5FU Recovery RNAseq Analysis
Emma M Pfortmiller
2025-02-10
Last updated: 2025-02-27
Checks: 7 0
Knit directory: Recovery_5FU/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20250217) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version b086ed1. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/Recovery_DE.Rmd) and HTML
(docs/Recovery_DE.html) files. If you’ve configured a
remote Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | b086ed1 | emmapfort | 2025-02-27 | wflow_publish(files = "analysis/Recovery_DE.Rmd") |
| html | 41aef30 | emmapfort | 2025-02-27 | Build site. |
| Rmd | e77e0d4 | emmapfort | 2025-02-27 | Make website cohesive and dataframes concise. |
Welcome to my research website.
── Attaching core tidyverse packages ──────────────────────── tidyverse 2.0.0 ──
✔ dplyr 1.1.4 ✔ readr 2.1.5
✔ forcats 1.0.0 ✔ stringr 1.5.1
✔ lubridate 1.9.3 ✔ tidyr 1.3.1
✔ purrr 1.0.2
── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()
ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errors
Loading required package: limma
Loading required package: ggrepel
Attaching package: 'PCAtools'
The following objects are masked from 'package:stats':
biplot, screeplot
Loading required package: affy
Loading required package: BiocGenerics
Attaching package: 'BiocGenerics'
The following object is masked from 'package:limma':
plotMA
The following objects are masked from 'package:lubridate':
intersect, setdiff, union
The following objects are masked from 'package:dplyr':
combine, intersect, setdiff, union
The following objects are masked from 'package:stats':
IQR, mad, sd, var, xtabs
The following objects are masked from 'package:base':
anyDuplicated, aperm, append, as.data.frame, basename, cbind,
colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
get, grep, grepl, intersect, is.unsorted, lapply, Map, mapply,
match, mget, order, paste, pmax, pmax.int, pmin, pmin.int,
Position, rank, rbind, Reduce, rownames, sapply, saveRDS, setdiff,
table, tapply, union, unique, unsplit, which.max, which.min
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Attaching package: 'affy'
The following object is masked from 'package:lubridate':
pm
Loading required package: EDASeq
Loading required package: ShortRead
Loading required package: BiocParallel
Loading required package: Biostrings
Loading required package: S4Vectors
Loading required package: stats4
Attaching package: 'S4Vectors'
The following objects are masked from 'package:lubridate':
second, second<-
The following objects are masked from 'package:dplyr':
first, rename
The following object is masked from 'package:tidyr':
expand
The following object is masked from 'package:utils':
findMatches
The following objects are masked from 'package:base':
expand.grid, I, unname
Loading required package: IRanges
Attaching package: 'IRanges'
The following object is masked from 'package:lubridate':
%within%
The following objects are masked from 'package:dplyr':
collapse, desc, slice
The following object is masked from 'package:purrr':
reduce
The following object is masked from 'package:grDevices':
windows
Loading required package: XVector
Attaching package: 'XVector'
The following object is masked from 'package:purrr':
compact
Loading required package: GenomeInfoDb
Attaching package: 'Biostrings'
The following object is masked from 'package:base':
strsplit
Loading required package: Rsamtools
Loading required package: GenomicRanges
Loading required package: GenomicAlignments
Loading required package: SummarizedExperiment
Loading required package: MatrixGenerics
Loading required package: matrixStats
Attaching package: 'matrixStats'
The following objects are masked from 'package:Biobase':
anyMissing, rowMedians
The following object is masked from 'package:dplyr':
count
Attaching package: 'MatrixGenerics'
The following objects are masked from 'package:matrixStats':
colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
