New Dominance Definition
Briana Mittleman
4/14/2020
Last updated: 2020-04-17
Checks: 7 0
Knit directory: Comparative_APA/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190902) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: code/chimp_log/
Ignored: code/human_log/
Ignored: data/.DS_Store
Ignored: data/TrialFiltersMeta.txt.sb-9845453e-R58Y0Q/
Ignored: data/mediation_prot/
Ignored: data/metadata_HCpanel.txt.sb-284518db-RGf0kd/
Ignored: data/metadata_HCpanel.txt.sb-a5794dd2-i594qs/
Ignored: output/.DS_Store
Untracked files:
Untracked: ._.DS_Store
Untracked: Chimp/
Untracked: Human/
Untracked: analysis/AREstabilityScores.Rmd
Untracked: analysis/CrossChimpThreePrime.Rmd
Untracked: analysis/DiffTop2SecondDom.Rmd
Untracked: analysis/DiffTransProtvsExpression.Rmd
Untracked: analysis/DiffUsedUTR.Rmd
Untracked: analysis/GvizPlots.Rmd
Untracked: analysis/HandC.TvN
Untracked: analysis/PhenotypeOverlap10.Rmd
Untracked: analysis/annotationBias.Rmd
Untracked: analysis/assessReadQual.Rmd
Untracked: analysis/diffExpressionPantro6.Rmd
Untracked: code/._AlignmentScores.sh
Untracked: code/._BothFCMM.sh
Untracked: code/._BothFCMMPrim.sh
Untracked: code/._BothFCnewOInclusive.sh
Untracked: code/._ChimpStarMM2.sh
Untracked: code/._ClassifyLeafviz.sh
Untracked: code/._ClosestorthoEx.sh
Untracked: code/._Config_chimp.yaml
Untracked: code/._Config_chimp_full.yaml
Untracked: code/._Config_human.yaml
Untracked: code/._ConvertJunc2Bed.sh
Untracked: code/._CountNucleotides.py
Untracked: code/._CrossMapChimpRNA.sh
Untracked: code/._CrossMapThreeprime.sh
Untracked: code/._DiffSplice.sh
Untracked: code/._DiffSplicePlots.sh
Untracked: code/._DiffSplicePlots_gencode.sh
Untracked: code/._DiffSplice_gencode.sh
Untracked: code/._DiffSplice_removebad.sh
Untracked: code/._Filter255MM.sh
Untracked: code/._FilterPrimSec.sh
Untracked: code/._FindIntronForDomPAS.sh
Untracked: code/._FindIntronForDomPAS_DF.sh
Untracked: code/._GetMAPQscore.py
Untracked: code/._GetSecondaryMap.py
Untracked: code/._Lift5perPAS.sh
Untracked: code/._LiftFinalChimpJunc2Human.sh
Untracked: code/._LiftOrthoPAS2chimp.sh
Untracked: code/._MapBadSamples.sh
Untracked: code/._MismatchNumbers.sh
Untracked: code/._PAS_ATTAAA.sh
Untracked: code/._PAS_ATTAAA_df.sh
Untracked: code/._PAS_seqExpanded.sh
Untracked: code/._PASsequences.sh
Untracked: code/._PASsequences_DF.sh
Untracked: code/._PlotNuclearUsagebySpecies.R
Untracked: code/._PlotNuclearUsagebySpecies_DF.R
Untracked: code/._QuantMergedClusters.sh
Untracked: code/._RNATranscriptDTplot.sh
Untracked: code/._ReverseLiftFilter.R
Untracked: code/._RunFixLeafCluster.sh
Untracked: code/._RunNegMCMediation.sh
Untracked: code/._RunNegMCMediationDF.sh
Untracked: code/._RunPosMCMediationDF.err
Untracked: code/._RunPosMCMediationDF.sh
Untracked: code/._SAF2Bed.py
Untracked: code/._Snakefile
Untracked: code/._SnakefilePAS
Untracked: code/._SnakefilePASfilt
Untracked: code/._SortIndexBadSamples.sh
Untracked: code/._StarMM2.sh
Untracked: code/._TestFC.sh
Untracked: code/._assignPeak2Intronicregion
Untracked: code/._assignPeak2Intronicregion.sh
Untracked: code/._bed215upbed.py
Untracked: code/._bed2Bedbothstrand.py
Untracked: code/._bed2SAF_gen.py
Untracked: code/._buildIndecpantro5
Untracked: code/._buildIndecpantro5.sh
Untracked: code/._buildLeafviz.sh
Untracked: code/._buildLeafviz_leadAnno.sh
Untracked: code/._buildStarIndex.sh
Untracked: code/._chimpChromprder.sh
Untracked: code/._chimpMultiCov.sh
Untracked: code/._chimpMultiCov255.sh
Untracked: code/._chimpMultiCovInclusive.sh
Untracked: code/._chooseSignalSite.py
Untracked: code/._cleanbed2saf.py
Untracked: code/._cluster.json
Untracked: code/._cluster2bed.py
Untracked: code/._clusterLiftReverse.sh
Untracked: code/._clusterLiftReverse_removebad.sh
Untracked: code/._clusterLiftprimary.sh
Untracked: code/._clusterLiftprimary_removebad.sh
Untracked: code/._converBam2Junc.sh
Untracked: code/._converBam2Junc_removeBad.sh
Untracked: code/._extraSnakefiltpas
Untracked: code/._extractPhyloReg.py
Untracked: code/._extractPhyloRegGene.py
Untracked: code/._extractPhylopGeneral.ph
Untracked: code/._extractPhylopGeneral.py
Untracked: code/._extractPhylopReg200down.py
Untracked: code/._extractPhylopReg200up.py
Untracked: code/._filter5percPAS.py
Untracked: code/._filterNumChroms.py
Untracked: code/._filterPASforMP.py
Untracked: code/._filterPostLift.py
Untracked: code/._filterPrimaryread.py
Untracked: code/._filterSecondaryread.py
Untracked: code/._fixExonFC.py
Untracked: code/._fixFCheadforExp.py
Untracked: code/._fixLeafCluster.py
Untracked: code/._fixLiftedJunc.py
Untracked: code/._fixUTRexonanno.py
Untracked: code/._formathg38Anno.py
Untracked: code/._formatpantro6Anno.py
Untracked: code/._getRNAseqMapStats.sh
Untracked: code/._hg19MapStats.sh
Untracked: code/._humanChromorder.sh
Untracked: code/._humanMultiCov.sh
Untracked: code/._humanMultiCov255.sh
Untracked: code/._humanMultiCov_inclusive.sh
Untracked: code/._intersectLiftedPAS.sh
Untracked: code/._liftJunctionFiles.sh
Untracked: code/._liftPAS19to38.sh
Untracked: code/._liftedchimpJunc2human.sh
Untracked: code/._makeNuclearDapaplots.sh
Untracked: code/._makeNuclearDapaplots_DF.sh
Untracked: code/._makeSamplyGroupsHuman_TvN.py
Untracked: code/._mapRNAseqhg19.sh
Untracked: code/._mapRNAseqhg19_newPipeline.sh
Untracked: code/._maphg19.sh
Untracked: code/._maphg19_subjunc.sh
Untracked: code/._mediation_test.R
Untracked: code/._mergeChimp3prime_inhg38.sh
Untracked: code/._mergeandBWRNAseq.sh
Untracked: code/._mergedBam2BW.sh
Untracked: code/._nameClusters.py
Untracked: code/._negativeMediation_montecarlo.R
Untracked: code/._negativeMediation_montecarloDF.R
Untracked: code/._numMultimap.py
Untracked: code/._overlapMMandOrthoexon.sh
Untracked: code/._overlapPASandOrthoexon.sh
Untracked: code/._overlapapaQTLPAS.sh
Untracked: code/._parseHg38.py
Untracked: code/._postiveMediation_montecarlo_DF.R
Untracked: code/._prepareCleanLiftedFC_5perc4LC.py
Untracked: code/._prepareLeafvizAnno.sh
Untracked: code/._preparePAS4lift.py
Untracked: code/._primaryLift.sh
Untracked: code/._processhg38exons.py
Untracked: code/._quantJunc.sh
Untracked: code/._quantJunc_TEST.sh
Untracked: code/._quantJunc_removeBad.sh
Untracked: code/._quantLiftedPASPrimary.sh
Untracked: code/._quantMerged_seperatly.sh
Untracked: code/._recLiftchim2human.sh
Untracked: code/._revLiftPAShg38to19.sh
Untracked: code/._reverseLift.sh
Untracked: code/._runCheckReverseLift.sh
Untracked: code/._runChimpDiffIso.sh
Untracked: code/._runCountNucleotides.sh
Untracked: code/._runFilterNumChroms.sh
Untracked: code/._runHumanDiffIso.sh
Untracked: code/._runNuclearDiffIso_DF.sh
Untracked: code/._runNuclearDifffIso.sh
Untracked: code/._runTotalDiffIso.sh
Untracked: code/._run_chimpverifybam.sh
Untracked: code/._run_verifyBam.sh
Untracked: code/._snakemake.batch
Untracked: code/._snakemakePAS.batch
Untracked: code/._snakemakePASchimp.batch
Untracked: code/._snakemakePAShuman.batch
Untracked: code/._snakemake_chimp.batch
Untracked: code/._snakemake_human.batch
Untracked: code/._snakemakefiltPAS.batch
Untracked: code/._snakemakefiltPAS_chimp
Untracked: code/._snakemakefiltPAS_chimp.sh
Untracked: code/._snakemakefiltPAS_human.sh
Untracked: code/._spliceSite2Fasta.py
Untracked: code/._submit-snakemake-chimp.sh
Untracked: code/._submit-snakemake-human.sh
Untracked: code/._submit-snakemakePAS-chimp.sh
Untracked: code/._submit-snakemakePAS-human.sh
Untracked: code/._submit-snakemakefiltPAS-chimp.sh
Untracked: code/._submit-snakemakefiltPAS-human.sh
Untracked: code/._subset_diffisopheno_Nuclear_HvC.py
Untracked: code/._subset_diffisopheno_Nuclear_HvC_DF.py
Untracked: code/._subset_diffisopheno_Total_HvC.py
Untracked: code/._threeprimeOrthoFC.sh
Untracked: code/._transcriptDTplotsNuclear.sh
Untracked: code/._verifyBam4973.sh
Untracked: code/._verifyBam4973inHuman.sh
Untracked: code/._wrap_chimpverifybam.sh
Untracked: code/._wrap_verifyBam.sh
Untracked: code/._writeMergecode.py
Untracked: code/.snakemake/
Untracked: code/ALLPAS_sequenceDF.err
Untracked: code/ALLPAS_sequenceDF.out
Untracked: code/AlignmentScores.err
Untracked: code/AlignmentScores.out
Untracked: code/AlignmentScores.sh
Untracked: code/BothFCMM.err
Untracked: code/BothFCMM.out
Untracked: code/BothFCMM.sh
Untracked: code/BothFCMMPrim.err
Untracked: code/BothFCMMPrim.out
Untracked: code/BothFCMMPrim.sh
Untracked: code/BothFCnewOInclusive.sh
Untracked: code/BothFCnewOInclusive.sh.err
Untracked: code/BothFCnewOInclusive.sh.out
Untracked: code/ChimpStarMM2.err
Untracked: code/ChimpStarMM2.out
Untracked: code/ChimpStarMM2.sh
Untracked: code/ClassifyLeafviz.sh
Untracked: code/ClosestorthoEx.err
Untracked: code/ClosestorthoEx.out
Untracked: code/ClosestorthoEx.sh
Untracked: code/Config_chimp.yaml
Untracked: code/Config_chimp_full.yaml
Untracked: code/Config_human.yaml
Untracked: code/ConvertJunc2Bed.err
Untracked: code/ConvertJunc2Bed.out
Untracked: code/ConvertJunc2Bed.sh
Untracked: code/CountNucleotides.py
Untracked: code/CrossMapChimpRNA.sh
Untracked: code/CrossMapThreeprime.sh
Untracked: code/CrossmapChimp3prime.err
Untracked: code/CrossmapChimp3prime.out
Untracked: code/CrossmapChimpRNA.err
Untracked: code/CrossmapChimpRNA.out
Untracked: code/DiffSplice.err
Untracked: code/DiffSplice.out
Untracked: code/DiffSplice.sh
Untracked: code/DiffSplicePlots.err
Untracked: code/DiffSplicePlots.out
Untracked: code/DiffSplicePlots.sh
Untracked: code/DiffSplicePlots_gencode.sh
Untracked: code/DiffSplice_gencode.sh
Untracked: code/DiffSplice_removebad.err
Untracked: code/DiffSplice_removebad.out
Untracked: code/DiffSplice_removebad.sh
Untracked: code/Filter255.err
Untracked: code/Filter255.out
Untracked: code/Filter255MM.sh
Untracked: code/FilterPrimSec.err
Untracked: code/FilterPrimSec.out
Untracked: code/FilterPrimSec.sh
Untracked: code/FilterReverseLift.err
Untracked: code/FilterReverseLift.out
Untracked: code/FindDomXCutoff.py
Untracked: code/FindIntronForDomPAS.err
Untracked: code/FindIntronForDomPAS.out
Untracked: code/FindIntronForDomPAS.sh
Untracked: code/FindIntronForDomPAS_DF.sh
Untracked: code/GencodeDiffSplice.err
Untracked: code/GencodeDiffSplice.out
Untracked: code/GetMAPQscore.py
Untracked: code/GetSecondaryMap.py
Untracked: code/GetTopminus2Usage.py
Untracked: code/HchromOrder.err
Untracked: code/HchromOrder.out
Untracked: code/IntersectMMandOrtho.err
Untracked: code/IntersectMMandOrtho.out
Untracked: code/IntersectPASandOrtho.err
Untracked: code/IntersectPASandOrtho.out
Untracked: code/JunctionLift.err
Untracked: code/JunctionLift.out
Untracked: code/JunctionLiftFinalChimp.err
Untracked: code/JunctionLiftFinalChimp.out
Untracked: code/Lift5perPAS.sh
Untracked: code/Lift5perPASbed.err
Untracked: code/Lift5perPASbed.out
Untracked: code/LiftClustersFirst.err
Untracked: code/LiftClustersFirst.out
Untracked: code/LiftClustersFirst_remove.err
Untracked: code/LiftClustersFirst_remove.out
Untracked: code/LiftClustersSecond.err
Untracked: code/LiftClustersSecond.out
Untracked: code/LiftClustersSecond_remove.err
Untracked: code/LiftClustersSecond_remove.out
Untracked: code/LiftFinalChimpJunc2Human.sh
Untracked: code/LiftOrthoPAS2chimp.sh
Untracked: code/LiftorthoPAS.err
Untracked: code/LiftorthoPASt.out
Untracked: code/Log.out
Untracked: code/MapBadSamples.err
Untracked: code/MapBadSamples.out
Untracked: code/MapBadSamples.sh
Untracked: code/MapStats.err
Untracked: code/MapStats.out
Untracked: code/MaxEntCode/
Untracked: code/MergeClusters.err
Untracked: code/MergeClusters.out
Untracked: code/MergeClusters.sh
Untracked: code/MismatchNumbers.err
Untracked: code/MismatchNumbers.out
Untracked: code/MismatchNumbers.sh
Untracked: code/PAS_ATTAAA.err
Untracked: code/PAS_ATTAAA.out
Untracked: code/PAS_ATTAAA.sh
Untracked: code/PAS_ATTAAADF.err
Untracked: code/PAS_ATTAAADF.out
Untracked: code/PAS_ATTAAA_df.sh
Untracked: code/PAS_seqExpanded.sh
Untracked: code/PAS_sequence.err
Untracked: code/PAS_sequence.out
Untracked: code/PAS_sequenceDF.err
Untracked: code/PAS_sequenceDF.out
Untracked: code/PASexpanded_sequenceDF.err
Untracked: code/PASexpanded_sequenceDF.out
Untracked: code/PASsequences.sh
Untracked: code/PASsequences_DF.sh
Untracked: code/PlotNuclearUsagebySpecies.R
Untracked: code/PlotNuclearUsagebySpecies_DF.R
Untracked: code/QuantMergeClusters
Untracked: code/QuantMergeClusters.err
Untracked: code/QuantMergeClusters.out
Untracked: code/QuantMergedClusters.sh
Untracked: code/RNATranscriptDTplot.err
Untracked: code/RNATranscriptDTplot.out
Untracked: code/RNATranscriptDTplot.sh
Untracked: code/Rev_liftoverPAShg19to38.err
Untracked: code/Rev_liftoverPAShg19to38.out
Untracked: code/ReverseLiftFilter.R
Untracked: code/RunFixCluster.err
Untracked: code/RunFixCluster.out
Untracked: code/RunFixLeafCluster.sh
Untracked: code/RunNegMCMediation.err
Untracked: code/RunNegMCMediation.sh
Untracked: code/RunNegMCMediationDF.err
Untracked: code/RunNegMCMediationDF.out
Untracked: code/RunNegMCMediationDF.sh
Untracked: code/RunNegMCMediationr.out
Untracked: code/RunNewDom.err
Untracked: code/RunNewDom.out
Untracked: code/RunPosMCMediation.err
Untracked: code/RunPosMCMediation.sh
Untracked: code/RunPosMCMediationDF.err
Untracked: code/RunPosMCMediationDF.out
Untracked: code/RunPosMCMediationDF.sh
Untracked: code/RunPosMCMediationr.out
Untracked: code/SAF215upbed_gen.py
Untracked: code/SAF2Bed.py
Untracked: code/Snakefile
Untracked: code/SnakefilePAS
Untracked: code/SnakefilePASfilt
Untracked: code/SortIndexBadSamples.err
Untracked: code/SortIndexBadSamples.out
Untracked: code/SortIndexBadSamples.sh
Untracked: code/StarMM2.err
Untracked: code/StarMM2.out
Untracked: code/StarMM2.sh
Untracked: code/TestFC.err
Untracked: code/TestFC.out
Untracked: code/TestFC.sh
Untracked: code/TotalTranscriptDTplot.err
Untracked: code/TotalTranscriptDTplot.out
Untracked: code/Upstream10Bases_general.py
Untracked: code/allPASSeq_df.sh
Untracked: code/apaQTLsnake.err
Untracked: code/apaQTLsnake.out
Untracked: code/apaQTLsnakePAS.err
Untracked: code/apaQTLsnakePAS.out
Untracked: code/apaQTLsnakePAShuman.err
Untracked: code/apaQTLsnakefiltPAS.err
Untracked: code/apaQTLsnakefiltPAS.out
Untracked: code/assignPeak2Intronicregion.err
Untracked: code/assignPeak2Intronicregion.out
Untracked: code/assignPeak2Intronicregion.sh
Untracked: code/bam2junc.err
Untracked: code/bam2junc.out
Untracked: code/bam2junc_remove.err
Untracked: code/bam2junc_remove.out
Untracked: code/bed215upbed.py
Untracked: code/bed2Bedbothstrand.py
Untracked: code/bed2SAF_gen.py
Untracked: code/bed2saf.py
Untracked: code/bg_to_cov.py
Untracked: code/buildIndecpantro5
Untracked: code/buildIndecpantro5.sh
Untracked: code/buildLeafviz.err
Untracked: code/buildLeafviz.out
Untracked: code/buildLeafviz.sh
Untracked: code/buildLeafviz_leadAnno.sh
Untracked: code/buildLeafviz_leafanno.err
Untracked: code/buildLeafviz_leafanno.out
Untracked: code/buildStarIndex.sh
Untracked: code/callPeaksYL.py
Untracked: code/chimpChromprder.sh
Untracked: code/chimpMultiCov.err
Untracked: code/chimpMultiCov.out
Untracked: code/chimpMultiCov.sh
Untracked: code/chimpMultiCov255.sh
Untracked: code/chimpMultiCovInclusive.err
Untracked: code/chimpMultiCovInclusive.out
Untracked: code/chimpMultiCovInclusive.sh
Untracked: code/chooseAnno2Bed.py
Untracked: code/chooseAnno2SAF.py
Untracked: code/chooseSignalSite.py
Untracked: code/chromOrder.err
Untracked: code/chromOrder.out
Untracked: code/classifyLeafviz.err
Untracked: code/classifyLeafviz.out
Untracked: code/cleanbed2saf.py
Untracked: code/cluster.json
Untracked: code/cluster2bed.py
Untracked: code/clusterLiftReverse.sh
Untracked: code/clusterLiftReverse_removebad.sh
Untracked: code/clusterLiftprimary.sh
Untracked: code/clusterLiftprimary_removebad.sh
Untracked: code/clusterPAS.json
Untracked: code/clusterfiltPAS.json
Untracked: code/comands2Mege.sh
Untracked: code/converBam2Junc.sh
Untracked: code/converBam2Junc_removeBad.sh
Untracked: code/convertNumeric.py
