech_factors <- metaData
tech_factors_sum <- tech_factors[,c(2:15)] %>% select(-CollectionDate)
p_comps <- 1:6
pc_cov_cor <- matrix(nrow = ncol(tech_factors_sum), ncol = length(p_comps),
dimnames = list(colnames(tech_factors_sum), colnames(x.pca$x)[p_comps]))
for (pc in p_comps) {
for (covariate in 1:ncol(tech_factors_sum)) {
lm_result <- lm(x.pca$x[, pc] ~ tech_factors_sum[, covariate])
r2 <- summary(lm_result)$r.squared
pc_cov_cor[covariate, pc] <- r2
}
}
pc_cov_pval <- matrix(nrow = ncol(tech_factors_sum), ncol = length(p_comps),
dimnames = list(colnames(tech_factors_sum), colnames(x.pca$x)[p_comps]))
for (pc in p_comps) {
for (covariate_2 in 1:ncol(tech_factors_sum)) {
lm_result_2 <- lm(x.pca$x[, pc] ~ tech_factors_sum[, covariate_2])
pval <- anova(lm_result_2)$'Pr(>F)'[1]
pc_cov_pval[covariate_2, pc] <- pval
}
}
PCs <- c("PC1", "PC2", "PC3", "PC4", "PC5", "PC6")
Tech_fac <- colnames(tech_factors_sum)
#Tech_fac <- c("Species", "Individual", "O2.", "Condition" , "Sex", "RIN" , "CO2", "Purity_high", "Purity_med" ,
#"Expt_Batch", "RNA_Batch", "Library_Batch", "Seq_pool", "Episomal_integration" )
heatmap.2(as.matrix(pc_cov_cor[Tech_fac,PCs]),col=brewer.pal(4, "Greens"), trace="none",
Rowv=FALSE, Colv=FALSE, key=T, main="Cor. PCs & tech factors", dendrogram="none",