colWeightedMeans, colWeightedMedians, colWeightedSds,
colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
rowWeightedSds, rowWeightedVars
The following object is masked from 'package:Biobase':
rowMedians
Attaching package: 'GenomicAlignments'
The following object is masked from 'package:dplyr':
last
Attaching package: 'ShortRead'
The following object is masked from 'package:affy':
intensity
The following object is masked from 'package:dplyr':
id
The following object is masked from 'package:purrr':
compose
The following object is masked from 'package:tibble':
viewThese samples (63 in total) have been aligned to the Ensembl 113 release of GRCh38 via subread, and counts have been generated using featureCounts with inbuilt hg38 genome
Data is located under 5FU Project on my computer All analysis saved in RDirectory Recovery_RNAseq
Individual 1 - 84-1 (9 samples)
Individual 2 - 87-1 (9 samples)
Individual 3 - 75-1 (9 samples)
Individual 4 - 75-1 (9 samples)
Individual 5 - 17-3 (9 samples)
Individual 6 - 90-1 (9 samples)
Individual 7 - 90-1 REP (9 samples)
Now that I have imported all of these counts files, I will make a matrix with all of the samples
First I’ll break them up by individual, and then combine them all together
Now that I’ve made individual matrices for each individual - I will combine them together into one large matrix
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
#Now that I've put together my matrix - I will do some QC checks
#now that I have all of my factors and my gene info, I will make some graphs to show the QC data
reads_by_sample <- c(tx_col)
fC_AllCounts %>%
ggplot(., aes (x = Conditions, y = Total_Align, fill = Treatment, group_by = Line))+
geom_col()+
geom_hline(aes(yintercept=20000000))+
scale_fill_manual(values=reads_by_sample)+
ggtitle(expression("Total number of reads by sample"))+
xlab("")+
ylab(expression("RNA-sequencing reads"))+
theme_bw()+
theme(plot.title = element_text(size = rel(2), hjust = 0.5),
axis.title = element_text(size = 15, color = "black"),
axis.ticks = element_line(linewidth = 1.5),
axis.line = element_line(linewidth = 1.5),
axis.text.y = element_text(size =10, color = "black", angle = 0, hjust = 0.8, vjust = 0.5),
axis.text.x = element_text(size =10, color = "black", angle = 90, hjust = 1, vjust = 0.2),
#strip.text.x = element_text(size = 15, color = "black", face = "bold"),
strip.text.y = element_text(color = "white"))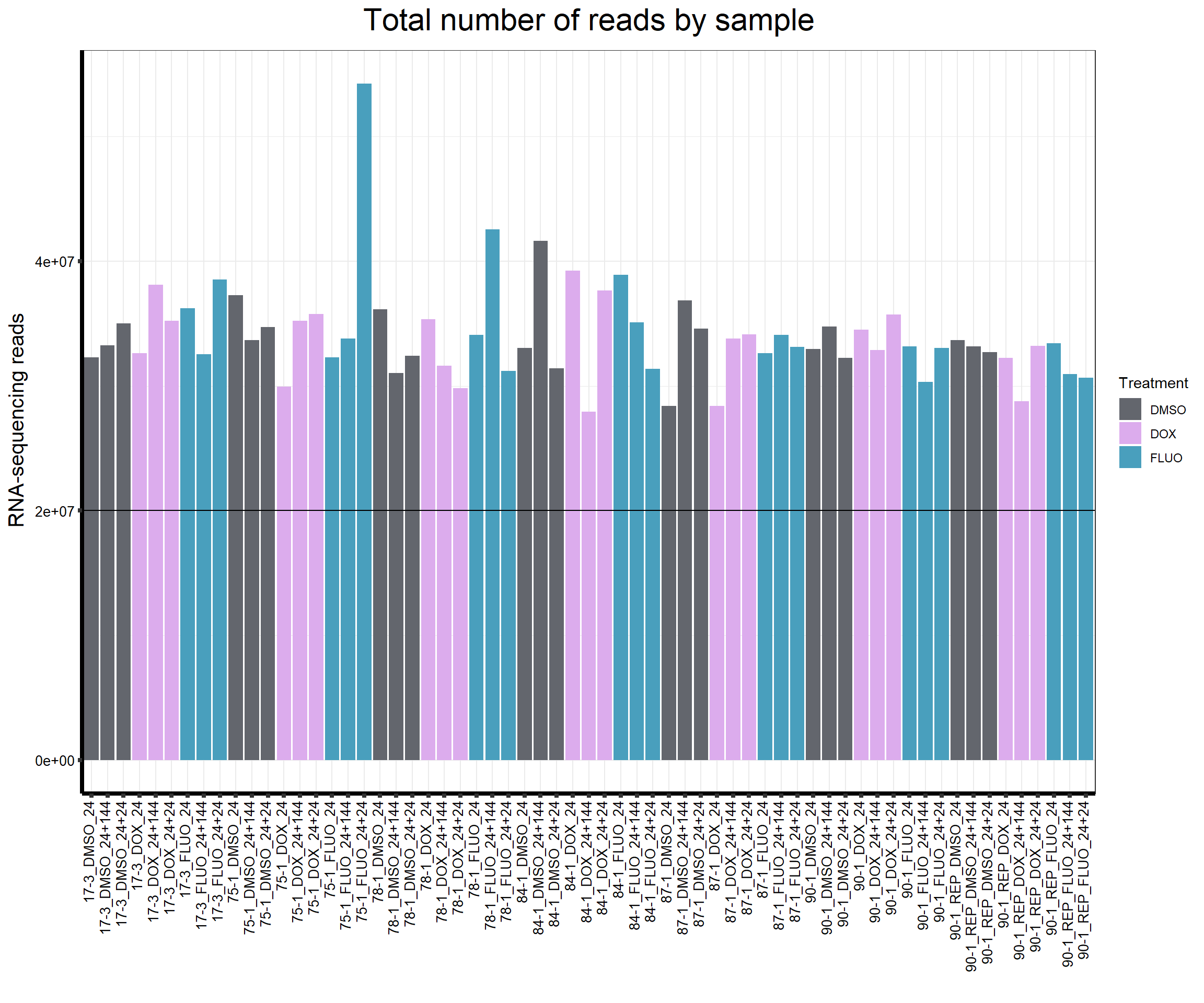
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
####Read Counts by Treatment####
fC_AllCounts %>%
ggplot(., aes (x =Treatment, y= Total_Align, fill = Treatment))+
geom_boxplot()+
scale_fill_manual(values=reads_by_sample)+
ggtitle(expression("Total number of reads by treatment"))+
xlab("")+
ylab(expression("RNA-sequencing reads"))+
theme_bw()+
theme(plot.title = element_text(size = rel(2), hjust = 0.5),
axis.title = element_text(size = 15, color = "black"),
axis.ticks = element_line(linewidth = 1.5),
axis.line = element_line(linewidth = 1.5),
axis.text.y = element_text(size =10, color = "black", angle = 0, hjust = 0.8, vjust = 0.5),
axis.text.x = element_text(size =10, color = "black", angle = 90, hjust = 1, vjust = 0.2),
#strip.text.x = element_text(size = 15, color = "black", face = "bold"),
strip.text.y = element_text(color = "white"))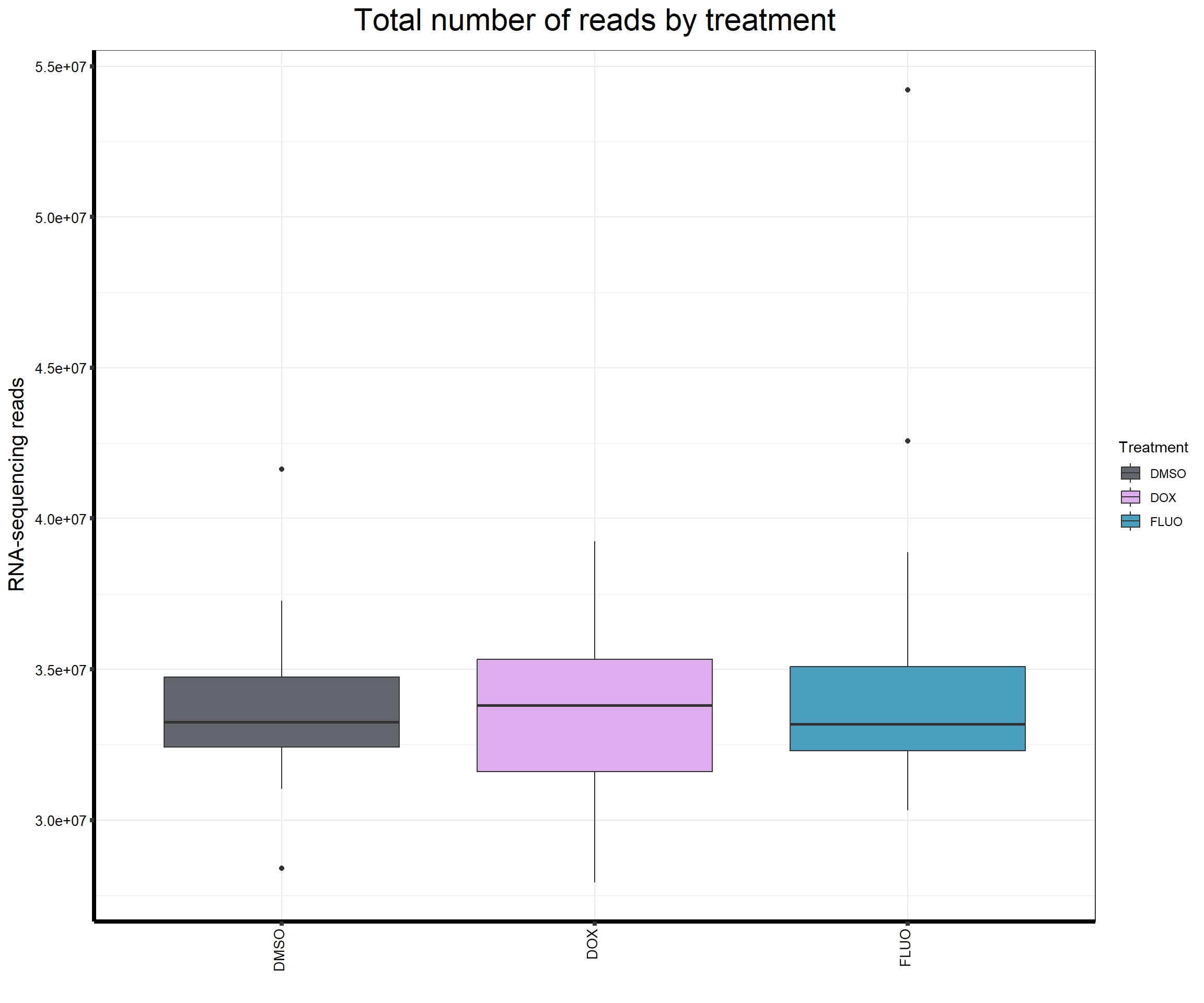
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
####Total Reads Per Individual####
fC_AllCounts %>%
ggplot(., aes (x =as.factor(Line), y=Total_Align))+
geom_boxplot(aes(fill=as.factor(Line)))+
scale_fill_brewer(palette = "Dark2", name = "Individual")+
ggtitle(expression("Total number of reads by individual"))+
xlab("")+
ylab(expression("RNA-sequencing reads"))+
theme_bw()+
theme(plot.title = element_text(size = rel(2), hjust = 0.5),
axis.title = element_text(size = 15, color = "black"),
axis.ticks = element_line(linewidth = 1.5),