Untracked: code/environment.yaml
Untracked: code/extraSnakefiltpas
Untracked: code/extractPhyloReg.py
Untracked: code/extractPhyloRegGene.py
Untracked: code/extractPhylopGeneral.py
Untracked: code/extractPhylopReg200down.py
Untracked: code/extractPhylopReg200up.py
Untracked: code/filter5perc.R
Untracked: code/filter5percPAS.py
Untracked: code/filter5percPheno.py
Untracked: code/filterBamforMP.pysam2_gen.py
Untracked: code/filterJuncChroms.err
Untracked: code/filterJuncChroms.out
Untracked: code/filterMissprimingInNuc10_gen.py
Untracked: code/filterNumChroms.py
Untracked: code/filterPASforMP.py
Untracked: code/filterPostLift.py
Untracked: code/filterPrimaryread.py
Untracked: code/filterSAFforMP_gen.py
Untracked: code/filterSecondaryread.py
Untracked: code/filterSortBedbyCleanedBed_gen.R
Untracked: code/filterpeaks.py
Untracked: code/fixExonFC.py
Untracked: code/fixFChead.py
Untracked: code/fixFChead_bothfrac.py
Untracked: code/fixFCheadforExp.py
Untracked: code/fixLeafCluster.py
Untracked: code/fixLiftedJunc.py
Untracked: code/fixUTRexonanno.py
Untracked: code/formathg38Anno.py
Untracked: code/generateStarIndex.err
Untracked: code/generateStarIndex.out
Untracked: code/generateStarIndexHuman.err
Untracked: code/generateStarIndexHuman.out
Untracked: code/getAlloverlap.py
Untracked: code/getRNAseqMapStats.sh
Untracked: code/hg19MapStats.err
Untracked: code/hg19MapStats.out
Untracked: code/hg19MapStats.sh
Untracked: code/humanChromorder.sh
Untracked: code/humanFiles
Untracked: code/humanMultiCov.err
Untracked: code/humanMultiCov.out
Untracked: code/humanMultiCov.sh
Untracked: code/humanMultiCov255.err
Untracked: code/humanMultiCov255.out
Untracked: code/humanMultiCov255.sh
Untracked: code/humanMultiCovInclusive.err
Untracked: code/humanMultiCovInclusive.out
Untracked: code/humanMultiCov_inclusive.sh
Untracked: code/intersectAnno.err
Untracked: code/intersectAnno.out
Untracked: code/intersectAnnoExt.err
Untracked: code/intersectAnnoExt.out
Untracked: code/intersectLiftedPAS.sh
Untracked: code/leafcutter_merge_regtools_redo.py
Untracked: code/liftJunctionFiles.sh
Untracked: code/liftPAS19to38.sh
Untracked: code/liftoverPAShg19to38.err
Untracked: code/liftoverPAShg19to38.out
Untracked: code/log/
Untracked: code/make5percPeakbed.py
Untracked: code/makeFileID.py
Untracked: code/makeNuclearDapaplots.sh
Untracked: code/makeNuclearDapaplots_DF.sh
Untracked: code/makeNuclearPlots.err
Untracked: code/makeNuclearPlots.out
Untracked: code/makeNuclearPlotsDF.err
Untracked: code/makeNuclearPlotsDF.out
Untracked: code/makePheno.py
Untracked: code/makeSamplyGroupsChimp_TvN.py
Untracked: code/makeSamplyGroupsHuman_TvN.py
Untracked: code/mapRNAseqhg19.sh
Untracked: code/mapRNAseqhg19_newPipeline.sh
Untracked: code/maphg19.err
Untracked: code/maphg19.out
Untracked: code/maphg19.sh
Untracked: code/maphg19_new.err
Untracked: code/maphg19_new.out
Untracked: code/maphg19_sub.err
Untracked: code/maphg19_sub.out
Untracked: code/maphg19_subjunc.sh
Untracked: code/mediation_test.R
Untracked: code/merge.err
Untracked: code/mergeChimp3prime_inhg38.sh
Untracked: code/merge_leafcutter_clusters_redo.py
Untracked: code/mergeandBWRNAseq.sh
Untracked: code/mergeandsort_ChimpinHuman.err
Untracked: code/mergeandsort_ChimpinHuman.out
Untracked: code/mergedBam2BW.sh
Untracked: code/mergedbam2bw.err
Untracked: code/mergedbam2bw.out
Untracked: code/mergedbamRNAand2bw.err
Untracked: code/mergedbamRNAand2bw.out
Untracked: code/nameClusters.py
Untracked: code/namePeaks.py
Untracked: code/negativeMediation_montecarlo.R
Untracked: code/negativeMediation_montecarloDF.R
Untracked: code/nuclearTranscriptDTplot.err
Untracked: code/nuclearTranscriptDTplot.out
Untracked: code/numMultimap.py
Untracked: code/overlapMMandOrthoexon.sh
Untracked: code/overlapPAS.err
Untracked: code/overlapPAS.out
Untracked: code/overlapPASandOrthoexon.sh
Untracked: code/overlapapaQTLPAS.sh
Untracked: code/overlapapaQTLPAS_extended.sh
Untracked: code/overlapapaQTLPAS_samples.sh
Untracked: code/parseHg38.py
Untracked: code/peak2PAS.py
Untracked: code/pheno2countonly.R
Untracked: code/postiveMediation_montecarlo.R
Untracked: code/postiveMediation_montecarlo_DF.R
Untracked: code/prepareAnnoLeafviz.err
Untracked: code/prepareAnnoLeafviz.out
Untracked: code/prepareCleanLiftedFC_5perc4LC.py
Untracked: code/prepareLeafvizAnno.sh
Untracked: code/preparePAS4lift.py
Untracked: code/prepare_phenotype_table.py
Untracked: code/primaryLift.err
Untracked: code/primaryLift.out
Untracked: code/primaryLift.sh
Untracked: code/processhg38exons.py
Untracked: code/quantJunc.sh
Untracked: code/quantJunc_TEST.sh
Untracked: code/quantJunc_removeBad.sh
Untracked: code/quantLiftedPAS.err
Untracked: code/quantLiftedPAS.out
Untracked: code/quantLiftedPAS.sh
Untracked: code/quantLiftedPASPrimary.err
Untracked: code/quantLiftedPASPrimary.out
Untracked: code/quantLiftedPASPrimary.sh
Untracked: code/quatJunc.err
Untracked: code/quatJunc.out
Untracked: code/recChimpback2Human.err
Untracked: code/recChimpback2Human.out
Untracked: code/recLiftchim2human.sh
Untracked: code/revLift.err
Untracked: code/revLift.out
Untracked: code/revLiftPAShg38to19.sh
Untracked: code/reverseLift.sh
Untracked: code/runCheckReverseLift.sh
Untracked: code/runChimpDiffIso.sh
Untracked: code/runChimpDiffIsoDF.sh
Untracked: code/runCountNucleotides.err
Untracked: code/runCountNucleotides.out
Untracked: code/runCountNucleotides.sh
Untracked: code/runCountNucleotidesPantro6.err
Untracked: code/runCountNucleotidesPantro6.out
Untracked: code/runCountNucleotides_pantro6.sh
Untracked: code/runFilterNumChroms.sh
Untracked: code/runHumanDiffIso.sh
Untracked: code/runHumanDiffIsoDF.sh
Untracked: code/runNewDom.sh
Untracked: code/runNuclearDiffIso_DF.sh
Untracked: code/runNuclearDifffIso.sh
Untracked: code/runTotalDiffIso.sh
Untracked: code/run_Chimpleafcutter_ds.err
Untracked: code/run_Chimpleafcutter_ds.out
Untracked: code/run_Chimpverifybam.err
Untracked: code/run_Chimpverifybam.out
Untracked: code/run_Humanleafcutter_dF.err
Untracked: code/run_Humanleafcutter_dF.out
Untracked: code/run_Humanleafcutter_ds.err
Untracked: code/run_Humanleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_ds.err
Untracked: code/run_Nuclearleafcutter_ds.out
Untracked: code/run_Nuclearleafcutter_dsDF.err
Untracked: code/run_Nuclearleafcutter_dsDF.out
Untracked: code/run_Totalleafcutter_ds.err
Untracked: code/run_Totalleafcutter_ds.out
Untracked: code/run_chimpverifybam.sh
Untracked: code/run_verifyBam.sh
Untracked: code/run_verifybam.err
Untracked: code/run_verifybam.out
Untracked: code/slurm-62824013.out
Untracked: code/slurm-62825841.out
Untracked: code/slurm-62826116.out
Untracked: code/slurm-64108209.out
Untracked: code/slurm-64108521.out
Untracked: code/slurm-64108557.out
Untracked: code/snakePASChimp.err
Untracked: code/snakePASChimp.out
Untracked: code/snakePAShuman.out
Untracked: code/snakemake.batch
Untracked: code/snakemakeChimp.err
Untracked: code/snakemakeChimp.out
Untracked: code/snakemakeHuman.err
Untracked: code/snakemakeHuman.out
Untracked: code/snakemakePAS.batch
Untracked: code/snakemakePASFiltChimp.err
Untracked: code/snakemakePASFiltChimp.out
Untracked: code/snakemakePASFiltHuman.err
Untracked: code/snakemakePASFiltHuman.out
Untracked: code/snakemakePAS_Human.batch
Untracked: code/snakemakePASchimp.batch
Untracked: code/snakemakePAShuman.batch
Untracked: code/snakemake_chimp.batch
Untracked: code/snakemake_human.batch
Untracked: code/snakemakefiltPAS.batch
Untracked: code/snakemakefiltPAS_chimp.sh
Untracked: code/snakemakefiltPAS_human.batch
Untracked: code/snakemakefiltPAS_human.sh
Untracked: code/spliceSite2Fasta.py
Untracked: code/submit-snakemake-chimp.sh
Untracked: code/submit-snakemake-human.sh
Untracked: code/submit-snakemakePAS-chimp.sh
Untracked: code/submit-snakemakePAS-human.sh
Untracked: code/submit-snakemakefiltPAS-chimp.sh
Untracked: code/submit-snakemakefiltPAS-human.sh
Untracked: code/subset_diffisopheno.py
Untracked: code/subset_diffisopheno_Chimp_tvN.py
Untracked: code/subset_diffisopheno_Chimp_tvN_DF.py
Untracked: code/subset_diffisopheno_Huma_tvN.py
Untracked: code/subset_diffisopheno_Huma_tvN_DF.py
Untracked: code/subset_diffisopheno_Nuclear_HvC.py
Untracked: code/subset_diffisopheno_Nuclear_HvC_DF.py
Untracked: code/subset_diffisopheno_Total_HvC.py
Untracked: code/test
Untracked: code/test.txt
Untracked: code/threeprimeOrthoFC.out
Untracked: code/threeprimeOrthoFC.sh
Untracked: code/threeprimeOrthoFCcd.err
Untracked: code/transcriptDTplotsNuclear.sh
Untracked: code/transcriptDTplotsTotal.sh
Untracked: code/verifyBam4973.sh
Untracked: code/verifyBam4973inHuman.sh
Untracked: code/verifybam4973.err
Untracked: code/verifybam4973.out
Untracked: code/verifybam4973HumanMap.err
Untracked: code/verifybam4973HumanMap.out
Untracked: code/wrap_Chimpverifybam.err
Untracked: code/wrap_Chimpverifybam.out
Untracked: code/wrap_chimpverifybam.sh
Untracked: code/wrap_verifyBam.sh
Untracked: code/wrap_verifybam.err
Untracked: code/wrap_verifybam.out
Untracked: code/writeMergecode.py
Untracked: data/._.DS_Store
Untracked: data/._HC_filenames.txt
Untracked: data/._HC_filenames.txt.sb-4426323c-IKIs0S
Untracked: data/._HC_filenames.xlsx
Untracked: data/._MapPantro6_meta.txt
Untracked: data/._MapPantro6_meta.txt.sb-a5794dd2-Cskmlm
Untracked: data/._MapPantro6_meta.xlsx
Untracked: data/._OppositeSpeciesMap.txt
Untracked: data/._OppositeSpeciesMap.txt.sb-a5794dd2-mayWJf
Untracked: data/._OppositeSpeciesMap.xlsx
Untracked: data/._RNASEQ_metadata.txt
Untracked: data/._RNASEQ_metadata.txt.sb-4426323c-TE4ns3
Untracked: data/._RNASEQ_metadata.txt.sb-51f67ae1-HXp7Gq
Untracked: data/._RNASEQ_metadata_2Removed.txt
Untracked: data/._RNASEQ_metadata_2Removed.txt.sb-4426323c-a4lBwx
Untracked: data/._RNASEQ_metadata_2Removed.xlsx
Untracked: data/._RNASEQ_metadata_stranded.txt
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-a5794dd2-D659m2
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-a5794dd2-ImNMoY
Untracked: data/._RNASEQ_metadata_stranded.txt.sb-e4bf31f0-ZGnGgl
Untracked: data/._RNASEQ_metadata_stranded.xlsx
Untracked: data/._TrialFiltersMeta.txt
Untracked: data/._TrialFiltersMeta.txt.sb-9845453e-R58Y0Q
Untracked: data/._metadata_HCpanel.txt
Untracked: data/._metadata_HCpanel.txt.sb-a3d92a2d-b9cYoF
Untracked: data/._metadata_HCpanel.txt.sb-a5794dd2-i594qs
Untracked: data/._metadata_HCpanel.txt.sb-f4823d1e-qihGek
Untracked: data/._metadata_HCpanel_frompantro5.xlsx
Untracked: data/._~$RNASEQ_metadata.xlsx
Untracked: data/._~$metadata_HCpanel.xlsx
Untracked: data/._.xlsx
Untracked: data/ARE_Elements/
Untracked: data/BaseComp/
Untracked: data/CleanLiftedPeaks_FC_primary/
Untracked: data/CompapaQTLpas/
Untracked: data/DNDS/
Untracked: data/DTmatrix/
Untracked: data/DiffExpression/
Untracked: data/DiffIso_Nuclear/
Untracked: data/DiffIso_Nuclear_DF/
Untracked: data/DiffIso_Total/
Untracked: data/DiffSplice/
Untracked: data/DiffSplice_liftedJunc/
Untracked: data/DiffSplice_removeBad/
Untracked: data/DomDefGreaterX/
Untracked: data/DominantPAS/
Untracked: data/DominantPAS_DF/
Untracked: data/EvalPantro5/
Untracked: data/HC_filenames.txt
Untracked: data/HC_filenames.xlsx
Untracked: data/Khan_prot/
Untracked: data/Li_eqtls/
Untracked: data/MapPantro6_meta.txt
Untracked: data/MapPantro6_meta.xlsx
Untracked: data/MapStats/
Untracked: data/NormalizedClusters/
Untracked: data/NuclearHvC/
Untracked: data/NuclearHvC_DF/
Untracked: data/OppositeSpeciesMap.txt
Untracked: data/OppositeSpeciesMap.xlsx
Untracked: data/OrthoExonBed/
Untracked: data/OverlapBenchmark/
Untracked: data/OverlappingPAS/
Untracked: data/PAS/
Untracked: data/PAS_SAF/
Untracked: data/PAS_doubleFilter/
Untracked: data/Peaks_5perc/
Untracked: data/Pheno_5perc/
Untracked: data/Pheno_5perc_DF_nuclear/
Untracked: data/Pheno_5perc_nuclear/
Untracked: data/Pheno_5perc_nuclear_old/
Untracked: data/Pheno_5perc_total/
Untracked: data/PhyloP/
Untracked: data/Pol2Chip/
Untracked: data/RNASEQ_metadata.txt
Untracked: data/RNASEQ_metadata_2Removed.txt
Untracked: data/RNASEQ_metadata_2Removed.xlsx
Untracked: data/RNASEQ_metadata_stranded.txt
Untracked: data/RNASEQ_metadata_stranded.txt.sb-e4bf31f0-ZGnGgl/
Untracked: data/RNASEQ_metadata_stranded.xlsx
Untracked: data/SignalSites/
Untracked: data/SignalSites_doublefilter/
Untracked: data/SpliceSite/
Untracked: data/TestAnnoBiasOE/
Untracked: data/TestMM2/
Untracked: data/TestMM2_AS/
Untracked: data/TestMM2_PrimaryRead/
Untracked: data/TestMM2_SeondaryRead/
Untracked: data/TestMM2_mismatch/
Untracked: data/TestMM2_quality/
Untracked: data/TestWithinMergePAS/
Untracked: data/Test_FC_methods/
Untracked: data/Threeprime2Ortho/
Untracked: data/TotalFractionPAS/
Untracked: data/TotalHvC/
Untracked: data/TrialFiltersMeta.txt
Untracked: data/TwoBadSampleAnalysis/
Untracked: data/Wang_ribo/
Untracked: data/apaQTLGenes/
Untracked: data/bioGRID/
Untracked: data/chainFiles/
Untracked: data/cleanPeaks_anno/
Untracked: data/cleanPeaks_byspecies/
Untracked: data/cleanPeaks_lifted/
Untracked: data/files4viz_nuclear/
Untracked: data/files4viz_nuclear_DF/
Untracked: data/gviz/
Untracked: data/leafviz/
Untracked: data/liftover_files/
Untracked: data/mediation/
Untracked: data/mediation_DF/
Untracked: data/metadata_HCpanel.txt
Untracked: data/metadata_HCpanel.xlsx
Untracked: data/metadata_HCpanel_extra.txt
Untracked: data/metadata_HCpanel_frompantro5.txt
Untracked: data/metadata_HCpanel_frompantro5.xlsx
Untracked: data/miRNA/
Untracked: data/multimap/
Untracked: data/orthoUTR/
Untracked: data/primaryLift/
Untracked: data/reverseLift/
Untracked: data/testQuant/
Untracked: data/~$RNASEQ_metadata.xlsx
Untracked: data/~$metadata_HCpanel.xlsx
Untracked: data/.xlsx
Untracked: output/._.DS_Store
Untracked: output/dtPlots/
Untracked: projectNotes.Rmd
Untracked: proteinModelSet.Rmd
Unstaged changes:
Modified: analysis/ExploredAPA.Rmd
Modified: analysis/MMExpreiment.Rmd
Modified: analysis/OppositeMap.Rmd
Modified: analysis/TotalDomStructure.Rmd
Modified: analysis/TotalVNuclearBothSpecies.Rmd
Modified: analysis/annotationInfo.Rmd
Modified: analysis/changeMisprimcut.Rmd
Modified: analysis/comp2apaQTLPAS.Rmd
Modified: analysis/correlationPhenos.Rmd
Modified: analysis/establishCutoffs.Rmd
Modified: analysis/investigatePantro5.Rmd
Modified: analysis/multiMap.Rmd
Modified: analysis/pol2.Rmd
Modified: analysis/speciesSpecific.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | afa2694 | brimittleman | 2020-04-17 | deal with ties |
| html | 385fe04 | brimittleman | 2020-04-16 | Build site. |
| Rmd | 4cc2f72 | brimittleman | 2020-04-16 | add prop plots and 1v1 lines |
| html | 71e0ab3 | brimittleman | 2020-04-15 | Build site. |
| Rmd | 8b2f73d | brimittleman | 2020-04-15 | add aggregate res |
| html | 38c6dc9 | brimittleman | 2020-04-14 | Build site. |
| Rmd | 2d0668e | brimittleman | 2020-04-14 | add new way to call dom |
library(workflowr)This is workflowr version 1.6.0
Run ?workflowr for help getting startedlibrary(gridExtra)
library(VennDiagram)Loading required package: gridLoading required package: futile.loggerlibrary(tidyverse)── Attaching packages ────────────────────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.1.1 ✔ purrr 0.3.2
✔ tibble 2.1.1 ✔ dplyr 0.8.0.1
✔ tidyr 0.8.3 ✔ stringr 1.3.1
✔ readr 1.3.1 ✔ forcats 0.3.0 ── Conflicts ───────────────────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::combine() masks gridExtra::combine()
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library(cowplot)
Attaching package: 'cowplot'The following object is masked from 'package:ggplot2':
ggsaveThe way I am calling dominance is a bit naive. I am looking for the most used PAS. It may be better to look for genes with actual dominance on one PAS, meaning a PAS is used X percent above the others in the gene. I will have a set of genes with 1 PAS, 1 domiant PAS, no dominant PAS. I can then look to see if these sets are te same or different across species.
