pH cluster analysis
David Stappard & Jens Daniel Müller
24 November, 2023
Last updated: 2023-11-24
Checks: 7 0
Knit directory: bgc_argo_r_argodata/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20211008) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version b24f211. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: output/
Untracked files:
Untracked: code/start_background_job_partial.R
Unstaged changes:
Deleted: code/start_background_job_core_load.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/pH_cluster_analysis.Rmd)
and HTML (docs/pH_cluster_analysis.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | efa9686 | ds2n19 | 2023-11-24 | Build site. |
| Rmd | ed9f431 | ds2n19 | 2023-11-24 | Cluster under surface extreme. |
| html | 80c16c2 | ds2n19 | 2023-11-15 | Build site. |
| Rmd | 3eba518 | ds2n19 | 2023-11-15 | Introduction of vertical alignment and cluster analysis to github website. |
Tasks
This markdown file carries out cluster analysis on previously created pH anomaly profiles.
The cluster_analysis_determine_k chunk is used to give an indication of an appropriate number of clusters and this can then set the option opt_num_clusters. Chunk cluster_analysis_cluster_details carried out the cluster analysis and the results are used in subsequent figures.
Set directories
location of pre-prepared data
Set options
Define options that are used to determine what analysis is done
# Options
# opt_analysis_type
# opt_analysis_type = 1 determine number of clusters to use
# opt_analysis_type = 2 do analysis based on identified number of clusters
opt_analysis_type <- 2
# opt_num_clusters
# How many clusters are used in the cluster analysis
opt_num_clusters <- c(7, 7, 7)
# What is the max depth of each profile_range
opt_max_depth <- c(614, 1225, 1600)
# Which profile range is used
opt_profile_range <- 3
# opt_measure
# opt_measure = 1 analysis is done using pH
# opt_measure = 2 analysis is done using h_plus
# opt_measure_label, opt_xlim and opt_xbreaks are associated formatting
opt_measure <- 2
if (opt_measure == 1){
opt_measure_label <- "pH anomaly"
opt_xlim <- c(-0.08, 0.08)
opt_xbreaks <- c(-0.08, -0.04, 0, 0.04, 0.08)
} else {
opt_measure_label <- expression("[H]"^"+" ~ "anomaly")
opt_xlim <- c(-2e-9, 2e-9)
opt_xbreaks <- c(-2e-9, -1e-9, 0, 1e-9, 2e-9)
}
# Chl-a formatting
opt_chla_measure_label <- expression("chlorophyll a ( mg m"^"-3"~")")
opt_chla_xlim <- c(-0.5, 2.0)
opt_chla_xbreaks <- c(-0.5, 0, 0.5, 1.0, 1.5, 2.0)
# oxygen formatting
opt_doxy_measure_label <- expression("dissolved oxygen ( µmol kg"^"-1"~")")
opt_doxy_xlim <- c(50, 350)
opt_doxy_xbreaks <- c(50, 100, 150, 200, 250, 300, 350)
# options relating to cluster analysis
opt_n_start <- 25
opt_max_iterations <- 500
opt_n_clusters <- 14 # Max number of clusters to try when determining optimal number of clusterstheme_set(theme_bw())
map <-
read_rds(paste(path_emlr_utilities,
"map_landmask_WOA18.rds",
sep = ""))Cluster analysis
Preperation
Prepare data for cluster analysis
# read data
pH_anomaly_va <- read_rds(file = paste0(path_argo_preprocessed, "/pH_anomaly_va.rds"))
# select profile based on profile_range and he appropriate max depth
pH_anomaly_va <- pH_anomaly_va %>%
filter(profile_range == opt_profile_range & depth <= opt_max_depth[opt_profile_range])
# Select target variable
if (opt_measure == 1) {
pH_anomaly_va_id <- pH_anomaly_va %>%
rename(
anomaly = anomaly_pH
)
} else {
pH_anomaly_va_id <- pH_anomaly_va %>%
rename(
anomaly = anomaly_h_plus
)
}
pH_anomaly_va_id <- pH_anomaly_va_id %>%
select(
file_id, depth, anomaly,
year, month, lat, lon
)
# wide table with each depth becoming a column
pH_anomaly_va_wide <- pH_anomaly_va_id %>%
select(file_id, depth, anomaly) %>%
pivot_wider(names_from = depth, values_from = anomaly)
# Drop any rows with missing values N/A caused by gaps in climatology data
pH_anomaly_va_wide <- pH_anomaly_va_wide %>%
drop_na()
# profile_id <- pH_anomaly_va_wide %>% select(file_id)
# Table for cluster analysis
# points <- pH_anomaly_va_wide %>%
# select(-c(file_id))
points <- pH_anomaly_va_wide %>%
column_to_rownames(var = "file_id")Number of clusters
if (opt_analysis_type == 1) {
# cluster analysis - What k? try between 1 and opt_n_clusters clusters
kclusts <-
tibble(k = 1:opt_n_clusters) %>%
mutate(
kclust = map(k, ~kmeans(points, .x, iter.max = opt_max_iterations, nstart = opt_n_start)),
tidied = map(kclust, tidy),
glanced = map(kclust, glance),
augmented = map(kclust, augment, points)
)
# cluster analysis data
clusters <-
kclusts %>%
unnest(cols = c(tidied))
assignments <-
kclusts %>%
unnest(cols = c(augmented))
clusterings <-
kclusts %>%
unnest(cols = c(glanced))
