Downstream analysis
Last updated: 2020-04-28
Checks: 5 2
Knit directory: lieb/
This reproducible R Markdown analysis was created with workflowr (version 1.6.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190717) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 16778b1. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: analysis/figure/
Ignored: analysis/pairwise_fitting_cache/
Ignored: analysis/running_mcmc_cache/
Ignored: data/.DS_Store
Ignored: output/.DS_Store
Ignored: output/chip_shiny/.DS_Store
Ignored: output/chip_shiny/Data/.DS_Store
Ignored: output/vision_shiny/.DS_Store
Ignored: output/vision_shiny/.Rhistory
Ignored: output/vision_shiny/Data/.DS_Store
Unstaged changes:
Modified: analysis/downstream.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/downstream.Rmd) and HTML (docs/downstream.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | bf87fa0 | hillarykoch | 2020-04-28 | add some downstream analysis |
| html | bf87fa0 | hillarykoch | 2020-04-28 | add some downstream analysis |
| Rmd | 30514e2 | hillarykoch | 2020-04-27 | update MCMC and outline downstrea, |
| html | 30514e2 | hillarykoch | 2020-04-27 | update MCMC and outline downstrea, |
| html | cc98d2b | hillarykoch | 2020-04-24 | replace cache and update to describe flex_mu |
| Rmd | 717e762 | hillarykoch | 2020-04-23 | add placeholder for downstream analyses |
| html | 717e762 | hillarykoch | 2020-04-23 | add placeholder for downstream analyses |
| Rmd | 2a75de4 | hillarykoch | 2020-02-20 | remove too-big header |
| html | 2a5b7c0 | hillarykoch | 2020-02-20 | Build site. |
| html | 3a9bf9d | hillarykoch | 2020-02-20 | Build site. |
| Rmd | a99d063 | hillarykoch | 2020-02-20 | add new page about downstream analysis |
| html | a99d063 | hillarykoch | 2020-02-20 | add new page about downstream analysis |
CLIMB’s output is effectively the posterior MCMC samples of the parameters of the normal mixture. These samples can be used for clustering/classification, visualizations, and statistical inference. Here, we show implementations of several of the analyses used in the CLIMB manuscript.
First, let’s load in the MCMC we briefly ran on simulated data in the running the MCMC section.
chain <- readRDS("output/chain.rds")Visualizing convergence
The first thing we should do make some trace plots for some of the parameters being estimated. This will help us decide when the chain has entered its stationary distribution. Here, we make plots of a mean, variance, covariance, and mixing weight.
# The mean in the first dimension of the third cluster
mu31 <- chain$mu_chains[[3]][,1]
# The variance of the first dimension of the second cluster
sigma21 <- chain$Sigma_chains[[2]][1,1,]
# The covariance between dimensions 2 and 3 in the fourth cluster
sigma423 <- chain$Sigma_chains[[4]][2,3,]
# The mixing weight of the seventh cluster
pi7 <- chain$prop_chain[,7]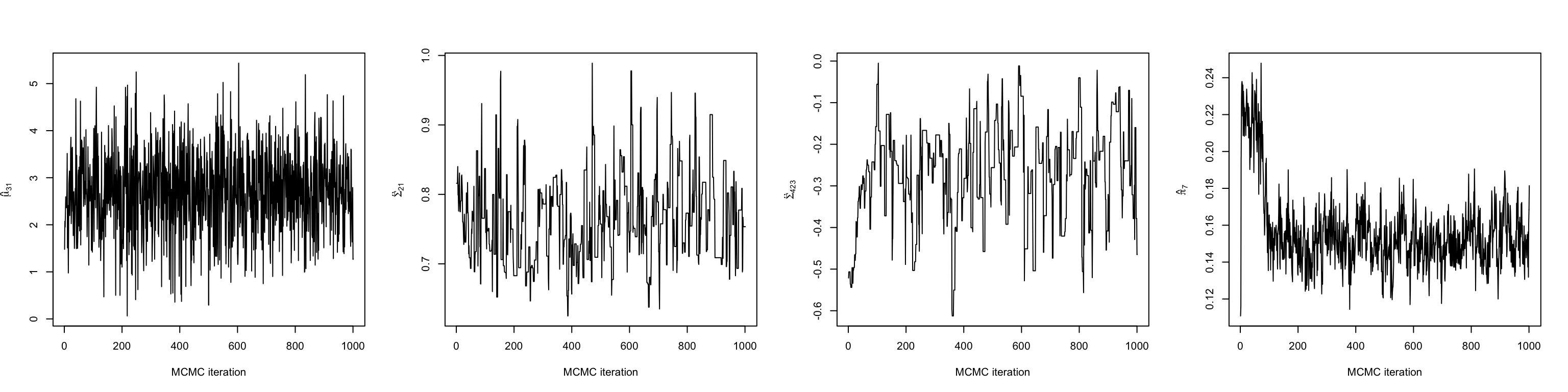
| Version | Author | Date |
|---|---|---|
| bf87fa0 | hillarykoch | 2020-04-28 |
Looking at a few of these trace plots reveals that the chain achieves stationary around 200 iterations in. Therefore, we discard the first 200 iterations as burn-in, and do our analyses on the remaining 800.
burnin <- 1:200Clustering dimensions and observations
CLIMB can provide a bi-clustering of analyzed data. Row clusters come from the estimated latent classes, of course, though these classes can even be grouped by their similarity.
