Differential gene expression analysis with DESeq2
2024-08-05
Last updated: 2024-08-05
Checks: 7 0
Knit directory: muse/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200712) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 9293da5. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: r_packages_4.3.3/
Ignored: r_packages_4.4.0/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/deseq2.Rmd) and HTML
(docs/deseq2.html) files. If you’ve configured a remote Git
repository (see ?wflow_git_remote), click on the hyperlinks
in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 9293da5 | Dave Tang | 2024-08-05 | Visualisations after a DGE analysis |
| html | 365890e | Dave Tang | 2024-08-05 | Build site. |
| Rmd | 3780bfa | Dave Tang | 2024-08-05 | A basic DESeq2 analysis |
| html | 1383bf7 | Dave Tang | 2024-08-05 | Build site. |
| Rmd | d5e67bc | Dave Tang | 2024-08-05 | Getting started with DESeq2 |
DESeq2 is used to:
Estimate variance-mean dependence in count data from high-throughput sequencing assays and test for differential expression based on a model using the negative binomial distribution.
Installation
Install using BiocManager::install().
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("DESeq2")We will use data from the pasilla package so install it too.
BiocManager::install("pasilla")Example data
Example dataset in the experiment data package {pasilla}.
fn <- system.file("extdata", "pasilla_gene_counts.tsv", package = "pasilla", mustWork = TRUE)
counts <- as.matrix(read.csv(fn, sep="\t", row.names = "gene_id"))
dim(counts)[1] 14599 7The matrix tallies the number of reads assigned for each gene in each sample.
tail(counts) untreated1 untreated2 untreated3 untreated4 treated1 treated2
FBgn0261570 3296 4910 2156 2060 5077 3069
FBgn0261571 0 0 0 0 1 0
FBgn0261572 4 13 4 11 7 3
FBgn0261573 2651 3653 1571 1612 3334 1848
FBgn0261574 6385 9318 3110 2819 10455 3508
FBgn0261575 6 53 1 3 42 3
treated3
FBgn0261570 3022
FBgn0261571 0
FBgn0261572 3
FBgn0261573 1908
FBgn0261574 3047
FBgn0261575 4Size factors
Estimate size factors.
DESeq2::estimateSizeFactorsForMatrix(counts)untreated1 untreated2 untreated3 untreated4 treated1 treated2 treated3
1.1382630 1.7930004 0.6495470 0.7516892 1.6355751 0.7612698 0.8326526 Mean-variance relationship
Variance versus mean for the (size factor adjusted)
counts data. The axes are logarithmic. Also shown are lines
through the origin with slopes 1 (green) and 2 (red).