key.title=NA, cexRow=0.9, cexCol=0.9)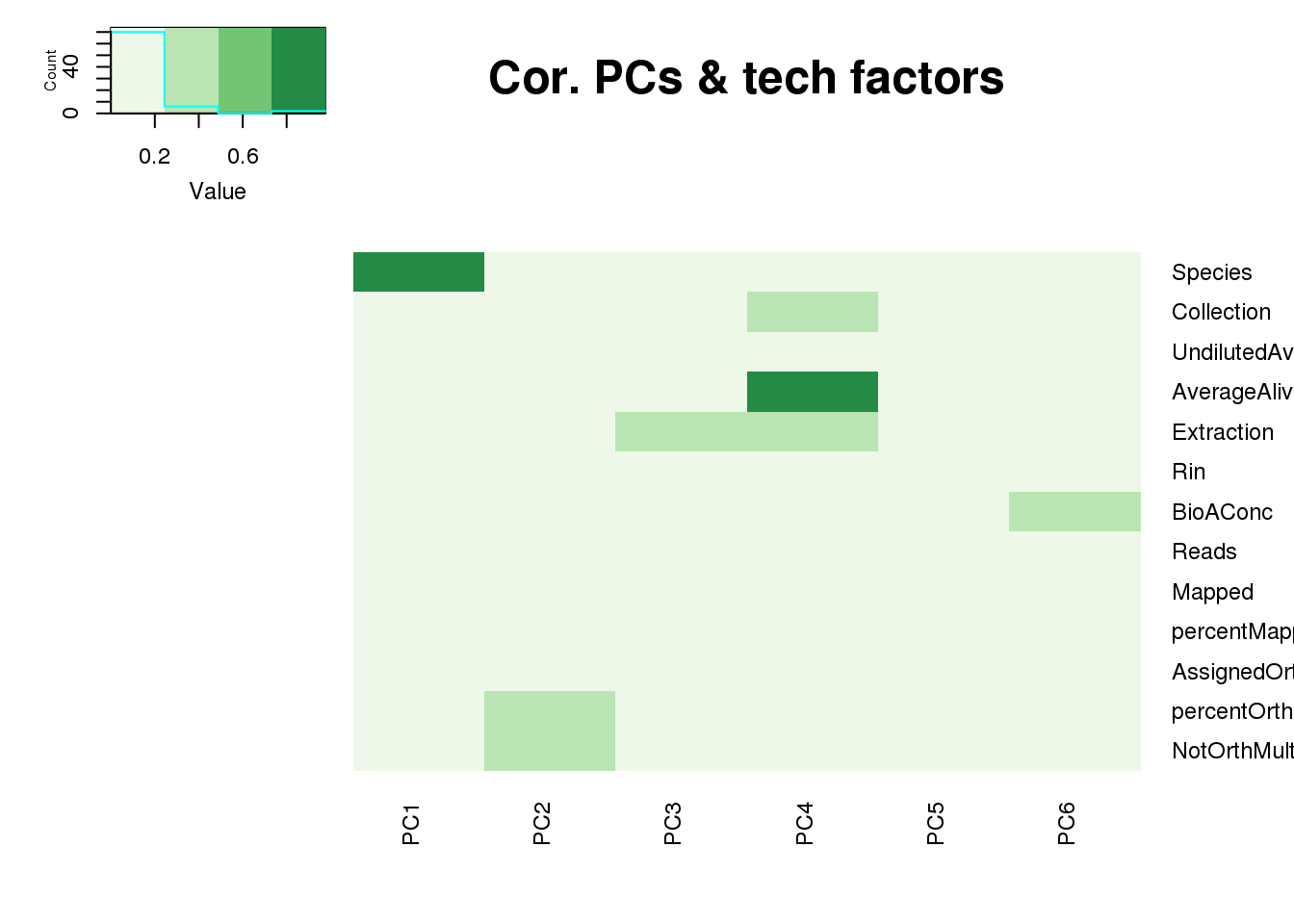
log10_pc_cov_pval <- -log(pc_cov_pval)
heatmap.2(as.matrix(log10_pc_cov_pval[Tech_fac,PCs]), col=brewer.pal(9, "Greens"), trace="none",
Rowv=FALSE, Colv=FALSE, key=T, main="-log10 pval of cor. PCs & tech factors", dendrogram="none",
key.title=NA, cexRow=0.9, cexCol=0.9)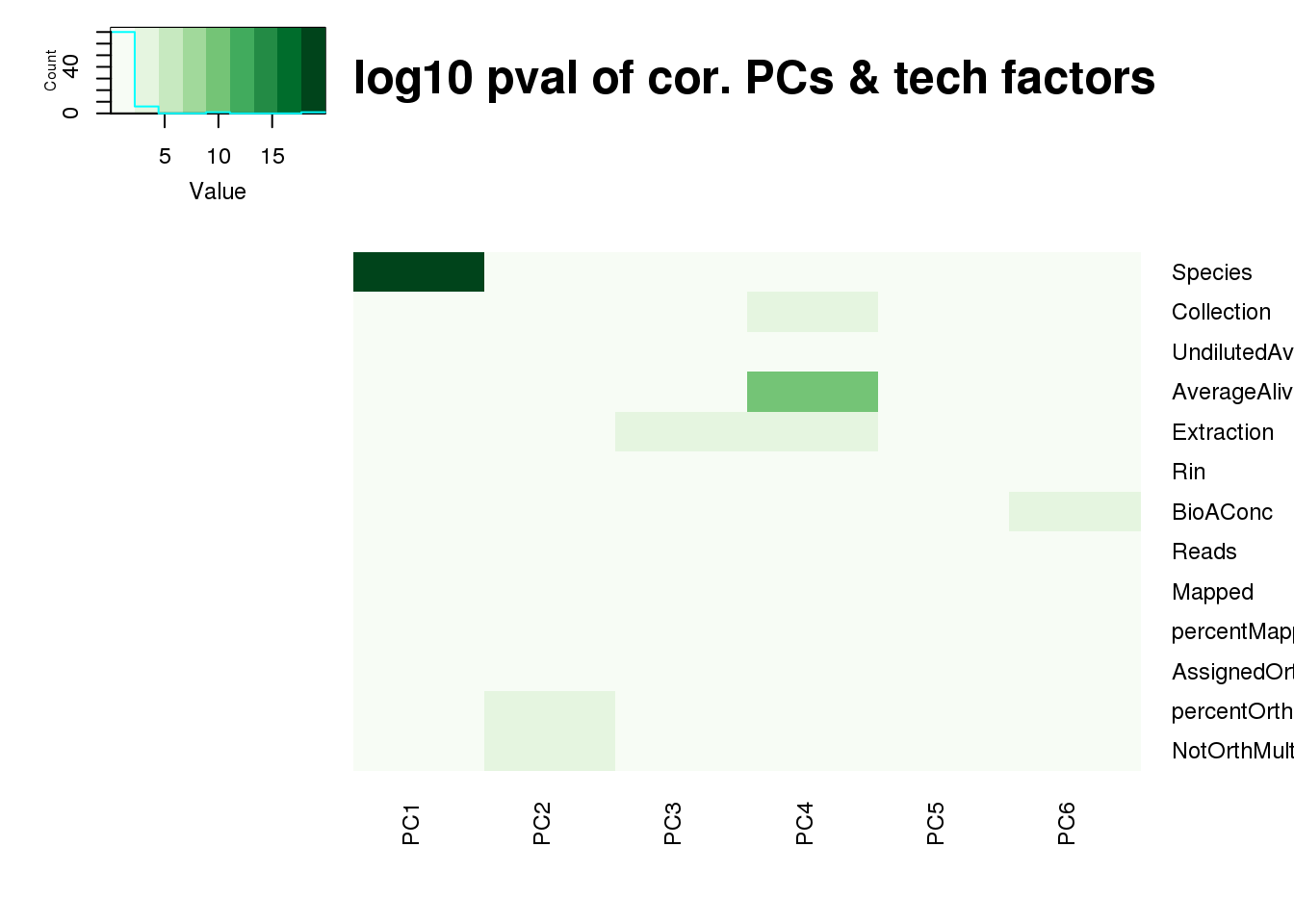
Test for DE
fit.cpm.voom = lmFit(cpm.voom, design, plot=T)
head(coef(fit.cpm.voom)) Chimp Human
ENSG00000217801 1.8181277 1.090565
ENSG00000186891 5.0847804 5.358102
ENSG00000186827 3.4803867 1.530705
ENSG00000078808 6.9813729 6.845371
ENSG00000176022 4.7019733 5.222465
ENSG00000184163 0.8714861 2.121312contr <- makeContrasts(Chimp - Human, levels = colnames(coef(fit.cpm.voom)))
contr Contrasts
Levels Chimp - Human
Chimp 1
Human -1tmp <- contrasts.fit(fit.cpm.voom, contr)
tmp <- eBayes(tmp)
top.table <- topTable(tmp, sort.by = "P", n = Inf)
head(top.table, 20) logFC AveExpr t P.Value adj.P.Val
ENSG00000133112 6.030526 8.579365 53.43162 6.149849e-16 4.099890e-12
ENSG00000204463 -6.207223 5.290526 -51.05863 1.072784e-15 4.099890e-12
ENSG00000205531 -5.980793 5.093652 -49.92171 1.413391e-15 4.099890e-12