CLIMB uses a distance based on class covariances to cluster the dimensions (columns). The function compute_distances_between_conditions will compute these pairwise distances between conditions based on model fit. This was used in the CLIMB manuscript to determine the relationship among hematopoietic cell populations based on CTCF binding patterns.
CLIMB’s estimated classes naturally cluster the rows. However, the function compute_distances_between_clusters will compute distances between estimated clusters as well. This distance is based on a symmetrified theoretical Kullback-Liebler divergence.
par(mfrow = c(1,2))
# The figure isn't as exciting with simulated data
col_distmat <- compute_distances_between_conditions(chain, burnin)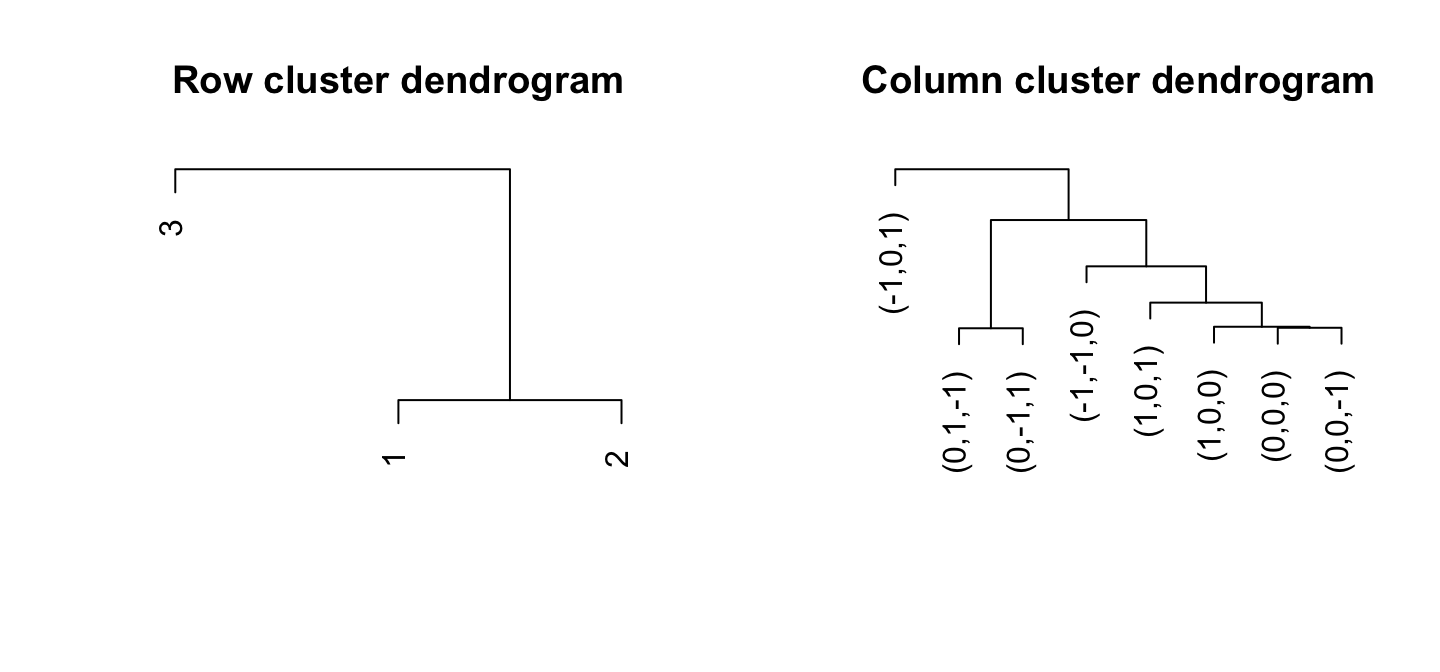
| Version | Author | Date |
|---|---|---|
| bf87fa0 | hillarykoch | 2020-04-28 |
These figures aren’t very thrilling on their own, but we can use them to make some nice heatmaps. CLIMB provides the function get_row_reordering to facilitate plotting the bi-clustering:
# Load back in the data
data("sim")
dat <- sim$data
# Get a row reordering for plotting
row_reordering <- get_row_reordering(row_clustering = hc_row, chain = chain, burnin = burnin)
# Melt (for ggplot2)
molten1 <-
reshape2::melt(data = dplyr::mutate(dat, "row" = row_reordering),
id.vars = "row")
# Rename the factor levels to match the dimensions
levels(molten1$variable) <- paste(1:3)[hc_col$order]
# Plot the bi-clustering heatmap, like in the CLIMB manuscript
p1 <- ggplot(data = molten1,
aes(x = variable, y = row, fill = value)) +
geom_raster() +
cowplot::theme_cowplot() +
theme(legend.position = "none", axis.ticks.y = element_blank(), axis.text.y = element_blank()) +
xlab("") + ylab("") +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = .5)) +
ggtitle("Bi-clustering\nheatmap") +
scale_fill_gradientn(colours=RColorBrewer::brewer.pal(9,"Greens"))
print(p1)
| Version | Author | Date |
|---|---|---|
| bf87fa0 | hillarykoch | 2020-04-28 |
Merging groups
CLIMB allows you to merge similar groups for the sake of visualization, for examble on the Genome Browser. In the CLIMB manuscript, we merged clusters of CTCF binding patterns to color peaks more in a more parsimonious set of 5 groups on the genome browser. We show how to do this for an arbitrary number of groups with CLIMB’s merge_classes function.