axis.line = element_line(linewidth = 1.5),
axis.text.y = element_text(size =10, color = "black", angle = 0, hjust = 0.8, vjust = 0.5),
axis.text.x = element_text(size =10, color = "black", angle = 0, hjust = 1),
#strip.text.x = element_text(size = 15, color = "black", face = "bold"),
strip.text.y = element_text(color = "white"))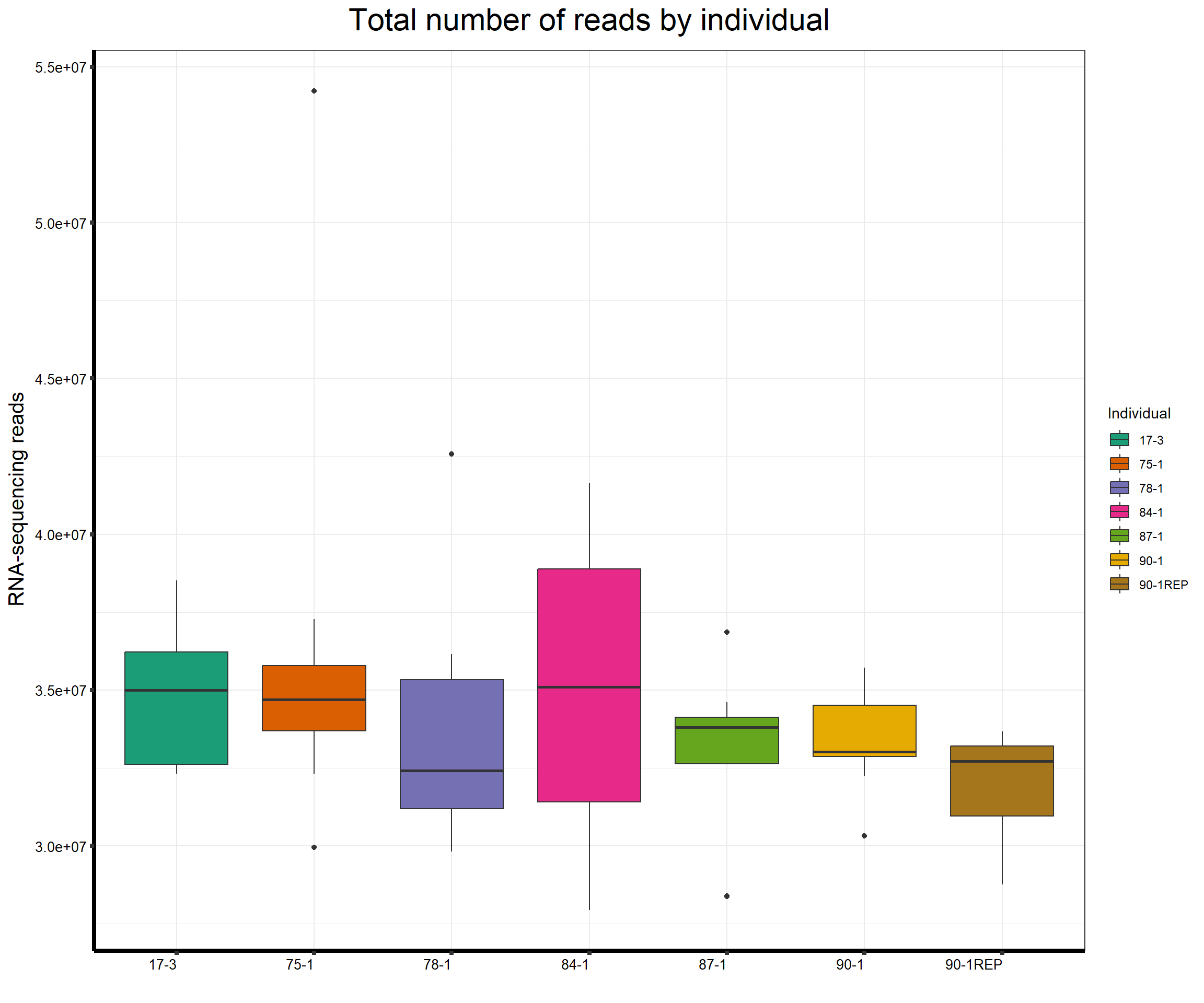
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
####Total Mapped Reads Per Drug####
reads_by_sample <- c(tx_col)
fC_AllCounts %>%
ggplot(., aes (x = Conditions, y = Assigned_Align, fill = Treatment, group_by = Line))+
geom_col()+
geom_hline(aes(yintercept=20000000))+
scale_fill_manual(values=reads_by_sample)+
ggtitle(expression("Total number of mapped reads by sample"))+
xlab("")+
ylab(expression("RNA-sequencing reads"))+
theme_bw()+
theme(plot.title = element_text(size = rel(2), hjust = 0.5),
axis.title = element_text(size = 15, color = "black"),
axis.ticks = element_line(linewidth = 1.5),
axis.line = element_line(linewidth = 1.5),
axis.text.y = element_text(size =10, color = "black", angle = 0, hjust = 0.8, vjust = 0.5),
axis.text.x = element_text(size =10, color = "black", angle = 90, hjust = 1, vjust = 0.2),
#strip.text.x = element_text(size = 15, color = "black", face = "bold"),
strip.text.y = element_text(color = "white"))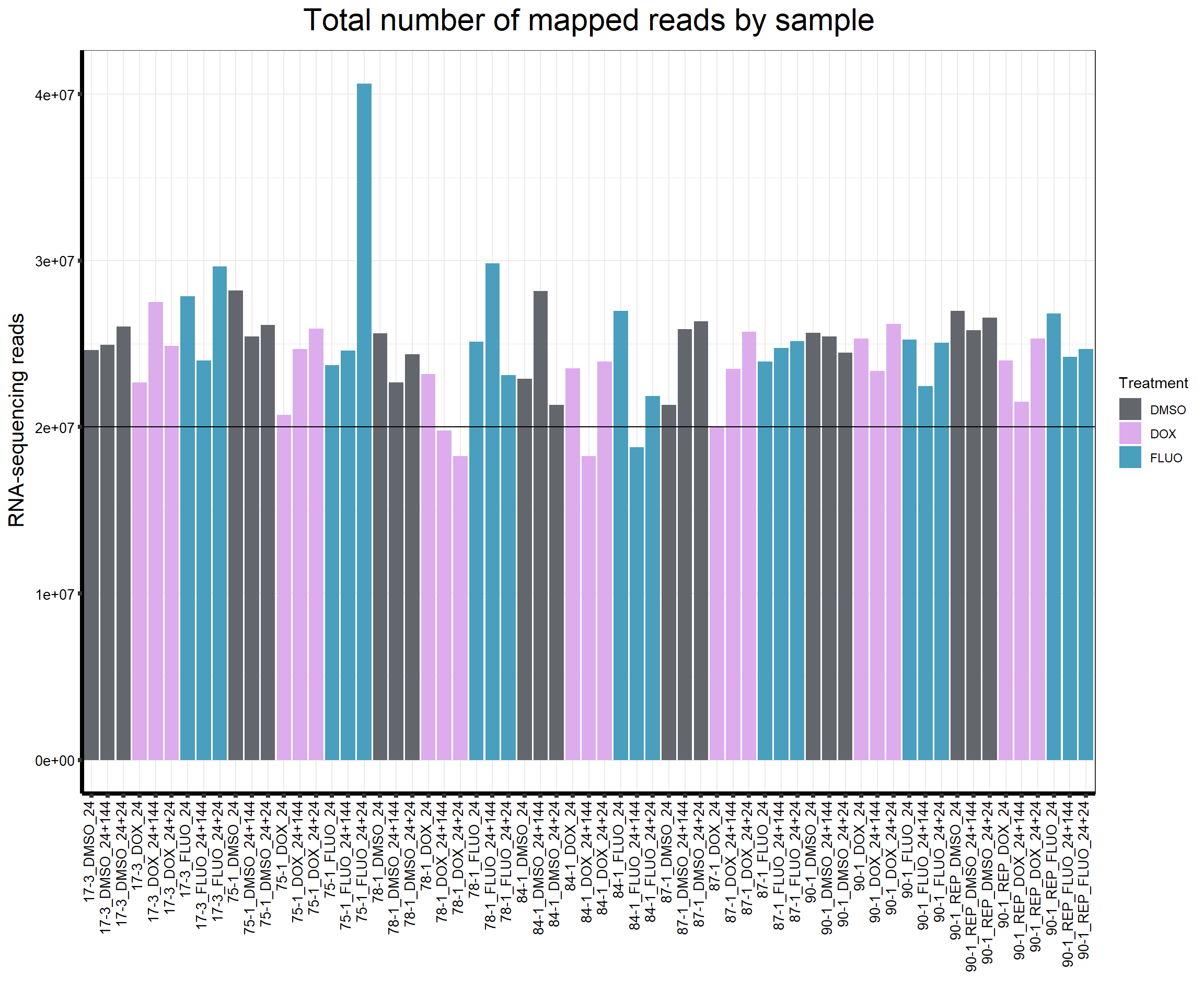
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
####Read Counts by Treatment####
fC_AllCounts %>%
ggplot(., aes (x =Treatment, y= Assigned_Align, fill = Treatment))+
geom_boxplot()+
scale_fill_manual(values=reads_by_sample)+
ggtitle(expression("Total number of mapped reads by treatment"))+
xlab("")+
ylab(expression("RNA-sequencing reads"))+
theme_bw()+
theme(plot.title = element_text(size = rel(2), hjust = 0.5),
axis.title = element_text(size = 15, color = "black"),
axis.ticks = element_line(linewidth = 1.5),
axis.line = element_line(linewidth = 1.5),
axis.text.y = element_text(size =10, color = "black", angle = 0, hjust = 0.8, vjust = 0.5),
axis.text.x = element_text(size =10, color = "black", angle = 90, hjust = 1, vjust = 0.2),
#strip.text.x = element_text(size = 15, color = "black", face = "bold"),
strip.text.y = element_text(color = "white"))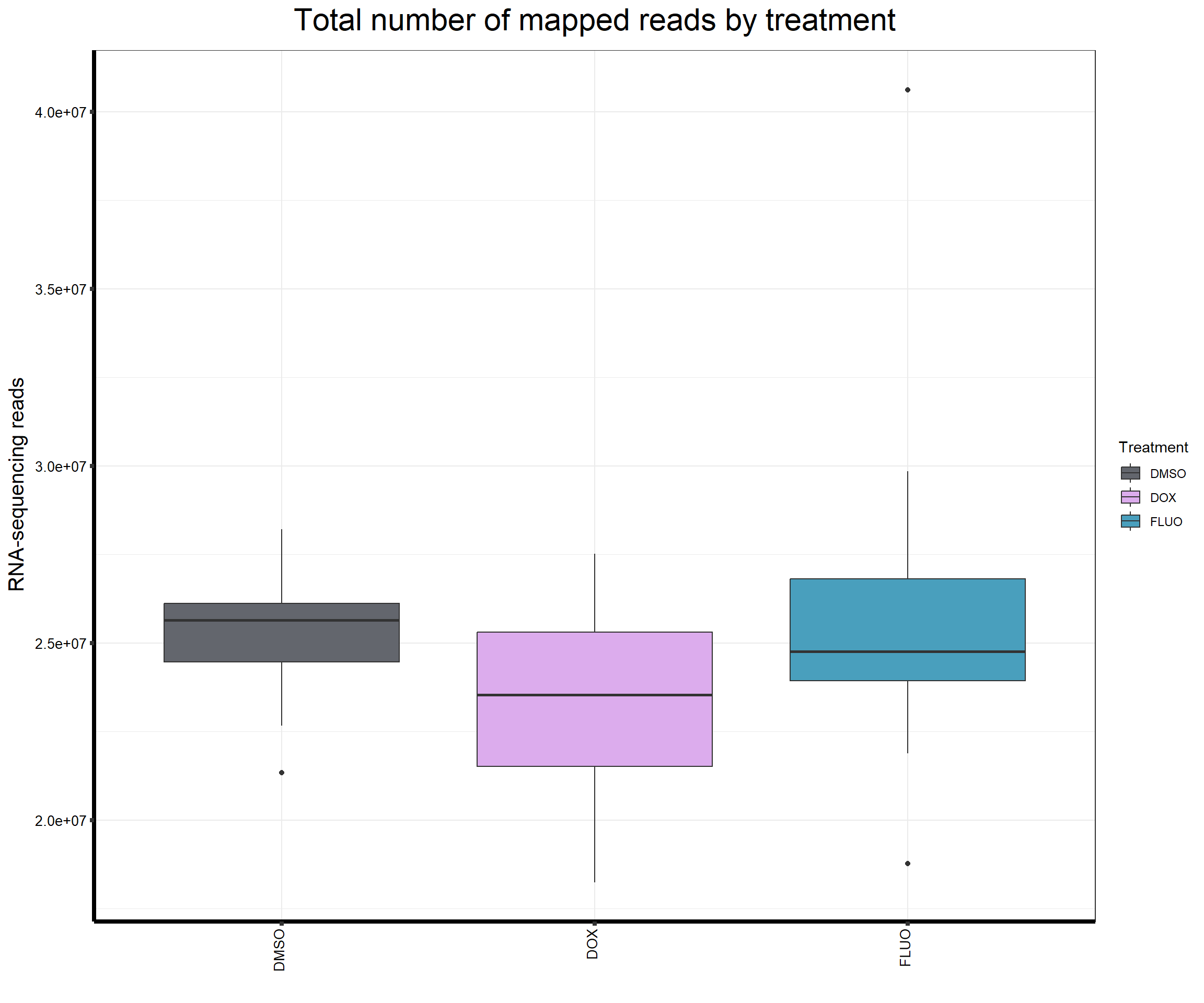
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
####Total Reads Per Individual####
fC_AllCounts %>%
ggplot(., aes (x =as.factor(Line), y=Assigned_Align))+
geom_boxplot(aes(fill=as.factor(Line)))+
scale_fill_brewer(palette = "Dark2", name = "Individual")+
ggtitle(expression("Total number of mapped reads by individual"))+
xlab("")+
ylab(expression("RNA-sequencing reads"))+
theme_bw()+
theme(plot.title = element_text(size = rel(2), hjust = 0.5),
axis.title = element_text(size = 15, color = "black"),
axis.ticks = element_line(linewidth = 1.5),
axis.line = element_line(linewidth = 1.5),
axis.text.y = element_text(size =10, color = "black", angle = 0, hjust = 0.8, vjust = 0.5),
axis.text.x = element_text(size =10, color = "black", angle = 0, hjust = 1),
#strip.text.x = element_text(size = 15, color = "black", face = "bold"),
strip.text.y = element_text(color = "white"))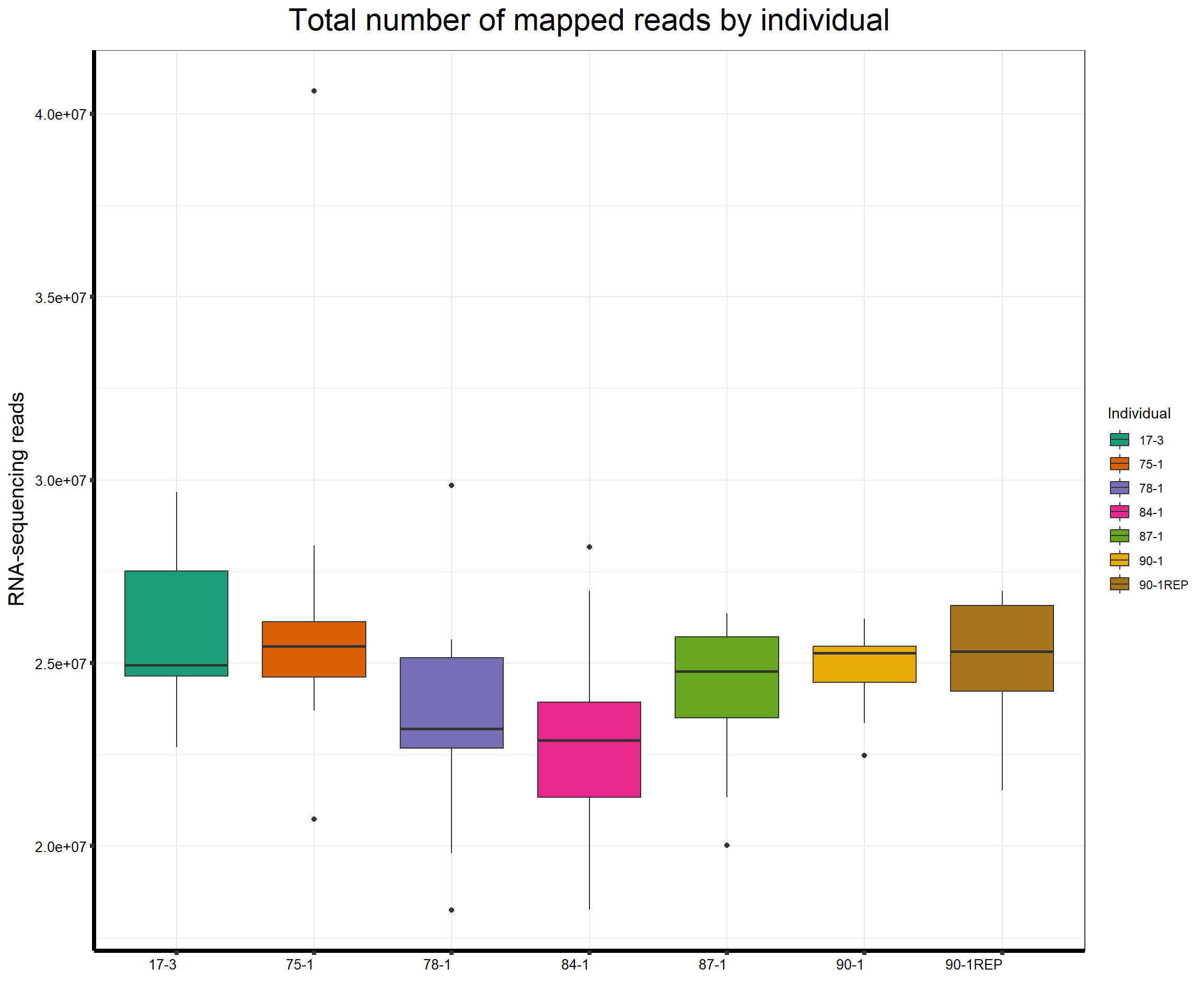
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
Now that I’ve made the final full matrix of all samples - 1. convert to log2cpm using cpm 2. filter for lowly expressed reads (rowMeans > 0)
boxplot(fC_Matrix_Full_cpm, xlab = "Conditions", ylab = expression("log"[2]* "counts per million"), ylim= c(-10, 20))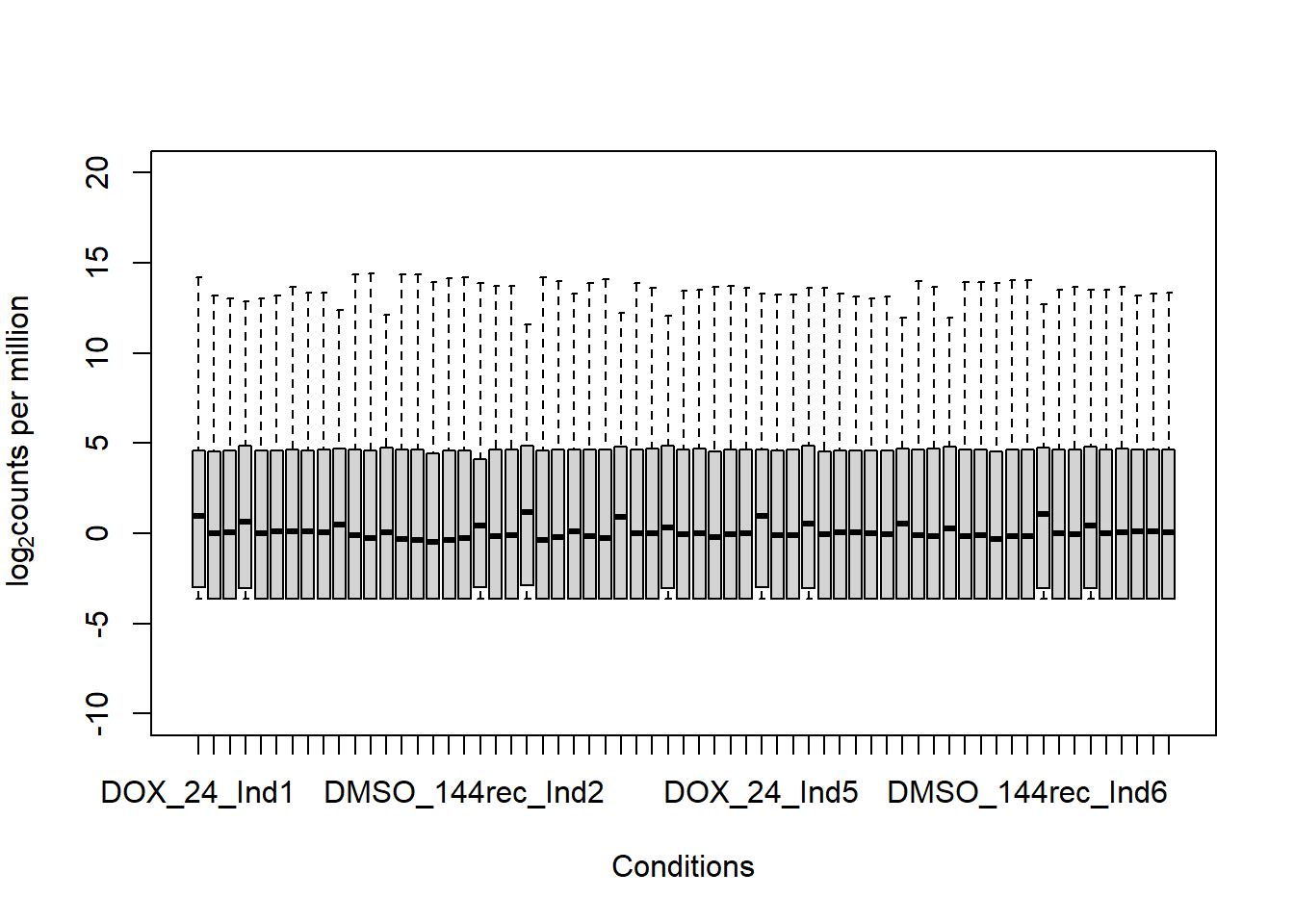
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
hist(fC_Matrix_Full_cpm, xlab = expression("log" [2]* "counts per million"))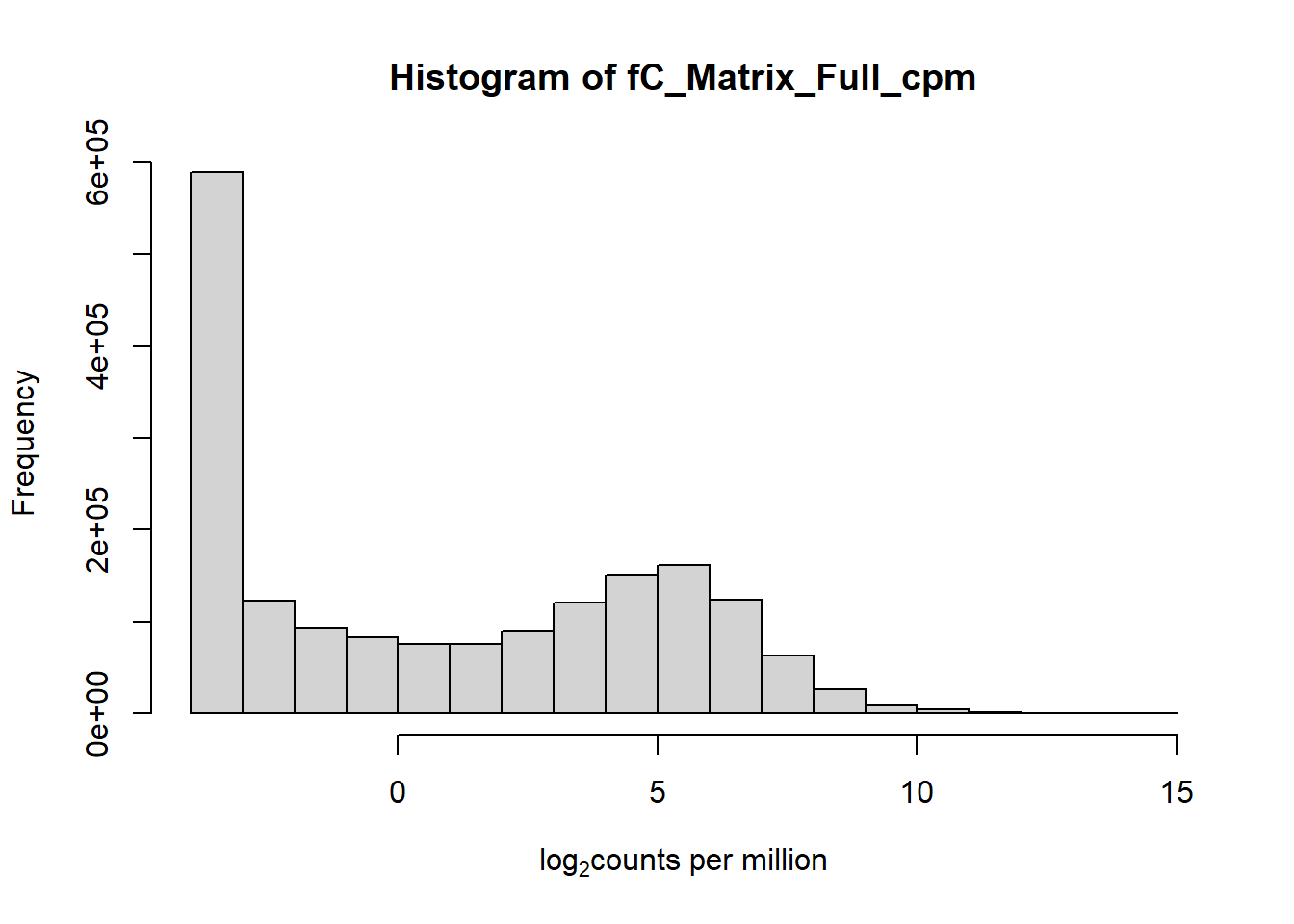
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
#write this to a csv just in case to save it
#write.csv(fC_Matrix_Full_cpm, "C:/Users/emmap/RDirectory/Recovery_RNAseq/Recovery_RNAseq/featureCounts_Concat_Matrix_AllSamples_cpm_EMP_250210.csv")
