DupSeq_QC_Lane11
Haider Inam
2022-08-22
Last updated: 2022-11-01
Checks: 7 0
Knit directory: duplex_sequencing_screen/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200402) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 9a33fee. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/Consensus_Data/.Rhistory
Ignored: data/Consensus_Data/Novogene_lane13/sample7/variant_caller_outputs/
Ignored: data/Consensus_Data/Novogene_lane13/sample8/variant_caller_outputs/
Ignored: data/Consensus_Data/Novogene_lane14/sample13/
Ignored: data/Consensus_Data/Novogene_lane14/sample14b/
Ignored: data/Consensus_Data/Novogene_lane14/sample1_combined/
Ignored: data/Consensus_Data/Novogene_lane14/sample8/variant_caller_outputs/
Ignored: data/Consensus_Data/Novogene_lane2/abl_ref/kraken2-master/newdir/
Ignored: data/Consensus_Data/Novogene_lane4/.ipynb_checkpoints/
Ignored: data/Consensus_Data/Novogene_lane4/GalaxyData/initialanalysis/
Ignored: data/Consensus_Data/Novogene_lane4/output/
Ignored: data/Consensus_Data/Novogene_lane6/
Ignored: data/Consensus_Data/Novogene_lane7/
Ignored: data/Consensus_Data/Ranomics_Pooled/RP4/
Ignored: data/Consensus_Data/Ranomics_Pooled/RP5/
Untracked files:
Untracked: data/Consensus_Data/Novogene_lane11/sample10/duplex_sum_ann-Haider’s MacBook Pro.csv
Untracked: data/Consensus_Data/Novogene_lane14/sample10_combined/variant_caller_outputs/duplex/
Unstaged changes:
Modified: analysis/lane14_comparisons.Rmd
Modified: analysis/variant_caller_2022.Rmd
Modified: data/.DS_Store
Modified: data/Consensus_Data/.DS_Store
Modified: data/Consensus_Data/Novogene_lane11/.DS_Store
Modified: data/Consensus_Data/Novogene_lane11/sample10/duplex_sum_ann.csv
Modified: data/Consensus_Data/Novogene_lane11/sample6/.DS_Store
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/DupSeq_QC_Lane11.Rmd) and
HTML (docs/DupSeq_QC_Lane11.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 9a33fee | haiderinam | 2022-11-01 | wflow_publish("analysis/DupSeq_QC_Lane11.Rmd") |
| Rmd | d358fe3 | haiderinam | 2022-10-29 | SSCS Error Analyses |
| html | d358fe3 | haiderinam | 2022-10-29 | SSCS Error Analyses |
| Rmd | 157f17b | haiderinam | 2022-10-24 | Lane 14 Data Added |
| Rmd | 097ad61 | haiderinam | 2022-09-29 | Lane 13 Analysis Added |
| html | 097ad61 | haiderinam | 2022-09-29 | Lane 13 Analysis Added |
| Rmd | fecc2e4 | haiderinam | 2022-08-23 | August 2022 Updates |
| Rmd | 7ed0af8 | haiderinam | 2022-08-22 | August 2022 Updates |
In the following chunks, I’m going to be doing some QC with the duplex sequencing data from novogene lane 11 This sequencing run had both EGFR and ABL data. The libraries are twist libraries.
I’m going to be looking closely at the following:
* Count distributions of EGFR vs ABL * How much coverage do we get with
a single sample of EGFR vs with two samples? Answer: 100k for 1 sample,
200k for two SSCS samples * What is the % on-target? * What is the % N?
* What is the % of split reads? * What is the % mouse?
- How much coverage do we get for DCS vs for SSCS? Answer: For a dupseq coverage of 40k, our SSCS coverage was 100k This is a very low % of SSCS reads that are orphaned. For schmitt’s adapter types, about 80-90% of the SSCS reads are orphaned.
- What is the error rate for SSCS vs for DCS? Answer:
- What is the % of mouse reads that we’re seeing for the samples? Answer: Mouse read %age is very low for EGFR (1% of the barcodes align to the mouse genome). For ABL, between 1 and 30% of the consensus reads align to the mouse genome
- How many unique mutants do we see for EGFR? What percent coverage is that? Answer: Combining two EGFR samples at the baseline and using SSCS counts, we achieve a coverage of 200k. At this coverage, we’re seeing 2340 out of 2700 mutants that twist’s QC data saw. That’s 90% of the mutants at baseline.
- What is the count distribution of the mutants in the data? aka how many times do we see each mutant? Answer:
- How many unique mutants do you see per 10k barcodes sequenced? How does downsampling influence the sequencing data output? Answer:
- How well do the mutant allele frequencies agree with twist’s MAFs?
- How many reads/consensus barcodes are taken up by each of the residues?
- What percent of the barcodes are taken up by the top 10 mutants? What about the top 50 mutants?
- Modify the function so that you’re not relying on ensembl’s VEP.
####Novogene Lane 12####
sample1=read.csv("data/Consensus_Data/Novogene_lane12/sample1/variant_caller_outputs/variants_unique_ann.csv")
sample1$sample="sample1"
sample3=read.csv("data/Consensus_Data/Novogene_lane12/sample3/variant_caller_outputs/variants_unique_ann.csv")
sample3$sample="sample3"
sample5=read.csv("data/Consensus_Data/Novogene_lane12/sample5/variant_caller_outputs/variants_unique_ann.csv")
sample5_all=read.csv("data/Consensus_Data/Novogene_lane12/sample5/variant_caller_outputs/variants_ann.csv")
sample5$sample="sample5"
sample7=read.csv("data/Consensus_Data/Novogene_lane12/sample7/variant_caller_outputs/variants_unique_ann.csv")
sample7$sample="sample7"
sample9=read.csv("data/Consensus_Data/Novogene_lane12/sample9/variant_caller_outputs/variants_unique_ann.csv")