I wrote a python script. I will test it with a small file.
mkdir ../data/DomDefGreaterX
head -n 1 ../data/PAS_doubleFilter/PAS_5perc_either_HumanCoord_BothUsage_meta_doubleFilter.txt > ../data/DomDefGreaterX/TestFile_ZSWIM7.txt
grep ZSWIM7 ../data/PAS_doubleFilter/PAS_5perc_either_HumanCoord_BothUsage_meta_doubleFilter.txt >> ../data/DomDefGreaterX/TestFile_ZSWIM7.txt
python FindDomXCutoff.py ../data/DomDefGreaterX/TestFile_ZSWIM7.txt ../data/DomDefGreaterX/TestDom_ZSWIM7.txt .3 Human
python FindDomXCutoff.py ../data/DomDefGreaterX/TestFile_ZSWIM7.txt ../data/DomDefGreaterX/TestDom_ZSWIM7.txt .3 Chimp
sbatch runNewDom.sh
Looks like this code works for both species. Not the most efficient but fine.
First I need the set of genes with 1 pas. These will not be included in the analysis. I added to the code to not include PAS that have 0 usage in that species.
MetaPAS=read.table("../data/PAS_doubleFilter/PAS_5perc_either_HumanCoord_BothUsage_meta_doubleFilter.txt", header = T, stringsAsFactors = F)
MetaCol=colnames(MetaPAS)
Human1PASGene= MetaPAS %>% filter(Human>0) %>% group_by(gene) %>% summarise(nPAS=n()) %>% filter(nPAS==1)
Chimp1PASGene= MetaPAS %>% filter(Chimp>0) %>% group_by(gene) %>% summarise(nPAS=n()) %>% filter(nPAS==1)
MetaPAS_human1= MetaPAS %>% filter(gene %in% Human1PASGene$gene, Human >0) %>% mutate(Set="Human1")
MetaPAS_chimp1= MetaPAS %>% filter(gene %in% Chimp1PASGene$gene, Chimp >0) %>% mutate(Set="Chimp1")Look at where these are.
OnePASboth=MetaPAS_human1 %>% bind_rows(MetaPAS_chimp1)
ggplot(OnePASboth, aes(x=loc, by=Set, fill=Set)) + geom_bar(position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="PAS location for genes with 1 PAS in each species")
Seperate results
I will run the code at different cutoffs from 10% different to 90% different. I can do this with a bash script.
I need to think about the best way to visualize this. I will start with 1 usage cutoff then move toward more. Start with the most stringent. For each one I will add the number of PAS in the gene origianlly:
90% difference
PASnum= MetaPAS %>% select(PAS, gene, Chimp, Human) %>% gather(Species, usage, -PAS, -gene) %>% filter(usage >0) %>% group_by(gene,Species) %>% summarise(nPAS=n())
PASnumHuman=PASnum %>% filter(Species=="Human") %>% select(-Species)
PASnumChimp=PASnum %>% filter(Species=="Chimp") %>% select(-Species)
HumanDom9=read.table("../data/DomDefGreaterX/Human_.9_dominantPAS.txt", col.names = MetaCol,stringsAsFactors = F) %>% mutate(set="Human9") %>% inner_join(PASnumHuman, by="gene")
nrow(HumanDom9)[1] 65ChimpDom9=read.table("../data/DomDefGreaterX/Chimp_.9_dominantPAS.txt", col.names = MetaCol,stringsAsFactors = F) %>% mutate(set="Chimp9") %>% inner_join(PASnumChimp, by="gene")
nrow(ChimpDom9)[1] 173Explore these:
BothDom9=HumanDom9 %>% bind_rows(ChimpDom9)
genloc9=ggplot(BothDom9, aes(x=loc, fill=set)) + geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.9",y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
genloc9
BothDom9_prop= BothDom9 %>% group_by(set, loc) %>% summarise(nLoc=n()) %>% ungroup() %>% group_by(set) %>% mutate(nSet=sum(nLoc), prop=nLoc/nSet)
proploc9=ggplot(BothDom9_prop, aes(x=loc,by=set, fill=set,y=prop)) + geom_bar(stat="identity",position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.9",y="Proportion",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")There are way more in chimp at this cutoff.
Look at how many genes have the same and different dominant at .9 difference:
HumanDom9_sm=HumanDom9 %>% select(PAS, gene, loc) %>% rename(HumanPAS=PAS, HumanLoc=loc)
ChimpDom9_sm=ChimpDom9 %>% select(PAS, gene, loc)%>% rename(ChimpPAS=PAS, ChimpLoc=loc)
both9sm=ChimpDom9_sm %>% inner_join(HumanDom9_sm, by="gene")
nrow(both9sm)[1] 0Look at the number of PAS in these genes:
None of the same genes are represented here.
ggplot(BothDom9, aes(x=nPAS, fill=set)) + geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="Number of PAS per gene with .9 difference",y="genes")
grid.newpage()
venn.plot9 <- draw.pairwise.venn(area1 = length(HumanDom9_sm$gene),
area2 = length(ChimpDom9_sm$gene),
cross.area = nrow(both9sm),
c("Human", "Chimp"), scaled = TRUE,
fill = c("orange", "green"),
cex = 1.5,
cat.cex = 1.5,
cat.pos = c(320, 25),
cat.dist = .05) 
Look at the location of the PAS not in gene.
HumanDom9_smLab= HumanDom9 %>% mutate(Set="Human")
ChimpDom9_smLab= ChimpDom9 %>% mutate(Set="Chimp")
BothDom9_smLab=HumanDom9_smLab %>% bind_rows(ChimpDom9_smLab)
diffgenes9=ggplot(BothDom9_smLab,aes(x=loc, fill=Set))+ geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2")+ labs(title="Non Matched: 0.9", y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
diffgenes9
BothDom9_smLab_prop=BothDom9_smLab%>% group_by(set, loc) %>% summarise(nLoc=n()) %>% ungroup() %>% group_by(set) %>% mutate(nSet=sum(nLoc), prop=nLoc/nSet)
propDiff9=ggplot(BothDom9_smLab_prop, aes(x=loc,by=set, fill=set,y=prop)) + geom_bar(stat="identity",position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.9: Different Genes",y="Proportion",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")80% difference
HumanDom8=read.table("../data/DomDefGreaterX/Human_.8_dominantPAS.txt", col.names = MetaCol,stringsAsFactors = F) %>% mutate(set="Human8") %>% inner_join(PASnumHuman, by="gene")
nrow(HumanDom8)[1] 318ChimpDom8=read.table("../data/DomDefGreaterX/Chimp_.8_dominantPAS.txt", col.names = MetaCol,stringsAsFactors = F) %>% mutate(set="Chimp8") %>% inner_join(PASnumChimp, by="gene")
nrow(ChimpDom8)[1] 571BothDom8=HumanDom8 %>% bind_rows(ChimpDom8)
genloc8=ggplot(BothDom8, aes(x=loc, fill=set)) + geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.8",y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
genloc8
HumanDom8_sm=HumanDom8 %>% select(PAS, gene, loc) %>% rename(HumanPAS=PAS, HumanLoc=loc)
ChimpDom8_sm=ChimpDom8 %>% select(PAS, gene, loc)%>% rename(ChimpPAS=PAS, ChimpLoc=loc)
both8sm=ChimpDom8_sm %>% inner_join(HumanDom8_sm, by="gene")
nrow(both8sm)[1] 192There are 192 genes that are the same.
grid.newpage()
venn.plot8 <- draw.pairwise.venn(area1 = length(HumanDom8_sm$gene),
area2 = length(ChimpDom8_sm$gene),
cross.area = nrow(both8sm),
c("Human", "Chimp"), scaled = TRUE,
fill = c("orange", "green"),
cex = 1.5,
cat.cex = 1.5,
cat.pos = c(320, 25),
cat.dist = .05) 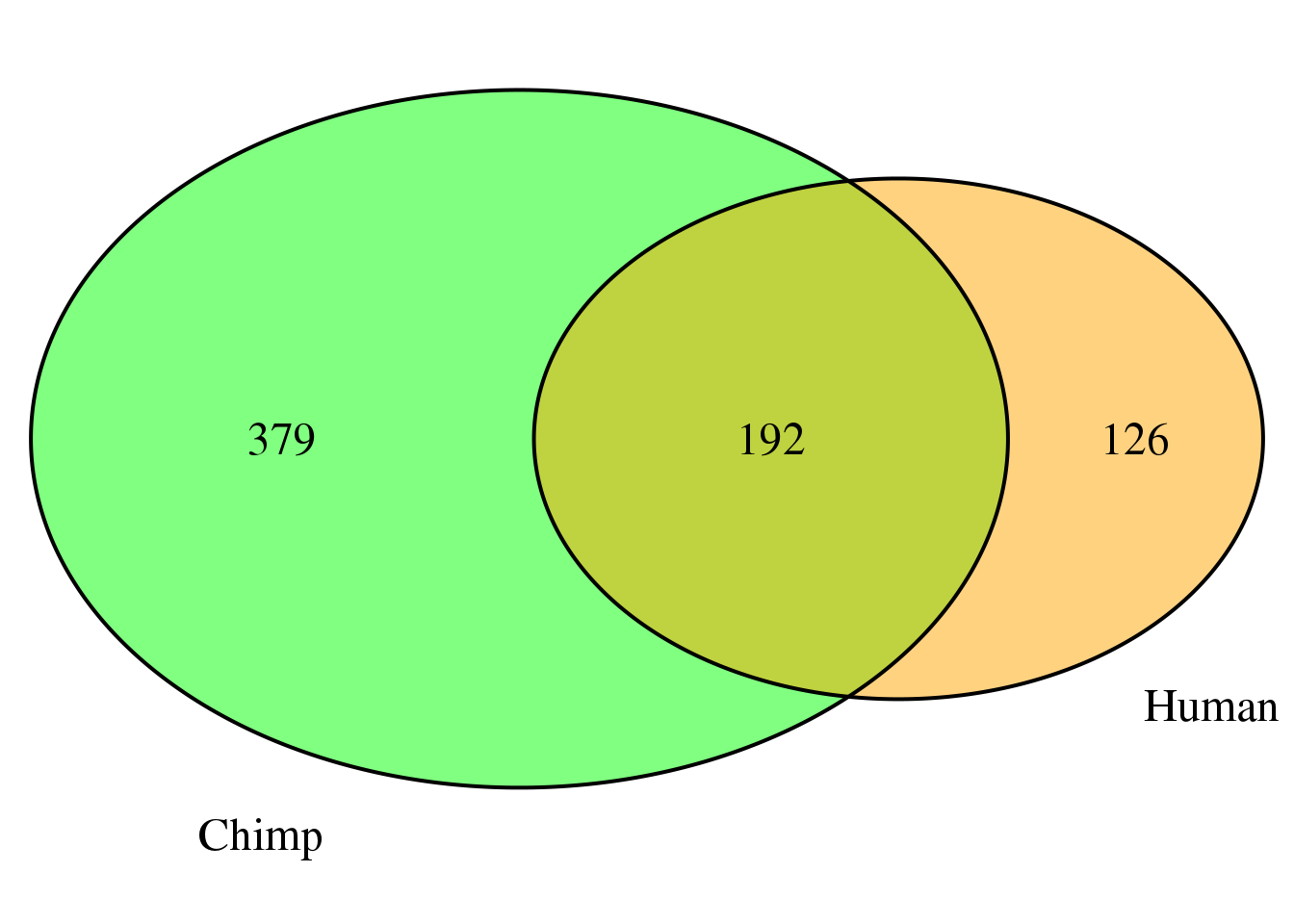
For these I can look to see if the PAS are the same or different:
both8sm_same= both8sm %>% filter(ChimpPAS==HumanPAS)
nrow(both8sm_same)[1] 189both8sm_diff= both8sm %>% filter(ChimpPAS!=HumanPAS)