#first check the number of variables in the cpm file
dim(fC_Matrix_Full_cpm)
[1] 28395 63
#[1] 28395 63
#now use rowMeans to filter out values with mean < 0 (this means rowMeans > 0 is what I'm left with) for each row/gene in the original file
rowMeans_All <- rowMeans(fC_Matrix_Full_cpm)
fC_Matrix_Full_cpm_filter <- fC_Matrix_Full_cpm[rowMeans_All > 0,]
dim(fC_Matrix_Full_cpm_filter)
[1] 14225 63
#[1] 14225 63
#with this I've filtered out a significant number of rows when filtering after cpm conversion - does this make it more balanced?
hist(fC_Matrix_Full_cpm_filter,
main = "Recovery All Individuals log2cpm Filtered",
xlab = expression("log"[2]*"counts per million"))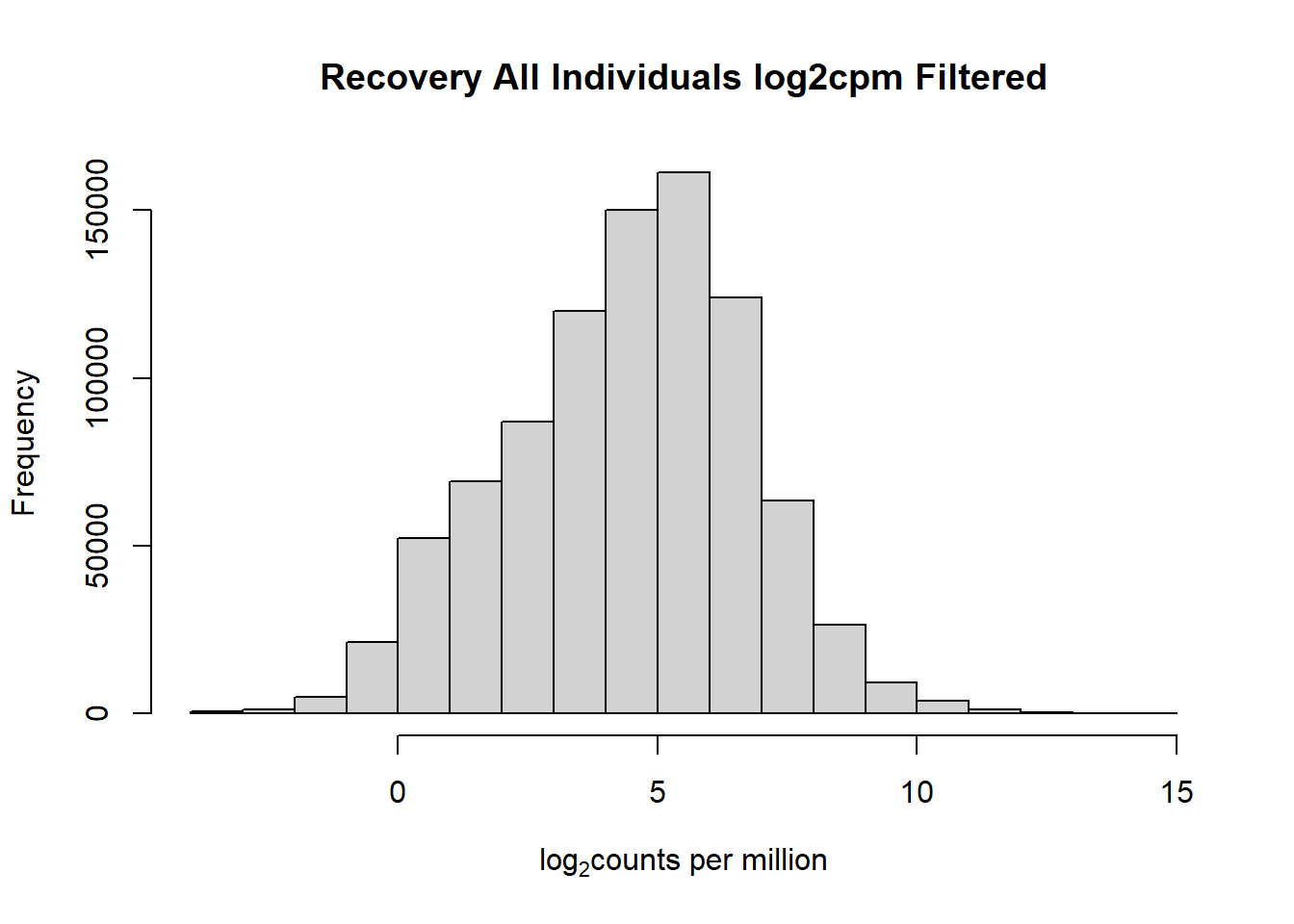
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
boxplot(fC_Matrix_Full_cpm_filter,
main = "Recovery All Individuals log2cpm Filtered",
xlab = "Conditions",
ylab = expression("log"[2]*"counts per million"),
ylim = c(-10,15))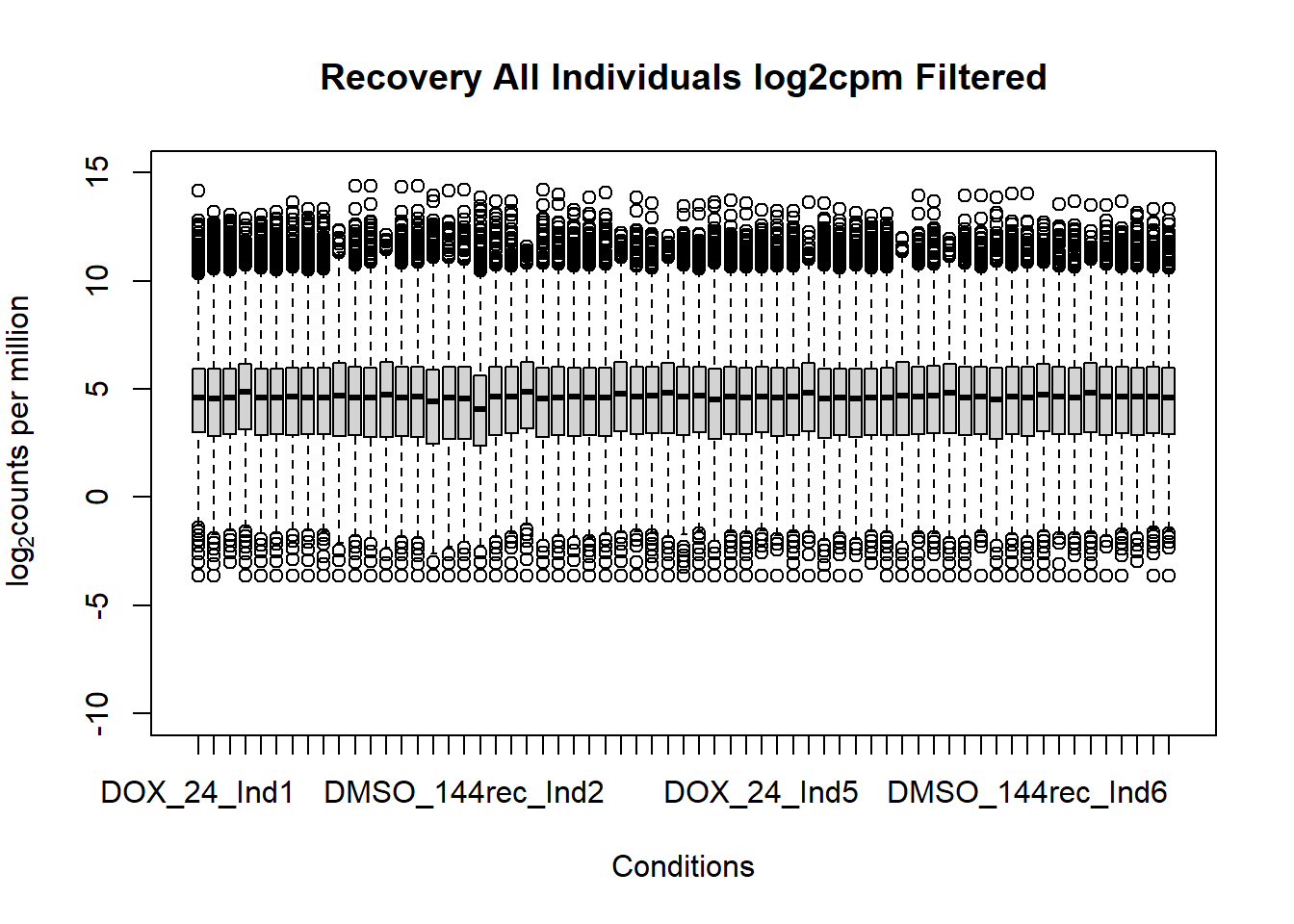
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
#write this to a csv to save
#write.csv(fC_Matrix_Full_cpm_filter, "C:/Users/emmap/RDirectory/Recovery_RNAseq/Recovery_RNAseq/featureCounts_Concat_Matrix_AllSamples_cpm_filtered_EMP_250210.csv")
####histogram of total counts, unfiltered####
hist(fC_Matrix_Full_cpm, main = "Recovery All Individuals log2cpm Unfiltered",
xlab = expression("log" [2]* "counts per million"))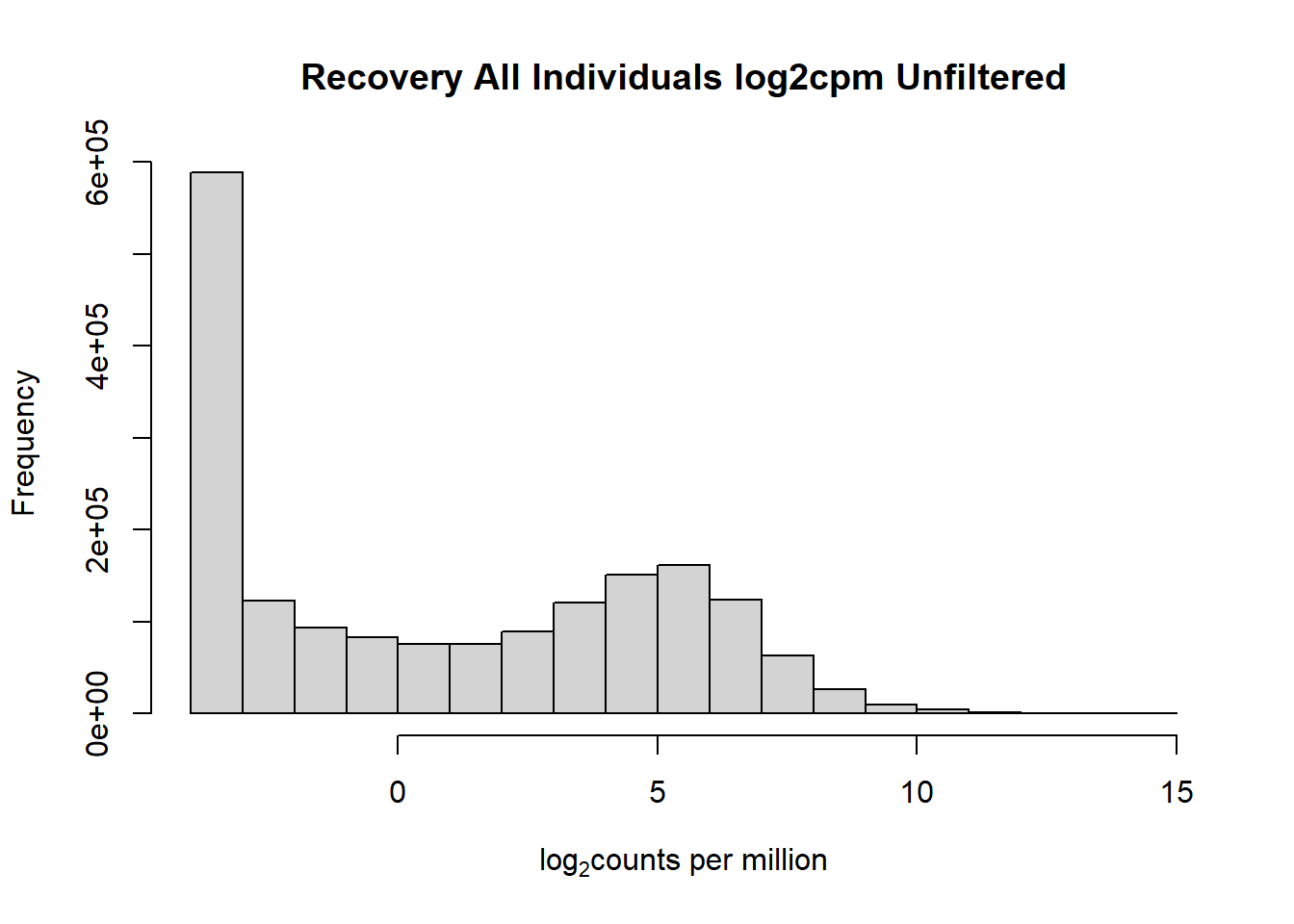
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
####histogram of total counts, filtered####
hist(fC_Matrix_Full_cpm_filter,
main = "Recovery All Individuals log2cpm Filtered rowMeans > 0",
xlab = expression("log"[2]*"counts per million"))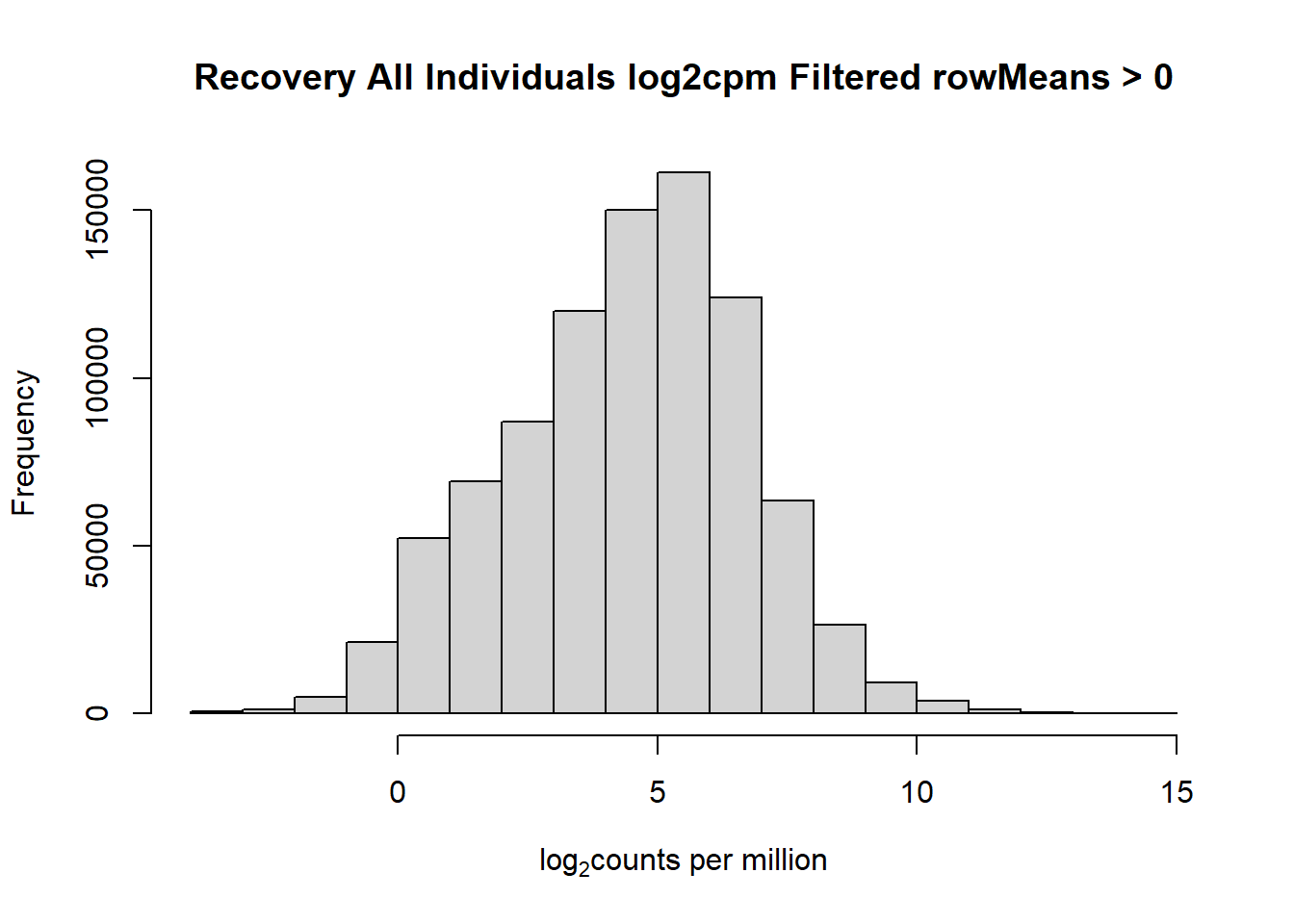
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
####boxplot log2cpm all samples####
boxplot(fC_Matrix_Full_cpm, xlab = "Conditions", ylab = expression("log"[2]* "counts per million"), ylim= c(-10, 20), main = "Boxplots of log2cpm per sample")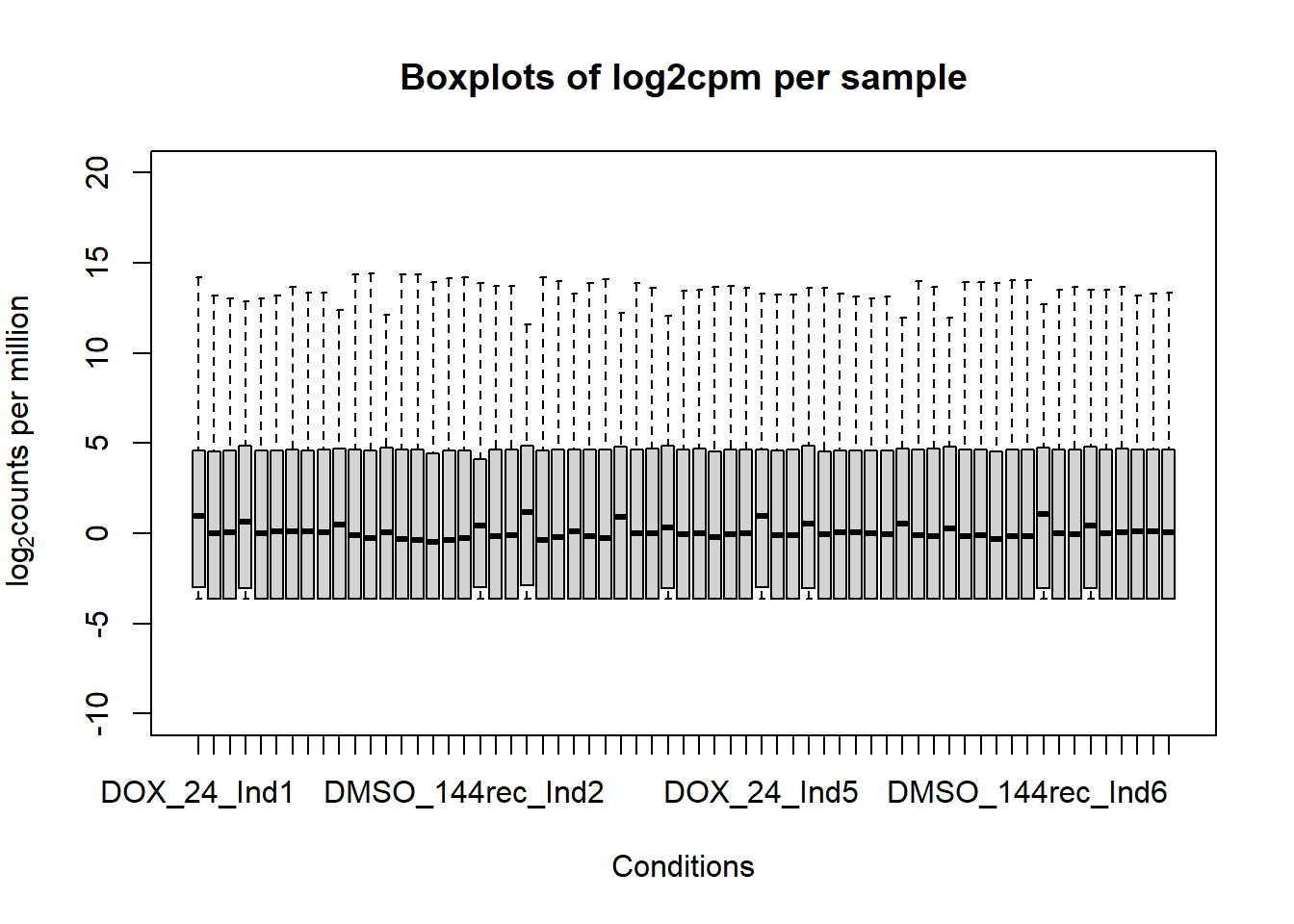
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
####boxplot log2cpm filtered all samples####
boxplot(fC_Matrix_Full_cpm_filter, xlab = "Conditions", ylab = expression("log"[2]* "counts per million"), ylim= c(-10, 20), main = "Boxplots of log2cpm Filtered rowMeans > 0 per sample")
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
Now that I’ve filtered the samples after log2cpm conversion, I can start to make heatmaps and PCA plots to look at my data
After trying and failing to get all three factors on one PCA plot - I will make iterations of each of these with two factors each
Now, I want to update these PCA plots to have treatment and time as the primary variables Individual will be a fill? Or somehow get some numbers labelled on the points.
PCA_data <- fC_Matrix_Full_cpm_filter %>%
prcomp(.) %>%
t()
PCA_data_test <- (prcomp(t(fC_Matrix_Full_cpm_filter), scale. = TRUE))
####now make an annotation for my PCA####
annot <- data.frame("samples" = colnames(fC_Matrix_Full_cpm_filter)) %>% separate_wider_delim(., cols = samples, names = c("Tx", "Time", "Ind"), delim = "_", cols_remove = FALSE) %>% unite(., col = "Tx_Time", Tx, Time, sep = "_", remove = FALSE)
#combine the prcomp matrix and annotation
annot_PCA_matrix <- PCA_data_test$x %>% cbind(., annot)
#now I can make a graph where I have filled values for individual! I have seven colors for seven individuals
# I have three fill values for three timepoints
#using annotation matrix above as well as annotated PCA matrix (annot_PCA_matrix)
#extra info for colors in the graph (fill parameter)
fill_col_ind <- c("#66C2A5", "#FC8D62", "#1F78B4", "#E78AC3", "#A6D854", "#FFD92A", "#8B3E9B")
fill_col_ind_dark <- c("#003F5C", "#45AE91", "#58508D", "#BC4099", "#8B3E9B", "#FF6361", "#FF2362")
fill_col_tx <- c("#63666D", "#DCACED", "#499FBD")