sf <- DESeq2::estimateSizeFactorsForMatrix(counts)
ncounts <- t(t(counts) / sf)
# untreated samples
uncounts <- ncounts[, grep("^untreated", colnames(ncounts)), drop = FALSE]
ggplot(
tibble(
mean = rowMeans(uncounts),
var = rowVars(uncounts)
),
aes(x = log1p(mean), y = log1p(var))
) +
geom_hex() +
coord_fixed() +
theme_minimal() +
theme(legend.position = "none") +
geom_abline(slope = 1:2, color = c("forestgreen", "red"))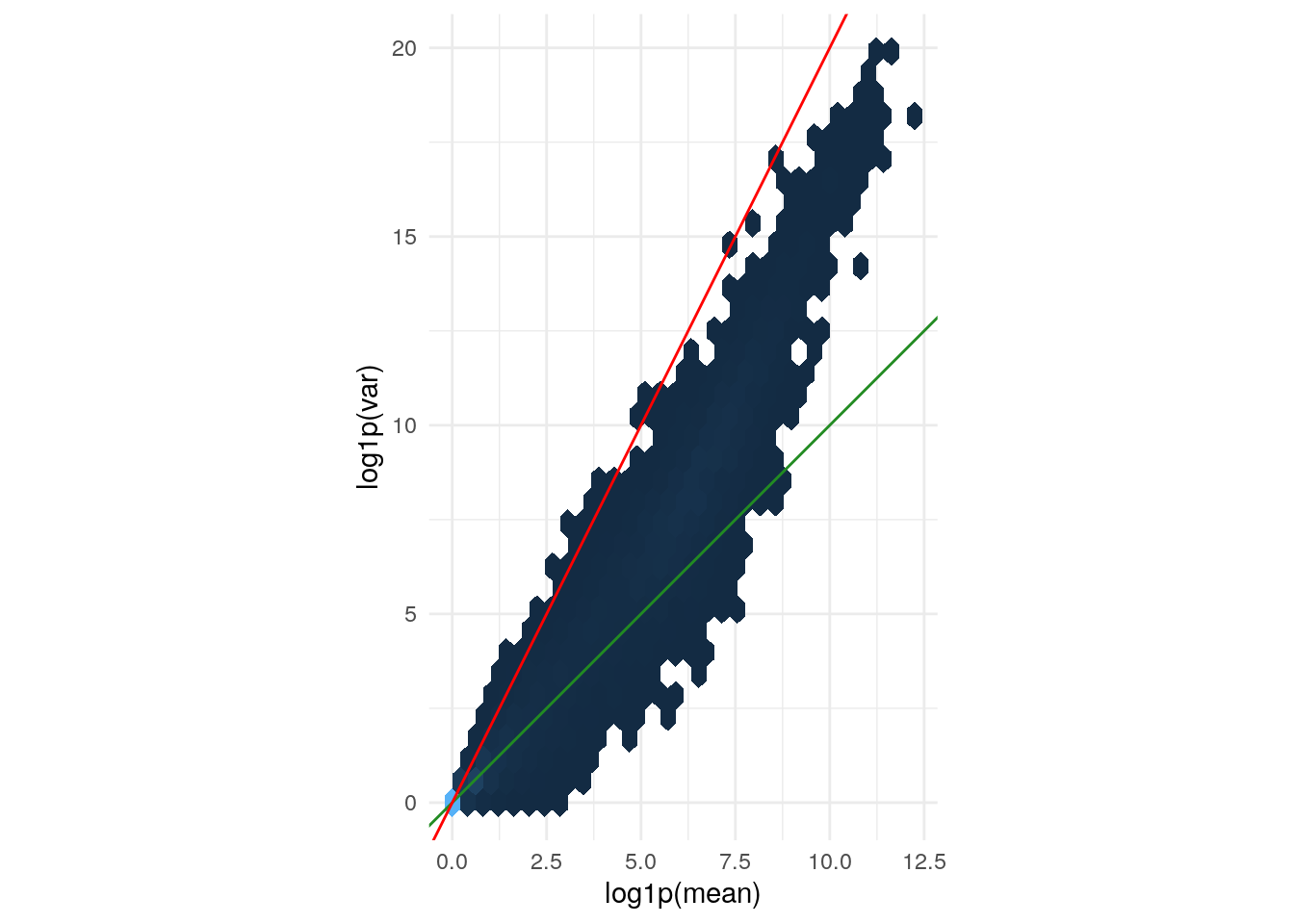
| Version | Author | Date |
|---|---|---|
| 1383bf7 | Dave Tang | 2024-08-05 |
The green line is what we expect if the variance equals the mean, as is the case for a Poisson-distributed random variable. This approximately fits the data in the lower range. The red line corresponds to the quadratic mean-variance relationship \(v = m^2\). We can see that in the upper range of the data, the quadratic relationship approximately fits the data.
A basic analysis
The {pasilla} data set is from an experiment on Drosophila melanogaster cell cultures that tested the effect of RNAi knockdown of the splicing factor pasilla on the cells’ transcriptome. There were two experimental conditions, termed untreated and treated in the column headers and they correspond to negative control and siRNA against pasilla. The experimental metadata of the seven samples in this dataset are loaded below.
annotationFile <- system.file("extdata", "pasilla_sample_annotation.csv", package = "pasilla", mustWork = TRUE)
pasillaSampleAnno <- readr::read_csv(annotationFile)Rows: 7 Columns: 6
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (4): file, condition, type, total number of reads
dbl (2): number of lanes, exon counts
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.pasillaSampleAnno# A tibble: 7 × 6
file condition type `number of lanes` total number of read…¹ `exon counts`
<chr> <chr> <chr> <dbl> <chr> <dbl>
1 treate… treated sing… 5 35158667 15679615
2 treate… treated pair… 2 12242535 (x2) 15620018
3 treate… treated pair… 2 12443664 (x2) 12733865
4 untrea… untreated sing… 2 17812866 14924838
5 untrea… untreated sing… 6 34284521 20764558
6 untrea… untreated pair… 2 10542625 (x2) 10283129
7 untrea… untreated pair… 2 12214974 (x2) 11653031
# ℹ abbreviated name: ¹`total number of reads`The dataset was produced in two batches, the first consisting of three sequencing libraries using single read sequencing and the second using using paired-end sequencing.