The output of a merge_classes call provides the means and mixing weights of the new parent groups. These means and weights are aggregated from each groups constituent classes. merged_z gives the new group assignments for each observations. The clustering obect is simply a call to stats::hclust().
# Merge into 5 parent groups
merged <- merge_classes(n_groups = 5, chain = chain, burnin = burnin)
# Check the structure of the merging
str(merged)List of 4
$ merged_z : int [1:1500] 5 2 2 2 2 2 2 2 2 2 ...
$ merged_mu : num [1:5, 1:3] 2.531 0.813 0 -2.501 -2.52 ...
$ merged_prop: num [1:5] 0.125 0.471 0.148 0.131 0.125
$ clustering :List of 7
..$ merge : int [1:7, 1:2] -3 -4 -2 -1 -8 1 -7 -6 -5 2 ...
..$ height : num [1:7] 0.00185 0.01065 0.02871 0.45571 1.09804 ...
..$ order : int [1:8] 7 3 6 8 1 2 4 5
..$ labels : chr [1:8] "1" "2" "4" "6" ...
..$ method : chr "complete"
..$ call : language hclust(d = as.dist(cluster_dist), method = "complete")
..$ dist.method: NULL
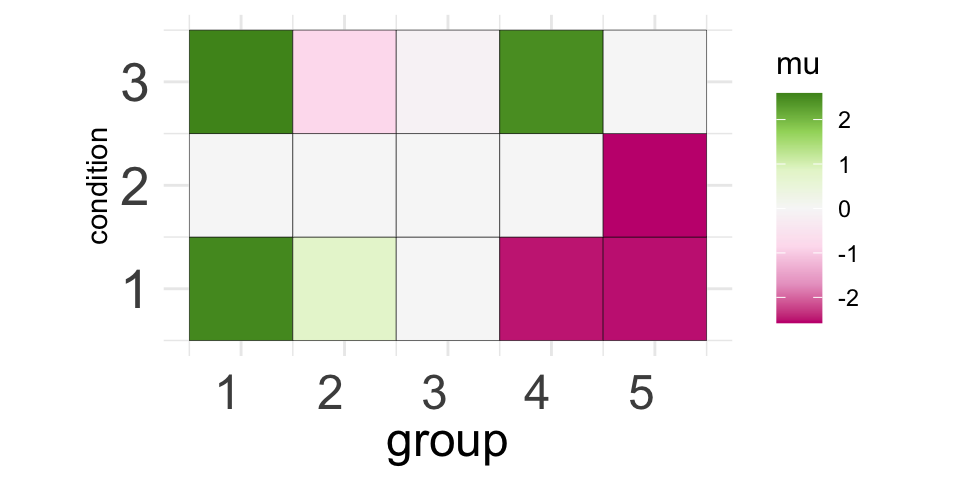
..- attr(*, "class")= chr "hclust"We can visualize the salient features of the new groups with a simple visualization of the merged group means:
library(RColorBrewer)
molten2 <- reshape2::melt(merged$merged_mu) %>%
`colnames<-` (c("group", "condition", "mu"))
p2 <- ggplot(data = molten2, aes(group, condition, fill = mu)) +
geom_tile(color = "black") +
scale_fill_distiller(palette = "PiYG", direction = 1) +
coord_fixed() +
theme_minimal() +
theme(axis.text.x = element_text(angle = 0, vjust = 0.5, hjust = 1, size = 18),
axis.text.y = element_text(size = 20),
axis.title.x = element_text(size = 18),
axis.ticks.y = element_blank(),
axis.ticks.x = element_blank(),
legend.title = element_text(size = 12))
print(p2)