sample9$sample="sample9"
sample3=sample3%>%
rowwise()%>%
mutate(ref_aa=strsplit(amino_acids,"/")[[1]][1],
alt_aa=strsplit(amino_acids,"/")[[1]][2])
sample3=sample3%>%mutate(maf=ct/depth)
sample3_simple=sample3%>%dplyr::select(alt_start_pos,protein_start,ref,alt,ref_aa,alt_aa,consequence_terms,ct,depth,maf)
sample5=sample5%>%
rowwise()%>%
mutate(ref_aa=strsplit(amino_acids,"/")[[1]][1],
alt_aa=strsplit(amino_acids,"/")[[1]][2])
sample5=sample5%>%mutate(maf=ct/depth)
sample5_simple=sample5%>%dplyr::select(alt_start_pos,protein_start,ref,alt,ref_aa,alt_aa,consequence_terms,ct,depth,maf)
samples_merged=merge(sample3_simple%>%filter(consequence_terms%in%"missense_variant"),sample5_simple%>%filter(consequence_terms%in%"missense_variant"),by=c("ref_aa","protein_start","alt_aa","ref","alt","alt_start_pos","consequence_terms"))
samples_merged=samples_merged%>%mutate(score=log2(maf.y/maf.x),score2=log2(ct.y/ct.x))
# a=samples_merged%>%filter(!ct.x%in%1)
samples_merged=samples_merged%>%mutate(resmuts=case_when(protein_start%in%253&alt_aa%in%"H"~T,
protein_start%in%255&alt_aa%in%"V"~T,
protein_start%in%486&alt_aa%in%"S"~T,
protein_start%in%396&alt_aa%in%"P"~T,
protein_start%in%255&alt_aa%in%"K"~T,
protein_start%in%315&alt_aa%in%"I"~T,
protein_start%in%252&alt_aa%in%"H"~T,
protein_start%in%253&alt_aa%in%"F"~T,
protein_start%in%250&alt_aa%in%"E"~T,
protein_start%in%359&alt_aa%in%"C"~T,
protein_start%in%351&alt_aa%in%"T"~T,
protein_start%in%355&alt_aa%in%"G"~T,
protein_start%in%317&alt_aa%in%"L"~T,
protein_start%in%359&alt_aa%in%"I"~T,
protein_start%in%355&alt_aa%in%"A"~T,
protein_start%in%459&alt_aa%in%"K"~T,
protein_start%in%276&alt_aa%in%"G"~T,
protein_start%in%299&alt_aa%in%"L"~T,
T~F))
ggplot(samples_merged%>%filter(!ct.x%in%1),aes(x=protein_start,y=score,color=resmuts))+geom_point()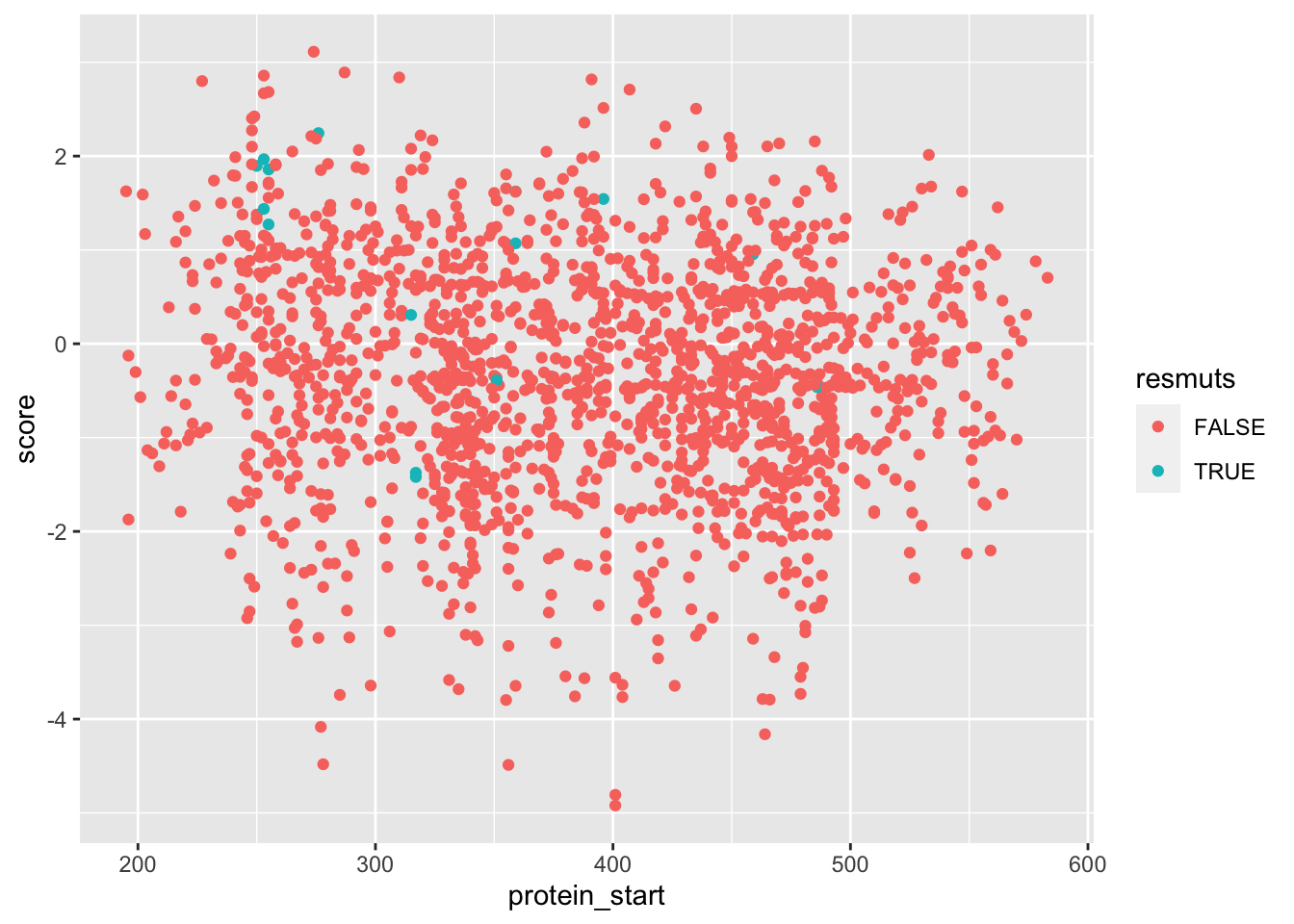
| Version | Author | Date |
|---|---|---|
| d358fe3 | haiderinam | 2022-10-29 |
# ggplot(samples_merged%>%filter(!ct.x%in%1),aes(x=protein_start,y=score2))+geom_point()
# sample3=sample3%>%rowwise()%>%mutate(a=c(protein_start,alt_aa))
#
# a=sample5_all%>%filter(protein_start==255,amino_acids%in%"E/K")
# sort(unique(a$codons))
#
# resmuts=sample5%>%filter(protein_start%in%c())
sample1=sample1%>%mutate(region=case_when(protein_start<250&protein_start>=242~1,
protein_start<258&protein_start>=250~2,
protein_start<266&protein_start>=258~3,
protein_start<274&protein_start>=266~4,
protein_start<282&protein_start>=274~5,
protein_start<290&protein_start>=282~6,
protein_start<298&protein_start>=290~7,
protein_start<306&protein_start>=298~8,
protein_start<314&protein_start>=306~9,
protein_start<322&protein_start>=314~10,
protein_start<330&protein_start>=322~11,
protein_start<338&protein_start>=330~12,
protein_start<346&protein_start>=338~13,
protein_start<354&protein_start>=346~14,
protein_start<362&protein_start>=354~15,
protein_start<370&protein_start>=362~16,
protein_start<378&protein_start>=370~17,
protein_start<386&protein_start>=378~18,
protein_start<394&protein_start>=386~19,
protein_start<402&protein_start>=394~20,
protein_start<410&protein_start>=402~21,
protein_start<418&protein_start>=410~22,
protein_start<426&protein_start>=418~23,
protein_start<434&protein_start>=426~24,
protein_start<442&protein_start>=434~25,
protein_start<450&protein_start>=442~26,
protein_start<458&protein_start>=450~27,
protein_start<466&protein_start>=458~28,
protein_start<474&protein_start>=466~29,
protein_start<482&protein_start>=474~30,
protein_start<490&protein_start>=482~31,
protein_start<498&protein_start>=490~32,
T~0))
library(RColorBrewer)
getPalette = colorRampPalette(brewer.pal(33, "Set2"))Warning in brewer.pal(33, "Set2"): n too large, allowed maximum for palette Set2 is 8
Returning the palette you asked for with that many colorssample1=sample1%>%rowwise()%>%mutate(ID=paste(protein_start,amino_acids,sep=""))
plotly=ggplot(sample1%>%filter(consequence_terms%in%"missense_variant",protein_start>=242,protein_start<=494),aes(x=protein_start,y=ct))+geom_col(color="black",aes(fill=factor(region)))+cleanup+scale_fill_manual(values=getPalette(33))
ggplotly(plotly)calls_sum=sample1%>%filter(consequence_terms%in%"missense_variant")%>%group_by(protein_start,region)%>%summarize(unique_mutants=n(),count=sum(ct))`summarise()` has grouped output by 'protein_start'. You can override using the `.groups` argument.# calls_sum=calls_sum
ggplot(calls_sum%>%filter(protein_start>=242,protein_start<=500),aes(x=protein_start,y=unique_mutants))+geom_col()+cleanup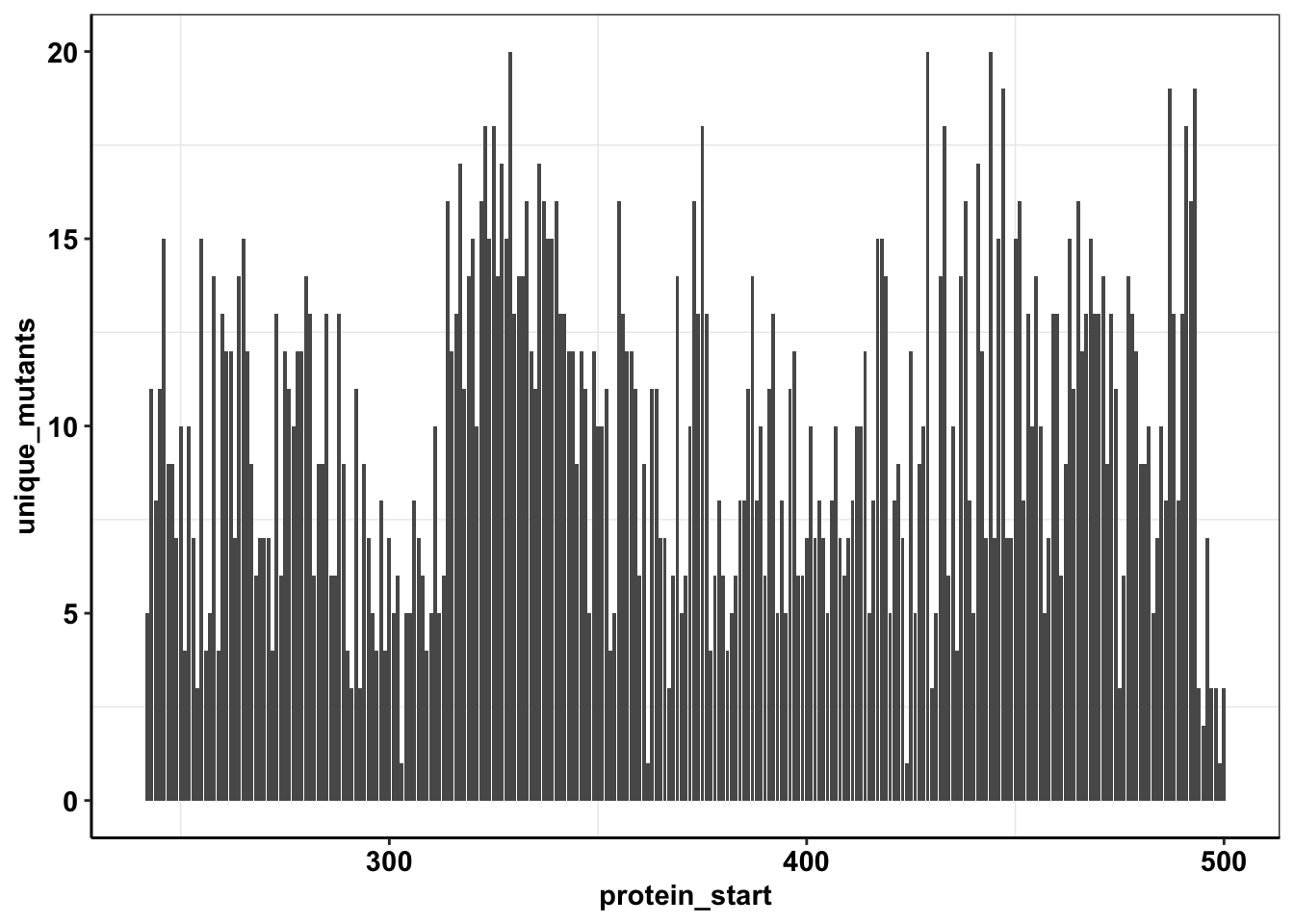
| Version | Author | Date |
|---|---|---|
| d358fe3 | haiderinam | 2022-10-29 |
ggplot(calls_sum%>%filter(protein_start>=242,protein_start<=500),aes(x=protein_start,y=count))+geom_col()+cleanup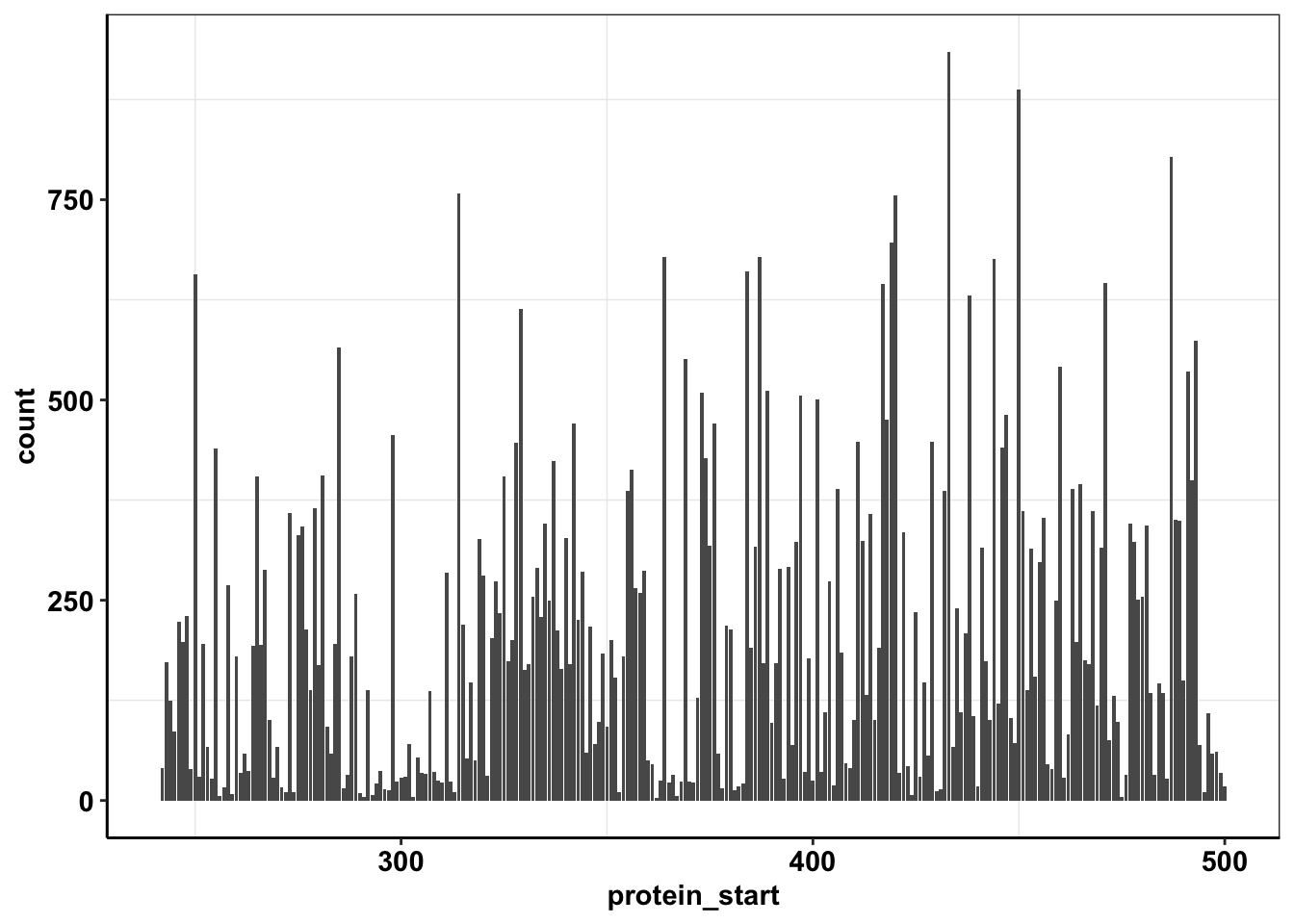
| Version | Author | Date |
|---|---|---|
| d358fe3 | haiderinam | 2022-10-29 |
plotly=ggplot(calls_sum%>%filter(protein_start>=242,protein_start<=500),aes(x=protein_start,y=count))+geom_col(color="black",aes(fill=factor(region)))+cleanup+scale_fill_manual(values=getPalette(33))
ggplotly(plotly)###The following plot is proof that I pooled in our iL3 independent regions very well.