ENSG00000105372 4.929862 7.949106 49.41909 1.599801e-15 4.099890e-12
ENSG00000142937 5.675315 8.225710 46.60155 3.281606e-15 6.727948e-12
ENSG00000145741 4.945314 6.308328 43.27471 8.118680e-15 1.387077e-11
ENSG00000142541 6.136435 6.806316 40.37221 1.896383e-14 2.777117e-11
ENSG00000100316 5.939356 8.286237 39.35544 2.589428e-14 3.318029e-11
ENSG00000072864 -6.888911 4.645283 -37.85395 4.162991e-14 4.741646e-11
ENSG00000183020 -4.839255 4.196150 -31.80094 3.478039e-13 3.565338e-10
ENSG00000071082 5.701659 6.557932 29.78081 7.722054e-13 7.196252e-10
ENSG00000147604 -6.185695 5.466412 -27.74841 1.820354e-12 1.555037e-09
ENSG00000116478 3.371308 5.843304 27.55764 1.979085e-12 1.560584e-09
ENSG00000186298 3.080858 6.879040 27.10999 2.413402e-12 1.767127e-09
ENSG00000198242 4.180714 4.991847 25.31780 5.520552e-12 3.772745e-09
ENSG00000105640 -2.840101 8.171072 -24.34587 8.856795e-12 5.619929e-09
ENSG00000197958 4.359628 7.193296 24.24324 9.319949e-12 5.619929e-09
ENSG00000198918 -5.732348 5.424144 -23.80278 1.162663e-11 6.621363e-09
ENSG00000198034 6.255072 7.138676 23.21900 1.568224e-11 8.460982e-09
ENSG00000167193 -4.243003 4.135098 -22.95159 1.802961e-11 9.241077e-09
B
ENSG00000133112 26.01466
ENSG00000204463 23.65724
ENSG00000205531 23.45459