nrow(both8sm_diff)[1] 3location for same:
same8=ggplot(both8sm_same,aes(x=ChimpLoc,fill=ChimpLoc)) +geom_bar(stat = "count") +scale_fill_brewer(palette = "Dark2")+ labs(title="Matched Gene Matched PAS .8", y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
same8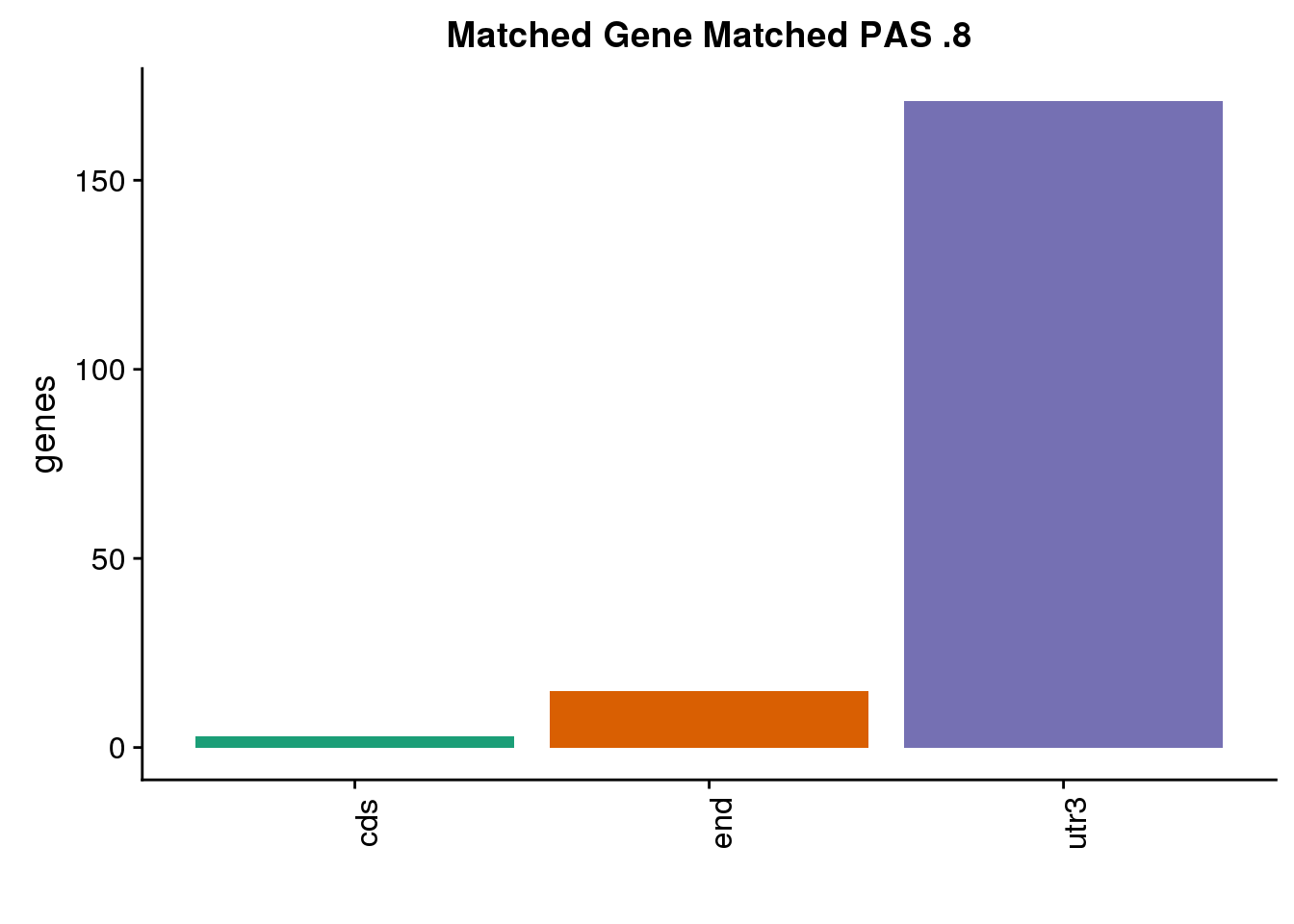
Only 3 are in different PAS
both8sm_diff ChimpPAS gene ChimpLoc HumanPAS HumanLoc
1 human21024 CKS1B utr3 human21027 end
2 chimp163919 IFNGR2 utr3 human215583 intron
3 human324647 NSUN5P1 utr5 human324646 utr5ggplot(BothDom8, aes(x=nPAS, fill=set)) + geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="Number of PAS per gene with .8 difference",y="genes")
HumanDom8_smLab= HumanDom8 %>% anti_join(ChimpDom8,by = "gene") %>% mutate(Set="Human")
ChimpDom8_smLab= ChimpDom8 %>% anti_join(HumanDom8,by = "gene") %>% mutate(Set="Chimp")
BothDom8_smLab=HumanDom8_smLab %>% bind_rows(ChimpDom8_smLab)
diffgenes8=ggplot(BothDom8_smLab,aes(x=loc, fill=Set))+ geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="Non Matched: 0.8", y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
diffgenes8
| Version | Author | Date |
|---|---|---|
| 385fe04 | brimittleman | 2020-04-16 |
BothDom8_prop= BothDom8 %>% group_by(set, loc) %>% summarise(nLoc=n()) %>% ungroup() %>% group_by(set) %>% mutate(nSet=sum(nLoc), prop=nLoc/nSet)
proploc8=ggplot(BothDom8_prop, aes(x=loc,by=set, fill=set,y=prop)) + geom_bar(stat="identity",position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.8",y="Proportion",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")BothDom8_smLab_prop=BothDom8_smLab%>% group_by(set, loc) %>% summarise(nLoc=n()) %>% ungroup() %>% group_by(set) %>% mutate(nSet=sum(nLoc), prop=nLoc/nSet)
propDiff8=ggplot(BothDom8_smLab_prop, aes(x=loc,by=set, fill=set,y=prop)) + geom_bar(stat="identity",position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.8: Different Genes",y="Proportion",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")70% difference
HumanDom7=read.table("../data/DomDefGreaterX/Human_.7_dominantPAS.txt", col.names = MetaCol,stringsAsFactors = F) %>% mutate(set="Human7") %>% inner_join(PASnumHuman, by="gene")
nrow(HumanDom7)[1] 637ChimpDom7=read.table("../data/DomDefGreaterX/Chimp_.7_dominantPAS.txt", col.names = MetaCol,stringsAsFactors = F) %>% mutate(set="Chimp7") %>% inner_join(PASnumChimp, by="gene")
nrow(ChimpDom7)[1] 988BothDom7=HumanDom7 %>% bind_rows(ChimpDom7)
genloc7=ggplot(BothDom7, aes(x=loc, fill=set)) + geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.7",y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
genloc7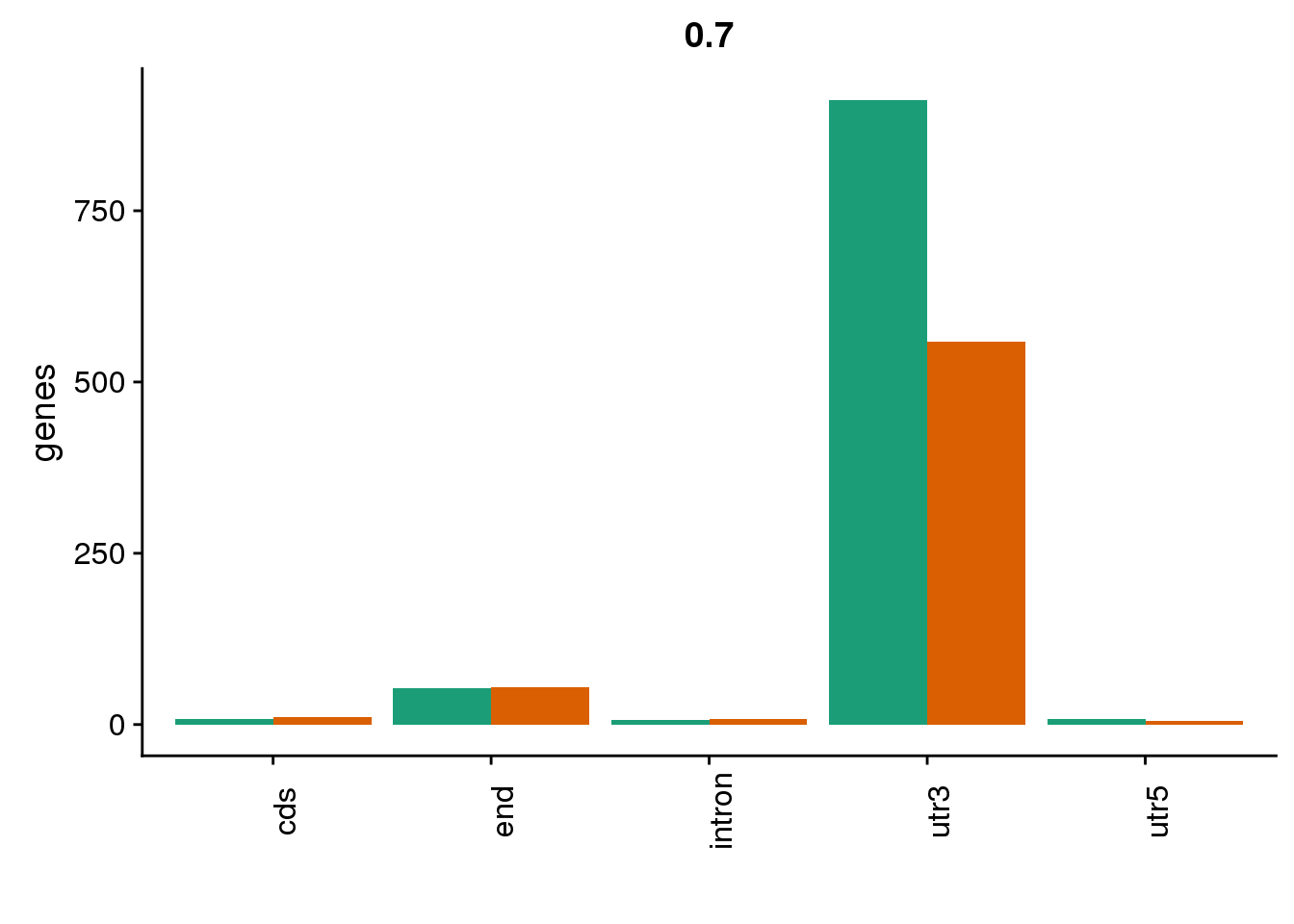
HumanDom7_sm=HumanDom7 %>% select(PAS, gene, loc) %>% rename(HumanPAS=PAS, HumanLoc=loc)
ChimpDom7_sm=ChimpDom7 %>% select(PAS, gene, loc)%>% rename(ChimpPAS=PAS, ChimpLoc=loc)
both7sm=ChimpDom7_sm %>% inner_join(HumanDom7_sm, by="gene")
nrow(both7sm)[1] 473grid.newpage()
venn.plot7 <- draw.pairwise.venn(area1 = length(HumanDom7_sm$gene),
area2 = length(ChimpDom7_sm$gene),
cross.area = nrow(both7sm),
c("Human", "Chimp"), scaled = TRUE,
fill = c("orange", "green"),
cex = 1.5,
cat.cex = 1.5,
cat.pos = c(320, 25),
cat.dist = .05) 
both7sm_same= both7sm %>% filter(ChimpPAS==HumanPAS)
nrow(both7sm_same)[1] 467both7sm_diff= both7sm %>% filter(ChimpPAS!=HumanPAS)
nrow(both7sm_diff)[1] 6location for same:
same7=ggplot(both7sm_same,aes(x=ChimpLoc,fill=ChimpLoc)) +geom_bar(stat = "count") +scale_fill_brewer(palette = "Dark2")+ labs(title="Matched Gene Matched PAS .7", y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
same7
both7sm_diff ChimpPAS gene ChimpLoc HumanPAS HumanLoc
1 human21024 CKS1B utr3 human21027 end
2 chimp137870 TMC8 utr3 human151981 utr3
3 human162402 MFSD12 utr3 human162401 utr3
4 chimp182091 MAT2A utr3 human184286 cds
5 chimp163919 IFNGR2 utr3 human215583 intron
6 human324647 NSUN5P1 utr5 human324646 utr5ggplot(BothDom7, aes(x=nPAS, fill=set)) + geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="Number of PAS per gene with .7 difference",y="genes")
HumanDom7_smLab= HumanDom7 %>% anti_join(ChimpDom7,by = "gene") %>% mutate(Set="Human")
ChimpDom7_smLab= ChimpDom7 %>% anti_join(HumanDom7,by = "gene") %>% mutate(Set="Chimp")
BothDom7_smLab=HumanDom7_smLab %>% bind_rows(ChimpDom7_smLab)
diffgenes7=ggplot(BothDom7_smLab,aes(x=loc, fill=Set))+ geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="Location of dominant PAS in genes \nnot matched between species: 0.7", y="genes")+ labs(title="Non Matched: 0.7", y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
diffgenes7
BothDom7_prop= BothDom7 %>% group_by(set, loc) %>% summarise(nLoc=n()) %>% ungroup() %>% group_by(set) %>% mutate(nSet=sum(nLoc), prop=nLoc/nSet)
proploc7=ggplot(BothDom7_prop, aes(x=loc,by=set, fill=set,y=prop)) + geom_bar(stat="identity",position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.7",y="Proportion",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")BothDom7_smLab_prop=BothDom7_smLab%>% group_by(set, loc) %>% summarise(nLoc=n()) %>% ungroup() %>% group_by(set) %>% mutate(nSet=sum(nLoc), prop=nLoc/nSet)
propDiff7=ggplot(BothDom7_smLab_prop, aes(x=loc,by=set, fill=set,y=prop)) + geom_bar(stat="identity",position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.7: Different Genes",y="Proportion",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")60% difference
HumanDom6=read.table("../data/DomDefGreaterX/Human_.6_dominantPAS.txt", col.names = MetaCol,stringsAsFactors = F) %>% mutate(set="Human6") %>% inner_join(PASnumHuman, by="gene")
nrow(HumanDom6)[1] 1014ChimpDom6=read.table("../data/DomDefGreaterX/Chimp_.6_dominantPAS.txt", col.names = MetaCol,stringsAsFactors = F) %>% mutate(set="Chimp6") %>% inner_join(PASnumChimp, by="gene")
nrow(ChimpDom6)[1] 1404BothDom6=HumanDom6 %>% bind_rows(ChimpDom6)
genloc6=ggplot(BothDom6, aes(x=loc, fill=set)) + geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.6",y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
genloc6
HumanDom6_sm=HumanDom6 %>% select(PAS, gene, loc) %>% rename(HumanPAS=PAS, HumanLoc=loc)
ChimpDom6_sm=ChimpDom6 %>% select(PAS, gene, loc)%>% rename(ChimpPAS=PAS, ChimpLoc=loc)
both6sm=ChimpDom6_sm %>% inner_join(HumanDom6_sm, by="gene")
nrow(both6sm)[1] 803grid.newpage()
venn.plot6 <- draw.pairwise.venn(area1 = length(HumanDom6_sm$gene),
area2 = length(ChimpDom6_sm$gene),
cross.area = nrow(both6sm),
c("Human", "Chimp"), scaled = TRUE,
fill = c("orange", "green"),
cex = 1.5,
cat.cex = 1.5,
cat.pos = c(320, 25),
cat.dist = .05) 
both6sm_same= both6sm %>% filter(ChimpPAS==HumanPAS)