#Now I have to switch the fill and color parameters as it's doing the opposite of what I wanted: need individuals to be the outline color (color), and tx to be the inside color (fill)
####PC1/PC2####
annot_PCA_matrix %>% ggplot(., aes(x=PC1, y=PC2, size = 10)) +
geom_point(aes(color = Ind, shape = Time, fill = Tx)) +
scale_shape_manual(values = c(21, 22, 24)) +
scale_fill_manual(values = fill_col_tx)+
scale_color_manual(values = c(fill_col_ind_dark))+
guides(fill=guide_legend(override.aes=list(shape=21)))+
guides(color=guide_legend(override.aes=list(shape=21)))+
guides(fill=guide_legend(override.aes=list(shape=21,fill=fill_col_tx,color=fill_col_tx)))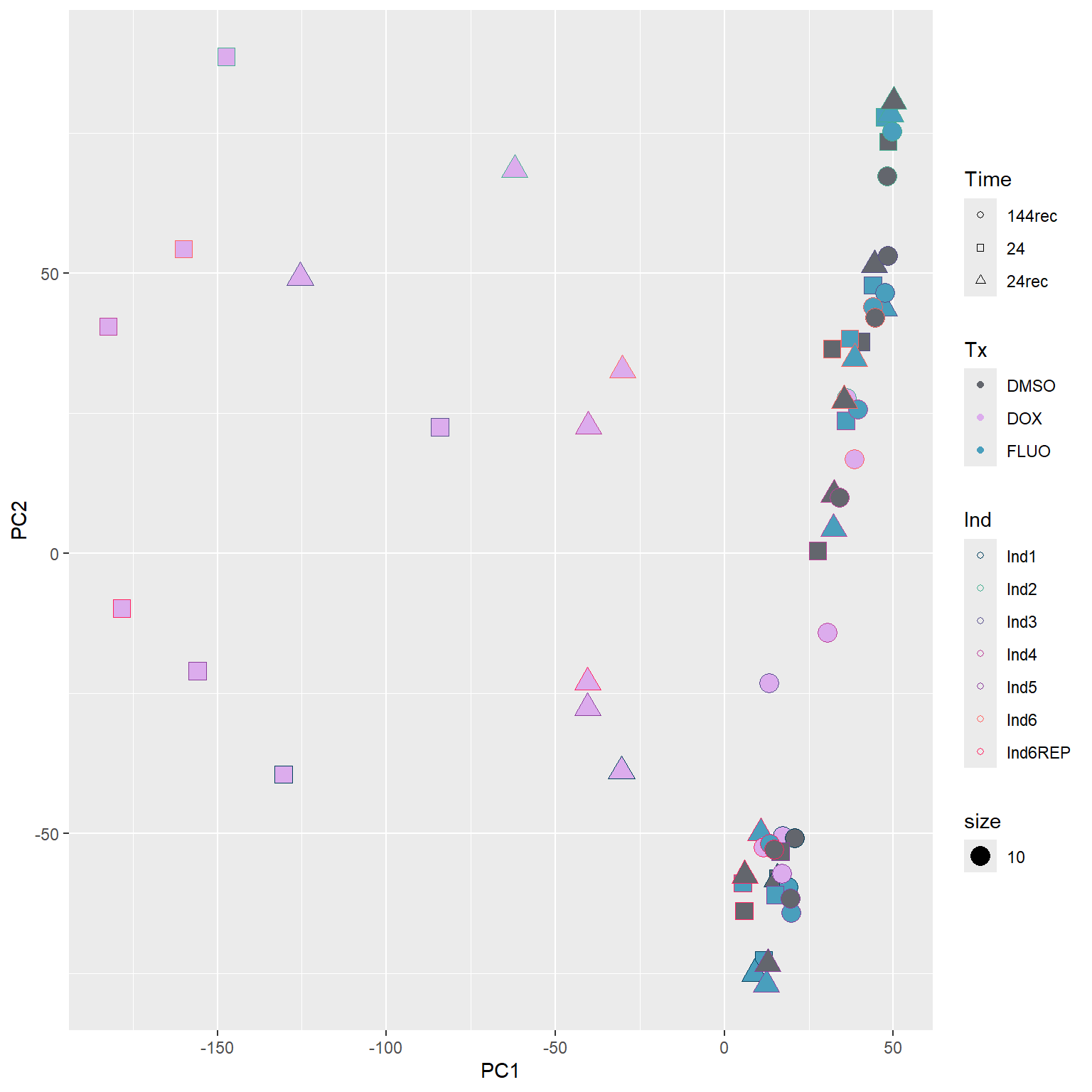
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
####PC2/PC3####
annot_PCA_matrix %>% ggplot(., aes(x=PC2, y=PC3)) +
geom_point(aes(color = Ind, shape = Time, fill = Tx, size = 10)) +
scale_shape_manual(values = c(21, 22, 24)) +
scale_fill_manual(values = fill_col_tx)+
scale_color_manual(values = c(fill_col_ind_dark))+
guides(fill=guide_legend(override.aes=list(shape=21)))+
guides(color=guide_legend(override.aes=list(shape=21)))+
guides(fill=guide_legend(override.aes=list(shape=21,fill=fill_col_tx,color=fill_col_tx)))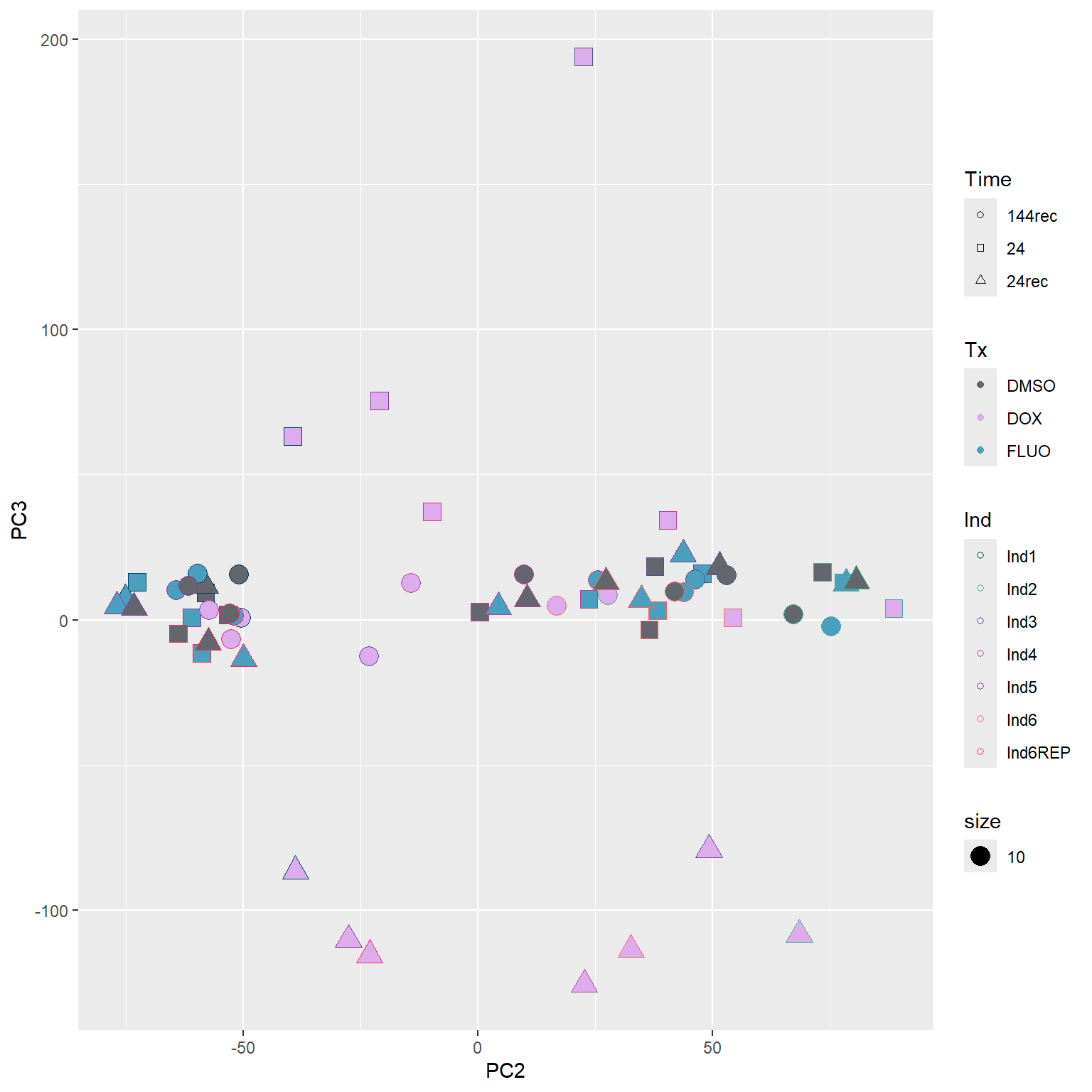
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
####PC3/PC4####
annot_PCA_matrix %>% ggplot(., aes(x=PC3, y=PC4)) +
geom_point(aes(color = Ind, shape = Time, fill = Tx, size = 10)) +
scale_shape_manual(values = c(21, 22, 24)) +
scale_fill_manual(values = fill_col_tx)+
scale_color_manual(values = c(fill_col_ind_dark))+
guides(fill=guide_legend(override.aes=list(shape=21)))+
guides(color=guide_legend(override.aes=list(shape=21)))+
guides(fill=guide_legend(override.aes=list(shape=21,fill=fill_col_tx,color=fill_col_tx)))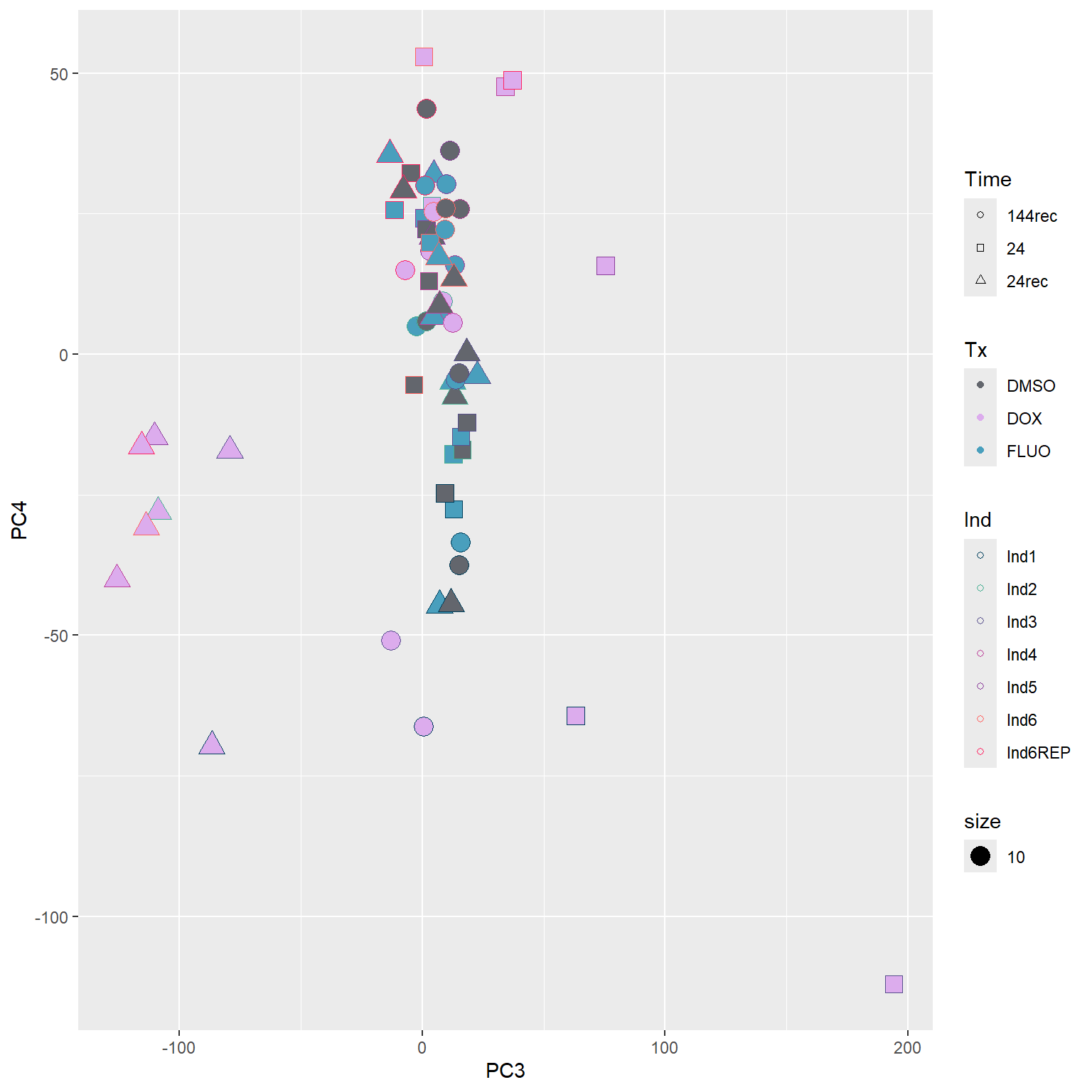
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
#correlation heatmap for all samples
####PEARSON FILTERED####
fC_Matrix_Full_cpm_filter_pearsoncor <-
cor(
fC_Matrix_Full_cpm_filter,
y = NULL,
use = "everything",
method = "pearson"
)
####SPEARMAN FILTERED####
fC_Matrix_Full_cpm_filter_spearmancor <-
cor(
fC_Matrix_Full_cpm_filter,
y = NULL,
use = "everything",
method = "spearman"
)
#Now let's graph these correlations
pheatmap(fC_Matrix_Full_cpm_filter_pearsoncor, border_color = "black", legend = TRUE, angle_col = 90, display_numbers = FALSE, number_color = "black", fontsize = 12, fontsize_number = 9)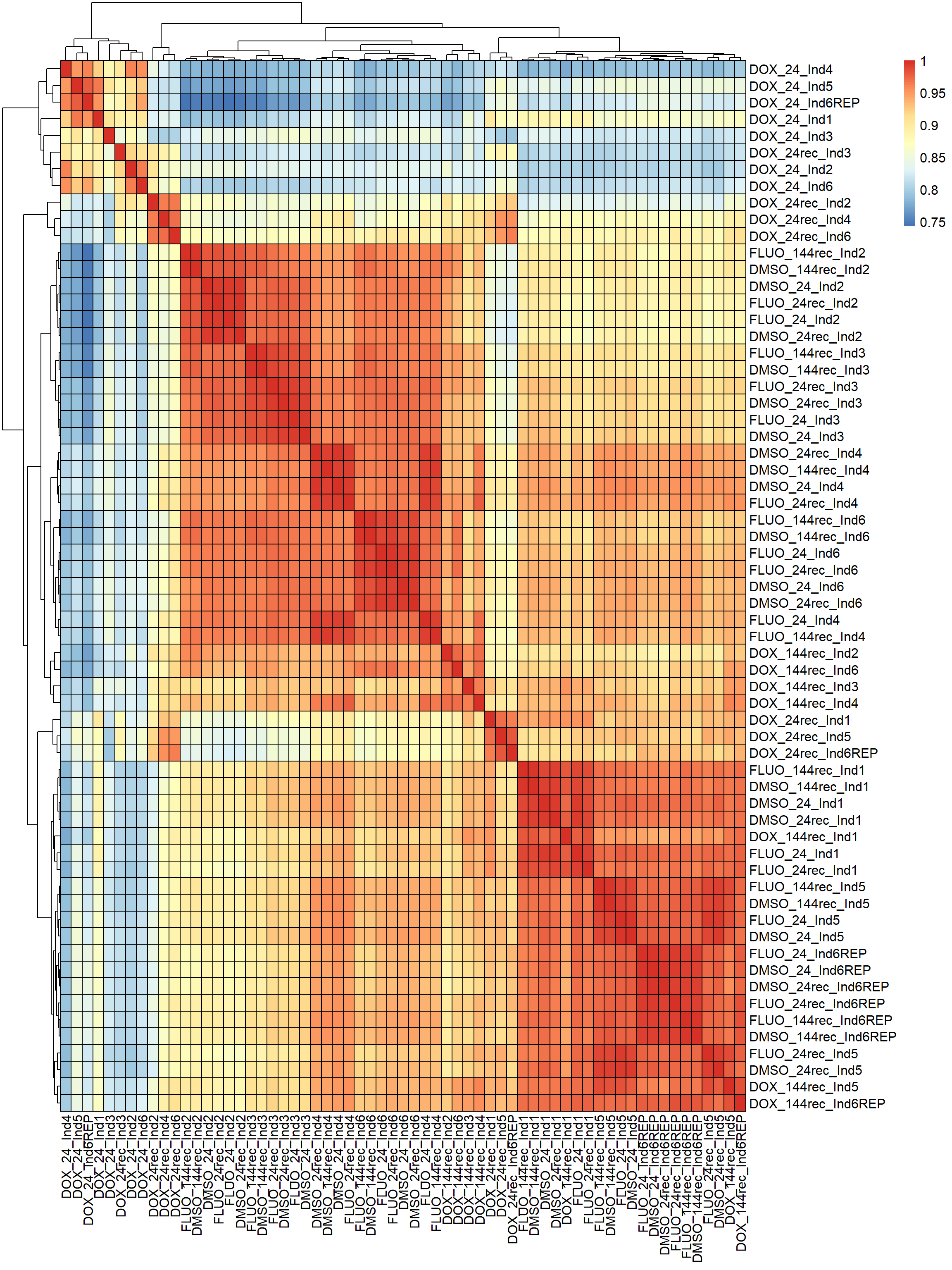
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
pheatmap(fC_Matrix_Full_cpm_filter_spearmancor, border_color = "black", legend = TRUE, angle_col = 90, display_numbers = FALSE, number_color = "black", fontsize = 12, fontsize_number = 9)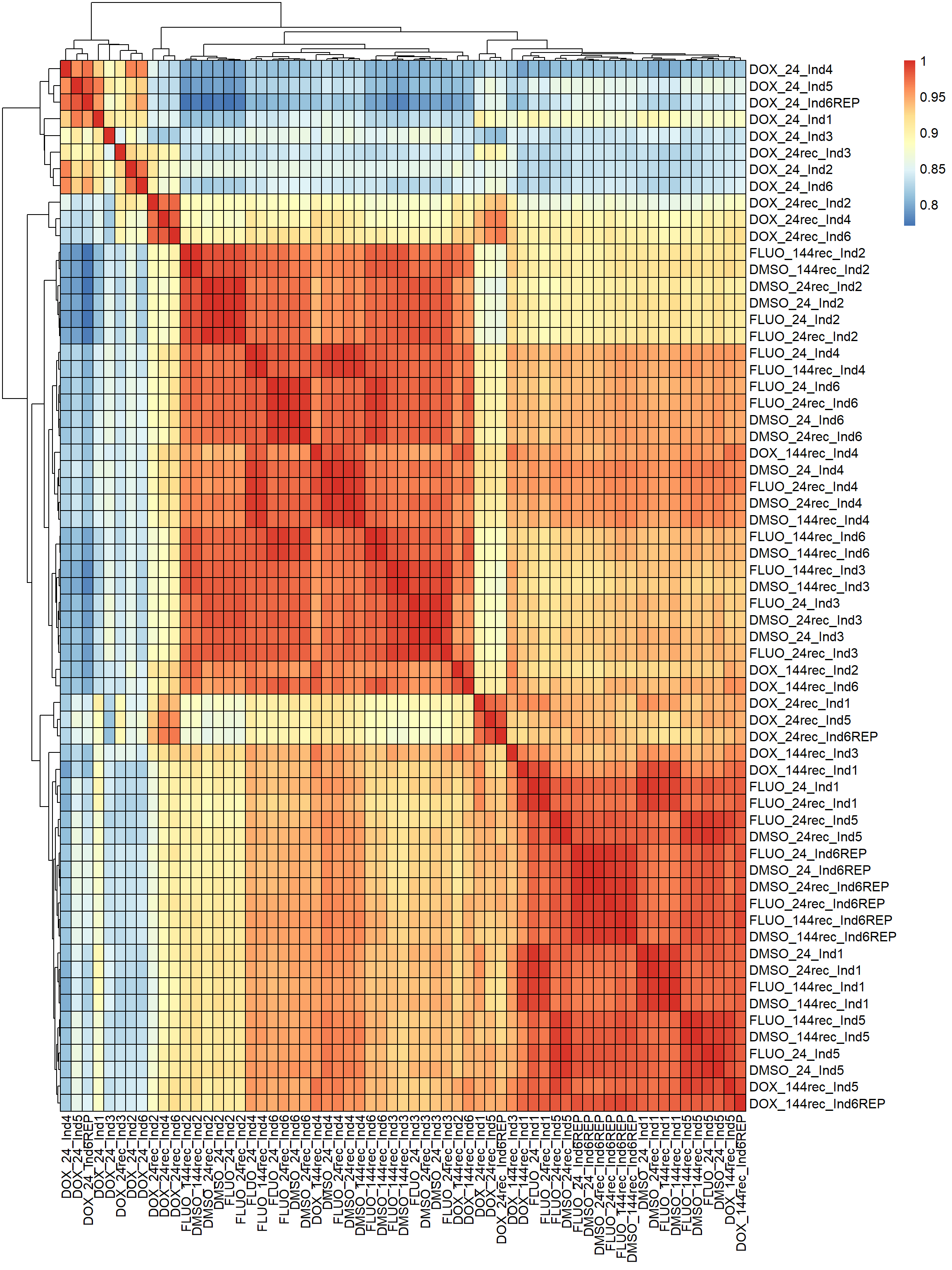
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
#now annotate these heatmaps
#Factor 1 - Individual
Individual <- as.factor(c(rep("Ind1", 9), rep("Ind2", 9), rep("Ind3", 9), rep("Ind4", 9), rep("Ind5", 9), rep("Ind6", 9), rep("Ind6REP", 9)))
#Factor 2 - Treatment
tx_factor <- c("DOX", "FLUO", "DMSO")
Tx <- as.factor(c(rep(tx_factor, 21)))
#view(Treatment)
#Factor 3 - Timepoint
time_factor <- c(rep("24", 3), rep("24rec", 3), rep("144rec", 3))
Time <- as.factor(c(rep(time_factor, 7)))
####annotation for colors####
annot_col_hm = list(Tx = c(DOX = "red", FLUO = "blue", DMSO = "black"),
Ind = c(Ind1 = "#66C2A5", Ind2 = "#FC8D62", Ind3 = "#1F78B4", Ind4 = "#E78AC3", Ind5 = "#A6D854", Ind6 = "#FFD92A", Ind6REP = "#8B3E9B"),
Time = c("24" = "#046A38", "24rec" = "#0050B5", "144rec" = "#B725AD"))
####annotation for values####
annot_list_hm <- data.frame(Individual = as.factor(c(rep("Ind1", 9), rep("Ind2", 9), rep("Ind3", 9), rep("Ind4", 9), rep("Ind5", 9), rep("Ind6", 9), rep("Ind6REP", 9))),
Tx = as.factor(c(rep(tx_factor, 21))),
Time = as.factor(c(rep(time_factor, 7))))
##add in the annotations from above into the dataframe