calls_sum_byregion=calls_sum%>%group_by(region)%>%summarize(unique_regions=n(),counts=sum(count))
getPalette = colorRampPalette(brewer.pal(33, "Set2"))Warning in brewer.pal(33, "Set2"): n too large, allowed maximum for palette Set2 is 8
Returning the palette you asked for with that many colorsplotly=ggplot(calls_sum_byregion%>%filter(!region%in%0),aes(x=region,y=counts))+geom_col(color="black",aes(fill=factor(region)))+cleanup+scale_fill_manual(values=getPalette(33))
ggplotly(plotly)highscore=samples_merged%>%filter(!ct.x%in%1,protein_start>=242,protein_start<=494)
highscore=highscore%>%mutate(subs_name=paste(ref_aa,protein_start,alt_aa,sep = ""))
plotly=ggplot(highscore,aes(x=reorder(subs_name,-score),y=score,fill=resmuts))+geom_col()+theme(axis.text.x=element_text(angle=90, hjust=1))+coord_flip()
ggplotly(plotly)a=highscore%>%filter(protein_start%in%"255")####Novogene Lane 11####
Sample 6
# getwd()
# sample5_all=read.csv("data/Consensus_Data/Novogene_lane11/sample5/variants_ann_sample5.csv")
sample5_sum=read.csv("data/Consensus_Data/Novogene_lane11/sample5/variant_caller_outputs/variants_unique_ann_sample5.csv")
# sample5_all=read.csv("data/Consensus_Data/Novogene_lane11/sample5/variants_ann_sample5.csv")
# a=sample5_all%>%filter(protein_start%in%903,ref%in%"T",alt%in%"C")
# sample6_all=read.csv("data/Consensus_Data/Novogene_lane11/sample6/variants_ann_sample5&6.csv")
sample6_sum=read.csv("data/Consensus_Data/Novogene_lane11/sample6/variant_caller_outputs/variants_unique_ann_sample6.csv")
sample5_simple=sample5_sum%>%dplyr::select(alt_start_pos,protein_start,ref,alt,consequence_terms,amino_acids,ct,depth)
sample5_simple$sample="sample5"
sample6_simple=sample6_sum%>%dplyr::select(alt_start_pos,protein_start,ref,alt,consequence_terms,amino_acids,ct,depth)
sample6_simple$sample="sample6"
sample56=rbind(sample5_simple,sample6_simple)
# a=sample56%>%filter(protein_start>=715,protein_start<=900,consequence_terms%in%"missense_variant")
plotly=ggplot(sample56%>%filter(protein_start>=715,protein_start<=900,consequence_terms%in%"missense_variant"),aes(x=protein_start,y=depth,fill=sample))+geom_col(position=position_dodge())+facet_wrap(~sample)+cleanup
ggplotly(plotly)Warning: `group_by_()` was deprecated in dplyr 0.7.0.
Please use `group_by()` instead.
See vignette('programming') for more helpsample56_merged=merge(sample5_simple,sample6_simple,by=c("alt_start_pos","protein_start","ref","alt","consequence_terms","amino_acids"),all.x = T)
# a=sample56_merged%>%filter(depth.x>=1000,protein_start>=715,protein_start<=870,consequence_terms%in%"missense_variant")
sample56_merged[sample56_merged$ct.y%in%NA,10]=1000
sample56_merged[sample56_merged$depth.y%in%NA,11]=1000
# sample56_merged=sample56_merged%>%
# rowwise()%>%
# mutate(ct.y=case_when(ct.y%in%NA~0,
# T~ct.y))
ggplot(sample56_merged%>%filter(depth.x>=1000,protein_start>=715,protein_start<=870,consequence_terms%in%"missense_variant"),aes(x=ct.x/depth.x,y=ct.y/depth.y))+
geom_point(color="black",shape=21,aes(fill=log10(ct.x)))+
scale_x_continuous(trans="log10")+
scale_y_continuous(trans="log10")+
cleanup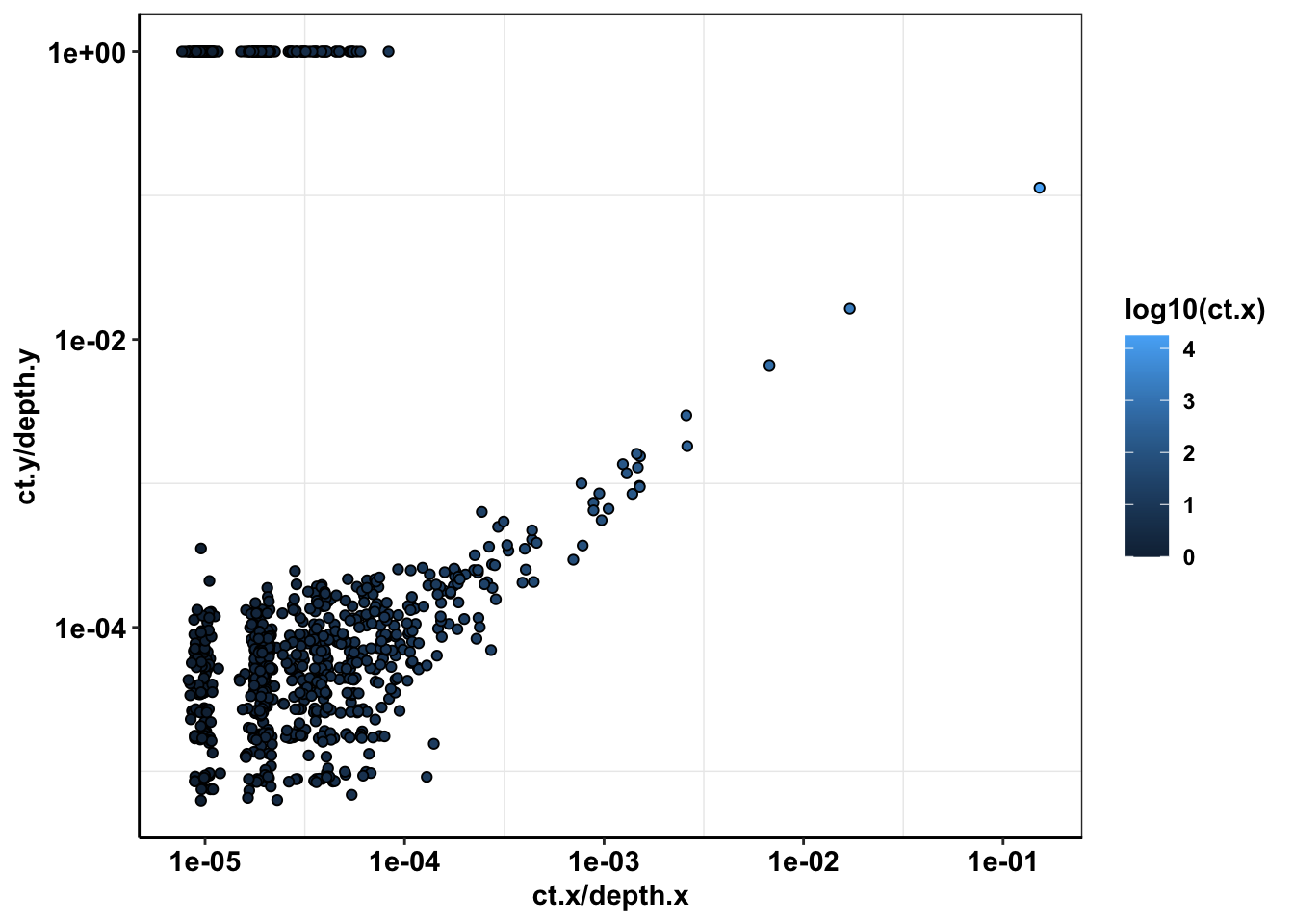
| Version | Author | Date |
|---|---|---|
| 097ad61 | haiderinam | 2022-09-29 |