ENSG00000105372 25.30811
ENSG00000142937 24.71006
ENSG00000145741 23.49375
ENSG00000142541 22.77665
ENSG00000100316 22.98772
ENSG00000072864 20.84202
ENSG00000183020 19.73654
ENSG00000071082 19.71403
ENSG00000147604 18.63627
ENSG00000116478 18.96720
ENSG00000186298 18.86337
ENSG00000198242 17.82095
ENSG00000105640 17.59772
ENSG00000197958 17.53671
ENSG00000198918 17.07923
ENSG00000198034 16.98147
ENSG00000167193 16.58857length(which(top.table$adj.P.Val < 0.05))[1] 3764Make a table to plot:
-log10(bh adjusted pval) vs logFC (log3 fold change)
top.table=top.table %>% mutate(Species=ifelse(logFC > 1 & adj.P.Val<.05, "Chimp", ifelse(logFC < -1 & adj.P.Val< .05, "Human", "Neither")))
ggplot(top.table, aes(x=logFC, y= -log10(adj.P.Val))) + geom_point(aes(col=Species), alpha=.3)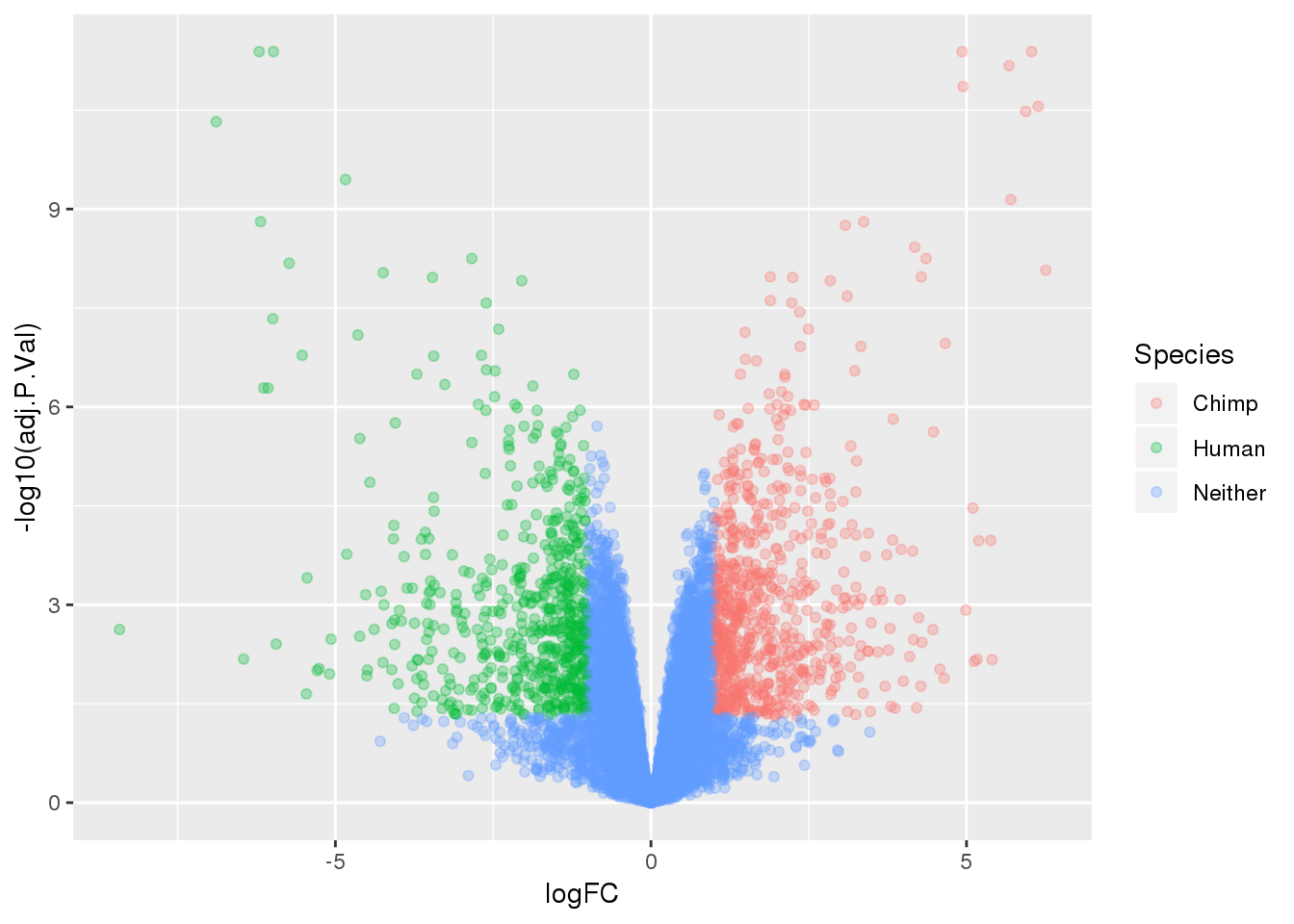
summary(decideTests(tmp)) Chimp - Human
Down 1883
NotSig 6487
Up 1881deGenes=as.data.frame(row.names(top.table[top.table$adj.P.Val < 0.05,]))
#mkdir ../data/DiffExpression
write.table(deGenes,"../data/DiffExpression/DE_genes.txt", col.names = F, row.names = F, quote = F)
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] grid stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] reshape2_1.4.3 RColorBrewer_1.1-2 VennDiagram_1.6.20