# What cluster works best?
clusterings %>%
ggplot(aes(k, tot.withinss)) +
geom_line() +
geom_point() +
scale_x_continuous(breaks = c(2, 4, 6, 8, 10, 12, 14))
}Cluster analysis
if (opt_analysis_type == 2) {
# caution: set.seed works only for one execution of the clustering
# How sensitive are our results to the acutal seed?
set.seed(1)
kclusts <-
tibble(k = opt_num_clusters[opt_profile_range]) %>%
mutate(
kclust = map(k, ~ kmeans(points, .x, iter.max = opt_max_iterations, nstart = opt_n_start)),
tidied = map(kclust, tidy),
glanced = map(kclust, glance),
augmented = map(kclust, augment, points)
)
profile_id <-
kclusts %>%
unnest(cols = c(augmented)) %>%
select(file_id = .rownames,
cluster = .cluster) %>%
mutate(file_id = as.numeric(file_id),
cluster = as.character(cluster))
# Add cluster to pH_anomaly_va
pH_anomaly_cluster <- full_join(pH_anomaly_va_id, profile_id)
# Note the null cluster results relate to the profiles that were removed
# from pH_anomaly_va_wide by the dron_na() function above.
pH_anomaly_cluster <- pH_anomaly_cluster %>%
filter(!is.na(cluster))
# Plot cluster mean
anomaly_cluster_mean <- pH_anomaly_cluster %>%
group_by(cluster, depth) %>%
summarise(
count_cluster = n(),
anomaly_mean = mean(anomaly, na.rm = TRUE),
anomaly_sd = sd(anomaly, na.rm = TRUE)
) %>%
ungroup()
anomaly_cluster_mean_year <- pH_anomaly_cluster %>%
group_by(cluster, depth, year) %>%
summarise(
count_cluster = n(),
anomaly_mean = mean(anomaly, na.rm = TRUE),
anomaly_sd = sd(anomaly, na.rm = TRUE)
) %>%
ungroup()
anomaly_year_mean <- pH_anomaly_cluster %>%
group_by(cluster, year) %>%