row.names(annot_list_hm) <- colnames(fC_Matrix_Full_cpm_filter_pearsoncor)
row.names(annot_list_hm) <- colnames(fC_Matrix_Full_cpm_filter_spearmancor)
####ANNOTATED HEATMAPS####
pheatmap(fC_Matrix_Full_cpm_filter_pearsoncor, border_color = "black", legend = TRUE, angle_col = 90, display_numbers = FALSE, number_color = "black", fontsize = 10, fontsize_number = 5, annotation_col = annot_list_hm, annotation_colors = annot_col_hm)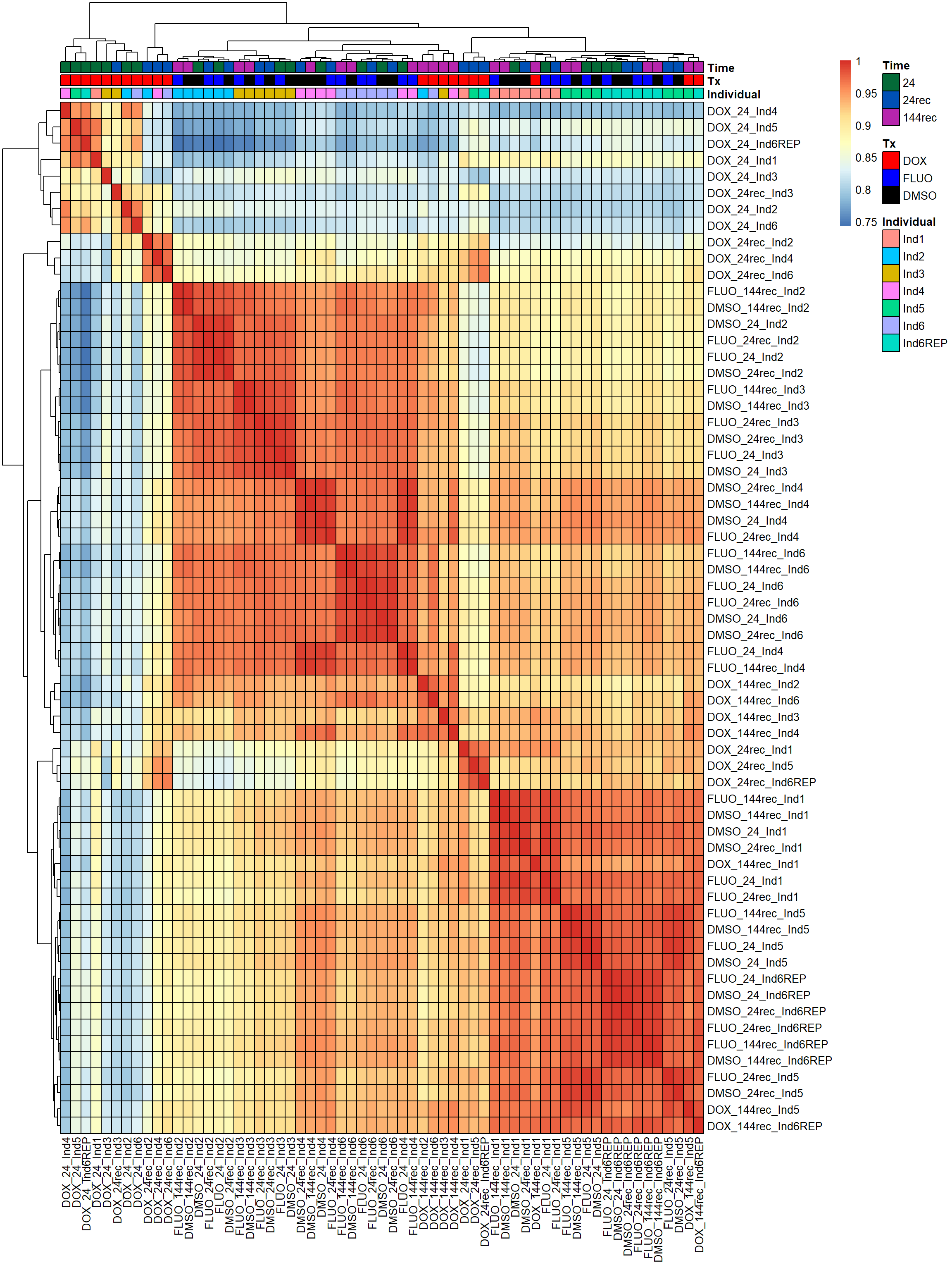
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
pheatmap(fC_Matrix_Full_cpm_filter_spearmancor, border_color = "black", legend = TRUE, angle_col = 90, display_numbers = FALSE, number_color = "black", fontsize = 10, fontsize_number = 5, annotation_col = annot_list_hm, annotation_colors = annot_col_hm)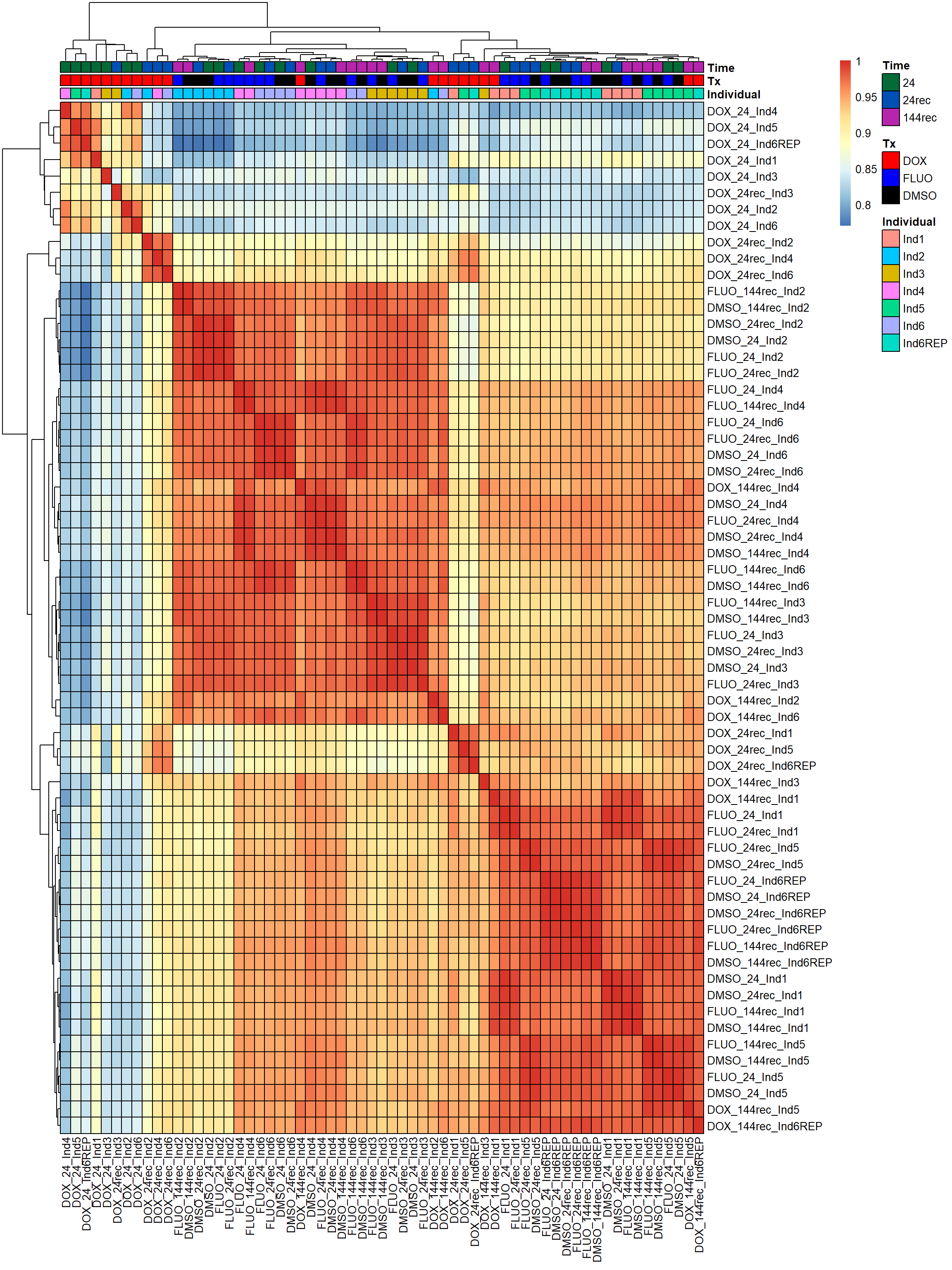
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
Now that I’ve finalized my QC checks, I will go ahead and begin the differential expression analysis pipeline.
# group <- c(rep(c(1, 2, 3, 4, 5, 6, 7, 8, 9), 6))
# group <- factor(group, levels = c("1", "2", "3", "4", "5", "6", "7", "8", "9"))
####rowMeans > 0 Filtering####
rowMeans_DE <- rowMeans(counts_DE)
counts_DE_filter <- counts_DE[rowMeans_DE > 0,]
dim(counts_DE_filter)
[1] 26445 54
group_1 <- rep(c("DOX_24","FLUO_24",
"DMSO_24",
"DOX_24rec",
"FLUO_24rec",
"DMSO_24rec",
"DOX_144rec",
"FLUO_144rec",
"DMSO_144rec"), 6)
dge <- DGEList.data.frame(counts = counts_DE_filter, group = group_1, genes = row.names(counts_DE_filter))
#check the inital file before norm factors are calculated
#dge$samples
#calculate the normalization factors with method TMM
dge_calc <- calcNormFactors(dge, method = "TMM")
#check final file after norm calculation
#dge_calc$samples
#after calculating norm factors, that column was changed.
#View(dge_calc)
#Pull out factors
snames <- data.frame("samples" = colnames(dge_calc)) %>% separate_wider_delim(., cols = samples, names = c("Treatment", "Time", "Individual"), delim = "_", cols_remove = FALSE)
#snames_list <- as.list(snames)
snames_time <- snames$Time
snames_tx <- snames$Treatment
snames_ind <- snames$Individual
#Create my model matrix
mm_r <- model.matrix(~0 + group_1)
p <- voom(dge_calc$counts, mm_r, plot = TRUE)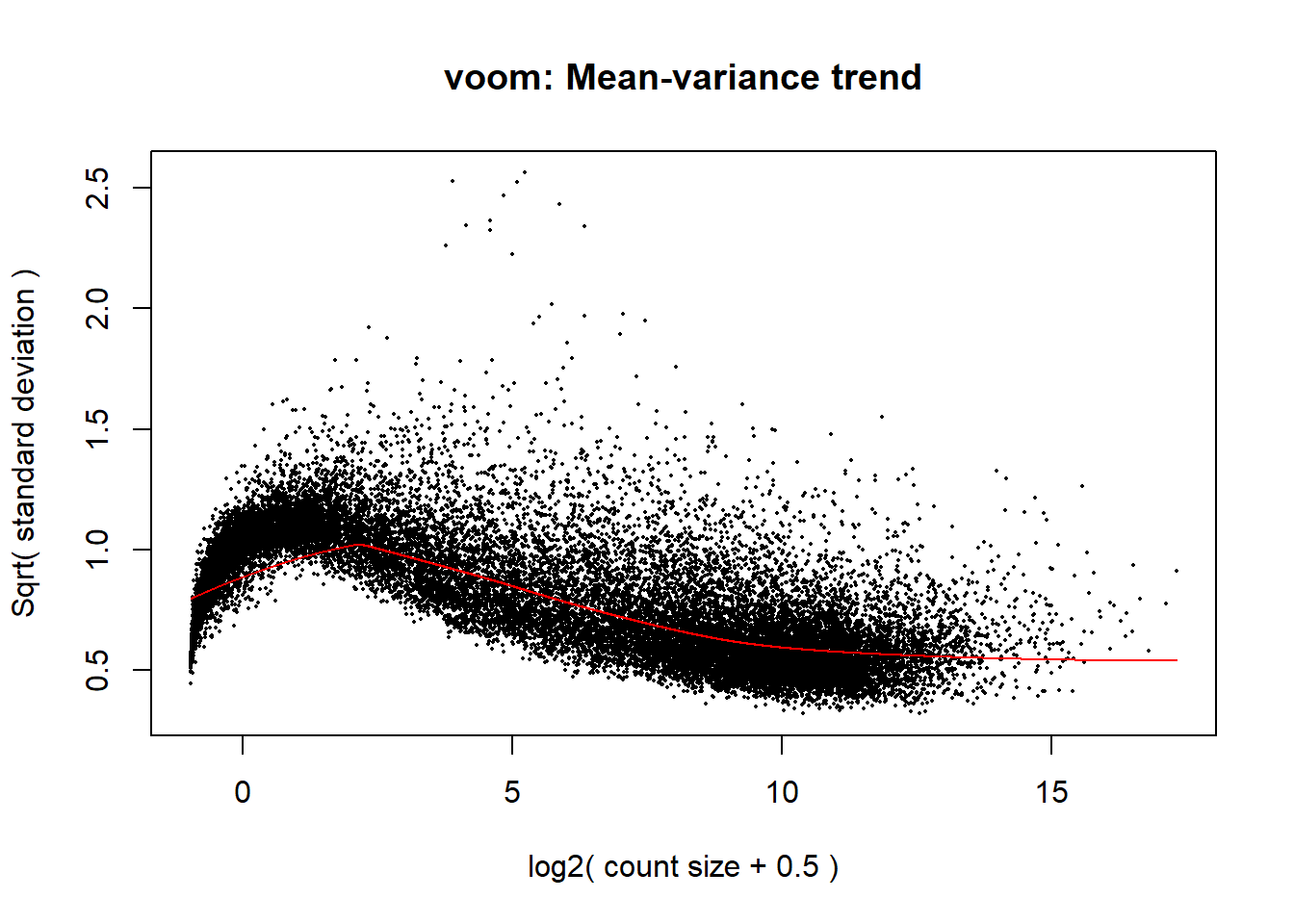
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
corfit <- duplicateCorrelation(p, mm_r, block = snames_ind)
v <- voom(dge_calc$counts, mm_r, block = snames_ind, correlation = corfit$consensus)
fit <- lmFit(v, mm_r, block = snames_ind, correlation = corfit$consensus)
#make sure to check which order the columns are in - otherwise they won't match right (it was moved into alphabetical and number order)
colnames(mm_r) <- c("DMSO_144rec","DMSO_24","DMSO_24rec","DOX_144rec","DOX_24","DOX_24rec","FLUO_144rec","FLUO_24","FLUO_24rec")
cm_r <- makeContrasts(
V.D24 = DOX_24 - DMSO_24,
V.F24 = FLUO_24 - DMSO_24,
V.D24r = DOX_24rec - DMSO_24rec,
V.F24r = FLUO_24rec - DMSO_24rec,
V.D144r = DOX_144rec - DMSO_144rec,
V.F144r = FLUO_144rec - DMSO_144rec,
levels = mm_r
)
vfit_r <- lmFit(p, mm_r)
vfit_r <- contrasts.fit(vfit_r, contrasts = cm_r)
efit2 <- eBayes(vfit_r)
results = decideTests(efit2)
summary(results)
V.D24 V.F24 V.D24r V.F24r V.D144r V.F144r
Down 4567 0 2083 0 288 0
NotSig 14521 26445 16339 26445 25296 26445
Up 7357 0 8023 0 861 0
#inbuilt annotation results rowMeans >0
# V.D24 V.F24 V.D24r V.F24r V.D144r V.F144r
# Down 4567 0 2083 0 288 0
# NotSig 14521 26445 16339 26445 25296 26445
# Up 7357 0 8023 0 861 0
## have no DE genes detected for 5FU, only DOX
####plot your voom####
voom_plot <- voom(dge_calc, mm_r, plot = TRUE)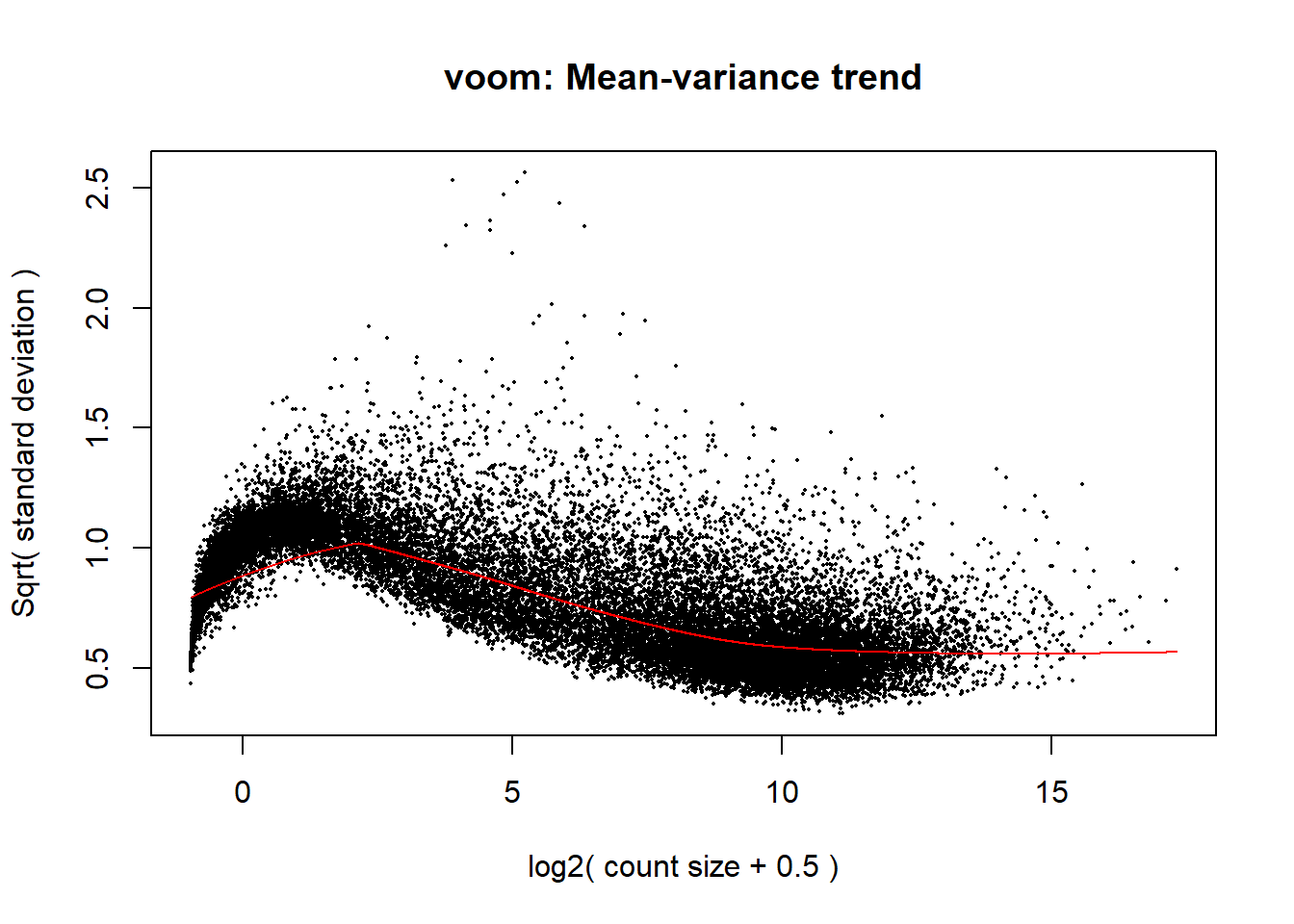
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
The voom plot for this with rowMeans > 0 has a dip in the front, meaning that there is likely some noise left over in my data. I am going to filter a bit stricter at rowMeans > 1 with the same process as above.