Create factors.
mutate(
pasillaSampleAnno,
condition = factor(condition, levels = c("untreated", "treated")),
type = factor(sub("-.*", "", type), levels = c("single", "paired"))
) -> pasillaSampleAnnoNote that the design is approximately balanced between the factor of
interest, condition, and the “nuisance factor”,
type.
with(pasillaSampleAnno, table(condition, type)) type
condition single paired
untreated 2 2
treated 1 2{DESeq2} uses DESeqDataSet objects and is an extension
of the SummarizedExperiment class in Bioconductor. We can
create a DESeqDataSet object using the constructor function
DESeqDataSetFromMatrix.
mt <- match(colnames(counts), sub("fb$", "", pasillaSampleAnno$file))
stopifnot(!any(is.na(mt)))
pasilla <- DESeqDataSetFromMatrix(
countData = counts,
colData = pasillaSampleAnno[mt, ],
design = ~ condition
)
class(pasilla)[1] "DESeqDataSet"
attr(,"package")
[1] "DESeq2"is(pasilla, "SummarizedExperiment")[1] TRUEAfter creating a DESeqDataSet we are ready to carry out
a differential expression analysis. The aim is to identify genes that
are differentially abundant between the treated and untreated cells. A
test that is conceptually similar to the t-test is used. A
choice of standard analysis steps are wrapped into a single function,
DESeq().
pasilla <- DESeq(pasilla)estimating size factorsestimating dispersionsgene-wise dispersion estimatesmean-dispersion relationshipfinal dispersion estimatesfitting model and testingThe DESeq() function is a wrapper that calls:
estimateSizeFactorsfor normalisationestimateDispersionsfor dispersion estimationnbinomWaldTestfor hypothesis tests for differential abundance
The test is between the two levels untreated and
treated of the factor condition, since this is
what was specified in the design: design = ~ condition.
res <- results(pasilla)
res[order(res$padj), ] |> head()log2 fold change (MLE): condition treated vs untreated
Wald test p-value: condition treated vs untreated
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
FBgn0039155 730.596 -4.61901 0.1687068 -27.3789 4.88599e-165
FBgn0025111 1501.411 2.89986 0.1269205 22.8479 1.53430e-115
FBgn0029167 3706.117 -2.19700 0.0969888 -22.6521 1.33042e-113
FBgn0003360 4343.035 -3.17967 0.1435264 -22.1539 9.56283e-109
FBgn0035085 638.233 -2.56041 0.1372952 -18.6490 1.28772e-77
FBgn0039827 261.916 -4.16252 0.2325888 -17.8965 1.25663e-71
padj
<numeric>
FBgn0039155 4.06661e-161
FBgn0025111 6.38497e-112
FBgn0029167 3.69104e-110
FBgn0003360 1.98979e-105
FBgn0035085 2.14354e-74
FBgn0039827 1.74316e-68Exploring the results
The first step after a differential expression analysis is to visualise the following plots:
- the histogram of p-values
- the MA plot
- an ordination plot
- a heatmap
Histogram of p-values of a differential expression analysis.”
ggplot(as(res, "data.frame"), aes(x = pvalue)) +
geom_histogram(binwidth = 0.01, fill = "Royalblue", boundary = 0) +
geom_hline(yintercept = 100, lty = 3) +
theme_minimal()Warning: Removed 2241 rows containing non-finite outside the scale range
(`stat_bin()`).
The p-value distribution shows a uniform background (non-DE genes) with values between 0 and 1 and a peak of small p-values at the left (DE genes). The ratio of the level of the background to the height of the peak provides us with a rough indication of the false discovery rate (FDR) that would be associated with calling the genes in the leftmost bin differentially expressed.
In this case, the leftmost bin contains 993 genes and the background level (dotted line) is around 100, so the FDR associated with calling all genes in the leftmost bin would be around 10%.
table(res$pvalue < 0.01)
FALSE TRUE
11365 993 A background distribution that is not uniform, but shows a tilted shape with an increase towards the right, tends to indicate batch effects, that is, underlying systematic variation that makes replicates look more different than expected.
The {DESeq2} package can produce the MA plot.
plotMA(pasilla, ylim = c(-2, 2))
{DESeq2} also has a function plotPCA() that can produce
PCA plots. PC1 is mostly aligned with the experimental covariate of
interest (untreated/treated), while PC2 is
roughly aligned with the sequencing protocol
(single/paired).
# transform using regularised logarithm
pas_rlog <- rlogTransformation(pasilla)
plotPCA(pas_rlog, intgroup=c("condition", "type")) + coord_fixed()using ntop=500 top features by varianceCoordinate system already present. Adding new coordinate system, which will
replace the existing one.