summarise(
count_cluster = n(),
anomaly_mean = mean(anomaly, na.rm = TRUE),
anomaly_sd = sd(anomaly, na.rm = TRUE)
) %>%
ungroup()
anomaly_year_mean <- anomaly_year_mean %>%
group_by(year) %>%
summarise(anomaly_mean = mean(anomaly_mean, na.rm = TRUE)) %>%
ungroup ()
# create figure
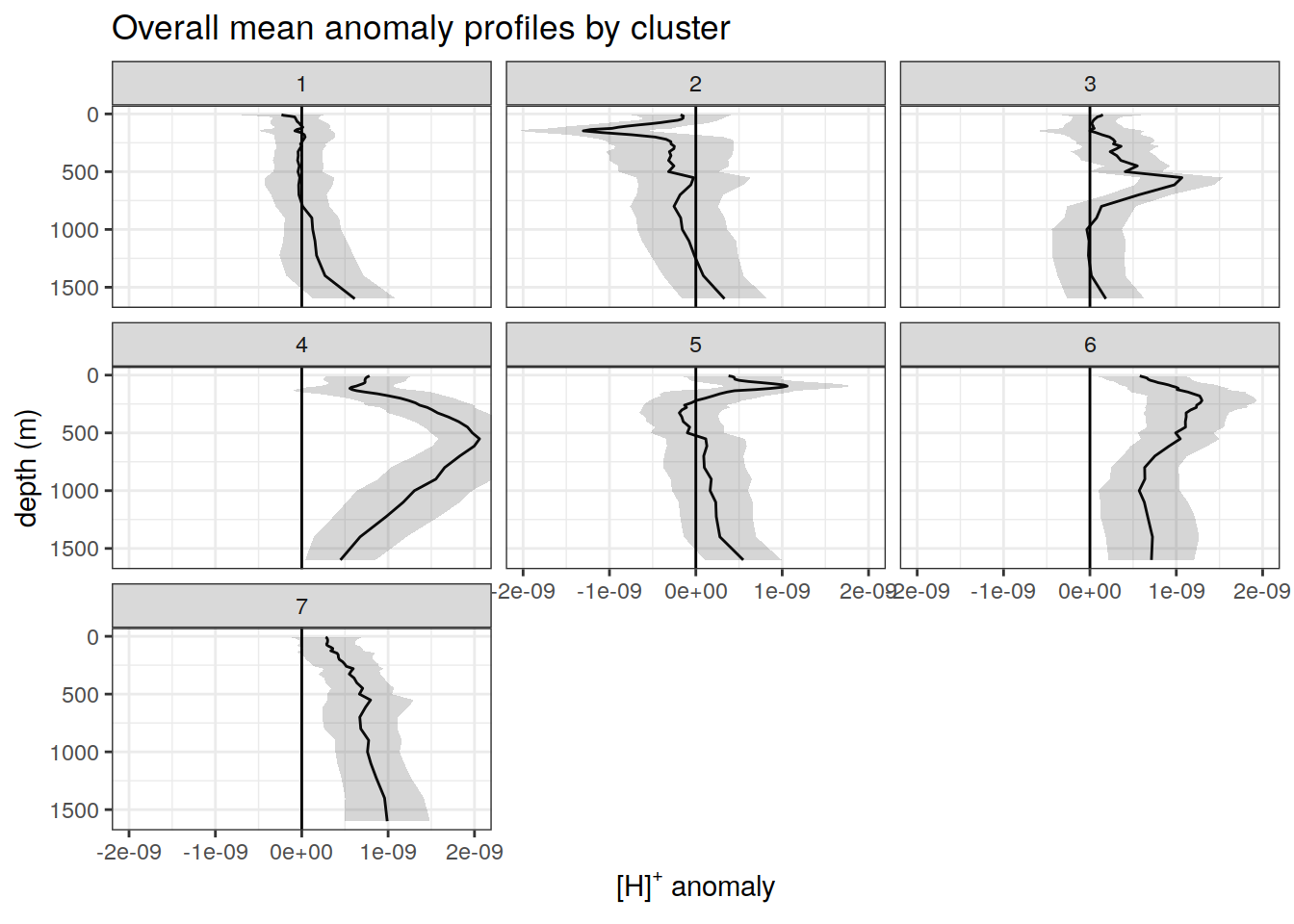
anomaly_cluster_mean %>%
ggplot() +
geom_path(aes(x = anomaly_mean,
y = depth)) +
geom_ribbon(aes(
xmax = anomaly_mean + anomaly_sd,
xmin = anomaly_mean - anomaly_sd,
y = depth
),
alpha = 0.2) +
geom_vline(xintercept = 0) +
scale_y_reverse() +
facet_wrap( ~ cluster) +
coord_cartesian(xlim = opt_xlim) +
scale_x_continuous(breaks = opt_xbreaks) +
labs(
title = paste0('Overall mean anomaly profiles by cluster'),
x = opt_measure_label,
y = 'depth (m)'
)
}
Cluster mean by year
if (opt_analysis_type == 2) {
anomaly_cluster_mean_year %>%
mutate(year = as.factor(year)) %>%
ggplot() +
geom_path(aes(x = anomaly_mean,
y = depth,
col = year)) +
geom_vline(xintercept = 0) +
scale_y_reverse() +
facet_wrap( ~ cluster) +
coord_cartesian(xlim = opt_xlim) +
scale_x_continuous(breaks = opt_xbreaks) +
scale_color_viridis_d() +
labs(
title = paste0('Overall mean anomaly profiles by cluster'),
x = opt_measure_label,
y = 'depth (m)'
)
}
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
Cluster climatology
# A nice short alternative, uses the package ggpmisc
anomaly_year_mean %>%
ggplot(aes(x = year,
y = anomaly_mean)) +
stat_poly_line() +
stat_poly_eq(use_label(c("eq", "R2","P", "n"))) +
geom_point()
| Version | Author | Date |
|---|---|---|
| 80c16c2 | ds2n19 | 2023-11-15 |