plotly=ggplot(sample5_sum%>%filter(protein_start>=715,protein_start<=870,consequence_terms%in%"missense_variant",ct<=50,type%in%"mnv"),aes(x=ct))+
geom_histogram(aes(y=..density..),color=1,fill="white",bins=50)+
geom_density(lwd = 1, colour = 4,fill = 4, alpha = 0.25)
ggplotly(plotly)plotly=ggplot(sample6_sum%>%filter(protein_start>=715,protein_start<=870,consequence_terms%in%"missense_variant",ct<=50,type%in%"mnv"),aes(x=ct))+
geom_histogram(aes(y=..density..),color=1,fill="white",bins=50)+
geom_density(lwd = 1, colour = 4,fill = 4, alpha = 0.25)
# scale_x_continuous(trans="log10")
ggplotly(plotly)# a=sample5_sum%>%filter(protein_start>=715,protein_start<=870,consequence_terms%in%"missense_variant",ct==1)
# nrow(a%>%filter(type%in%"mnv"))
# b=sample6_sum%>%filter(protein_start>=715,protein_start<=870,consequence_terms%in%"missense_variant",ct==1)
# nrow(b%>%filter(type%in%"mnv"))
#For sample 5, out of the mutants that are only seen once, 34% are mnvs (a lot of SNP errors). So a lot of the mutants that we are seeing at a coverage of 1 are false. This means that for sample 5, the distribution of mutants is centered >1
#For sample 6, out of the mutants that are only seen once, 62% are mnvs (low SNP errors)
calls_sum=sample5_sum%>%filter(consequence_terms%in%"missense_variant")%>%group_by(protein_start)%>%summarize(unique_mutants=n(),count=sum(ct))
ggplot(calls_sum%>%filter(protein_start>=715,protein_start<=870),aes(x=protein_start,y=unique_mutants))+geom_col()+cleanup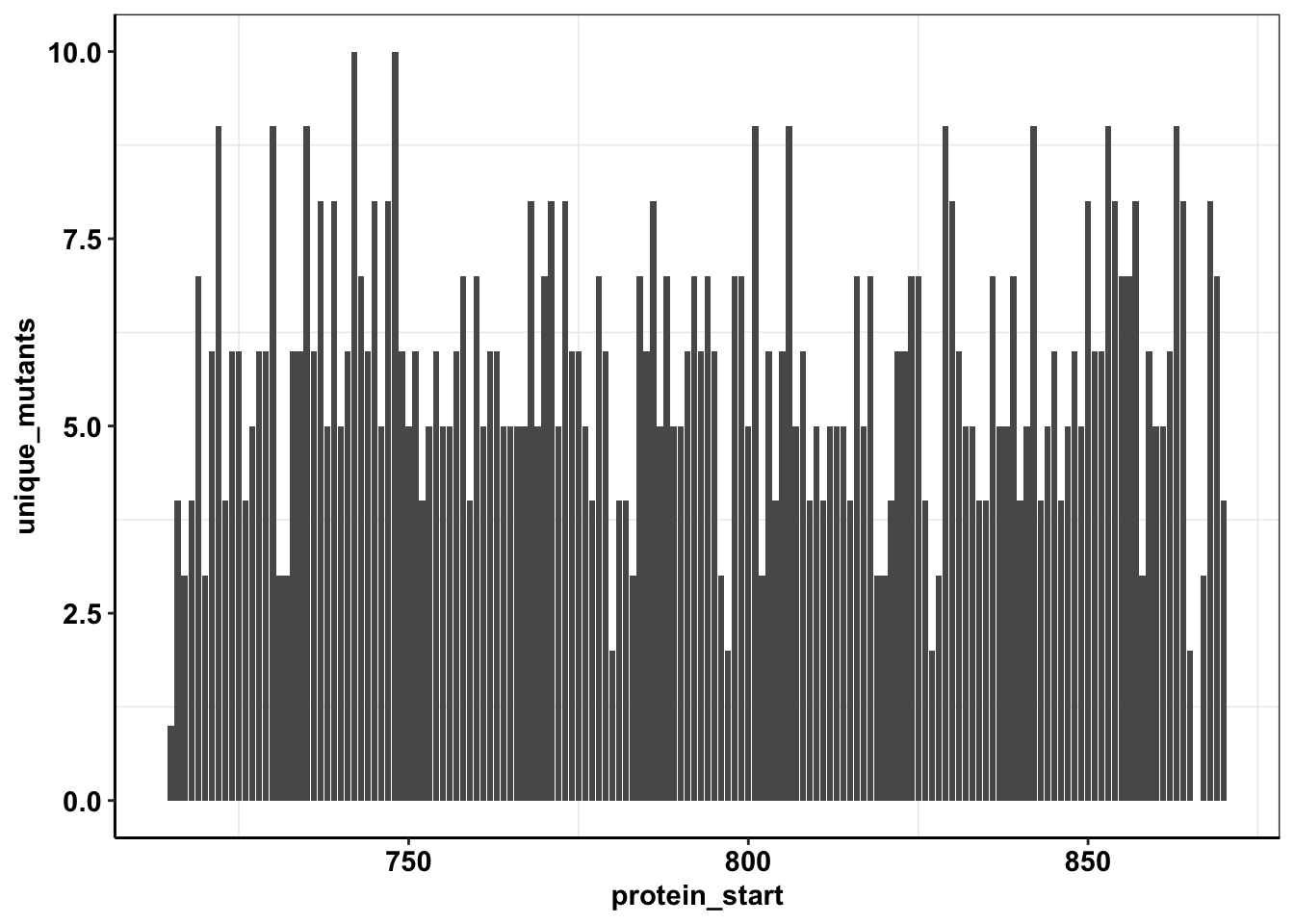
| Version | Author | Date |
|---|---|---|
| 097ad61 | haiderinam | 2022-09-29 |
ggplot(calls_sum%>%filter(protein_start>=715,protein_start<=870),aes(x=protein_start,y=count))+geom_col()+cleanup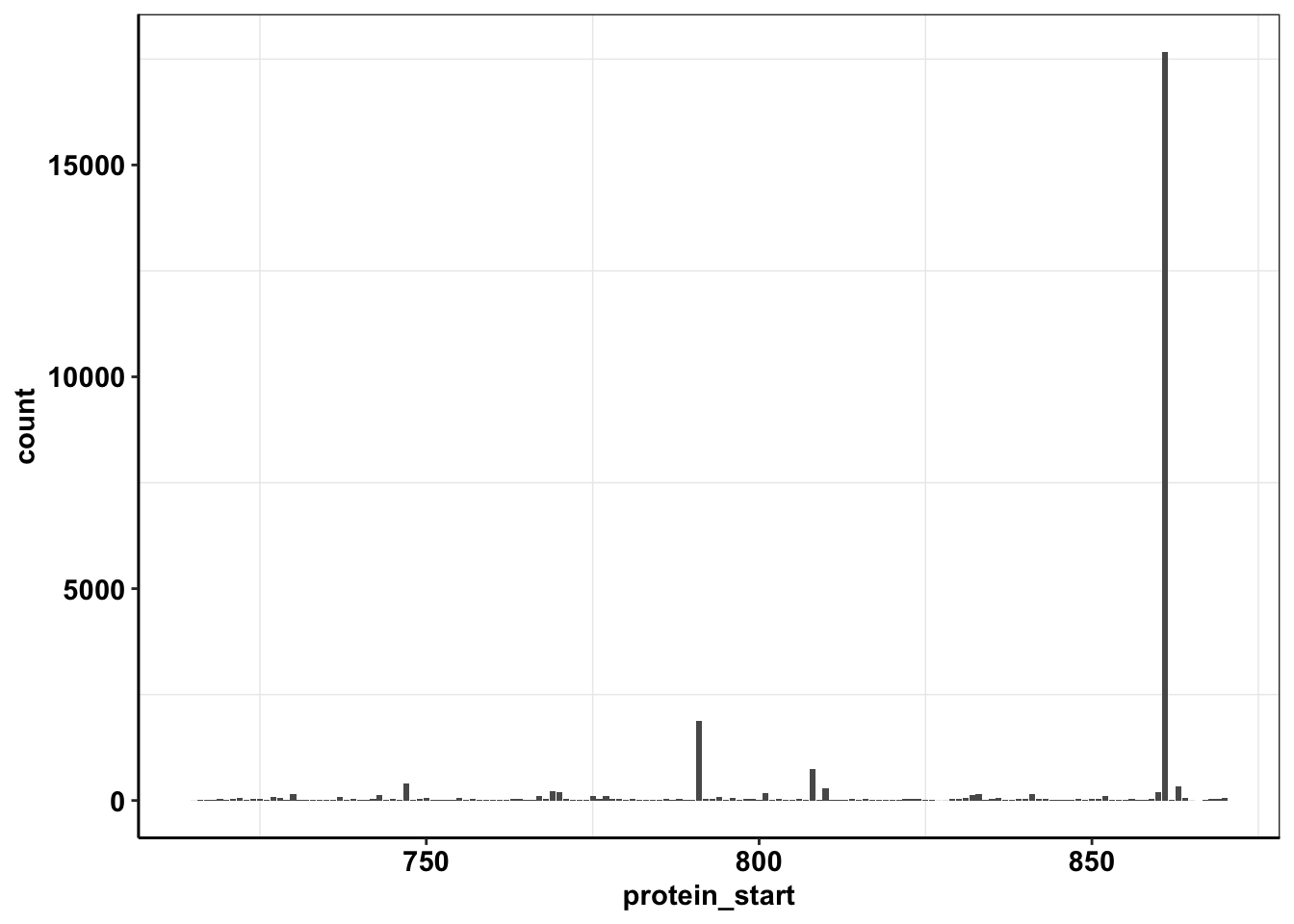
| Version | Author | Date |
|---|---|---|
| 097ad61 | haiderinam | 2022-09-29 |
calls_sum=sample6_sum%>%filter(consequence_terms%in%"missense_variant")%>%group_by(protein_start)%>%summarize(unique_mutants=n(),count=sum(ct))
ggplot(calls_sum%>%filter(protein_start>=715,protein_start<=870),aes(x=protein_start,y=unique_mutants))+geom_col()+cleanup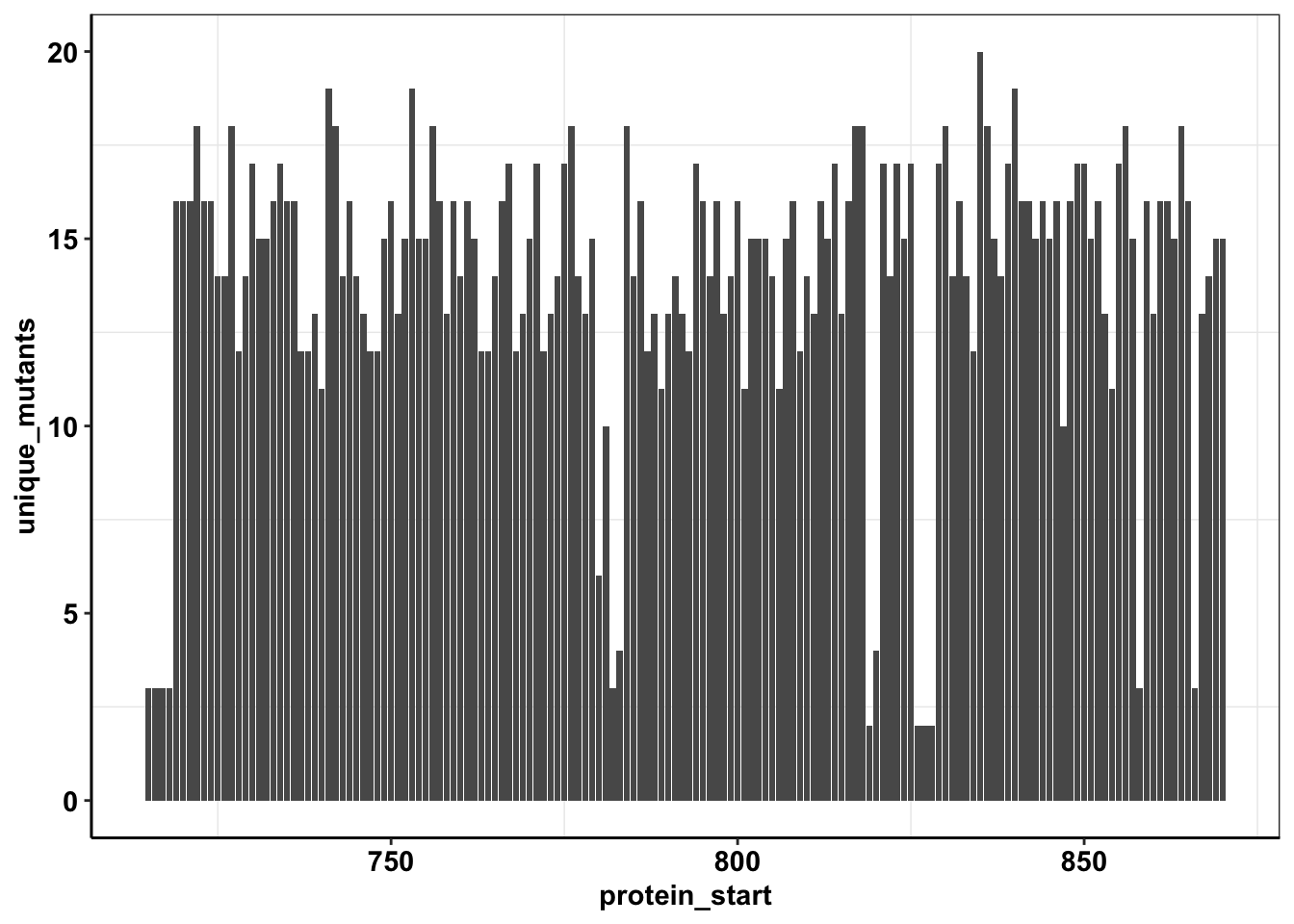
| Version | Author | Date |
|---|---|---|
| 097ad61 | haiderinam | 2022-09-29 |
ggplot(calls_sum%>%filter(protein_start>=715,protein_start<=870),aes(x=protein_start,y=count))+geom_col()+cleanup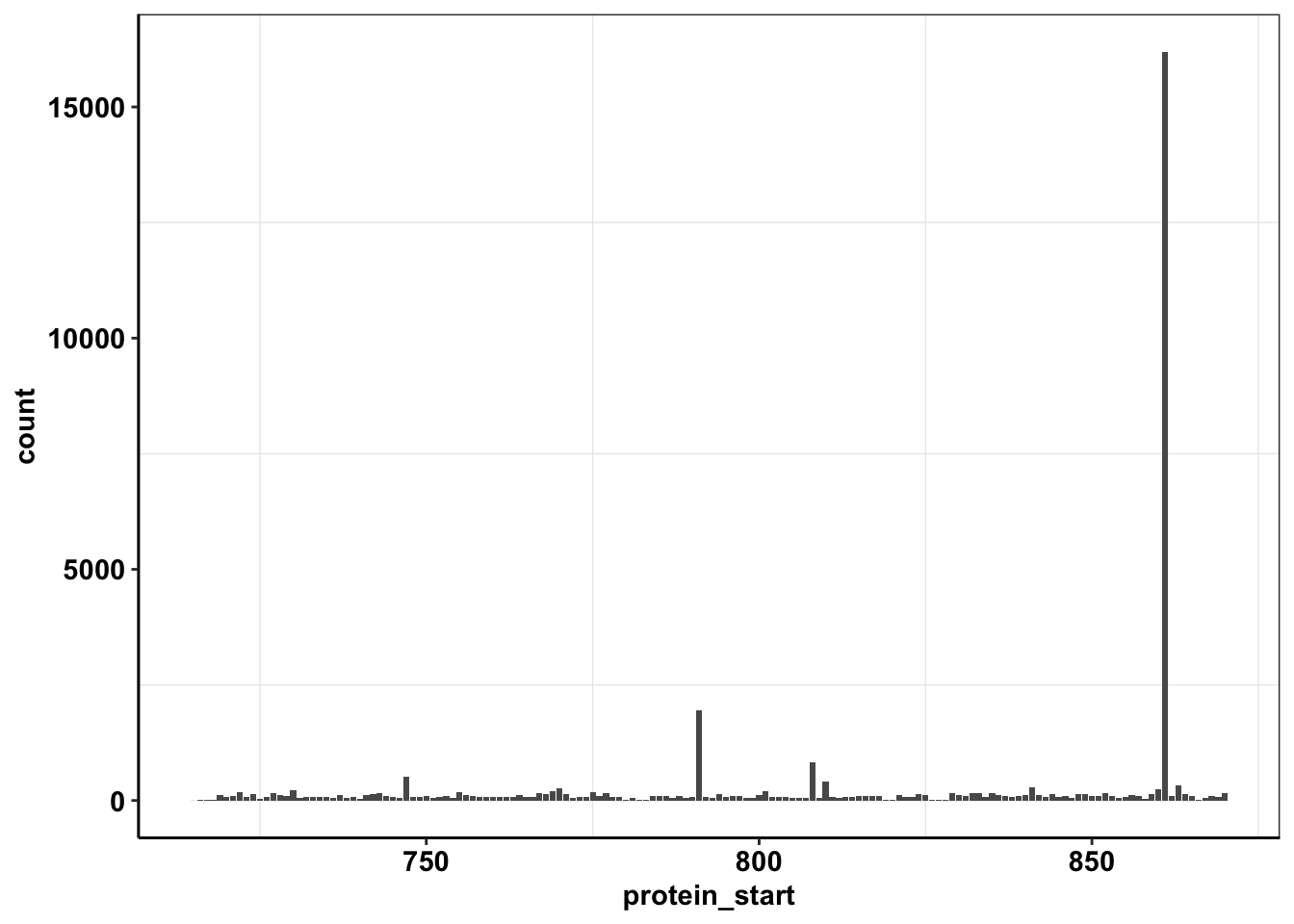
| Version | Author | Date |
|---|---|---|
| 097ad61 | haiderinam | 2022-09-29 |
ggplot(calls_sum%>%filter(protein_start>=715,protein_start<=870,!protein_start%in%861),aes(x=protein_start,y=count))+geom_col()+cleanup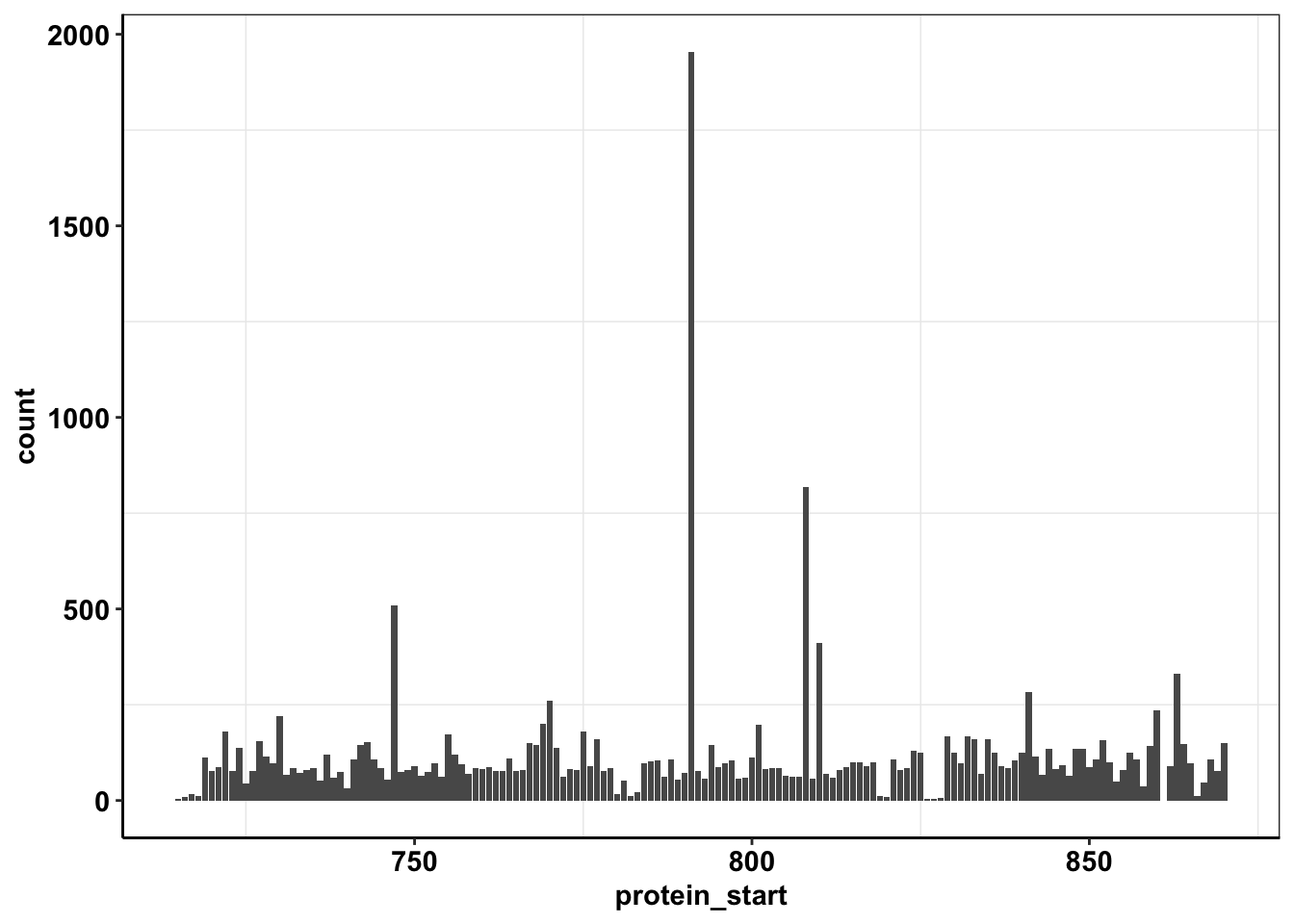
| Version | Author | Date |
|---|---|---|
| 097ad61 | haiderinam | 2022-09-29 |
ggplot(calls_sum%>%filter(protein_start>=715,protein_start<=870,!protein_start%in%c(861,791)),aes(x=protein_start,y=count))+geom_col()+cleanup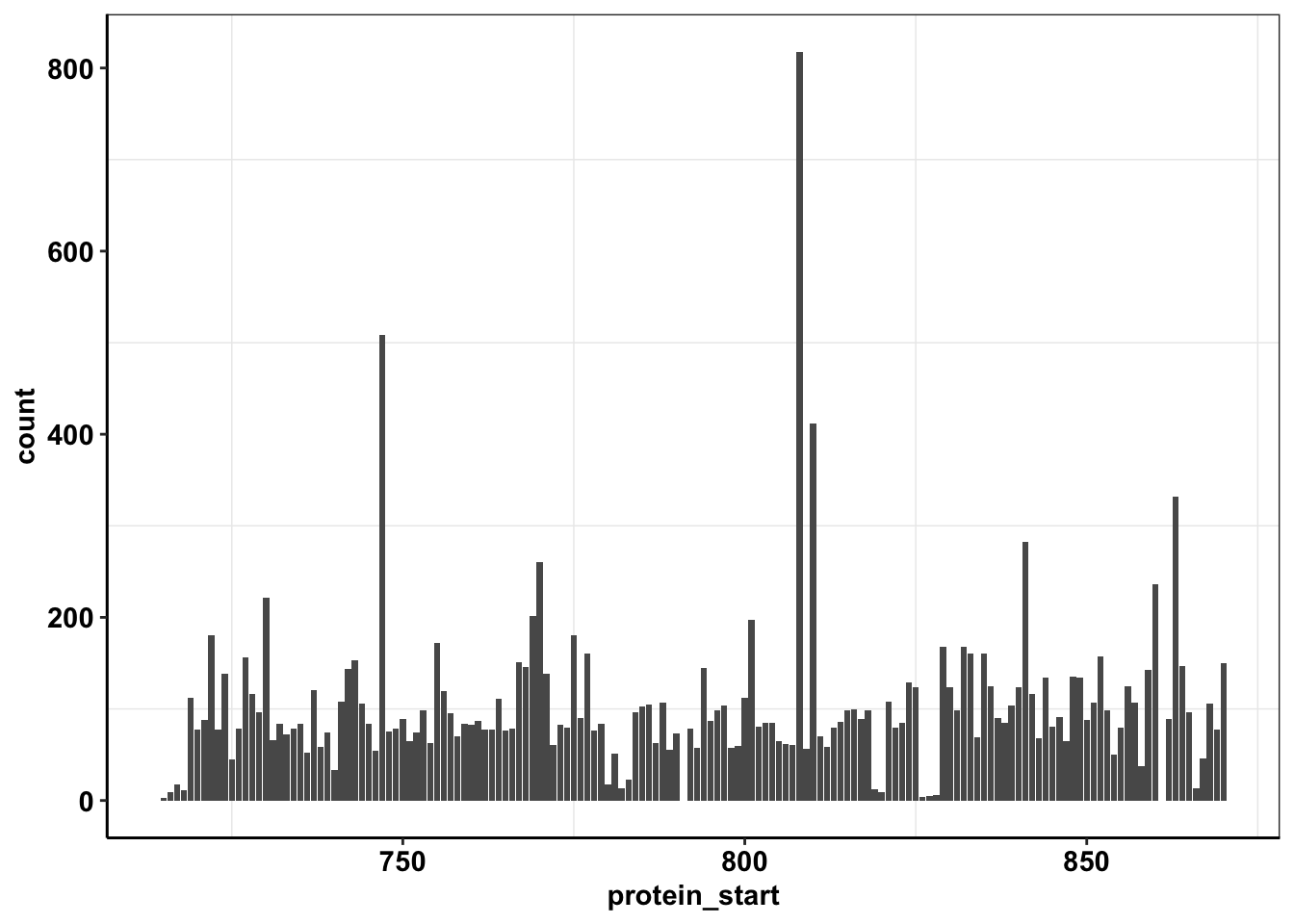
| Version | Author | Date |
|---|---|---|
| 097ad61 | haiderinam | 2022-09-29 |
# a=sample5_sum%>%filter(consequence_terms%in%"missense_variant",protein_start>=715,protein_start<=870)
# sum(a$ct)Sample 6
calls_missense=calls_missense%>%
filter(protein_start==protein_end,consequence_terms%in%"missense_variant")%>%
rowwise()%>%
mutate(ref_aa=strsplit(amino_acids,"/")[[1]][1],
alt_aa=strsplit(amino_acids,"/")[[1]][2])
ggplot(calls_missense,aes(x=protein_start,y=alt_aa))+