nrow(both6sm_same)[1] 796both6sm_diff= both6sm %>% filter(ChimpPAS!=HumanPAS)
nrow(both6sm_diff)[1] 7location for same:
same6=ggplot(both6sm_same,aes(x=ChimpLoc,fill=ChimpLoc)) +geom_bar(stat = "count") +scale_fill_brewer(palette = "Dark2")+ labs(title="Matched Gene Matched PAS .6", y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
same6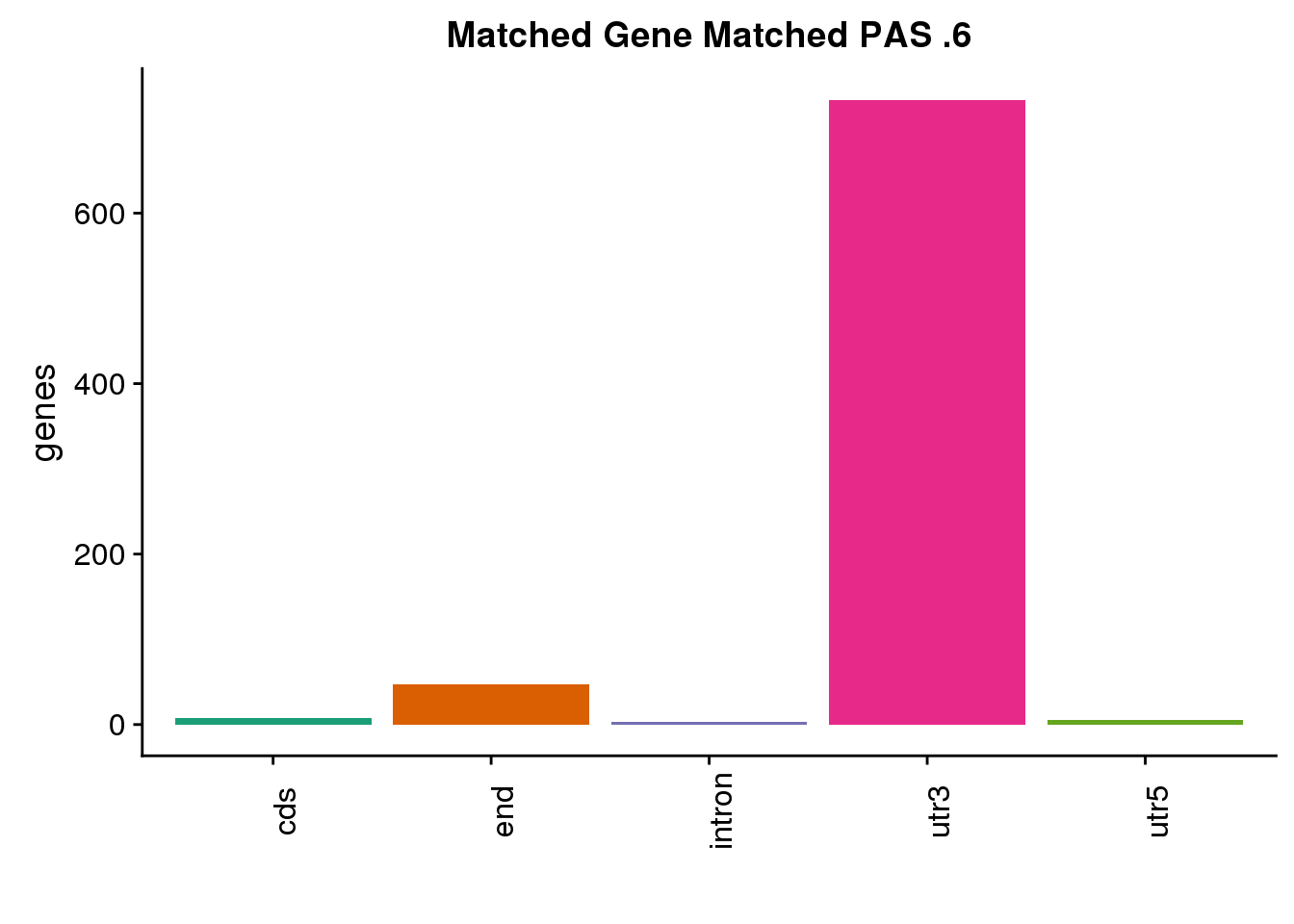
both6sm_diff ChimpPAS gene ChimpLoc HumanPAS HumanLoc
1 human21024 CKS1B utr3 human21027 end
2 human60067 FTH1 utr3 human60068 cds
3 chimp137870 TMC8 utr3 human151981 utr3
4 human162402 MFSD12 utr3 human162401 utr3
5 chimp182091 MAT2A utr3 human184286 cds
6 chimp163919 IFNGR2 utr3 human215583 intron
7 human324647 NSUN5P1 utr5 human324646 utr5ggplot(BothDom6, aes(x=nPAS, fill=set)) + geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="Number of PAS per gene with .6 difference",y="genes")
HumanDom6_smLab= HumanDom6 %>% anti_join(ChimpDom6,by = "gene") %>% mutate(Set="Human")
ChimpDom6_smLab= ChimpDom6 %>% anti_join(HumanDom6,by = "gene") %>% mutate(Set="Chimp")
BothDom6_smLab=HumanDom6_smLab %>% bind_rows(ChimpDom6_smLab)
diffgenes6=ggplot(BothDom6_smLab,aes(x=loc, fill=Set))+ geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="Non Matched: 0.6", y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
diffgenes6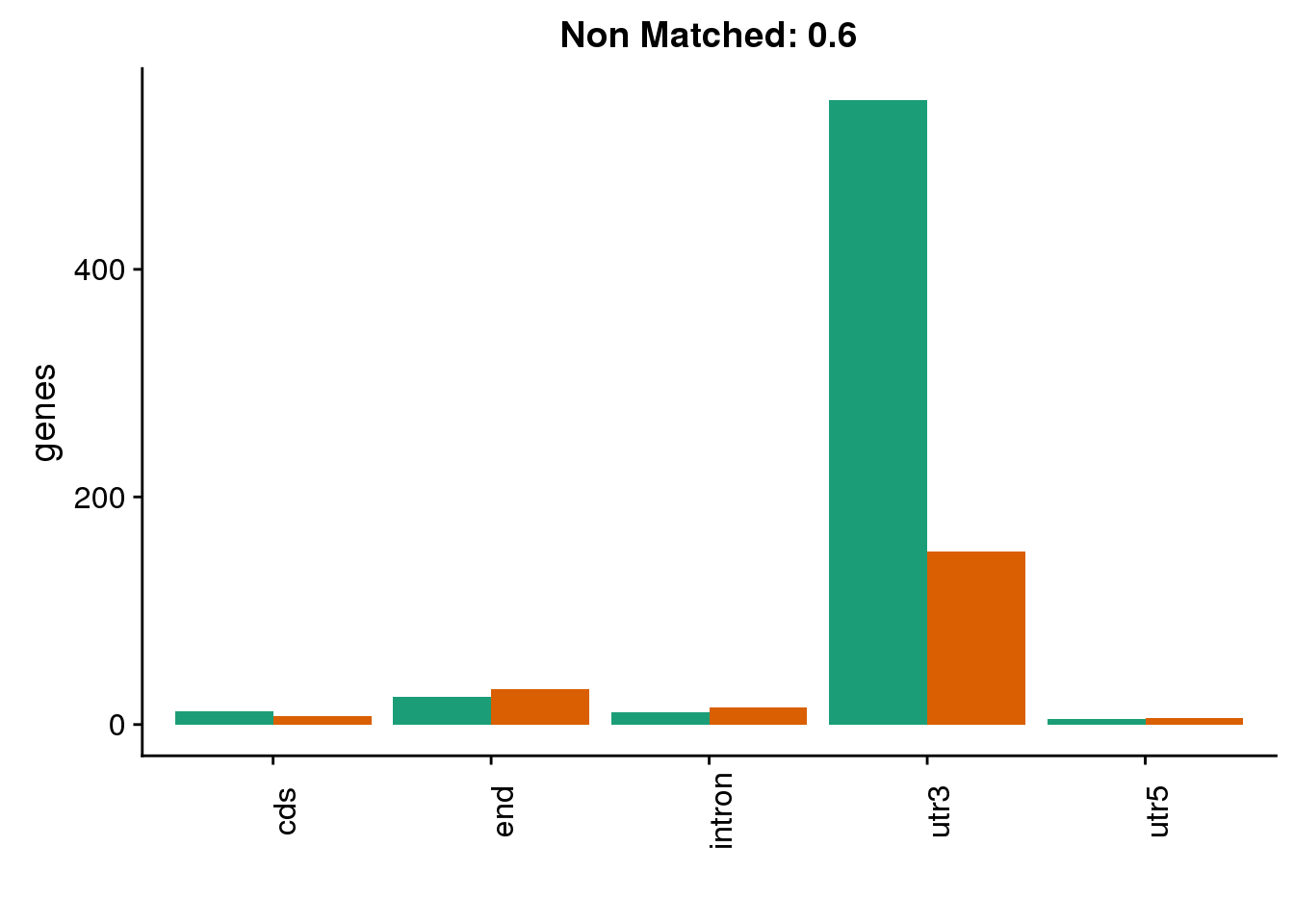
BothDom6_prop= BothDom6 %>% group_by(set, loc) %>% summarise(nLoc=n()) %>% ungroup() %>% group_by(set) %>% mutate(nSet=sum(nLoc), prop=nLoc/nSet)
proploc6=ggplot(BothDom6_prop, aes(x=loc,by=set, fill=set,y=prop)) + geom_bar(stat="identity",position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.6",y="Proportion",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")BothDom6_smLab_prop=BothDom6_smLab%>% group_by(set, loc) %>% summarise(nLoc=n()) %>% ungroup() %>% group_by(set) %>% mutate(nSet=sum(nLoc), prop=nLoc/nSet)
propDiff6=ggplot(BothDom6_smLab_prop, aes(x=loc,by=set, fill=set,y=prop)) + geom_bar(stat="identity",position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.6: Different Genes",y="Proportion",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")50% difference
HumanDom5=read.table("../data/DomDefGreaterX/Human_.5_dominantPAS.txt", col.names = MetaCol,stringsAsFactors = F) %>% mutate(set="Human5") %>% inner_join(PASnumHuman, by="gene")
nrow(HumanDom5)[1] 1404ChimpDom5=read.table("../data/DomDefGreaterX/Chimp_.5_dominantPAS.txt", col.names = MetaCol,stringsAsFactors = F) %>% mutate(set="Chimp5") %>% inner_join(PASnumChimp, by="gene")
nrow(ChimpDom5)[1] 1908BothDom5=HumanDom5 %>% bind_rows(ChimpDom5)
genloc5=ggplot(BothDom5, aes(x=loc, fill=set)) + geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.5",y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
genloc5
HumanDom5_sm=HumanDom5 %>% select(PAS, gene, loc) %>% rename(HumanPAS=PAS, HumanLoc=loc)
ChimpDom5_sm=ChimpDom5 %>% select(PAS, gene, loc)%>% rename(ChimpPAS=PAS, ChimpLoc=loc)
both5sm=ChimpDom5_sm %>% inner_join(HumanDom5_sm, by="gene")
nrow(both5sm)[1] 1160grid.newpage()
venn.plot5 <- draw.pairwise.venn(area1 = length(HumanDom5_sm$gene),
area2 = length(ChimpDom5_sm$gene),
cross.area = nrow(both5sm),
c("Human", "Chimp"), scaled = TRUE,
fill = c("orange", "green"),
cex = 1.5,
cat.cex = 1.5,
cat.pos = c(320, 25),
cat.dist = .05) 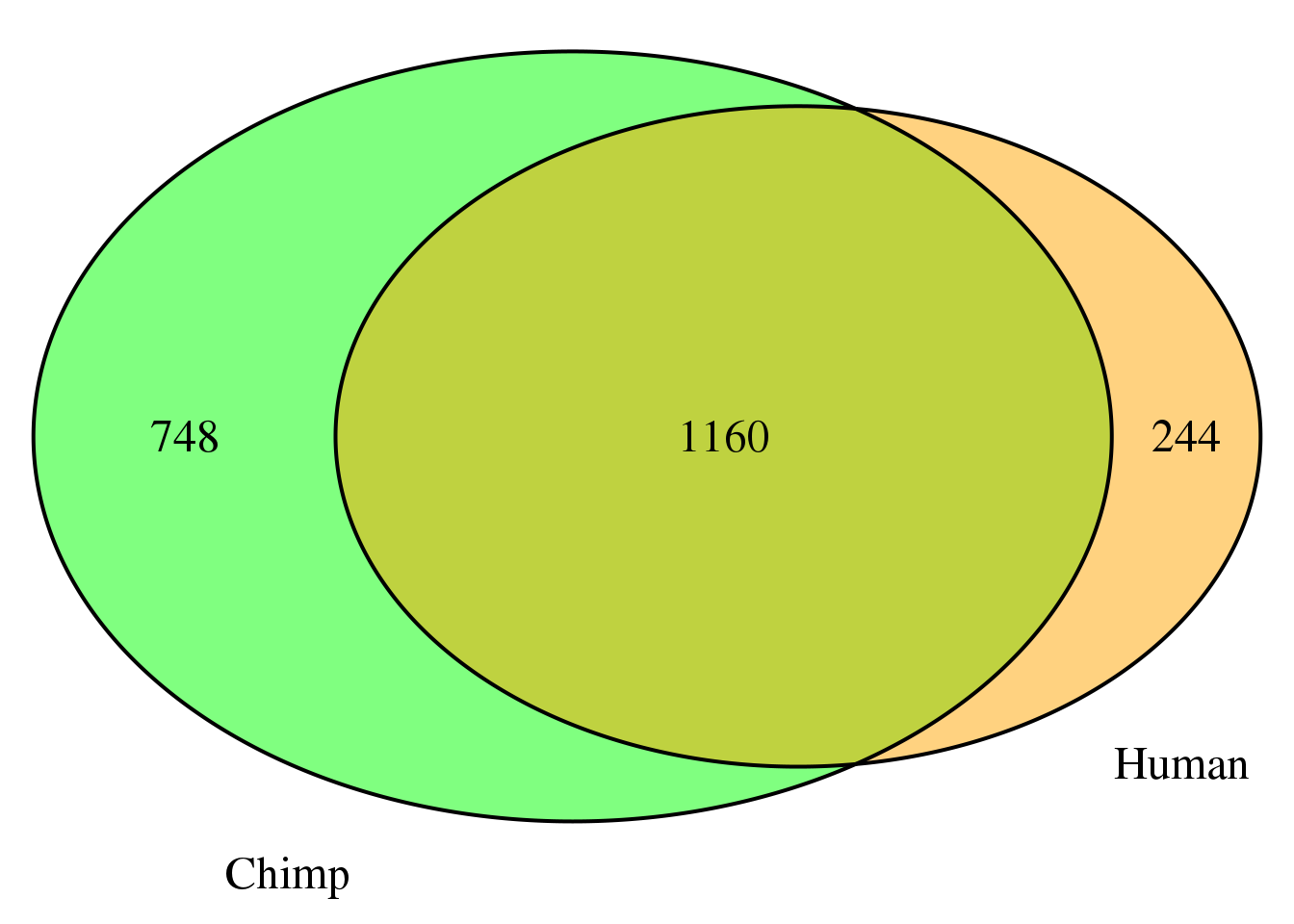
both5sm_same= both5sm %>% filter(ChimpPAS==HumanPAS)
nrow(both5sm_same)[1] 1145both5sm_diff= both5sm %>% filter(ChimpPAS!=HumanPAS)
nrow(both5sm_diff)[1] 15location for same:
same5=ggplot(both5sm_same,aes(x=ChimpLoc,fill=ChimpLoc)) +geom_bar(stat = "count") +scale_fill_brewer(palette = "Dark2")+ labs(title="Matched Gene Matched PAS .5", y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
same5
both5sm_diff ChimpPAS gene ChimpLoc HumanPAS HumanLoc
1 human21024 CKS1B utr3 human21027 end
2 human48359 PGAM1 utr3 human48357 intron
3 human60067 FTH1 utr3 human60068 cds
4 human68171 DDX6 utr3 human68160 utr3
5 chimp137870 TMC8 utr3 human151981 utr3
6 human162402 MFSD12 utr3 human162401 utr3
7 chimp182091 MAT2A utr3 human184286 cds
8 human205442 RBCK1 utr3 human205434 utr3
9 chimp163919 IFNGR2 utr3 human215583 intron
10 chimp165747 LSS utr3 human217562 intron
11 human225614 RPL32 utr3 human225615 utr3
12 human250766 NCBP2 utr3 chimp224588 cds
13 human324647 NSUN5P1 utr5 human324646 utr5
14 chimp308202 FABP5 utr3 human345717 cds
15 human366498 SLC27A4 intron human366500 utr3ggplot(BothDom5, aes(x=nPAS, fill=set)) + geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="Number of PAS per gene with .5 difference",y="genes")
HumanDom5_smLab= HumanDom5 %>% anti_join(ChimpDom5,by = "gene") %>% mutate(Set="Human")
ChimpDom5_smLab= ChimpDom5 %>% anti_join(HumanDom5,by = "gene") %>% mutate(Set="Chimp")
BothDom5_smLab=HumanDom5_smLab %>% bind_rows(ChimpDom5_smLab)
diffgenes5=ggplot(BothDom5_smLab,aes(x=loc, fill=Set))+ geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="Non Matched: 0.5", y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
diffgenes5
BothDom5_prop= BothDom5 %>% group_by(set, loc) %>% summarise(nLoc=n()) %>% ungroup() %>% group_by(set) %>% mutate(nSet=sum(nLoc), prop=nLoc/nSet)
proploc5=ggplot(BothDom5_prop, aes(x=loc,by=set, fill=set,y=prop)) + geom_bar(stat="identity",position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.5",y="Proportion",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")BothDom5_smLab_prop=BothDom5_smLab%>% group_by(set, loc) %>% summarise(nLoc=n()) %>% ungroup() %>% group_by(set) %>% mutate(nSet=sum(nLoc), prop=nLoc/nSet)
propDiff5=ggplot(BothDom5_smLab_prop, aes(x=loc,by=set, fill=set,y=prop)) + geom_bar(stat="identity",position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.5: Different Genes",y="Proportion",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")40% difference
HumanDom4=read.table("../data/DomDefGreaterX/Human_.4_dominantPAS.txt", col.names = MetaCol,stringsAsFactors = F) %>% mutate(set="Human4") %>% inner_join(PASnumHuman, by="gene")
nrow(HumanDom4)[1] 1833ChimpDom4=read.table("../data/DomDefGreaterX/Chimp_.4_dominantPAS.txt", col.names = MetaCol,stringsAsFactors = F) %>% mutate(set="Chimp4") %>% inner_join(PASnumChimp, by="gene")
nrow(ChimpDom4)[1] 2478BothDom4=HumanDom4 %>% bind_rows(ChimpDom4)
genloc4=ggplot(BothDom4, aes(x=loc, fill=set)) + geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.4",y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
genloc4
HumanDom4_sm=HumanDom4 %>% select(PAS, gene, loc) %>% rename(HumanPAS=PAS, HumanLoc=loc)
