Phase-specific scores for Leng et al. 2015
Joyce Hsiao
Last updated: 2018-02-01
Code version: 4bc41dd
Overview/Results
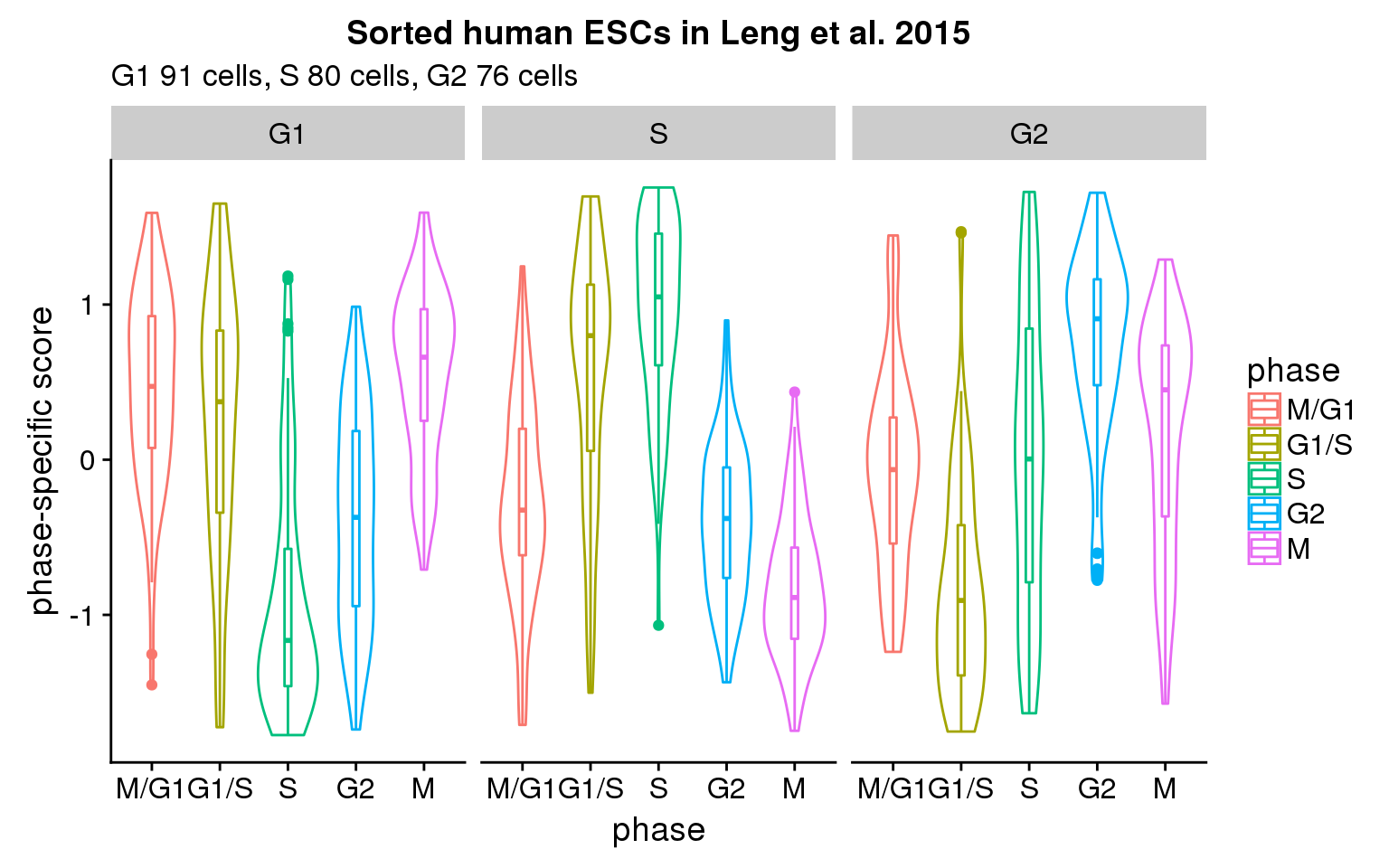
Consider the fucci-expression human ESC in Leng et al. 2015
What are the expression profiles of the sorted cells given the phase-specific scores in Macoscko et al. 2015?
- G1 cells score high on M/G1, G1/S, and M.
- S cells score high on G1/S, and S.
- G2 cells score high oh G2 and M.