geom_tile(color="black",aes(fill=(ct/depth)))+
scale_x_continuous(name="Position along EGFR Kinase",expand=c(0,0),limits=c(717,870))+
scale_y_discrete(name="Amino Acid Substitution",expand=c(0,0))+
scale_fill_continuous(name="Allele \nFrequency",trans="log10")+
cleanup+
theme(legend.position = "none")
# a=calls_missense%>%filter(protein_start>=715,protein_start<=880)
# ggsave("egfr_heatmap_sample5.pdf",width=6,height=4,units = "in",useDingbats=F)
ggplot(calls_missense_sum,aes(x=protein_start,y=count))+geom_col()+scale_x_continuous(limits=c(710,875))SAMPLE 2
library("plotly")
sample2_sum=read.csv("data/Consensus_Data/Novogene_lane11/sample2/archive/variants_unique_ann_sample2.csv",header = T,stringsAsFactors = F)[-1]
sample2_sum$consensus="sscs"
sample2_sum=sample2_sum%>%
# filter(protein_start==protein_end,consequence_terms%in%"missense_variant")%>%
rowwise()%>%
mutate(ref_aa=strsplit(amino_acids,"/")[[1]][1],
alt_aa=strsplit(amino_acids,"/")[[1]][2])
sample2_all=read.csv("data/Consensus_Data/Novogene_lane11/sample2/archive/variants_ann_sample2.csv",header = T,stringsAsFactors = F)[-1]
sample2_all$consensus="sscs"
dcs_sum=read.csv("data/Consensus_Data/Novogene_lane11/sample2/archive/duplex_sum_ann.csv",header = T,stringsAsFactors = F)[-1]
dcs_sum$consensus="dcs"
sample2_all=sample2_all%>%
# filter(protein_start==protein_end,consequence_terms%in%"missense_variant")%>%
rowwise()%>%
mutate(ref_aa=strsplit(amino_acids,"/")[[1]][1],
alt_aa=strsplit(amino_acids,"/")[[1]][2])
# a=sample2_all%>%filter(protein_start%in%493,amino_acids%in%"F")
sample2_sum
dcs_sum=dcs_sum%>%
filter(protein_start==protein_end,consequence_terms%in%"missense_variant")%>%
rowwise()%>%
mutate(ref_aa=strsplit(amino_acids,"/")[[1]][1],
alt_aa=strsplit(amino_acids,"/")[[1]][2])
ggplot(sample2_sum,aes(x=protein_start,y=alt_aa))+
geom_tile(color="black",aes(fill=ct))+
scale_x_continuous(expand=c(0,0),limits=c(242,333))+
scale_y_discrete(expand=c(0,0))
all_sum=rbind(sample2_sum,dcs_sum)
plotly=ggplot(all_sum,aes(x=ct,fill=consensus,alpha=0.5))+
geom_density(position=position_dodge(),bins=100)+
scale_x_continuous(limits=c(0,100))
# scale_x_continuous(trans="log10")
ggplotly(plotly)
plotly=ggplot(all_sum,aes(x=ct,fill=consensus,alpha=0.5))+
geom_density(position=position_dodge(),bins=100)+
# scale_x_continuous(limits=c(0,100))+
scale_x_continuous(trans="log10")
ggplotly(plotly)
ggplot(all_sum,aes(x=protein_start,y=alt_aa))+
geom_tile(color="black",aes(fill=ct))+
facet_grid(~consensus)+
scale_x_continuous(expand=c(0,0),limits=c(241,333))+
scale_y_discrete(expand=c(0,0))+
scale_fill_gradient(na.value = "black",low="red",high="blue",name="Count")
# a=sscs_sum%>%filter(protein_start>=242,protein_start<=289)
# a=a%>%group_by(protein_start)%>%summarize(count=n())
calls_sum=sample2_sum%>%filter(consequence_terms%in%"missense_variant")%>%group_by(protein_start)%>%summarize(unique_mutants=n(),count=sum(ct))
ggplot(calls_sum%>%filter(protein_start>=242,protein_start<=500),aes(x=protein_start,y=count))+geom_col()+cleanup
ggplot(calls_sum%>%filter(protein_start>=242,protein_start<=500),aes(x=protein_start,y=unique_mutants))+geom_col()+cleanup
# a=sample2_sum%>%filter(consequence_terms%in%"missense_variant",protein_start>=242,protein_start<=500)
# sum(a$ct)Lane 11 SAMPLE 10 and 11
###########Sample 10############