####rowMeans >1####
rowMeans_DE <- rowMeans(counts_DE)
counts_DE_filter1 <- counts_DE[rowMeans_DE > 1,]
dim(counts_DE_filter1)
[1] 20443 54
dge1 <- DGEList.data.frame(counts = counts_DE_filter1, group = group_1, genes = row.names(counts_DE_filter1))
#check the inital file before norm factors are calculated
#dge1$samples
#calculate the normalization factors with method TMM
dge1_calc <- calcNormFactors(dge1, method = "TMM")
#check final file after norm calculation
#dge1_calc$samples
#View(dge1_calc)
#Pull out factors
snames1 <- data.frame("samples" = colnames(dge1_calc)) %>% separate_wider_delim(., cols = samples, names = c("Treatment", "Time", "Individual"), delim = "_", cols_remove = FALSE)
#snames_list <- as.list(snames)
snames1_time <- snames1$Time
snames1_tx <- snames1$Treatment
snames1_ind <- snames1$Individual
#Create my model matrix
mm_r1 <- model.matrix(~0 + group_1)
p1 <- voom(dge1_calc$counts, mm_r1, plot = TRUE)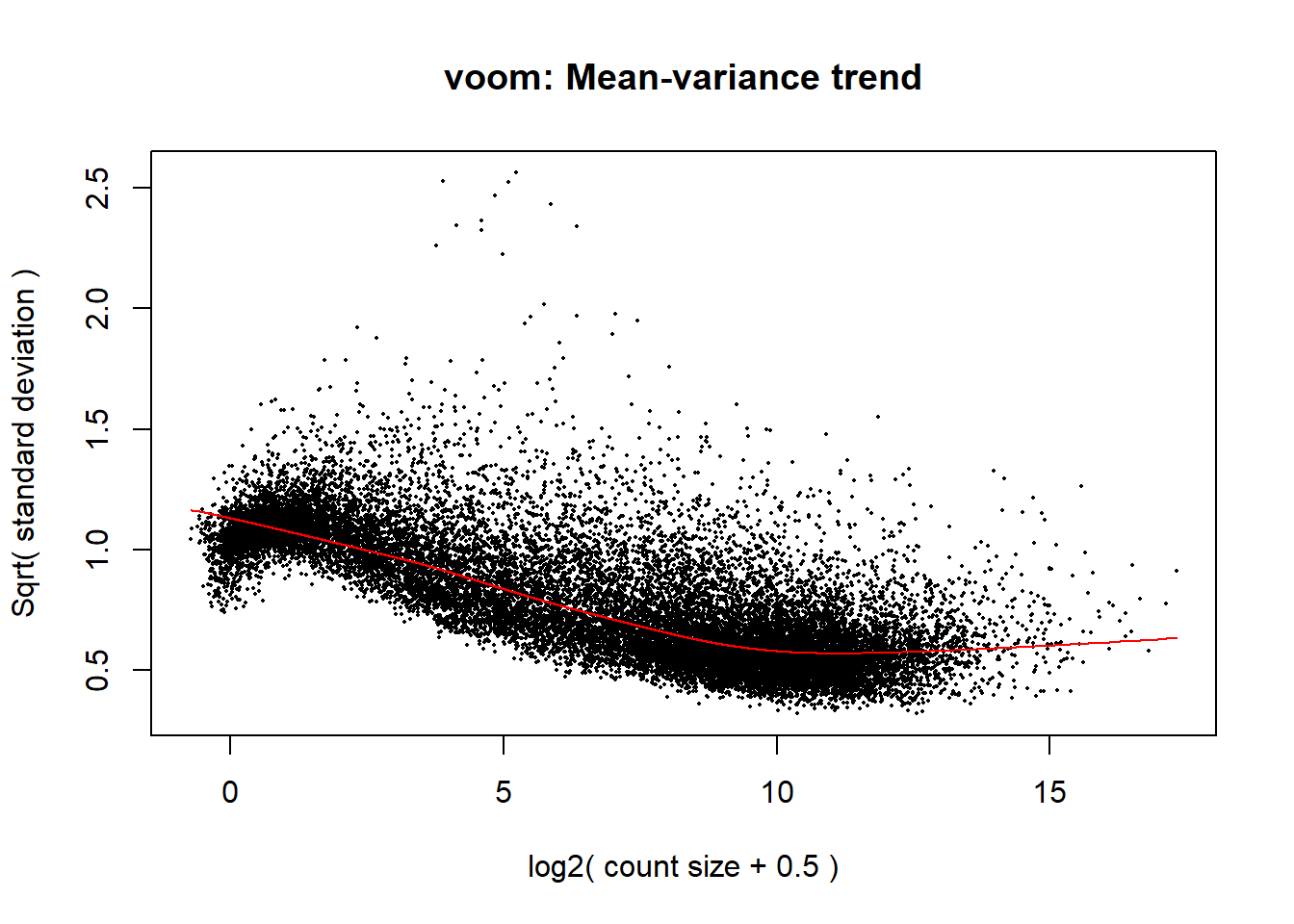
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
corfit1 <- duplicateCorrelation(p1, mm_r1, block = snames1_ind)
v1 <- voom(dge1_calc$counts, mm_r1, block = snames1_ind, correlation = corfit1$consensus)
fit1 <- lmFit(v1, mm_r1, block = snames1_ind, correlation = corfit1$consensus)
#make sure to check which order the columns are in - otherwise they won't match right (it was moved into alphabetical and number order)
colnames(mm_r1) <- c("DMSO_144rec","DMSO_24","DMSO_24rec","DOX_144rec","DOX_24","DOX_24rec","FLUO_144rec","FLUO_24","FLUO_24rec")
cm_r1 <- makeContrasts(
V.D24 = DOX_24 - DMSO_24,
V.F24 = FLUO_24 - DMSO_24,
V.D24r = DOX_24rec - DMSO_24rec,
V.F24r = FLUO_24rec - DMSO_24rec,
V.D144r = DOX_144rec - DMSO_144rec,
V.F144r = FLUO_144rec - DMSO_144rec,
levels = mm_r1
)
vfit_r1 <- lmFit(p1, mm_r1)
vfit_r1 <- contrasts.fit(vfit_r1, contrasts = cm_r1)
efit4 <- eBayes(vfit_r1)
results1 = decideTests(efit4)
summary(results1)
V.D24 V.F24 V.D24r V.F24r V.D144r V.F144r
Down 4640 0 2120 0 263 0
NotSig 9460 20443 11485 20443 19838 20443
Up 6343 0 6838 0 342 0
#inbuilt annotation results rowMeans >0
# V.D24 V.F24 V.D24r V.F24r V.D144r V.F144r
# Down 4567 0 2083 0 288 0
# NotSig 14521 26445 16339 26445 25296 26445
# Up 7357 0 8023 0 861 0
## have no DE genes detected for 5FU, only DOX
####plot your voom####
voom_plot1 <- voom(dge1_calc, mm_r1, plot = TRUE)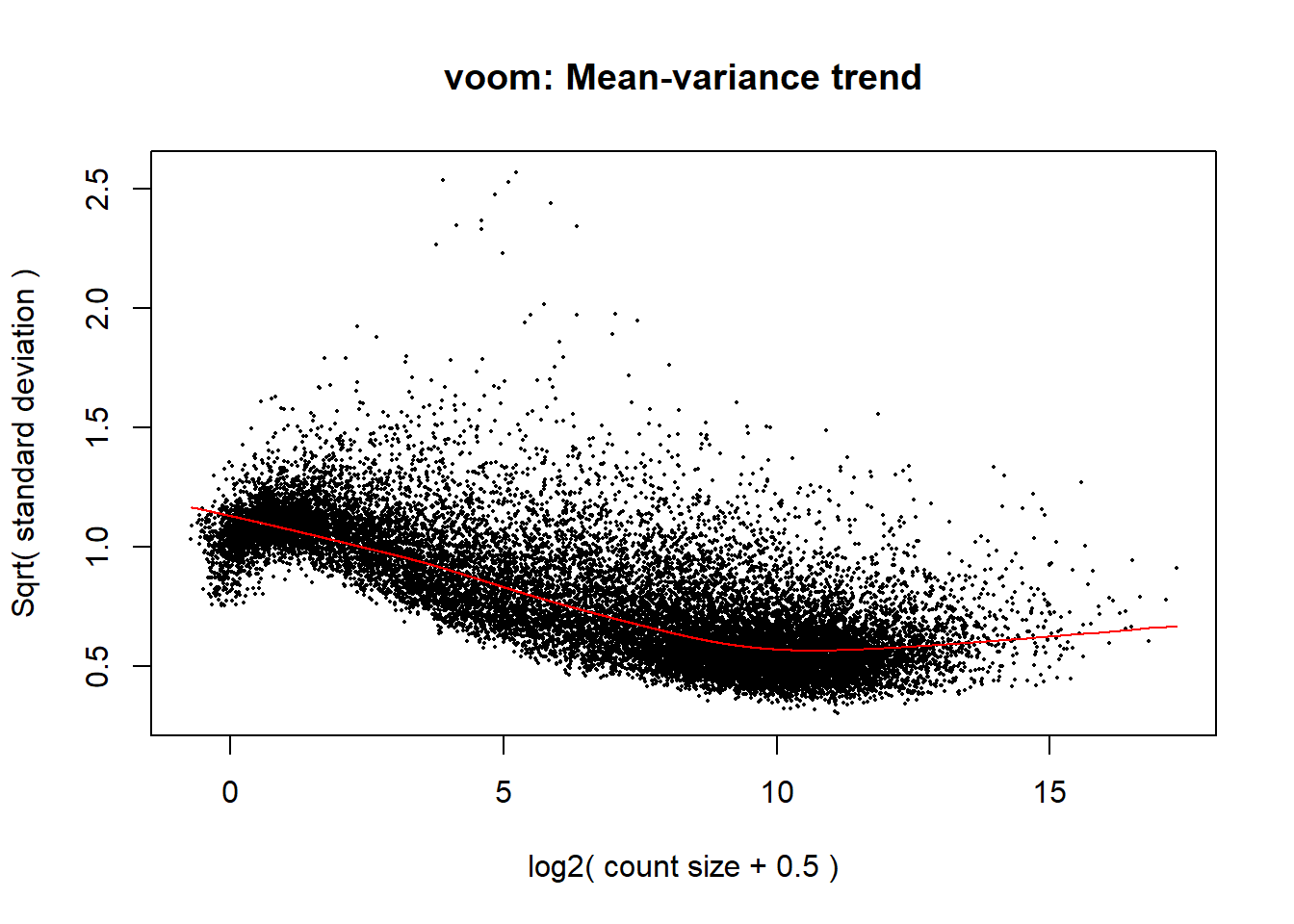
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
This looks much better! No more noise in the front. I could potentially filter at rowMeans >0.5, but I will leave that for now.
Now I can go ahead and make toptables for safekeeping and later analysis
top.table_V.D24 <- topTable(fit = efit4, coef = "V.D24", number = nrow(dge_calc), adjust.method = "BH", p.value = 1, sort.by = "none")
head(top.table_V.D24)
top.table_V.F24 <- topTable(fit = efit4, coef = "V.F24", number = nrow(dge_calc), adjust.method = "BH", p.value = 1, sort.by = "none")
head(top.table_V.F24)
top.table_V.D24r <- topTable(fit = efit4, coef = "V.D24r", number = nrow(dge_calc), adjust.method = "BH", p.value = 1, sort.by = "none")
head(top.table_V.D24r)
top.table_V.F24r <- topTable(fit = efit4, coef = "V.F24r", number = nrow(dge_calc), adjust.method = "BH", p.value = 1, sort.by = "none")
head(top.table_V.F24r)
top.table_V.D144r <- topTable(fit = efit4, coef = "V.D144r", number = nrow(dge_calc), adjust.method = "BH", p.value = 1, sort.by = "none")
head(top.table_V.D144r)
top.table_V.F144r <- topTable(fit = efit4, coef = "V.F144r", number = nrow(dge_calc), adjust.method = "BH", p.value = 1, sort.by = "none")
head(top.table_V.F144r)
Volcano Plots
generate_volcano_plot <- function(toptable, title) {
# Add Significance Labels
toptable$Significance <- "Not Significant"
toptable$Significance[toptable$logFC > 0 & toptable$adj.P.Val < 0.05] <- "Upregulated"
toptable$Significance[toptable$logFC < 0 & toptable$adj.P.Val < 0.05] <- "Downregulated"
# Generate Volcano Plot
ggplot(toptable, aes(x = logFC, y = -log10(P.Value), color = Significance)) +
geom_point(alpha = 0.4, size = 2) +
scale_color_manual(values = c("Downregulated" = "red", "Upregulated" = "blue", "Not Significant" = "gray")) +
xlim(-5, 5) +
labs(title = title, x = "log2 Fold Change", y = "-log10 P-value") +
theme(legend.position = "none",
plot.title = element_text(size = rel(1.5), hjust = 0.5),
axis.title = element_text(size = rel(1.25))) +
theme_bw()
}
#now that I've made a function, I can make volcano plots for each of my comparisons (6 total)
volcano_plots <- list(
"V.D24" = generate_volcano_plot(top.table_V.D24, "Volcano Plot DOX 24hr (adj P-val<0.05)"),
"V.F24" = generate_volcano_plot(top.table_V.F24, "Volcano Plot 5FU 24hr (adj P-val<0.05)"),
"V.D24r" = generate_volcano_plot(top.table_V.D24r, "Volcano Plot DOX 24hr Recovery (adj P-val<0.05)"),
"V.F24r" = generate_volcano_plot(top.table_V.F24r, "Volcano Plot 5FU 24hr Recovery (adj P-val<0.05)"),
"V.D144r" = generate_volcano_plot(top.table_V.D144r, "Volcano Plot DOX 6d Recovery (adj P-val<0.05)"),
"V.F144r" = generate_volcano_plot(top.table_V.F144r, "Volcano Plot 5FU 6d Recovery (adj P-val<0.05)")
)
# Display each volcano plot
for (plot_name in names(volcano_plots)) {
print(volcano_plots[[plot_name]])
}
Warning: Removed 344 rows containing missing values or values outside the scale range
(`geom_point()`).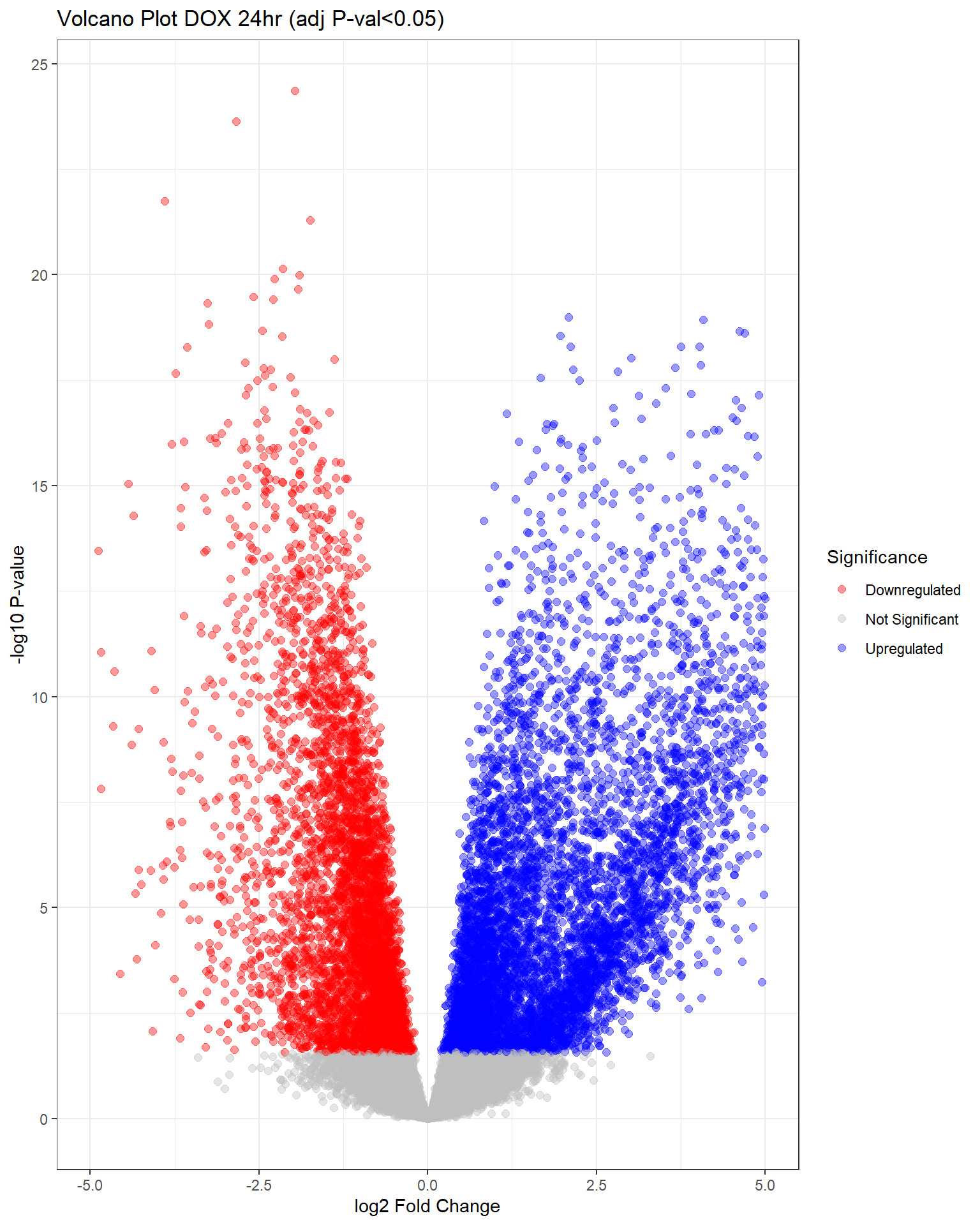
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
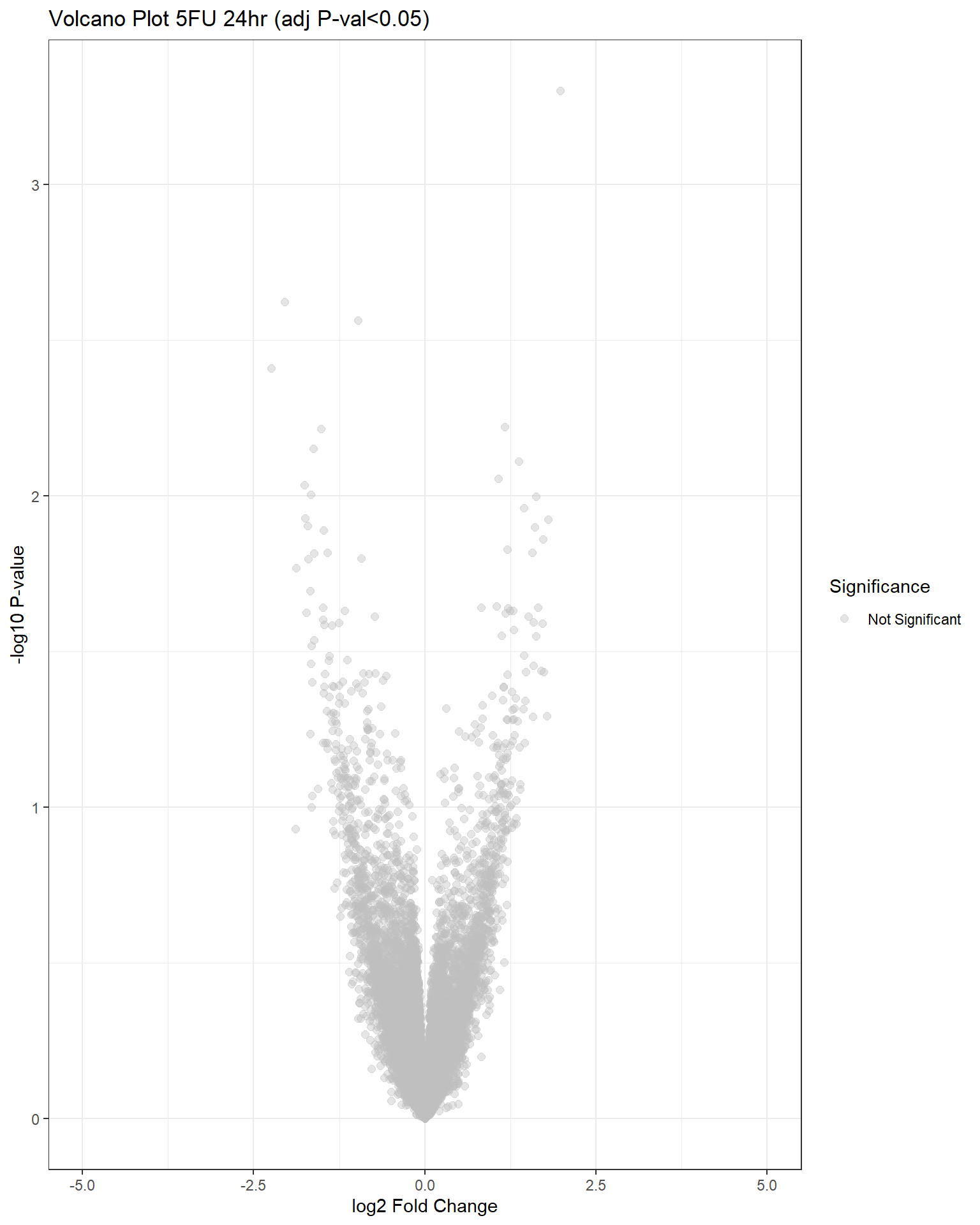
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
Warning: Removed 66 rows containing missing values or values outside the scale range
(`geom_point()`).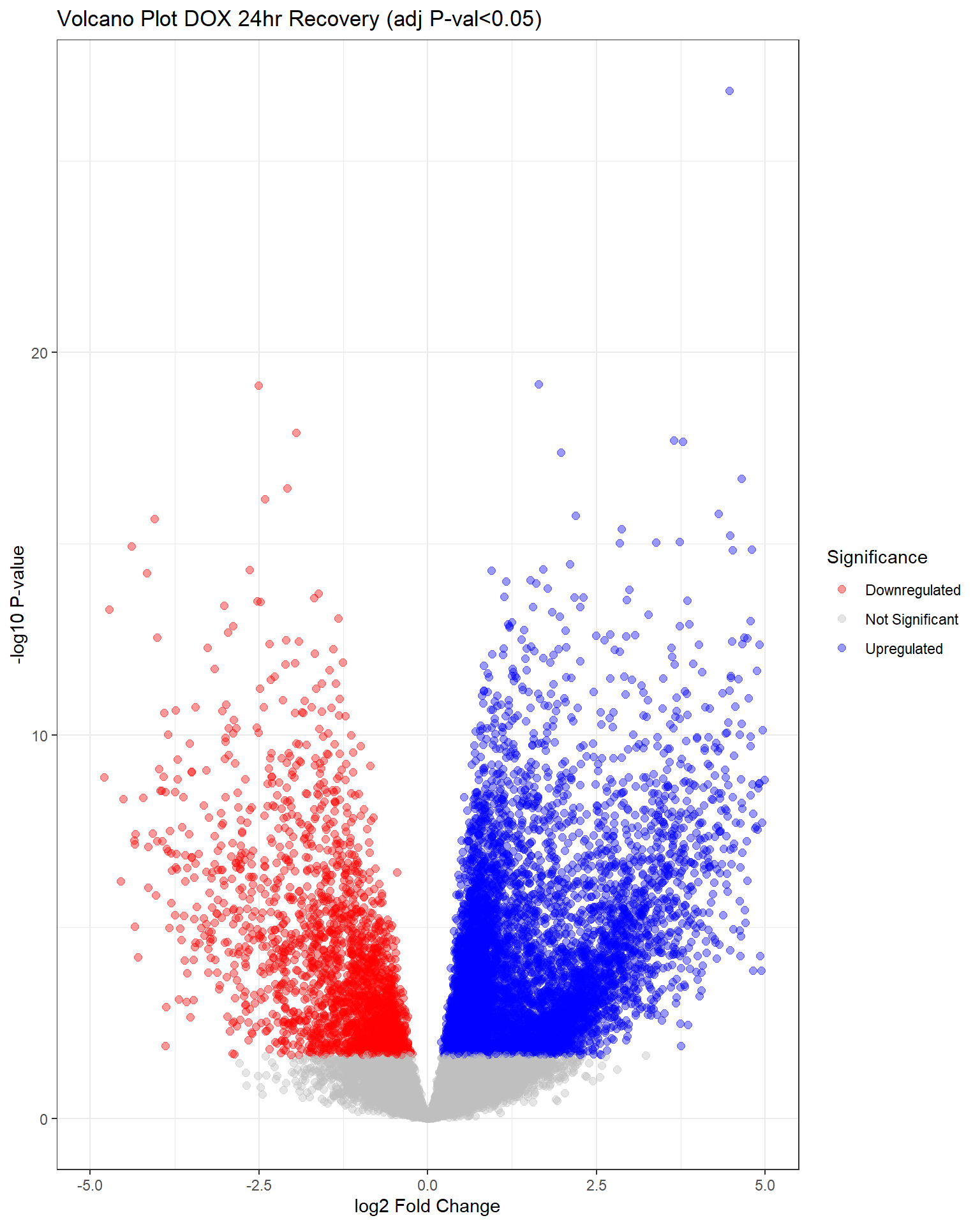
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
Warning: Removed 23 rows containing missing values or values outside the scale range
(`geom_point()`).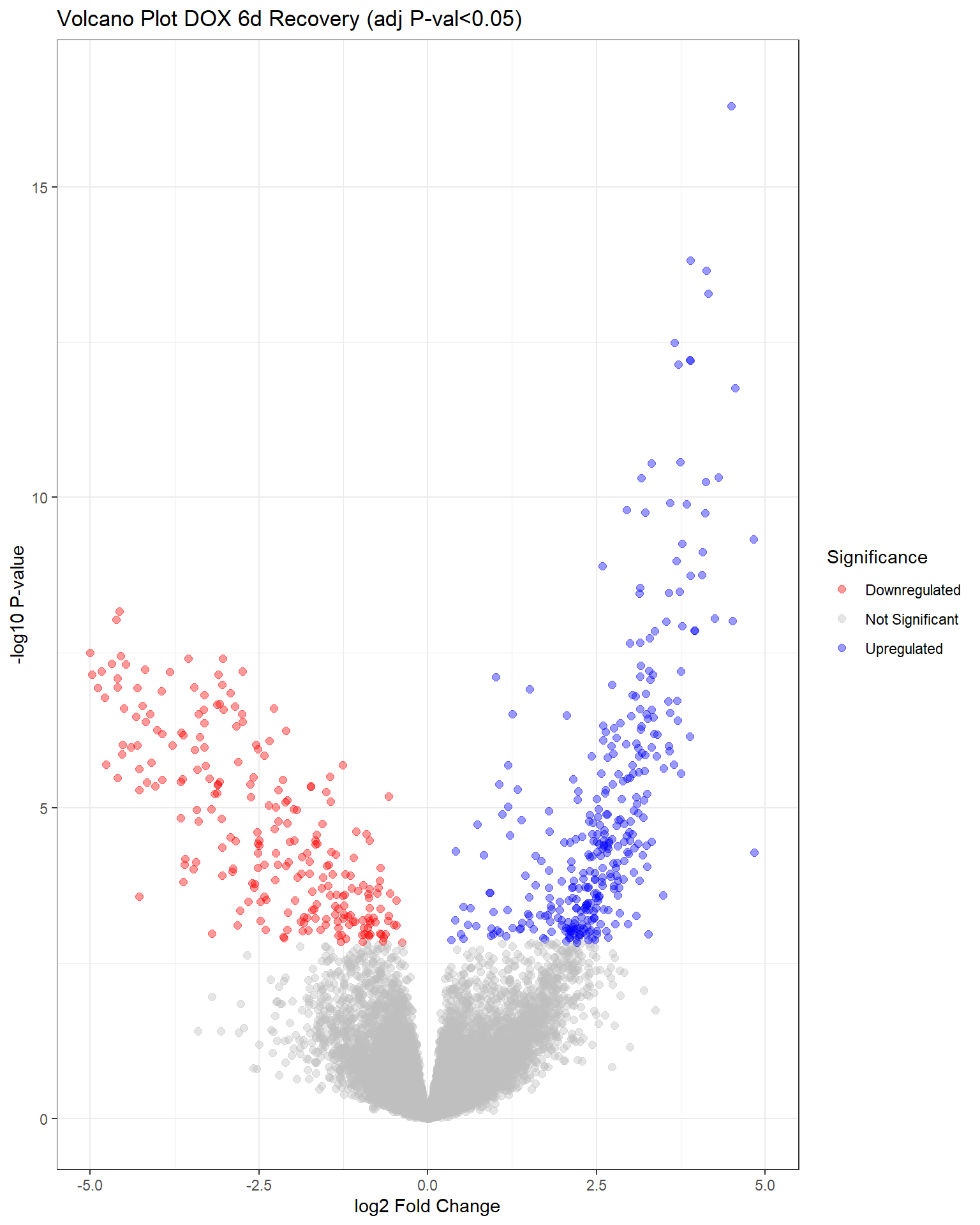