Cluster by year
count of each cluster by year
if (opt_analysis_type == 2) {
# Determine profile count by cluster and year
cluster_by_year <- pH_anomaly_cluster %>%
count(cluster, year,
name = "count_cluster")
year_min <- min(cluster_by_year$year)
year_max <- max(cluster_by_year$year)
# create figure
cluster_by_year %>%
ggplot(aes(x = year, y = count_cluster, col = cluster, group=cluster))+
geom_point() +
geom_line() +
scale_x_continuous(breaks = seq(year_min, year_max, 2)) +
scale_color_brewer(palette = 'Dark2')+
labs(x = 'year',
y = 'number of profiles',
col = 'cluster',
title = 'Count of profiles by year and cluster')
}
| Version | Author | Date |
|---|---|---|
| 80c16c2 | ds2n19 | 2023-11-15 |
Cluster by month
count of each cluster by month of year
if (opt_analysis_type == 2) {
# Determine profile count by cluster and year
cluster_by_year <- pH_anomaly_cluster %>%
count(cluster, month,
name = "count_cluster")
# create figure
cluster_by_year %>%
ggplot(aes(x = month, y = count_cluster, col = cluster, group=cluster))+
geom_point() +
geom_line() +
scale_x_continuous(breaks = seq(1, 12, 2)) +
scale_color_brewer(palette = 'Dark2')+
labs(x = 'month',
y = 'number of profiles',
col = 'cluster',
title = 'Count of profiles by month and cluster')
}
| Version | Author | Date |
|---|---|---|
| 80c16c2 | ds2n19 | 2023-11-15 |
Cluster spatial
location of each cluster on map, spatial analysis
if (opt_analysis_type == 2) {
# create figure
map +
geom_tile(data = pH_anomaly_cluster,
aes(x = lon,
y = lat,
fill = cluster))+
lims(y = c(-85, -30))+
scale_fill_brewer(palette = 'Dark2')+