sscs_sum=read.csv("data/Consensus_Data/Novogene_lane11/sample10/sscs_sum_ann.csv",header = T,stringsAsFactors = F)[-1]
sscs_sum=sscs_sum%>%
filter(protein_start==protein_end,consequence_terms%in%"missense_variant")%>%
rowwise()%>%
mutate(ref_aa=strsplit(amino_acids,"/")[[1]][1],
alt_aa=strsplit(amino_acids,"/")[[1]][2])
ggplot(sscs_sum,aes(x=protein_start,y=alt_aa))+
geom_tile(color="black",aes(fill=ct))+
scale_x_continuous(expand=c(0,0),limits=c(242,330))+
scale_y_discrete(expand=c(0,0))
###########Sample 8############
sscs_sum=read.csv("data/Consensus_Data/Novogene_lane11/sample8/sscs_sum_ann.csv",header = T,stringsAsFactors = F)[-1]
sscs_sum=sscs_sum%>%
filter(protein_start==protein_end,consequence_terms%in%"missense_variant")%>%
rowwise()%>%
mutate(ref_aa=strsplit(amino_acids,"/")[[1]][1],
alt_aa=strsplit(amino_acids,"/")[[1]][2])
ggplot(sscs_sum,aes(x=protein_start,y=alt_aa))+
geom_tile(color="black",aes(fill=ct))+
scale_x_continuous(expand=c(0,0),limits=c(242,330))+
scale_y_discrete(expand=c(0,0))Lane 11 SAMPLE 7
###########Sample 10############
sscs_sum=read.csv("data/Consensus_Data/Novogene_lane11/sample7/archive/sscs_sum_ann.csv",header = T,stringsAsFactors = F)[-1]
sscs_sum=sscs_sum%>%
filter(protein_start==protein_end,consequence_terms%in%"missense_variant")%>%
rowwise()%>%
mutate(ref_aa=strsplit(amino_acids,"/")[[1]][1],
alt_aa=strsplit(amino_acids,"/")[[1]][2])
ggplot(sscs_sum,aes(x=protein_start,y=alt_aa))+
geom_tile(color="black",aes(fill=ct))+
# scale_x_continuous(expand=c(0,0),limits=c(242,330))+
scale_x_continuous(expand=c(0,0))+
scale_y_discrete(expand=c(0,0))
###########Sample 8############
sscs_sum=read.csv("data/Consensus_Data/Novogene_lane11/sample8/sscs_sum_ann.csv",header = T,stringsAsFactors = F)[-1]
sscs_sum=sscs_sum%>%
filter(protein_start==protein_end,consequence_terms%in%"missense_variant")%>%
rowwise()%>%
mutate(ref_aa=strsplit(amino_acids,"/")[[1]][1],
alt_aa=strsplit(amino_acids,"/")[[1]][2])
ggplot(sscs_sum,aes(x=protein_start,y=alt_aa))+
geom_tile(color="black",aes(fill=ct))+
scale_x_continuous(expand=c(0,0),limits=c(242,330))+
scale_y_discrete(expand=c(0,0))Plotting the correlation of allele frequencies of stuff that I see in sample 2 vs in sample 7
samplex=read.csv("data/Consensus_Data/Novogene_lane14/sample15/variant_caller_outputs/variants_unique_ann.csv",header=T,stringsAsFactors = F)
samplex=samplex%>%
rowwise()%>%
mutate(ref_aa=strsplit(amino_acids,"/")[[1]][1],
alt_aa=strsplit(amino_acids,"/")[[1]][2])
samplex=samplex%>%mutate(maf=ct/depth)
samplex_simple=samplex%>%dplyr::select(alt_start_pos,protein_start,ref,alt,ref_aa,alt_aa,consequence_terms,ct,depth,maf)
sampley=read.csv("data/Consensus_Data/Novogene_lane14/sample17/variant_caller_outputs/variants_unique_ann.csv",header=T,stringsAsFactors = F)
sampley=sampley%>%
rowwise()%>%
mutate(ref_aa=strsplit(amino_acids,"/")[[1]][1],
alt_aa=strsplit(amino_acids,"/")[[1]][2])
sampley=sampley%>%mutate(maf=ct/depth)
sampley_simple=sampley%>%dplyr::select(alt_start_pos,protein_start,ref,alt,ref_aa,alt_aa,consequence_terms,ct,depth,maf)
samples_xy=merge(samplex_simple%>%filter(consequence_terms%in%"missense_variant"),sampley_simple%>%filter(consequence_terms%in%"missense_variant"),by=c("ref_aa","protein_start","alt_aa","ref","alt","alt_start_pos","consequence_terms"),all = T)