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
| Version | Author | Date |
|---|---|---|
| 41aef30 | emmapfort | 2025-02-27 |
Now that I’ve gone through and figured out the basics of DE - let’s remove unwanted variation using ruv This uses my replicates to figure out covariates in the data and remove variation occurring due to batch effects or other effects We’re seeing a batch effect from RNA extraction (all RNAseq library prep done in a single batch) occurring here in PCA and cor. heatmaps rather than individuals clustering together.
# #counts need to be integer values and in a numeric matrix for ruv
# counts_DE_matrix <- as.matrix(counts_DE_filter1)
# dim(counts_DE_matrix)
# #[1] 20443 54 - new inbuilt
# #this does NOT contain the replicate samples, save for later
#
# ##this file contains the replicates that I can pull from
# fC_Matrix_cpm_filter_ruv <- as.matrix (fC_Matrix_Full_cpm_filter)
#
# #create a matrix specifying the replicates
# reps <- as.data.frame(fC_Matrix_Full_cpm_filter) %>%
# dplyr::select(contains("Ind6"))
# dim(reps)
# #[1] 14225 18
# reps_mat <- as.matrix(reps)
#
# #using the documentation do this:
# #controls: gene-by-sample numeric matrix containing counts
# controls <- rownames(fC_Matrix_Full_cpm_filter)
#
# #I have replicates for Individual 6, which I can compare using this method
# #They are matched as following:
# ##col46:55, 47:56, 48:57, 49:58, 50:59, 51:60, 52:61, 53:62, 54:63
# ###this is the scIdx argument for reps, the cIdx argument is for genes
# #differences <- matrix(data = c(1, 10, 2, 11, 3, 12, 4, 13, 5, 14, 6, 15, 7, 16, 8, 17, 9, 18), byrow = TRUE, nrow = 9)
# differences_full <- matrix(data = c(46, 55, 47, 56, 48 , 57, 49, 58, 50 , 59, 51, 60, 52, 61, 53, 62, 54, 63), byrow = TRUE, nrow = 9)
#
#
# signature(x = "matrix", cIdx = "ANY", k = "numeric", scIdx = "matrix")
#
#
# seqRUVs <- RUVs(rep, controls, k=3, differences_full, isLog = TRUE)
#
# pData(counts_DE)
# head(seqRUVs)
#
#
# ###now try putting this in your matrix?
# design <- model.matrix(~0 + group_1, seqRUVs)
#
#
#
#
# ####try it exactly like the documentation and filter afterwards?####
# genes <- rownames(fC_Matrix_Full)[grep("^"), rownames(fC_Matrix_Full)]
# spikes <- rownames()
#
# ####leaving off here EMP 25/02/26####
sessionInfo()R version 4.4.2 (2024-10-31 ucrt)
Platform: x86_64-w64-mingw32/x64
Running under: Windows 11 x64 (build 22000)
Matrix products: default
locale:
[1] LC_COLLATE=English_United States.utf8
[2] LC_CTYPE=English_United States.utf8
[3] LC_MONETARY=English_United States.utf8
[4] LC_NUMERIC=C
[5] LC_TIME=English_United States.utf8
time zone: America/Chicago
tzcode source: internal
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] RUVSeq_1.40.0 EDASeq_2.40.0
[3] ShortRead_1.64.0 GenomicAlignments_1.42.0
[5] SummarizedExperiment_1.36.0 MatrixGenerics_1.18.1
[7] matrixStats_1.4.1 Rsamtools_2.22.0
[9] GenomicRanges_1.58.0 Biostrings_2.74.0
[11] GenomeInfoDb_1.42.3 XVector_0.46.0
[13] IRanges_2.40.0 S4Vectors_0.44.0
[15] BiocParallel_1.40.0 biomaRt_2.62.1
[17] RColorBrewer_1.1-3 Cormotif_1.52.0
[19] affy_1.84.0 Biobase_2.66.0
[21] BiocGenerics_0.52.0 PCAtools_2.18.0
[23] ggrepel_0.9.6 ggfortify_0.4.17
[25] pheatmap_1.0.12 edgeR_4.4.0
[27] limma_3.62.1 readxl_1.4.3
[29] edgebundleR_0.1.4 lubridate_1.9.3
[31] forcats_1.0.0 stringr_1.5.1
[33] dplyr_1.1.4 purrr_1.0.2
[35] readr_2.1.5 tidyr_1.3.1
[37] tidyverse_2.0.0 tibble_3.2.1
[39] hrbrthemes_0.8.7 reshape2_1.4.4
[41] ggplot2_3.5.1 workflowr_1.7.1
loaded via a namespace (and not attached):
[1] later_1.4.1 BiocIO_1.16.0
[3] bitops_1.0-9 filelock_1.0.3
[5] R.oo_1.27.0 cellranger_1.1.0
[7] preprocessCore_1.68.0 XML_3.99-0.18
[9] lifecycle_1.0.4 httr2_1.1.0
[11] pwalign_1.2.0 rprojroot_2.0.4
[13] vroom_1.6.5 MASS_7.3-61
[15] processx_3.8.5 lattice_0.22-6
[17] magrittr_2.0.3 sass_0.4.9
[19] rmarkdown_2.29 jquerylib_0.1.4
[21] yaml_2.3.10 httpuv_1.6.15
[23] cowplot_1.1.3 DBI_1.2.3
[25] abind_1.4-8 zlibbioc_1.52.0
[27] R.utils_2.12.3 RCurl_1.98-1.16
[29] rappdirs_0.3.3 git2r_0.35.0
[31] gdtools_0.4.1 GenomeInfoDbData_1.2.13
[33] irlba_2.3.5.1 dqrng_0.4.1
[35] DelayedMatrixStats_1.28.1 codetools_0.2-20
[37] DelayedArray_0.32.0 xml2_1.3.6
[39] tidyselect_1.2.1 farver_2.1.2
[41] UCSC.utils_1.2.0 ScaledMatrix_1.14.0
[43] BiocFileCache_2.14.0 jsonlite_1.8.9
[45] systemfonts_1.1.0 tools_4.4.2
[47] progress_1.2.3 Rcpp_1.0.13-1
[49] glue_1.8.0 gridExtra_2.3
[51] Rttf2pt1_1.3.12 SparseArray_1.6.0
[53] xfun_0.49 withr_3.0.2
[55] BiocManager_1.30.25 fastmap_1.2.0
[57] latticeExtra_0.6-30 callr_3.7.6
[59] digest_0.6.37 rsvd_1.0.5
[61] timechange_0.3.0 R6_2.6.1
[63] mime_0.12 colorspace_2.1-1
[65] jpeg_0.1-10 RSQLite_2.3.8
[67] R.methodsS3_1.8.2 generics_0.1.3
[69] fontLiberation_0.1.0 rtracklayer_1.66.0
[71] prettyunits_1.2.0 httr_1.4.7
[73] htmlwidgets_1.6.4 S4Arrays_1.6.0
[75] whisker_0.4.1 pkgconfig_2.0.3
[77] gtable_0.3.6 blob_1.2.4
[79] hwriter_1.3.2.1 htmltools_0.5.8.1
[81] fontBitstreamVera_0.1.1 scales_1.3.0
[83] png_0.1-8 knitr_1.49
[85] rstudioapi_0.17.1 tzdb_0.4.0
[87] rjson_0.2.23 curl_6.0.1
[89] cachem_1.1.0 parallel_4.4.2
[91] extrafont_0.19 AnnotationDbi_1.68.0
[93] restfulr_0.0.15 pillar_1.10.1
[95] grid_4.4.2 vctrs_0.6.5
[97] promises_1.3.2 BiocSingular_1.22.0
[99] dbplyr_2.5.0 beachmat_2.22.0
[101] xtable_1.8-4 extrafontdb_1.0
[103] evaluate_1.0.3 GenomicFeatures_1.58.0
[105] cli_3.6.3 locfit_1.5-9.10
[107] compiler_4.4.2 rlang_1.1.4
[109] crayon_1.5.3 labeling_0.4.3
[111] aroma.light_3.36.0 interp_1.1-6
[113] ps_1.8.1 getPass_0.2-4
[115] plyr_1.8.9 fs_1.6.5
[117] stringi_1.8.4 deldir_2.0-4
[119] munsell_0.5.1 fontquiver_0.2.1
[121] Matrix_1.7-1 hms_1.1.3
[123] sparseMatrixStats_1.18.0 bit64_4.5.2
[125] KEGGREST_1.46.0 statmod_1.5.0
[127] shiny_1.10.0 igraph_2.1.1
[129] memoise_2.0.1 affyio_1.76.0
[131] bslib_0.9.0 bit_4.5.0