ChimpDom4_sm=ChimpDom4 %>% select(PAS, gene, loc)%>% rename(ChimpPAS=PAS, ChimpLoc=loc)
both4sm=ChimpDom4_sm %>% inner_join(HumanDom4_sm, by="gene")
nrow(both4sm)[1] 1565grid.newpage()
venn.plot4 <- draw.pairwise.venn(area1 = length(HumanDom4_sm$gene),
area2 = length(ChimpDom4_sm$gene),
cross.area = nrow(both4sm),
c("Human", "Chimp"), scaled = TRUE,
fill = c("orange", "green"),
cex = 1.5,
cat.cex = 1.5,
cat.pos = c(320, 25),
cat.dist = .05) 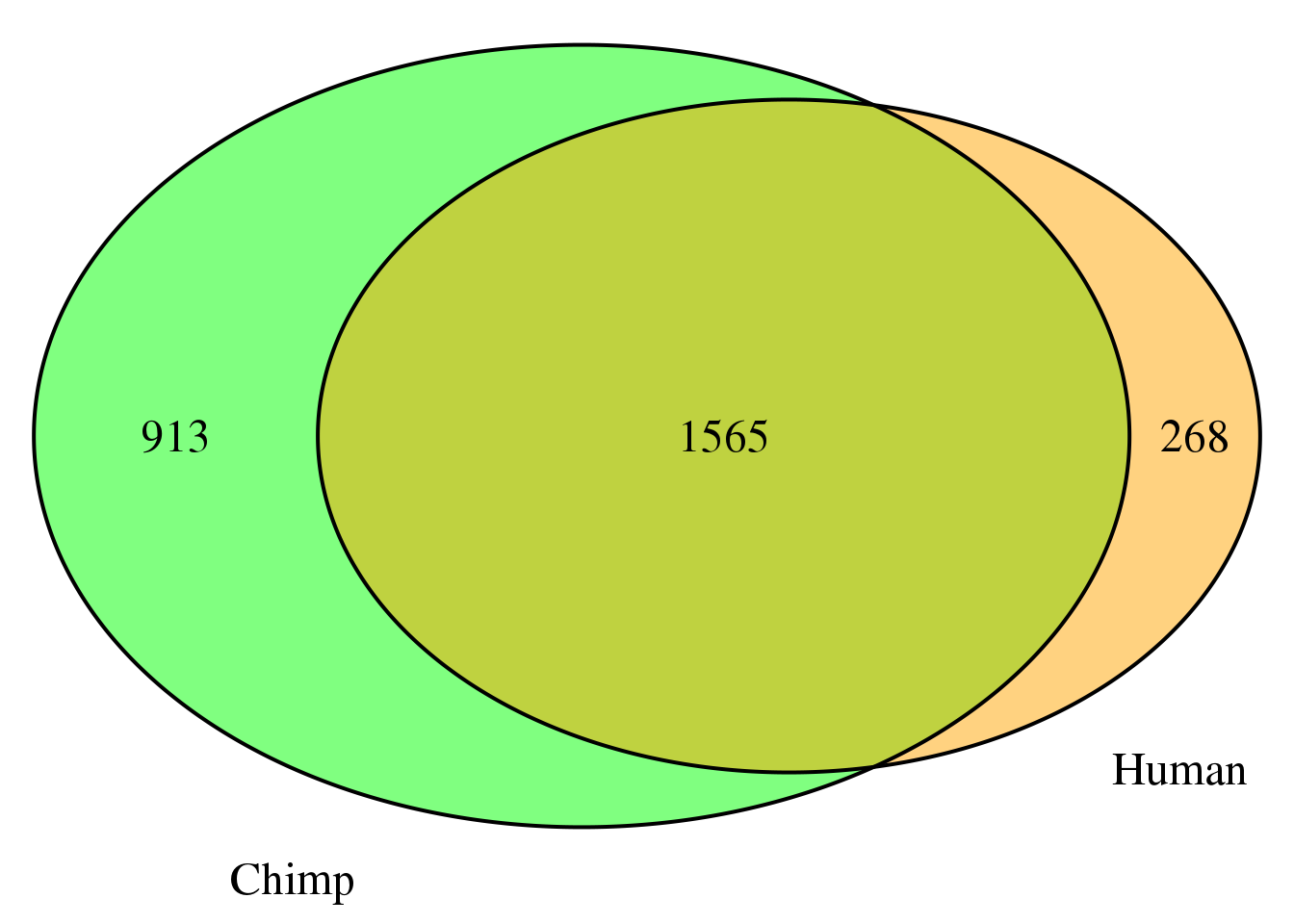
both4sm_same= both4sm %>% filter(ChimpPAS==HumanPAS)
nrow(both4sm_same)[1] 1543both4sm_diff= both4sm %>% filter(ChimpPAS!=HumanPAS)
nrow(both4sm_diff)[1] 22location for same:
same4=ggplot(both4sm_same,aes(x=ChimpLoc,fill=ChimpLoc)) +geom_bar(stat = "count") +scale_fill_brewer(palette = "Dark2")+ labs(title="Matched Gene Matched PAS .4", y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
same4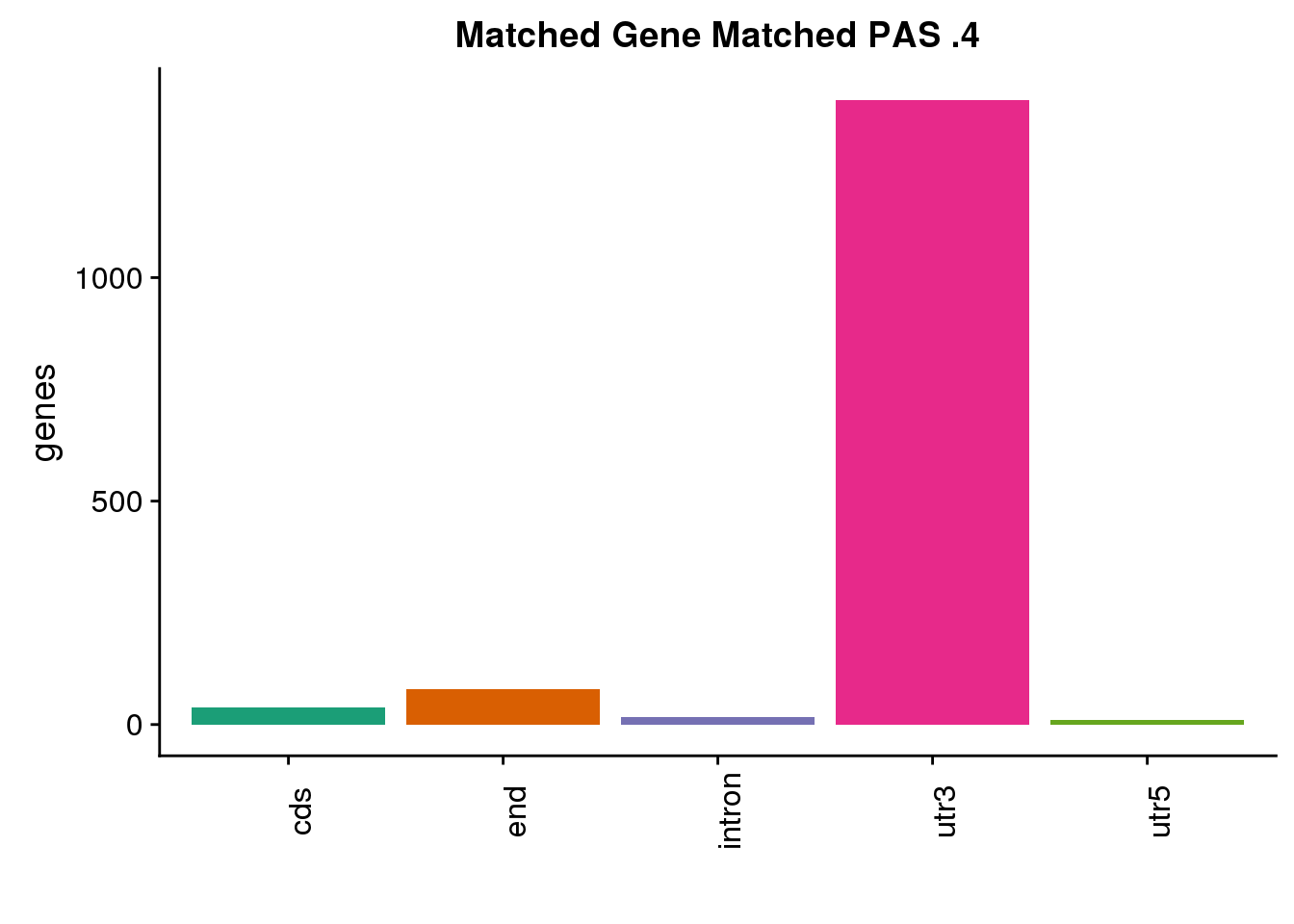
22 is enough to start to plot:
both4sm_diff_g= both4sm_diff %>% select(gene, HumanLoc, ChimpLoc) %>% gather('Species', 'Loc', -gene)
diffDom4=ggplot(both4sm_diff_g, aes(x=Loc, fill=Species))+ geom_bar(stat="count",position = "dodge") +scale_fill_brewer(palette = "Dark2")+ labs(y='Genes', title="Different Dominant PAS: 0.4",x="") +theme(axis.text.x = element_text(angle = 90),legend.position = "none")
diffDom4
ggplot(BothDom4, aes(x=nPAS, fill=set)) + geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="Number of PAS per gene with .4 difference",y="genes")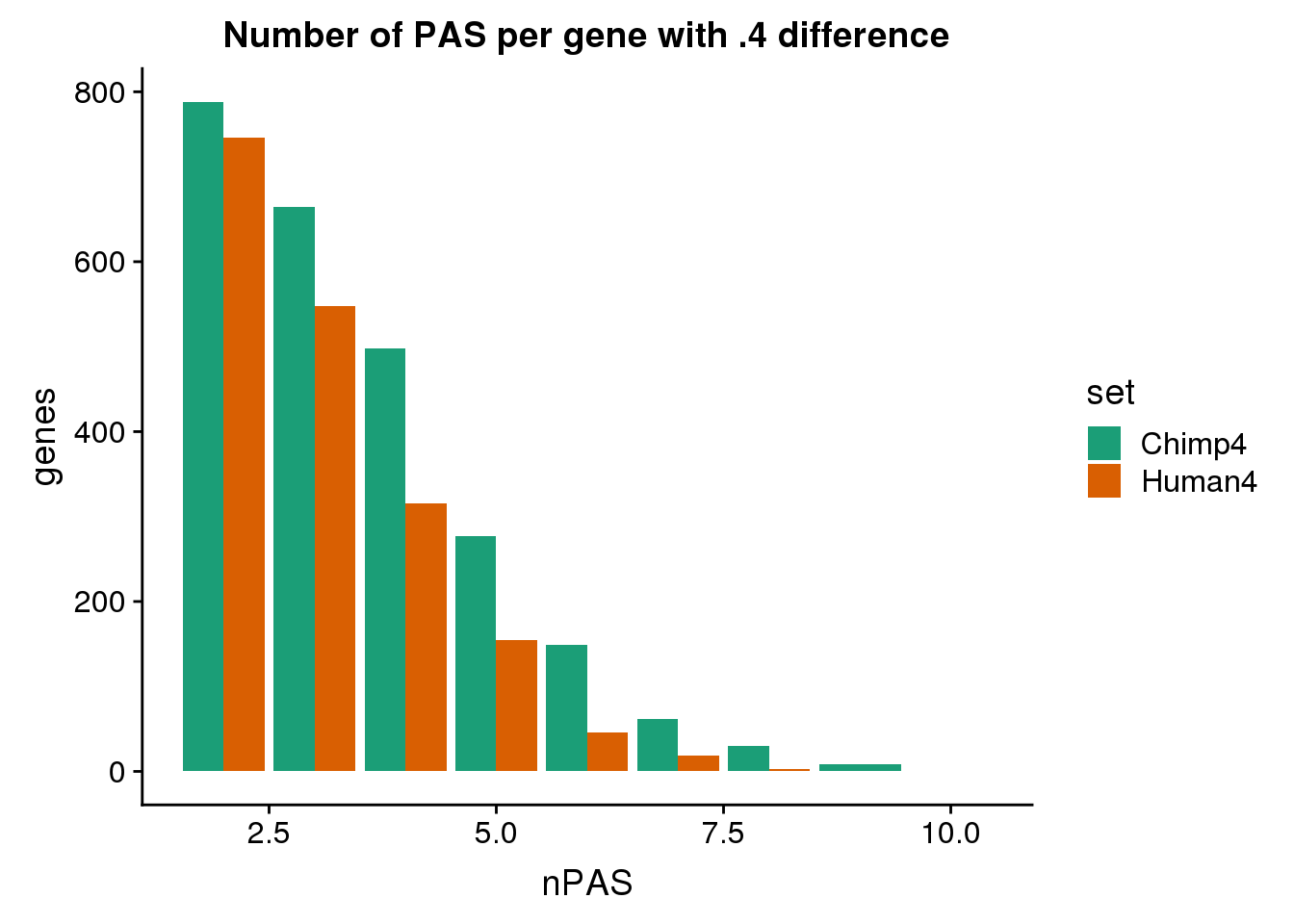
HumanDom4_smLab= HumanDom4 %>% anti_join(ChimpDom4,by = "gene") %>% mutate(Set="Human")
ChimpDom4_smLab= ChimpDom4 %>% anti_join(HumanDom4,by = "gene") %>% mutate(Set="Chimp")
BothDom4_smLab=HumanDom4_smLab %>% bind_rows(ChimpDom4_smLab)
diffgenes4=ggplot(BothDom4_smLab,aes(x=loc, fill=Set))+ geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="Non Matched: 0.4", y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
diffgenes4
| Version | Author | Date |
|---|---|---|
| 385fe04 | brimittleman | 2020-04-16 |
BothDom4_prop= BothDom4 %>% group_by(set, loc) %>% summarise(nLoc=n()) %>% ungroup() %>% group_by(set) %>% mutate(nSet=sum(nLoc), prop=nLoc/nSet)
proploc4=ggplot(BothDom4_prop, aes(x=loc,by=set, fill=set,y=prop)) + geom_bar(stat="identity",position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.4",y="Proportion",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")BothDom4_smLab_prop=BothDom4_smLab%>% group_by(set, loc) %>% summarise(nLoc=n()) %>% ungroup() %>% group_by(set) %>% mutate(nSet=sum(nLoc), prop=nLoc/nSet)
propDiff4=ggplot(BothDom4_smLab_prop, aes(x=loc,by=set, fill=set,y=prop)) + geom_bar(stat="identity",position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.4: Different Genes",y="Proportion",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")30% difference
HumanDom3=read.table("../data/DomDefGreaterX/Human_.3_dominantPAS.txt", col.names = MetaCol,stringsAsFactors = F) %>% mutate(set="Human3") %>% inner_join(PASnumHuman, by="gene")
nrow(HumanDom3)[1] 2403ChimpDom3=read.table("../data/DomDefGreaterX/Chimp_.3_dominantPAS.txt", col.names = MetaCol,stringsAsFactors = F) %>% mutate(set="Chimp3") %>% inner_join(PASnumChimp, by="gene")
nrow(ChimpDom3)[1] 3127BothDom3=HumanDom3 %>% bind_rows(ChimpDom3)
genloc3=ggplot(BothDom3, aes(x=loc, fill=set)) + geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.3",y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
genloc3
HumanDom3_sm=HumanDom3 %>% select(PAS, gene, loc) %>% rename(HumanPAS=PAS, HumanLoc=loc)
ChimpDom3_sm=ChimpDom3 %>% select(PAS, gene, loc)%>% rename(ChimpPAS=PAS, ChimpLoc=loc)
both3sm=ChimpDom3_sm %>% inner_join(HumanDom3_sm, by="gene")
nrow(both3sm)[1] 2069grid.newpage()
venn.plot3 <- draw.pairwise.venn(area1 = length(HumanDom3_sm$gene),
area2 = length(ChimpDom3_sm$gene),
cross.area = nrow(both3sm),
c("Human", "Chimp"), scaled = TRUE,
fill = c("orange", "green"),
cex = 1.5,
cat.cex = 1.5,
cat.pos = c(320, 25),
cat.dist = .05) 
both3sm_same= both3sm %>% filter(ChimpPAS==HumanPAS)
nrow(both3sm_same)[1] 2031both3sm_diff= both3sm %>% filter(ChimpPAS!=HumanPAS)
nrow(both3sm_diff)[1] 38location for same:
same3=ggplot(both3sm_same,aes(x=ChimpLoc,fill=ChimpLoc)) +geom_bar(stat = "count") +scale_fill_brewer(palette = "Dark2")+ labs(title="Matched Gene Matched PAS .3", y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
same3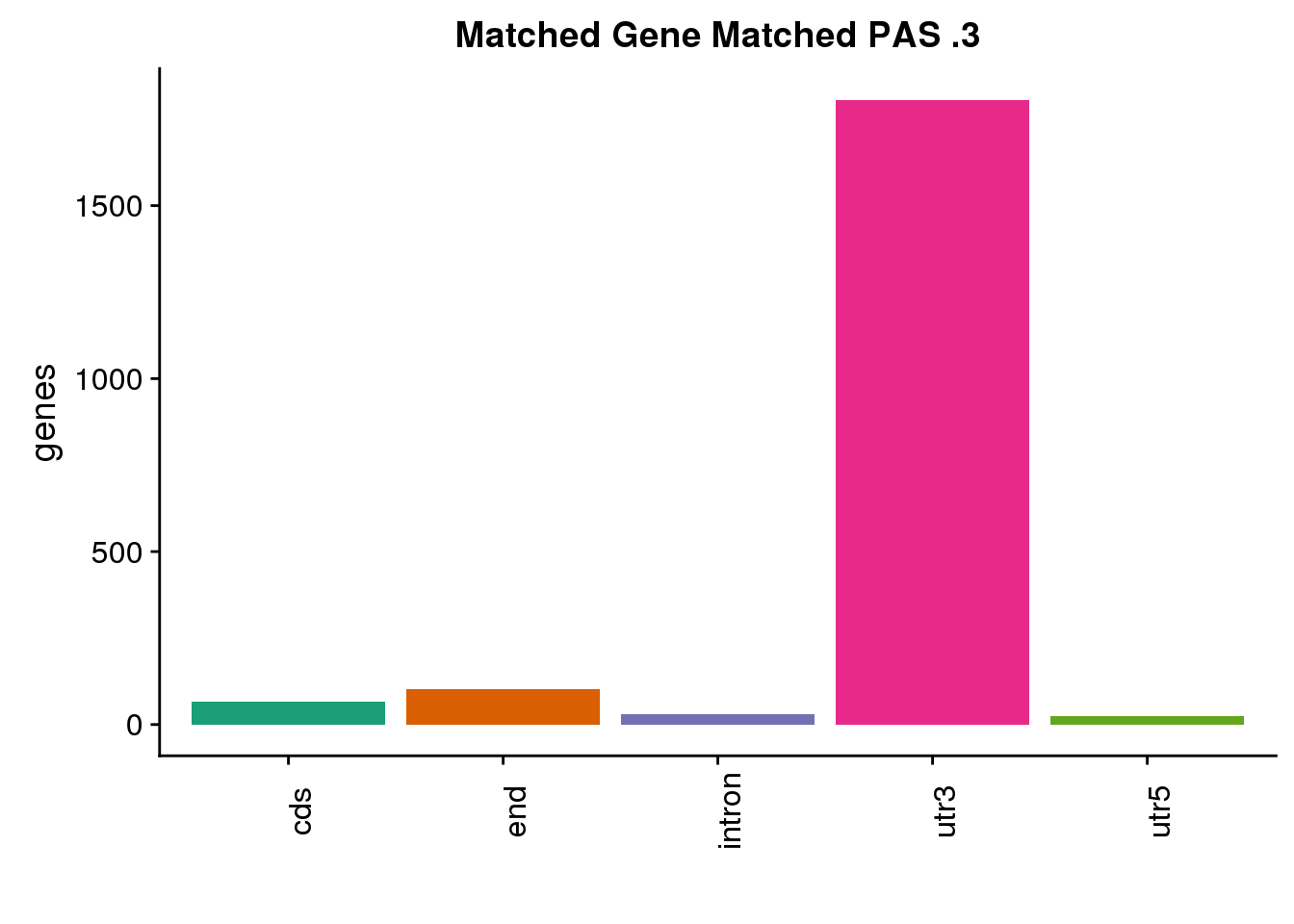
both3sm_diff_g= both3sm_diff %>% select(gene, HumanLoc, ChimpLoc) %>% gather('Species', 'Loc', -gene)
diffDom3=ggplot(both3sm_diff_g, aes(x=Loc, fill=Species))+ geom_bar(stat="count",position = "dodge") +scale_fill_brewer(palette = "Dark2")+ labs(y='Genes', title="Different Dominant PAS: 0.3",x="") +theme(axis.text.x = element_text(angle = 90),legend.position = "none")
diffDom3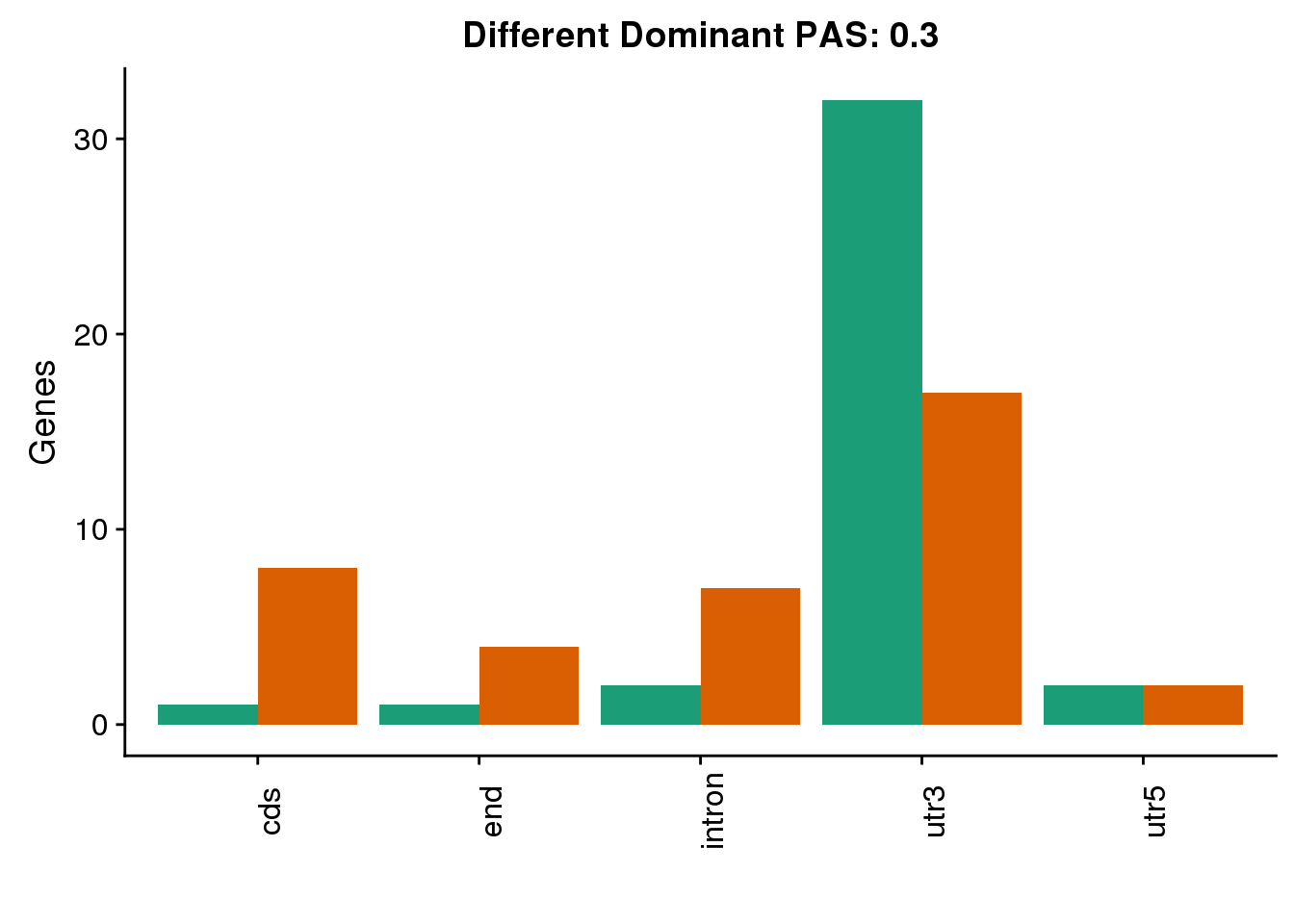
| Version | Author | Date |
|---|---|---|
| 385fe04 | brimittleman | 2020-04-16 |
ggplot(BothDom3, aes(x=nPAS, fill=set)) + geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="Number of PAS per gene with .3 difference",y="genes")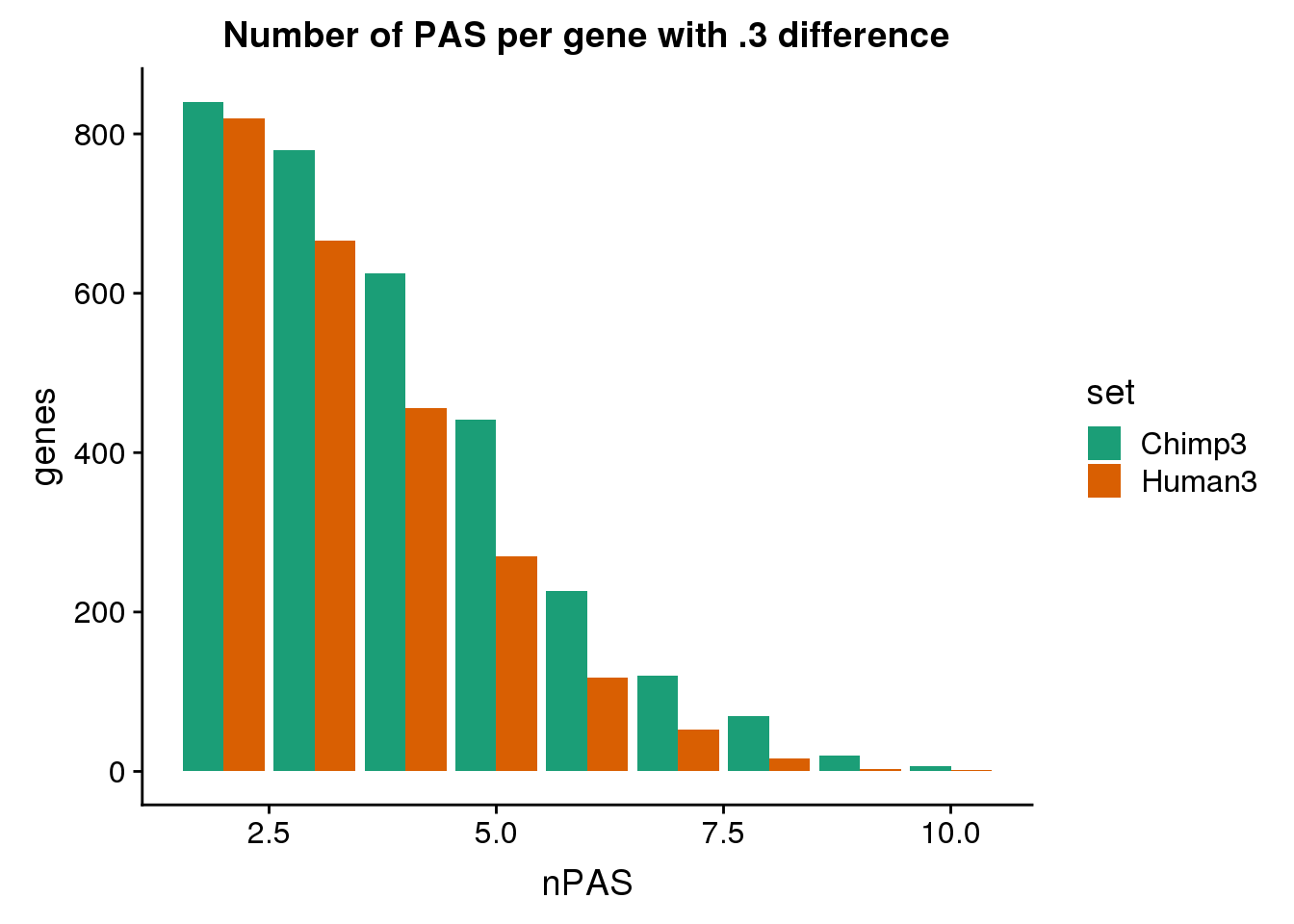
HumanDom3_smLab= HumanDom3 %>% anti_join(ChimpDom3,by = "gene") %>% mutate(Set="Human")
ChimpDom3_smLab= ChimpDom3 %>% anti_join(HumanDom3,by = "gene") %>% mutate(Set="Chimp")
BothDom3_smLab=HumanDom3_smLab %>% bind_rows(ChimpDom3_smLab)
diffgenes3=ggplot(BothDom3_smLab,aes(x=loc, fill=Set))+ geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="Non Matched: 0.3", y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
diffgenes3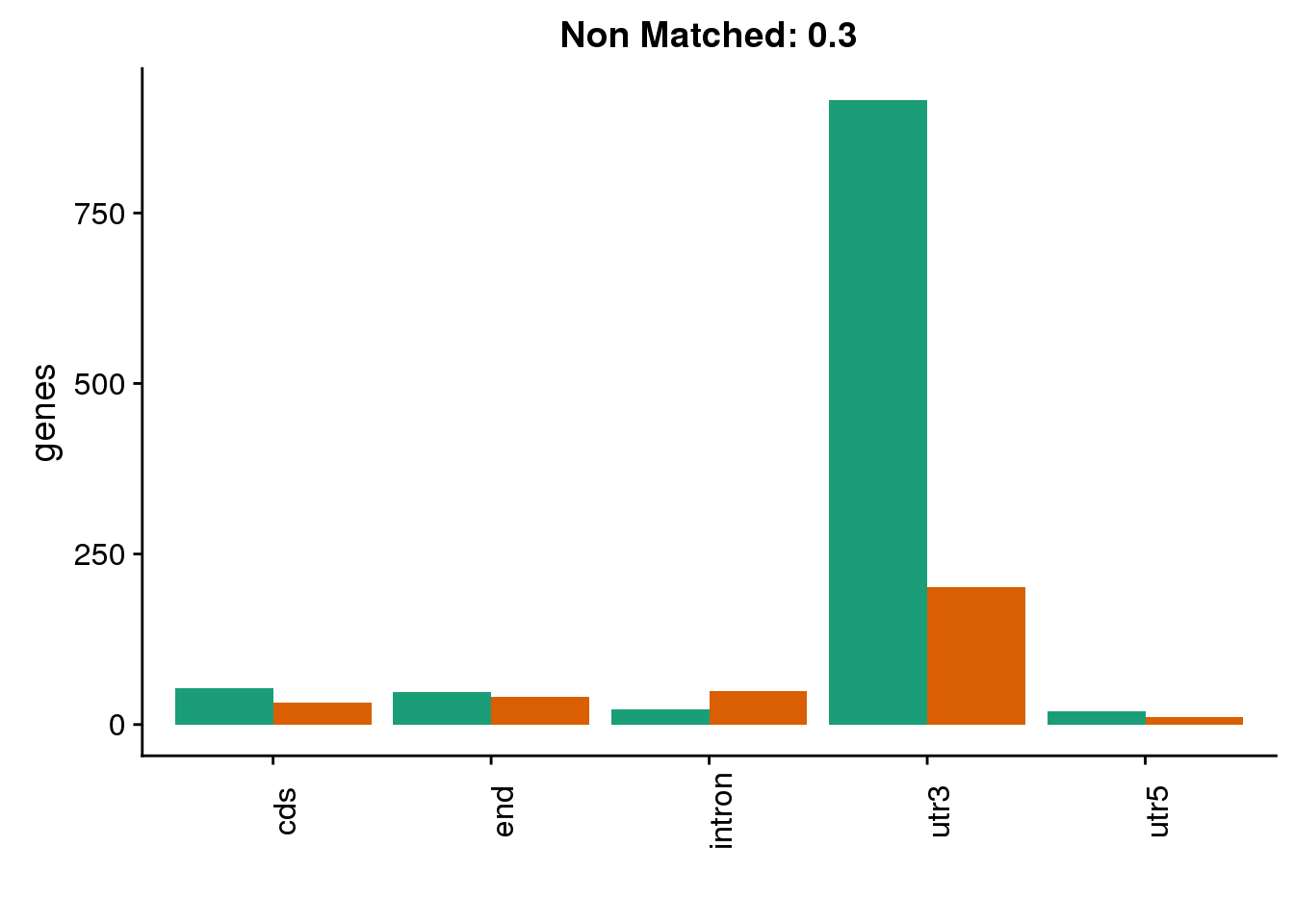
| Version | Author | Date |
|---|---|---|
| 385fe04 | brimittleman | 2020-04-16 |
BothDom3_prop= BothDom3 %>% group_by(set, loc) %>% summarise(nLoc=n()) %>% ungroup() %>% group_by(set) %>% mutate(nSet=sum(nLoc), prop=nLoc/nSet)
proploc3=ggplot(BothDom3_prop, aes(x=loc,by=set, fill=set,y=prop)) + geom_bar(stat="identity",position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.3",y="Proportion",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")BothDom3_prop= BothDom3 %>% group_by(set, loc) %>% summarise(nLoc=n()) %>% ungroup() %>% group_by(set) %>% mutate(nSet=sum(nLoc), prop=nLoc/nSet)
proploc3=ggplot(BothDom3_prop, aes(x=loc,by=set, fill=set,y=prop)) + geom_bar(stat="identity",position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.3",y="Proportion",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")BothDom3_smLab_prop=BothDom3_smLab%>% group_by(set, loc) %>% summarise(nLoc=n()) %>% ungroup() %>% group_by(set) %>% mutate(nSet=sum(nLoc), prop=nLoc/nSet)