plotly=ggplot(samples_xy%>%filter(protein_start>=242,protein_start<=492),aes(x=maf.x,y=maf.y))+
geom_point(color="black",shape=21,aes(fill=ct.x))+
scale_x_continuous(trans="log")+
scale_y_continuous(trans="log")+
geom_abline()+
theme_bw()
ggplotly(plotly)Warning in L$marker$color[idx] <- aes2plotly(data, params, "fill")[idx]: number
of items to replace is not a multiple of replacement length# cor(samples_xy$maf.x,samples_xy$maf.y)source("code/merge_samples.R")
lane12sortedsamples=merge_samples("Novogene_lane11/Sample1","Novogene_lane11/Sample2")
lane12sortedsamples=merge_samples(lane12sortedsamples,"Novogene_lane11/Sample3")
lane12sortedsamples=merge_samples(lane12sortedsamples,"Novogene_lane11/Sample4")
lane12sortedsamples=lane12sortedsamples%>%
rowwise()%>%
mutate(ref_aa=strsplit(amino_acids,"/")[[1]][1],
alt_aa=strsplit(amino_acids,"/")[[1]][2])
lane12sortedsamples=lane12sortedsamples%>%mutate(maf=ct/depth)
# samplex_simple=samplex%>%dplyr::select(alt_start_pos,protein_start,ref,alt,ref_aa,alt_aa,consequence_terms,ct,depth,maf)
# sampley=sampley%>%
# rowwise()%>%
# mutate(ref_aa=strsplit(amino_acids,"/")[[1]][1],
# alt_aa=strsplit(amino_acids,"/")[[1]][2])
# sampley=sampley%>%mutate(maf=ct/depth)
# sampley_simple=sampley%>%dplyr::select(alt_start_pos,protein_start,ref,alt,ref_aa,alt_aa,consequence_terms,ct,depth,maf)
ggplot(lane12sortedsamples%>%
filter(nchar(alt_aa)%in%1,
consequence_terms%in%"missense_variant",
protein_start>=242,
protein_start<=282),
aes(x=protein_start,y=alt_aa,fill=maf))+
geom_tile()+
theme_bw()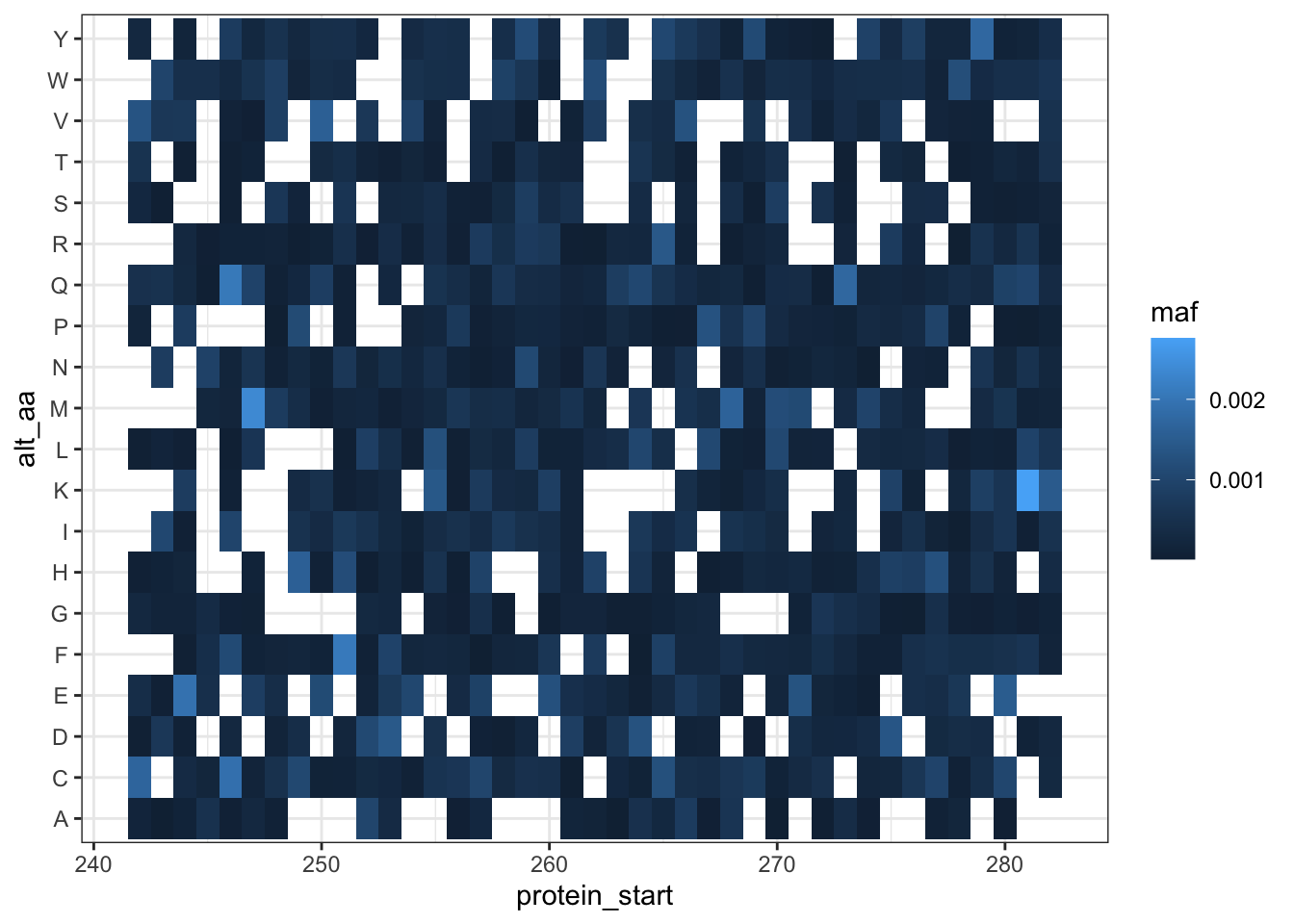
| Version | Author | Date |
|---|---|---|
| d358fe3 | haiderinam | 2022-10-29 |
ggplot(lane12sortedsamples)
| Version | Author | Date |
|---|---|---|
| d358fe3 | haiderinam | 2022-10-29 |
#To do: error plot where red lines outside mutagenized rgn
#plot how many variants do you see with 1 sample vs 2 vs 3 etc
# a=lane12sortedsamples%>%
# filter(nchar(alt_aa)%in%1,
# consequence_terms%in%"missense_variant",
# protein_start>=242,
# protein_start<300)
qc_data=read.csv("data/QC_Data_Cloning/TwistQC-ABL252residues-Q-146958.csv",header = T,stringsAsFactors = F)
qc_data=qc_data%>%filter(!AA.Position%in%NA)
ggplot(qc_data,aes(x=AA.Position,y=variant_aa,fill=variant_maf))+
geom_tile()+
theme_bw()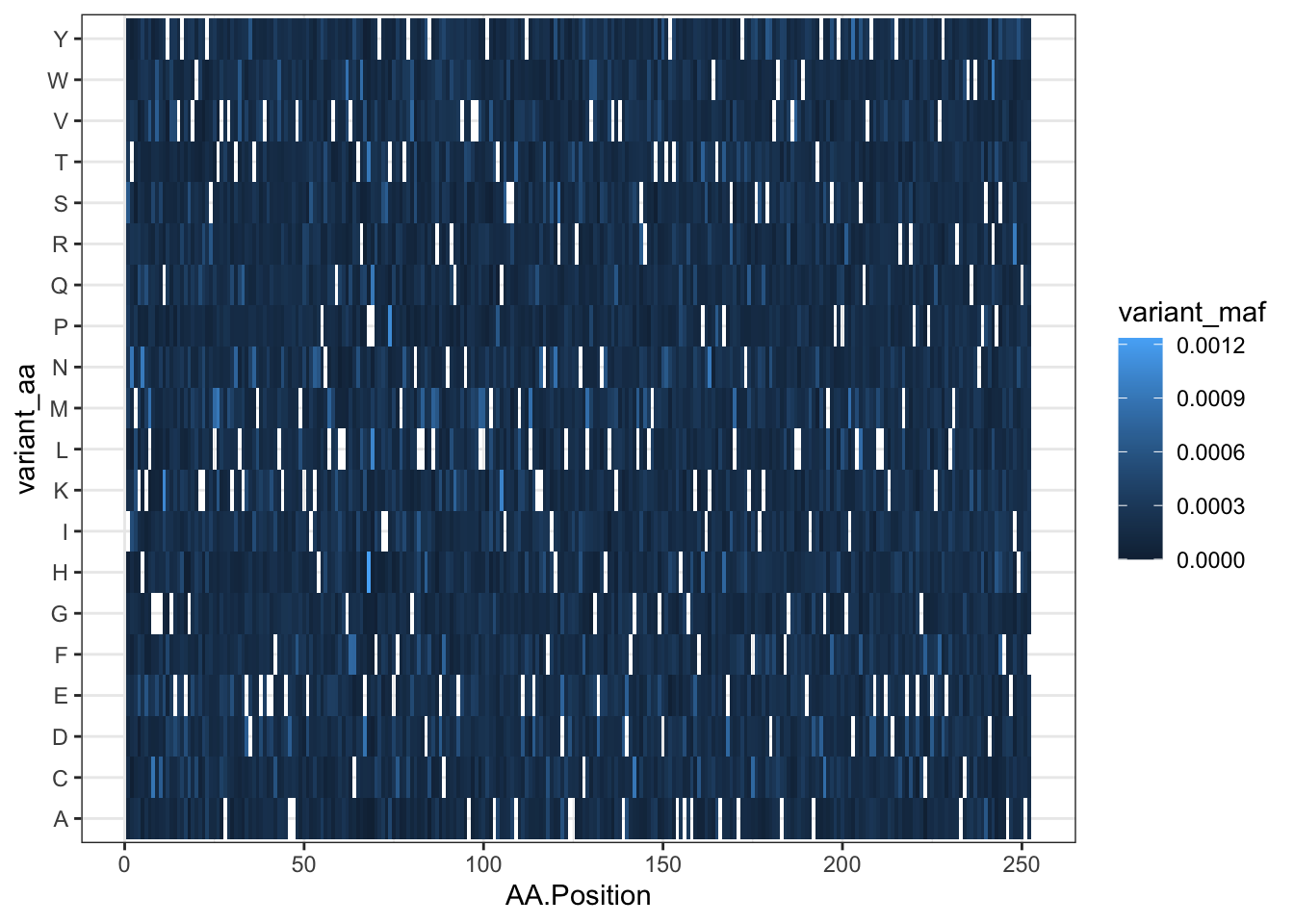
| Version | Author | Date |
|---|---|---|
| d358fe3 | haiderinam | 2022-10-29 |
qc_data_formerge=qc_data%>%filter(AA.Position<60)
lane12sorted_formerge=lane12sortedsamples%>%filter(nchar(alt_aa)%in%1,
consequence_terms%in%"missense_variant",
protein_start>=242,
protein_start<302)%>%
dplyr::select(wt_aa=ref_aa,
variant_aa=alt_aa,
protein_start,
variant_maf=maf,
ct)%>%
mutate(AA.Position=protein_start-241)
qc_data_merged=merge(qc_data,lane12sorted_formerge,by=c("AA.Position","wt_aa","variant_aa"),all.x = T)
qc_data_merged$detected="Yes"
qc_data_merged[qc_data_merged$variant_maf.y%in%NA,"detected"]="No"
qc_data_merged[qc_data_merged$variant_maf.y%in%NA,"variant_maf.y"]=1e-5
# With this plot, we can see that most of the variants are within 4 fold of each other in both the twist library and with our librayr
qc_data_merged[qc_data_merged$ct%in%NA,"ct"]=0
qc_data_merged$counts="0"