Heatmap using the regularised logarithm (rlog) transformed data of the 30 most variable genes.
library("pheatmap")
wanted <- order(rowMeans(assay(pas_rlog)), decreasing = TRUE)[1:30]
pheatmap(
assay(pas_rlog)[wanted, ],
scale = "row",
annotation_col = as.data.frame(
colData(pas_rlog)[, c("condition", "type")]
)
)
Note the clustering of the sequencing protocol and this highlights that our differential expression analysis was probably too naive and that we should adjust for the “nuisance” factor.
sessionInfo()R version 4.4.0 (2024-04-24)
Platform: x86_64-pc-linux-gnu
Running under: Ubuntu 22.04.4 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: Etc/UTC
tzcode source: system (glibc)
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] pheatmap_1.0.12 hexbin_1.28.3
[3] pasilla_1.32.0 DEXSeq_1.50.0
[5] RColorBrewer_1.1-3 AnnotationDbi_1.66.0
[7] BiocParallel_1.38.0 DESeq2_1.44.0
[9] SummarizedExperiment_1.34.0 Biobase_2.64.0
[11] MatrixGenerics_1.16.0 matrixStats_1.3.0
[13] GenomicRanges_1.56.1 GenomeInfoDb_1.40.1
[15] IRanges_2.38.1 S4Vectors_0.42.1
[17] BiocGenerics_0.50.0 lubridate_1.9.3
[19] forcats_1.0.0 stringr_1.5.1
[21] dplyr_1.1.4 purrr_1.0.2
[23] readr_2.1.5 tidyr_1.3.1
[25] tibble_3.2.1 ggplot2_3.5.1
[27] tidyverse_2.0.0 workflowr_1.7.1
loaded via a namespace (and not attached):
[1] rstudioapi_0.16.0 jsonlite_1.8.8 magrittr_2.0.3
[4] farver_2.1.2 rmarkdown_2.27 fs_1.6.4
[7] zlibbioc_1.50.0 vctrs_0.6.5 memoise_2.0.1
[10] Rsamtools_2.20.0 htmltools_0.5.8.1 S4Arrays_1.4.1
[13] progress_1.2.3 curl_5.2.1 SparseArray_1.4.8
[16] sass_0.4.9 bslib_0.7.0 httr2_1.0.1
[19] cachem_1.1.0 whisker_0.4.1 lifecycle_1.0.4
[22] pkgconfig_2.0.3 Matrix_1.7-0 R6_2.5.1
[25] fastmap_1.2.0 GenomeInfoDbData_1.2.12 digest_0.6.35
[28] colorspace_2.1-0 ps_1.7.6 rprojroot_2.0.4
[31] geneplotter_1.82.0 RSQLite_2.3.7 hwriter_1.3.2.1
[34] filelock_1.0.3 labeling_0.4.3 fansi_1.0.6
[37] timechange_0.3.0 httr_1.4.7 abind_1.4-5
[40] compiler_4.4.0 bit64_4.0.5 withr_3.0.0
[43] DBI_1.2.3 highr_0.11 biomaRt_2.60.1
[46] rappdirs_0.3.3 DelayedArray_0.30.1 tools_4.4.0
[49] httpuv_1.6.15 glue_1.7.0 callr_3.7.6
[52] promises_1.3.0 grid_4.4.0 getPass_0.2-4
[55] generics_0.1.3 gtable_0.3.5 tzdb_0.4.0
[58] hms_1.1.3 xml2_1.3.6 utf8_1.2.4
[61] XVector_0.44.0 pillar_1.9.0 vroom_1.6.5
[64] genefilter_1.86.0 later_1.3.2 splines_4.4.0
[67] BiocFileCache_2.12.0 lattice_0.22-6 survival_3.5-8
[70] bit_4.0.5 annotate_1.82.0 tidyselect_1.2.1
[73] locfit_1.5-9.9 Biostrings_2.72.1 knitr_1.47
[76] git2r_0.33.0 xfun_0.44 statmod_1.5.0
[79] stringi_1.8.4 UCSC.utils_1.0.0 yaml_2.3.8
[82] evaluate_0.24.0 codetools_0.2-20 cli_3.6.2
[85] xtable_1.8-4 munsell_0.5.1 processx_3.8.4
[88] jquerylib_0.1.4 Rcpp_1.0.12 dbplyr_2.5.0
[91] png_0.1-8 XML_3.99-0.16.1 parallel_4.4.0
[94] blob_1.2.4 prettyunits_1.2.0 bitops_1.0-7
[97] scales_1.3.0 crayon_1.5.2 rlang_1.1.4
[100] KEGGREST_1.44.1