propDiff3=ggplot(BothDom3_smLab_prop, aes(x=loc,by=set, fill=set,y=prop)) + geom_bar(stat="identity",position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.3: Different Genes",y="Proportion",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")20% difference
HumanDom2=read.table("../data/DomDefGreaterX/Human_.2_dominantPAS.txt", col.names = MetaCol,stringsAsFactors = F) %>% mutate(set="Human2") %>% inner_join(PASnumHuman, by="gene")
nrow(HumanDom2)[1] 3183ChimpDom2=read.table("../data/DomDefGreaterX/Chimp_.2_dominantPAS.txt", col.names = MetaCol,stringsAsFactors = F) %>% mutate(set="Chimp2") %>% inner_join(PASnumChimp, by="gene")
nrow(ChimpDom2)[1] 3969BothDom2=HumanDom2 %>% bind_rows(ChimpDom2)
genloc2=ggplot(BothDom2, aes(x=loc, fill=set)) + geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.2",y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
genloc2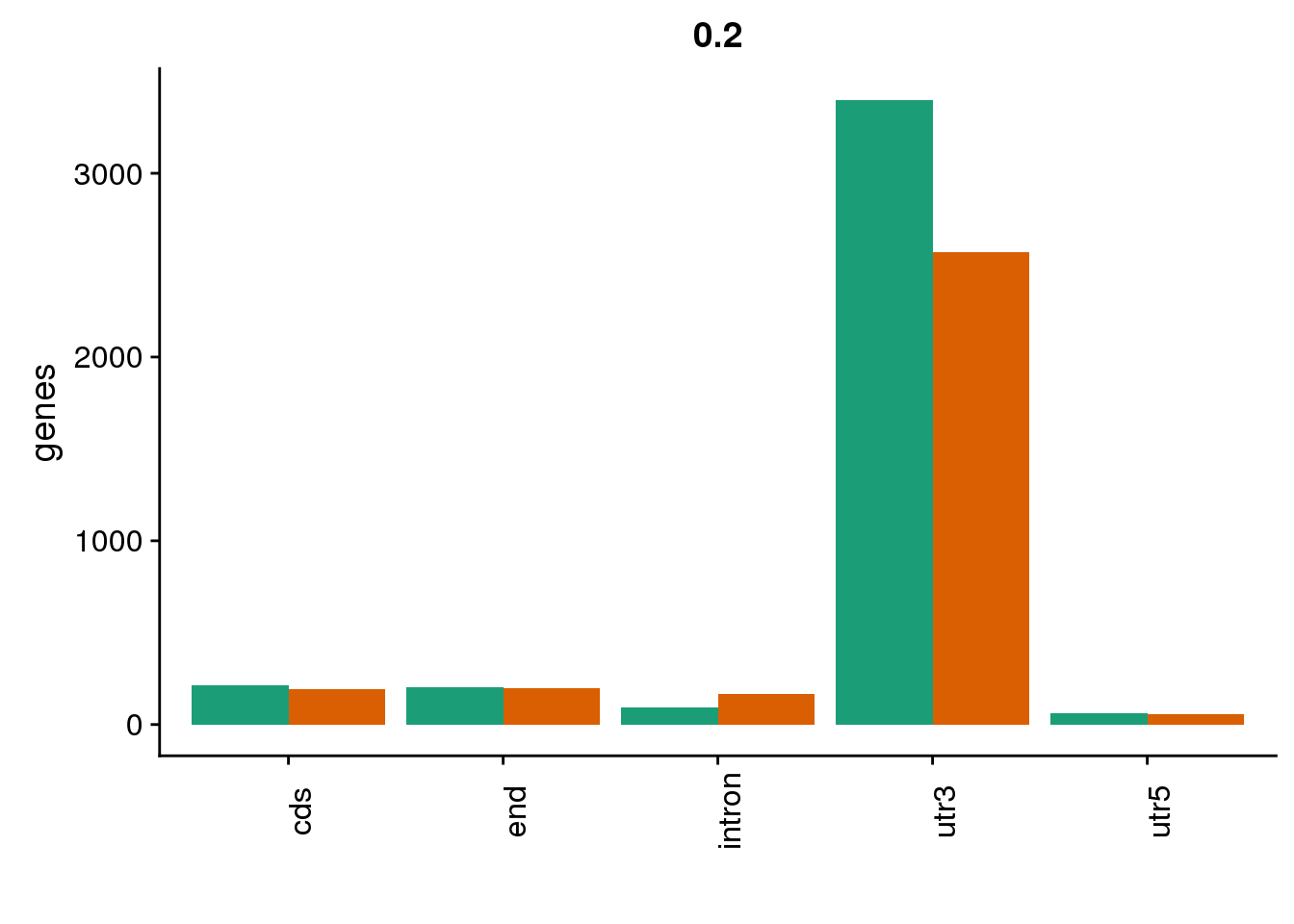
HumanDom2_sm=HumanDom2 %>% select(PAS, gene, loc) %>% rename(HumanPAS=PAS, HumanLoc=loc)
ChimpDom2_sm=ChimpDom2 %>% select(PAS, gene, loc)%>% rename(ChimpPAS=PAS, ChimpLoc=loc)
both2sm=ChimpDom2_sm %>% inner_join(HumanDom2_sm, by="gene")
nrow(both2sm)[1] 2711grid.newpage()
venn.plot2 <- draw.pairwise.venn(area1 = length(HumanDom2_sm$gene),
area2 = length(ChimpDom2_sm$gene),
cross.area = nrow(both2sm),
c("Human", "Chimp"), scaled = TRUE,
fill = c("orange", "green"),
cex = 1.5,
cat.cex = 1.5,
cat.pos = c(320, 25),
cat.dist = .05) 
both2sm_same= both2sm %>% filter(ChimpPAS==HumanPAS)
nrow(both2sm_same)[1] 2634both2sm_diff= both2sm %>% filter(ChimpPAS!=HumanPAS)
nrow(both2sm_diff)[1] 77location for same:
same2=ggplot(both2sm_same,aes(x=ChimpLoc,fill=ChimpLoc)) +geom_bar(stat = "count") +scale_fill_brewer(palette = "Dark2")+ labs(title="Matched Gene Matched PAS .2", y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
same2
both2sm_diff_g= both2sm_diff %>% select(gene, HumanLoc, ChimpLoc) %>% gather('Species', 'Loc', -gene)
diffDom2=ggplot(both2sm_diff_g, aes(x=Loc, fill=Species))+ geom_bar(stat="count",position = "dodge") +scale_fill_brewer(palette = "Dark2")+ labs(y='Genes', title="Different Dominant PAS: 0.2",x="") +theme(axis.text.x = element_text(angle = 90),legend.position = "none")
diffDom2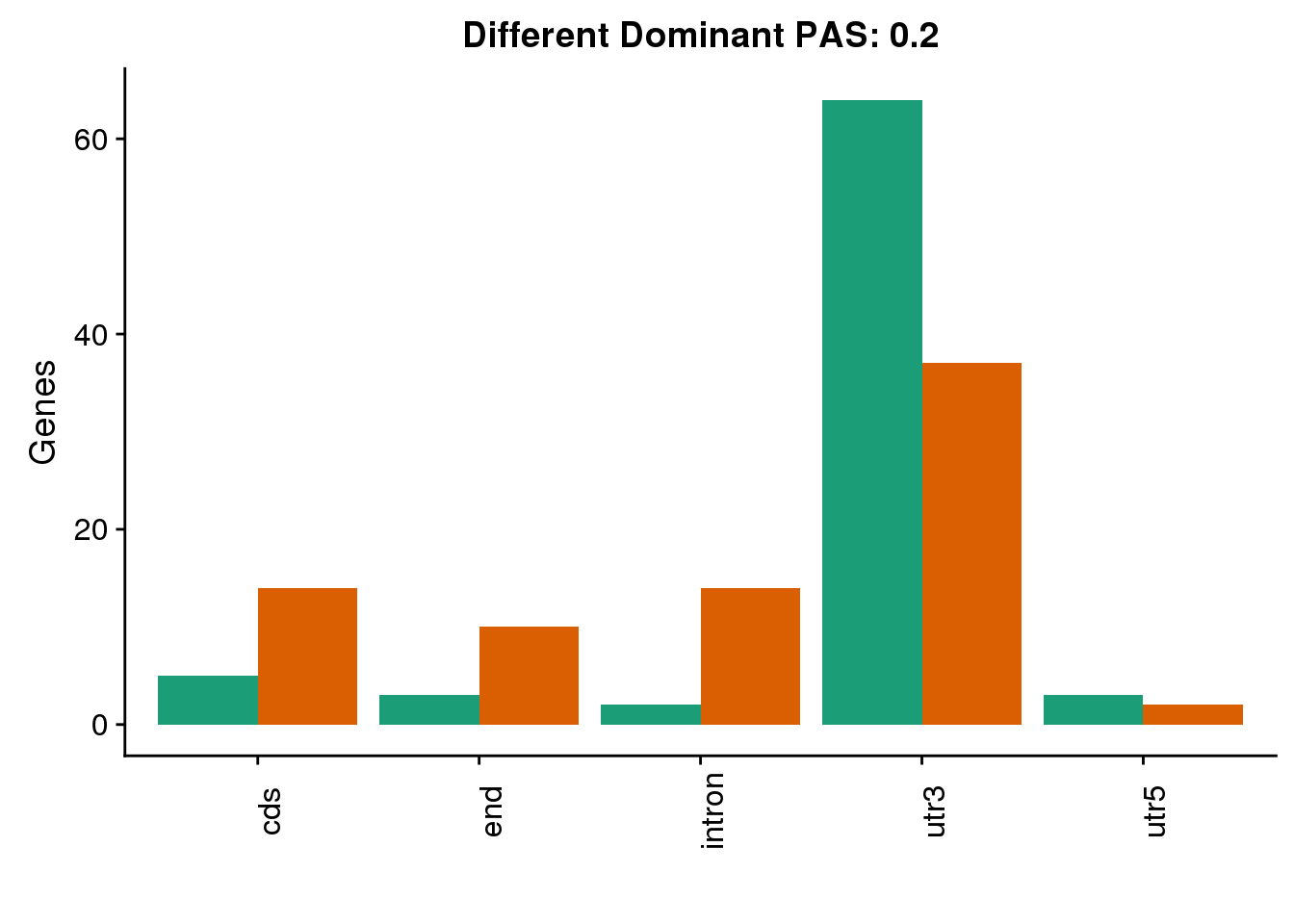
ggplot(BothDom2, aes(x=nPAS, fill=set)) + geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="Number of PAS per gene with .2 difference",y="genes")
| Version | Author | Date |
|---|---|---|
| 385fe04 | brimittleman | 2020-04-16 |
HumanDom2_smLab= HumanDom2 %>% anti_join(ChimpDom2,by = "gene") %>% mutate(Set="Human")
ChimpDom2_smLab= ChimpDom2 %>% anti_join(HumanDom2,by = "gene") %>% mutate(Set="Chimp")
BothDom2_smLab=HumanDom2_smLab %>% bind_rows(ChimpDom2_smLab)
diffgenes2=ggplot(BothDom2_smLab,aes(x=loc, fill=Set))+ geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="Non Matched: 0.2", y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
diffgenes2
| Version | Author | Date |
|---|---|---|
| 385fe04 | brimittleman | 2020-04-16 |
BothDom2_prop= BothDom2 %>% group_by(set, loc) %>% summarise(nLoc=n()) %>% ungroup() %>% group_by(set) %>% mutate(nSet=sum(nLoc), prop=nLoc/nSet)
proploc2=ggplot(BothDom2_prop, aes(x=loc,by=set, fill=set,y=prop)) + geom_bar(stat="identity",position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.2",y="Proportion",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")BothDom2_smLab_prop=BothDom2_smLab%>% group_by(set, loc) %>% summarise(nLoc=n()) %>% ungroup() %>% group_by(set) %>% mutate(nSet=sum(nLoc), prop=nLoc/nSet)
propDiff2=ggplot(BothDom2_smLab_prop, aes(x=loc,by=set, fill=set,y=prop)) + geom_bar(stat="identity",position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.2: Different Genes",y="Proportion",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")10% difference
HumanDom1=read.table("../data/DomDefGreaterX/Human_.1_dominantPAS.txt", col.names = MetaCol,stringsAsFactors = F) %>% mutate(set="Human1") %>% inner_join(PASnumHuman, by="gene")
nrow(HumanDom1)[1] 4417ChimpDom1=read.table("../data/DomDefGreaterX/Chimp_.1_dominantPAS.txt", col.names = MetaCol,stringsAsFactors = F) %>% mutate(set="Chimp1") %>% inner_join(PASnumChimp, by="gene")
nrow(ChimpDom1)[1] 5254BothDom1=HumanDom1 %>% bind_rows(ChimpDom1)
genloc1=ggplot(BothDom1, aes(x=loc, fill=set)) + geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.1",y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
genloc1
| Version | Author | Date |
|---|---|---|
| 385fe04 | brimittleman | 2020-04-16 |
HumanDom1_sm=HumanDom1 %>% select(PAS, gene, loc) %>% rename(HumanPAS=PAS, HumanLoc=loc)
ChimpDom1_sm=ChimpDom1 %>% select(PAS, gene, loc)%>% rename(ChimpPAS=PAS, ChimpLoc=loc)
both1sm=ChimpDom1_sm %>% inner_join(HumanDom1_sm, by="gene")
nrow(both1sm)[1] 3814grid.newpage()
venn.plot1 <- draw.pairwise.venn(area1 = length(HumanDom1_sm$gene),
area2 = length(ChimpDom1_sm$gene),
cross.area = nrow(both1sm),
c("Human", "Chimp"), scaled = TRUE,
fill = c("orange", "green"),
cex = 1.5,
cat.cex = 1.5,
cat.pos = c(320, 25),
cat.dist = .05) 
| Version | Author | Date |
|---|---|---|
| 385fe04 | brimittleman | 2020-04-16 |
both1sm_same= both1sm %>% filter(ChimpPAS==HumanPAS)
nrow(both1sm_same)[1] 3542both1sm_diff= both1sm %>% filter(ChimpPAS!=HumanPAS)
nrow(both1sm_diff)[1] 272location for same:
same1=ggplot(both1sm_same,aes(x=ChimpLoc,fill=ChimpLoc)) +geom_bar(stat = "count") +scale_fill_brewer(palette = "Dark2")+ labs(title="Matched Gene Matched PAS .1", y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
same1
| Version | Author | Date |
|---|---|---|
| 385fe04 | brimittleman | 2020-04-16 |
both1sm_diff_g= both1sm_diff %>% select(gene, HumanLoc, ChimpLoc) %>% gather('Species', 'Loc', -gene)
diffDom1=ggplot(both1sm_diff_g, aes(x=Loc, fill=Species))+ geom_bar(stat="count",position = "dodge") +scale_fill_brewer(palette = "Dark2")+ labs(y='PAS', title="Location for Different Dominant PAS at 10% difference") +labs(y='Genes', title="Different Dominant PAS: 0.1",x="") +theme(axis.text.x = element_text(angle = 90),legend.position = "none")
diffDom1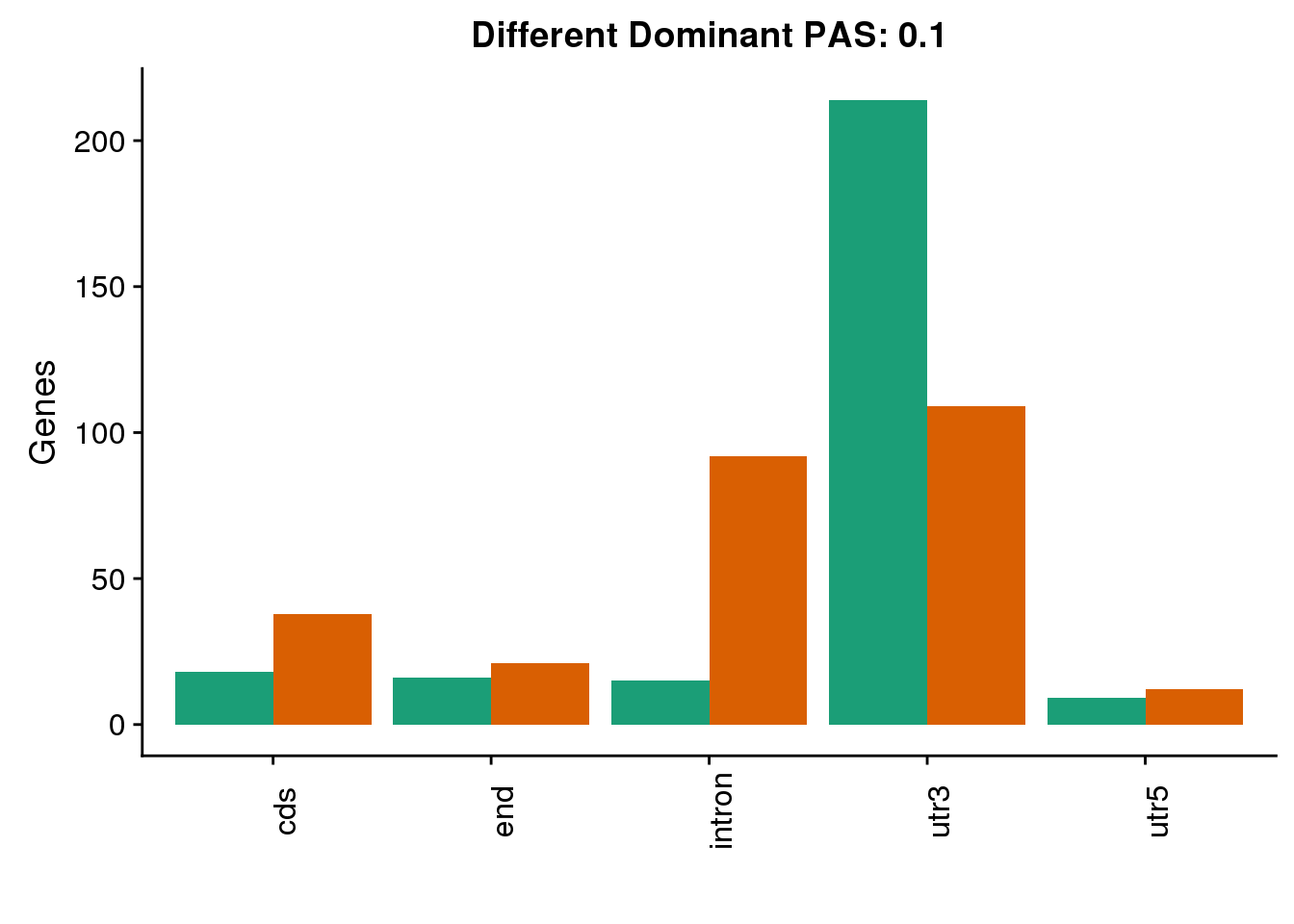
| Version | Author | Date |
|---|---|---|
| 385fe04 | brimittleman | 2020-04-16 |
ggplot(BothDom1, aes(x=nPAS, fill=set)) + geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="Number of PAS per gene with .1 difference",y="genes")
| Version | Author | Date |
|---|---|---|
| 385fe04 | brimittleman | 2020-04-16 |