qc_data_merged[qc_data_merged$ct%in%1,"counts"]="1"
qc_data_merged[qc_data_merged$ct%in%2,"counts"]="2"
qc_data_merged[qc_data_merged$ct%in%3,"counts"]="3"
qc_data_merged[qc_data_merged$ct>=4,"counts"]=">3"
plotly=ggplot(qc_data_merged,aes(x=variant_maf.x,y=variant_maf.y,color=ct))+
geom_point()+
scale_x_continuous(trans="log2")+
scale_y_continuous(trans="log2")+
theme_bw()
ggplotly(plotly)Warning: Transformation introduced infinite values in continuous x-axisplotly=ggplot(qc_data_merged,aes(x=variant_maf.x,y=variant_maf.y))+
geom_point(color="black",shape=21,aes(fill=counts))+
scale_x_continuous(trans="log2")+
scale_y_continuous(trans="log2")+
theme_bw()
ggplotly(plotly)Warning: Transformation introduced infinite values in continuous x-axisa=qc_data_merged%>%filter(AA.Position<=49,detected%in%"No")
#With the following plot, we can see that of the variants that we don't see in our library, they're not the low AF samples.
ggplot(qc_data_merged%>%filter(AA.Position<=49),aes(x=detected,y=variant_maf.x,fill=detected))+
geom_violin()+
scale_y_continuous(trans="log10")+
theme_bw()+
geom_jitter(shape=16, position=position_jitter(0.2))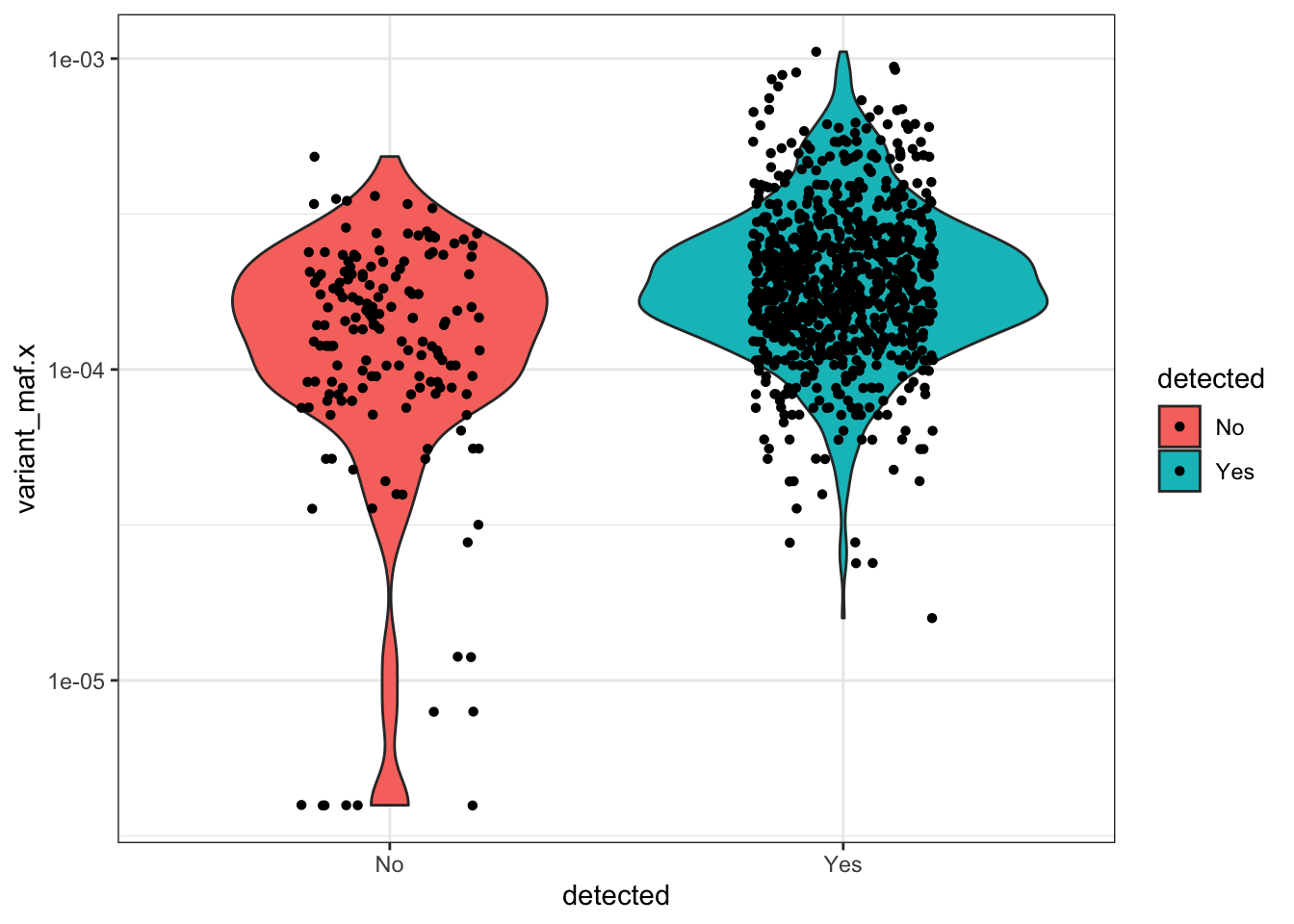
| Version | Author | Date |
|---|---|---|
| d358fe3 | haiderinam | 2022-10-29 |
# ggplot(qc_data_merged%>%filter(variant_maf.x>0),aes(x=variant_maf.x,y=variant_maf.y))+
# stat_density_2d(aes(fill=..level..),geom = "polygon", colour="white")+
# scale_fill_continuous(type="viridis")+
# theme_bw()+
# scale_x_continuous(trans="log")+
# scale_y_continuous(trans="log")
# a=qc_data_merged%>%filter(ct>=10)
cor(qc_data_merged$variant_maf.x,qc_data_merged$variant_maf.y)[1] 0.122601# cor(a$variant_maf.x,a$variant_maf.y)#Numbers: Between AAs 242 and 291, twist made 909 QC-confirmed mutants, we detected 839, or 92% of those. #Numbers: Between AAs 242 and 291, twist made 999 mutants overall (including QC-failed mutants), we detected 839, or 82% of those.
sessionInfo()R version 4.0.0 (2020-04-24)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS 10.16
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRblas.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] parallel stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] RColorBrewer_1.1-2 doParallel_1.0.15 iterators_1.0.12 foreach_1.5.0
[5] tictoc_1.0 plotly_4.9.2.1 ggplot2_3.3.3 dplyr_1.0.6
[9] stringr_1.4.0
loaded via a namespace (and not attached):
[1] tidyselect_1.1.0 xfun_0.31 bslib_0.3.1 purrr_0.3.4
[5] colorspace_1.4-1 vctrs_0.3.8 generics_0.0.2 htmltools_0.5.2
[9] viridisLite_0.3.0 yaml_2.2.1 utf8_1.1.4 rlang_0.4.11
[13] jquerylib_0.1.4 later_1.0.0 pillar_1.6.1 glue_1.4.1
[17] withr_2.4.2 DBI_1.1.0 lifecycle_1.0.0 munsell_0.5.0
[21] gtable_0.3.0 workflowr_1.6.2 htmlwidgets_1.5.1 codetools_0.2-16
[25] evaluate_0.14 labeling_0.3 knitr_1.28 fastmap_1.1.0
[29] crosstalk_1.1.0.1 httpuv_1.5.2 fansi_0.4.1 Rcpp_1.0.4.6
[33] promises_1.1.0 backports_1.1.7 scales_1.1.1 jsonlite_1.7.2
[37] farver_2.0.3 fs_1.4.1 digest_0.6.25 stringi_1.7.5
[41] rprojroot_1.3-2 grid_4.0.0 tools_4.0.0 magrittr_2.0.1
[45] sass_0.4.1 lazyeval_0.2.2 tibble_3.1.2 crayon_1.4.1
[49] whisker_0.4 tidyr_1.1.3 pkgconfig_2.0.3 ellipsis_0.3.2
[53] data.table_1.12.8 assertthat_0.2.1 rmarkdown_2.14 httr_1.4.2
[57] R6_2.4.1 git2r_0.27.1 compiler_4.0.0