[4] futile.logger_1.4.3 R.utils_2.7.0 R.oo_1.22.0
[7] R.methodsS3_1.7.1 edgeR_3.24.0 limma_3.38.2
[10] gplots_3.0.1 scales_1.0.0 forcats_0.3.0
[13] stringr_1.3.1 dplyr_0.8.0.1 purrr_0.3.2
[16] readr_1.3.1 tidyr_0.8.3 tibble_2.1.1
[19] ggplot2_3.1.1 tidyverse_1.2.1 workflowr_1.5.0
loaded via a namespace (and not attached):
[1] Rcpp_1.0.2 locfit_1.5-9.1 lubridate_1.7.4
[4] lattice_0.20-38 gtools_3.8.1 assertthat_0.2.0
[7] rprojroot_1.3-2 digest_0.6.18 R6_2.3.0
[10] cellranger_1.1.0 plyr_1.8.4 futile.options_1.0.1
[13] backports_1.1.2 evaluate_0.12 httr_1.3.1
[16] pillar_1.3.1 rlang_0.4.0 lazyeval_0.2.1
[19] readxl_1.1.0 rstudioapi_0.10 gdata_2.18.0
[22] whisker_0.3-2 rmarkdown_1.10 labeling_0.3
[25] munsell_0.5.0 broom_0.5.1 compiler_3.5.1
[28] httpuv_1.4.5 modelr_0.1.2 pkgconfig_2.0.2
[31] htmltools_0.3.6 tidyselect_0.2.5 crayon_1.3.4
[34] withr_2.1.2 later_0.7.5 bitops_1.0-6
[37] nlme_3.1-137 jsonlite_1.6 gtable_0.2.0
[40] formatR_1.5 git2r_0.26.1 magrittr_1.5
[43] KernSmooth_2.23-15 cli_1.1.0 stringi_1.2.4
[46] fs_1.3.1 promises_1.0.1 xml2_1.2.0
[49] generics_0.0.2 lambda.r_1.2.3 tools_3.5.1
[52] glue_1.3.0 hms_0.4.2 yaml_2.2.0
[55] colorspace_1.3-2 caTools_1.17.1.1 rvest_0.3.2
[58] knitr_1.20 haven_1.1.2