HumanDom1_smLab= HumanDom1 %>% anti_join(ChimpDom1,by = "gene") %>% mutate(Set="Human")
ChimpDom1_smLab= ChimpDom1 %>% anti_join(HumanDom1,by = "gene") %>% mutate(Set="Chimp")
BothDom1_smLab=HumanDom1_smLab %>% bind_rows(ChimpDom1_smLab)
diffgenes1=ggplot(BothDom1_smLab,aes(x=loc, fill=Set))+ geom_bar(stat="count", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="Non Matched: 0.1", y="genes",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")
diffgenes1
| Version | Author | Date |
|---|---|---|
| 385fe04 | brimittleman | 2020-04-16 |
BothDom1_prop= BothDom1 %>% group_by(set, loc) %>% summarise(nLoc=n()) %>% ungroup() %>% group_by(set) %>% mutate(nSet=sum(nLoc), prop=nLoc/nSet)
proploc1=ggplot(BothDom1_prop, aes(x=loc,by=set, fill=set,y=prop)) + geom_bar(stat="identity",position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.1",y="Proportion",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")BothDom1_smLab_prop=BothDom1_smLab%>% group_by(set, loc) %>% summarise(nLoc=n()) %>% ungroup() %>% group_by(set) %>% mutate(nSet=sum(nLoc), prop=nLoc/nSet)
propDiff1=ggplot(BothDom1_smLab_prop, aes(x=loc,by=set, fill=set,y=prop)) + geom_bar(stat="identity",position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="0.1: Different Genes",y="Proportion",x="")+theme(axis.text.x = element_text(angle = 90),legend.position = "none")Aggregate results:
Next step will be to aggregate these plots in a way that is both easier to visualize but also easier to draw conclusions from.
First plot the number of genes with a dominant PAS at each cutoff.
Cutoffs=seq(0.1,.9, .1)
Human=c(nrow(HumanDom1),nrow(HumanDom2),nrow(HumanDom3),nrow(HumanDom4),nrow(HumanDom5),nrow(HumanDom6),nrow(HumanDom7),nrow(HumanDom8),nrow(HumanDom9))
Chimp=c(nrow(ChimpDom1),nrow(ChimpDom2),nrow(ChimpDom3),nrow(ChimpDom4),nrow(ChimpDom5),nrow(ChimpDom6),nrow(ChimpDom7),nrow(ChimpDom8),nrow(ChimpDom9))
DomNumsDF=as.data.frame(cbind(Cutoffs, Human, Chimp)) %>% gather("Species", "DominantGenes", -Cutoffs)
DomNumsDF$Cutoffs= as.factor(DomNumsDF$Cutoffs)
ggplot(DomNumsDF, aes(x=Cutoffs, fill=Species, y=DominantGenes)) + geom_bar(stat="identity", position = "dodge") + scale_fill_brewer(palette = "Dark2") + labs(title="Number of Genes with a Dominant PAS",x="Difference in usage to be called dominant")
| Version | Author | Date |
|---|---|---|
| 385fe04 | brimittleman | 2020-04-16 |
General location:
genloctiles <- ggdraw() +
draw_label(
"Location of Dominant PAS",
fontface = 'bold',
x = 0,
hjust = 0
)
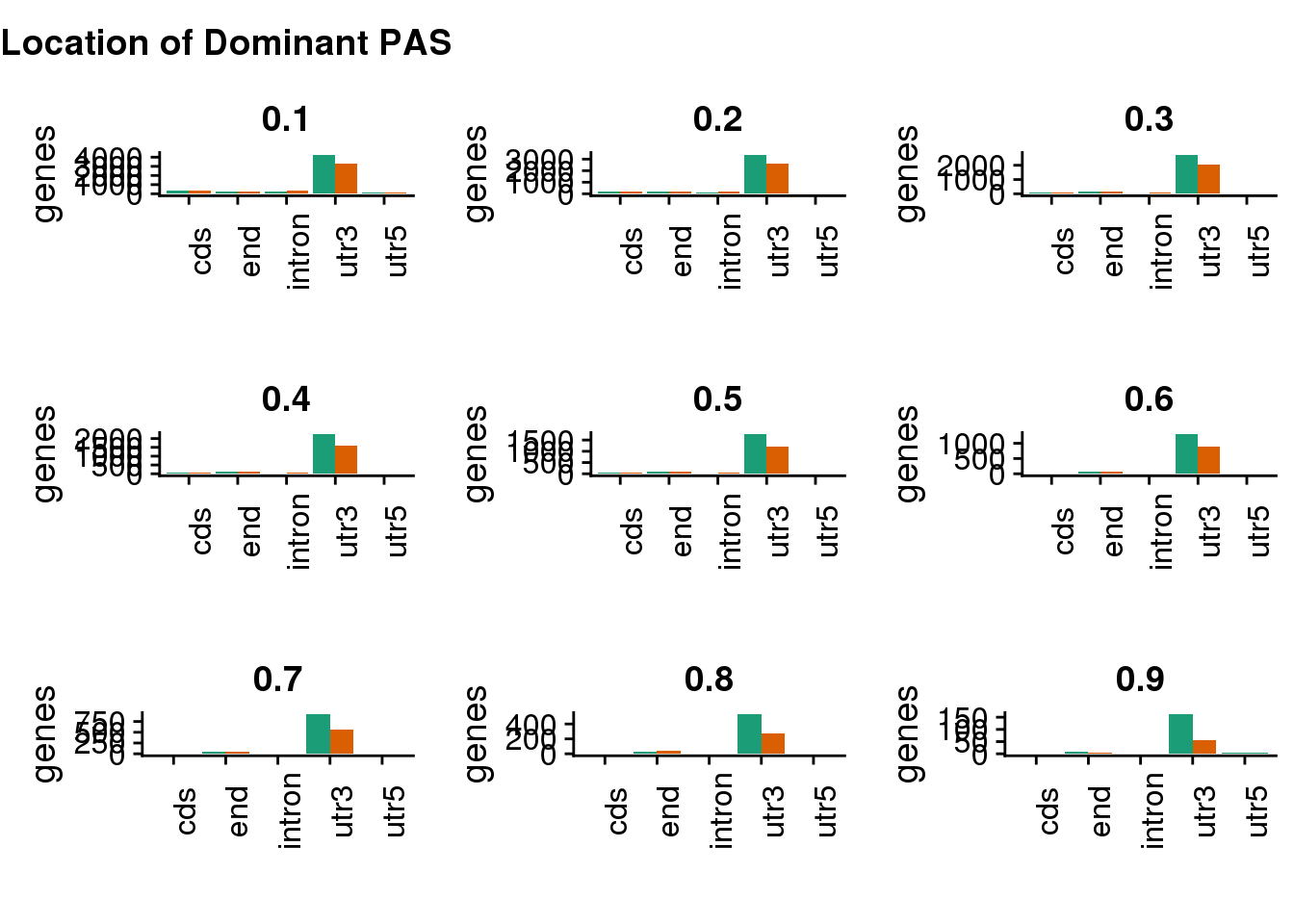
genloc=plot_grid(genloc1,genloc2,genloc3,genloc4,genloc5,genloc6,genloc7,genloc8,genloc9)
plot_grid(
genloctiles, genloc,
ncol = 1,
# rel_heights values control vertical title margins
rel_heights = c(0.1, 1)
)
| Version | Author | Date |
|---|---|---|
| 385fe04 | brimittleman | 2020-04-16 |
This shows this pattern is robust to the cutoff.
Proportion:
proploctiles <- ggdraw() +
draw_label(
"Location of Dominant PAS by proportion",
fontface = 'bold',
x = 0,
hjust = 0
)
proploc=plot_grid(proploc9,proploc8,proploc7,proploc6,proploc5,proploc4,proploc3,proploc2,proploc1)
plot_grid(
proploctiles, proploc,
ncol = 1,
# rel_heights values control vertical title margins
rel_heights = c(0.1, 1)
)
| Version | Author | Date |
|---|---|---|
| 385fe04 | brimittleman | 2020-04-16 |
Plot the joint genes.
diffdomtiles <- ggdraw() +
draw_label(
"Location of Dominant PAS in genes with different Dominant PAS",
fontface = 'bold',
x = 0,
hjust = 0
)
diffdom=plot_grid(diffDom1,diffDom2,diffDom3,diffDom4)
plot_grid(
diffdomtiles, diffdom,
ncol = 1,
# rel_heights values control vertical title margins
rel_heights = c(0.1, 1)
)
| Version | Author | Date |
|---|---|---|
| 385fe04 | brimittleman | 2020-04-16 |
This is also robust to cutoff.
Location when they do not share the same gene.
diffgenes9
diffgenestiles <- ggdraw() +
draw_label(
"Location of Dominant PAS in genes with a dominant PAS only in 1 species",
fontface = 'bold',
x = 0,
hjust = 0
)
diffgenes=plot_grid(diffgenes1,diffgenes2,diffgenes3,diffgenes4,diffgenes5,diffgenes6,diffgenes7,diffgenes8,diffgenes9)
plot_grid(
diffgenestiles, diffgenes,
ncol = 1,
# rel_heights values control vertical title margins
rel_heights = c(0.1, 1)
)
| Version | Author | Date |
|---|---|---|
| 385fe04 | brimittleman | 2020-04-16 |
#propDiff1
diffgenesProptiles <- ggdraw() +
draw_label(
"Location of Dominant PAS in genes with a dominant PAS only in 1 species- Proportion",
fontface = 'bold',
x = 0,
hjust = 0
)
diffPropgenes=plot_grid(propDiff1,propDiff2,propDiff3,propDiff4,propDiff5,propDiff6,propDiff7,propDiff8,propDiff9)
plot_grid(
diffgenesProptiles, diffPropgenes,
ncol = 1,
# rel_heights values control vertical title margins
rel_heights = c(0.1, 1)
)
| Version | Author | Date |
|---|---|---|
| 385fe04 | brimittleman | 2020-04-16 |
This is robust as well.
Look at the overlapping genes with the same PAS
#same1
SAMESAMEtiles <- ggdraw() +
draw_label(
"Location of Dominant PAS in genes with the same dominant PAS",
fontface = 'bold',
x = 0,
hjust = 0
)
SAMESAME=plot_grid(same1,same2,same3,same4,same5,same6,same7,same8)
plot_grid(
SAMESAMEtiles, SAMESAME,
ncol = 1,
# rel_heights values control vertical title margins
rel_heights = c(0.1, 1)
)
| Version | Author | Date |
|---|---|---|
| 385fe04 | brimittleman | 2020-04-16 |
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] grid stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] cowplot_0.9.4 forcats_0.3.0 stringr_1.3.1
[4] dplyr_0.8.0.1 purrr_0.3.2 readr_1.3.1
[7] tidyr_0.8.3 tibble_2.1.1 ggplot2_3.1.1
[10] tidyverse_1.2.1 VennDiagram_1.6.20 futile.logger_1.4.3
[13] gridExtra_2.3 workflowr_1.6.0
loaded via a namespace (and not attached):
[1] tidyselect_0.2.5 haven_1.1.2 lattice_0.20-38
[4] colorspace_1.3-2 generics_0.0.2 htmltools_0.3.6
[7] yaml_2.2.0 rlang_0.4.0 later_0.7.5
[10] pillar_1.3.1 withr_2.1.2 glue_1.3.0
[13] RColorBrewer_1.1-2 lambda.r_1.2.3 modelr_0.1.2
[16] readxl_1.1.0 plyr_1.8.4 cellranger_1.1.0
[19] munsell_0.5.0 gtable_0.2.0 rvest_0.3.2
[22] evaluate_0.12 labeling_0.3 knitr_1.20
[25] httpuv_1.4.5 broom_0.5.1 Rcpp_1.0.2
[28] promises_1.0.1 backports_1.1.2 scales_1.0.0
[31] formatR_1.5 jsonlite_1.6 fs_1.3.1
[34] hms_0.4.2 digest_0.6.18 stringi_1.2.4
[37] rprojroot_1.3-2 cli_1.1.0 tools_3.5.1
[40] magrittr_1.5 lazyeval_0.2.1 futile.options_1.0.1
[43] crayon_1.3.4 whisker_0.3-2 pkgconfig_2.0.2
[46] xml2_1.2.0 lubridate_1.7.4 rstudioapi_0.10
[49] assertthat_0.2.0 rmarkdown_1.10 httr_1.3.1
[52] R6_2.3.0 nlme_3.1-137 git2r_0.26.1
[55] compiler_3.5.1