Compared to the preliminary analysis of our data (here), we observe this pattern in some individuals but not all of the individuals. What’s clear is that phase-specific scores along are not adequate in predicting cell cycle phase. Importantly, there appears to be a pattern in average expression levels within each cell state (fucci-assigned). G1 cells peaks at M/G1 and G1/S, S cells peaks at S, while G2 cells peaks at G2.
- 123 genes were identified as variable along cell cycle phase.
Data and packages
Packages
library(Biobase)
library(ggplot2)
library(cowplot)
library(data.table)
library(tidyr)Load the normalized and filtered data from Leng et al 2015.
#install_github("jhsiao999/singleCellRNASeqHumanLengESC")
library("singleCellRNASeqHumanLengESC")
data("HumanLengESC")
counts <- exprs(HumanLengESC)
libsize <- colSums(counts)
cpm <- t(t(counts)*(10^6)/libsize)
log2cpm <- log2(cpm+1)
pdata <- pData(HumanLengESC)
table(pData(HumanLengESC)$cell_state)
G1 G2 H1 S
91 76 213 80 select fucci-expression cells
cpm <- cpm[,pdata$cell_state != "H1"]
log2cpm <- log2cpm[,pdata$cell_state != "H1"]
pdata <- pdata[pdata$cell_state != "H1", ]filter genes
genes_to_include <- which(rowMeans(cpm)>1)
log2cpm <- log2cpm[genes_to_include,]import cell cycle genes info
cellcycle <- readRDS("../data/cellcycle-genes-previous-studies/rds/macosko-2017.rds")
which_cc <- which(rownames(log2cpm) %in% cellcycle$hgnc)subset genes to include only cell cycle genes
log2cpm_cc <- log2cpm[which_cc, ]
cc_genes <- cellcycle[which(cellcycle$hgnc %in% rownames(log2cpm)),]compute phase-specific scores
cc_scores <- lapply(1:uniqueN(cc_genes$phase), function(i) {
ph <- unique(cc_genes$phase)[i]
df_sub <- log2cpm_cc[rownames(log2cpm_cc) %in% cc_genes$hgnc[cc_genes$phase == ph],]
mn <- colMeans(df_sub)
cc <- cor(t(rbind(mn, df_sub)))
cc_mean <- cc[-1,1]
genes_cc <- names(cc_mean)[which(cc_mean > .3)]
scores_raw <- colMeans(df_sub[rownames(df_sub) %in% genes_cc,])
scores_z <- scale(scores_raw)
return(list(scores_z=scores_z, ngenes = length(genes_cc)))
})
names(cc_scores) <- unique(cc_genes$phase)
ngenes <- sapply(cc_scores, function(x) x[[2]])
print(ngenes) S G1/S M/G1 M G2
10 6 27 46 34 scores <- do.call(cbind, lapply(cc_scores, function(x) x[[1]]))
colnames(scores) <- unique(cc_genes$phase)
scores_z <- t(apply(scores, 1, scale))
colnames(scores_z) <- unique(cc_genes$phase)
scores_z <- as.data.frame(scores_z)
scores_z_long <- gather(scores_z, key=phase, value=scores)
scores_z_long$sample_id <- rep(rownames(scores_z), ncol(scores_z))
scores_z_long$cell_state <- pdata$cell_state[match(scores_z_long$sample_id, rownames(pdata))]Analysis
Session information
R version 3.4.1 (2017-06-30)
Platform: x86_64-redhat-linux-gnu (64-bit)
Running under: Scientific Linux 7.2 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /usr/lib64/R/lib/libRblas.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] singleCellRNASeqHumanLengESC_0.99.0 tidyr_0.7.2
[3] data.table_1.10.4-3 cowplot_0.9.2
[5] ggplot2_2.2.1 Biobase_2.38.0
[7] BiocGenerics_0.24.0
loaded via a namespace (and not attached):
[1] Rcpp_0.12.14 knitr_1.17 magrittr_1.5 munsell_0.4.3
[5] colorspace_1.3-2 rlang_0.1.4 stringr_1.2.0 plyr_1.8.4
[9] tools_3.4.1 grid_3.4.1 gtable_0.2.0 git2r_0.20.0
[13] htmltools_0.3.6 yaml_2.1.16 lazyeval_0.2.1 rprojroot_1.3-1
[17] digest_0.6.13 tibble_1.3.4 purrr_0.2.4 glue_1.2.0
[21] evaluate_0.10.1 rmarkdown_1.8 labeling_0.3 stringi_1.1.6
[25] compiler_3.4.1 scales_0.5.0 backports_1.1.2 This R Markdown site was created with workflowr