labs(title = 'cluster spatial distribution')
}
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
location of each cluster on separate maps, spatial analysis
if (opt_analysis_type == 2) {
# create figure
map +
geom_tile(data = pH_anomaly_cluster,
aes(x = lon,
y = lat,
fill = cluster)) +
lims(y = c(-85, -30)) +
scale_fill_brewer(palette = 'Dark2') +
facet_wrap( ~ cluster, ncol = 2) +
labs(title = 'cluster spatial distribution')
}
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
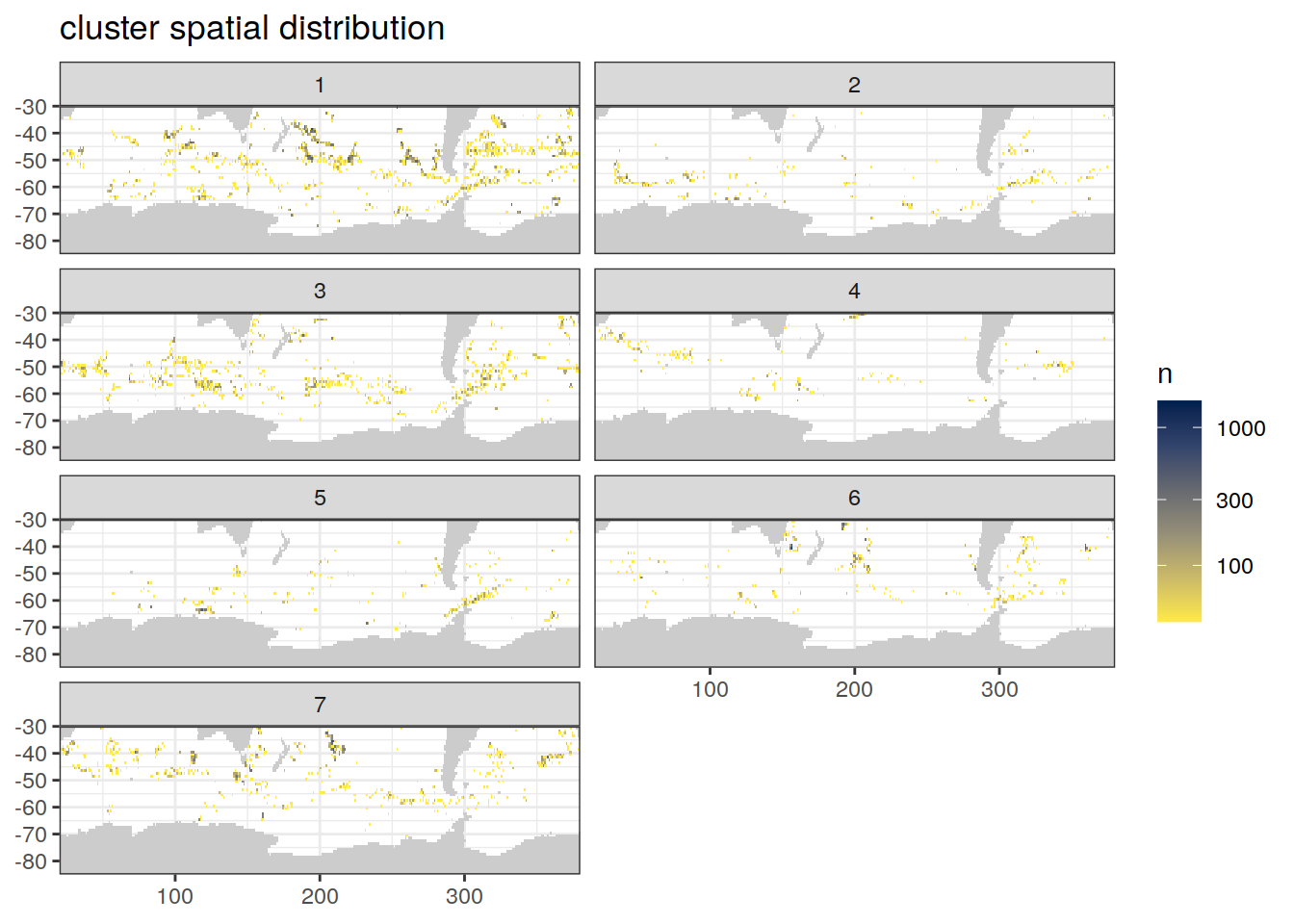
count of measurements for each cluster on separate maps, spatial analysis
if (opt_analysis_type == 2) {
# create figure
map +
geom_tile(data = pH_anomaly_cluster %>%
count(lat, lon, cluster),
aes(x = lon,
y = lat,
fill = n)) +
lims(y = c(-85, -30)) +
scale_fill_viridis_c(option = "cividis", direction = -1, trans = "log10") +
facet_wrap( ~ cluster, ncol = 2) +
labs(title = 'cluster spatial distribution')
}
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
Cluster spatial year
location of each cluster on map, spatial analysis by year
if (opt_analysis_type == 2) {
# create figure
pH_anomaly_cluster %>%
group_split(year) %>%
map(
~ map +
geom_tile(data = .x,
aes(
x = lon,
y = lat,
fill = cluster
)) +
#lims(y = c(-85, -30))+
scale_fill_brewer(palette = 'Dark2') +
facet_wrap( ~ cluster, ncol = 2) +
labs(title = paste0(
'cluster spatial distribution ', unique(.x$year)
))
)
}[[1]]
| Version | Author | Date |
|---|---|---|
| 80c16c2 | ds2n19 | 2023-11-15 |
[[2]]
| Version | Author | Date |
|---|---|---|
| 80c16c2 | ds2n19 | 2023-11-15 |
[[3]]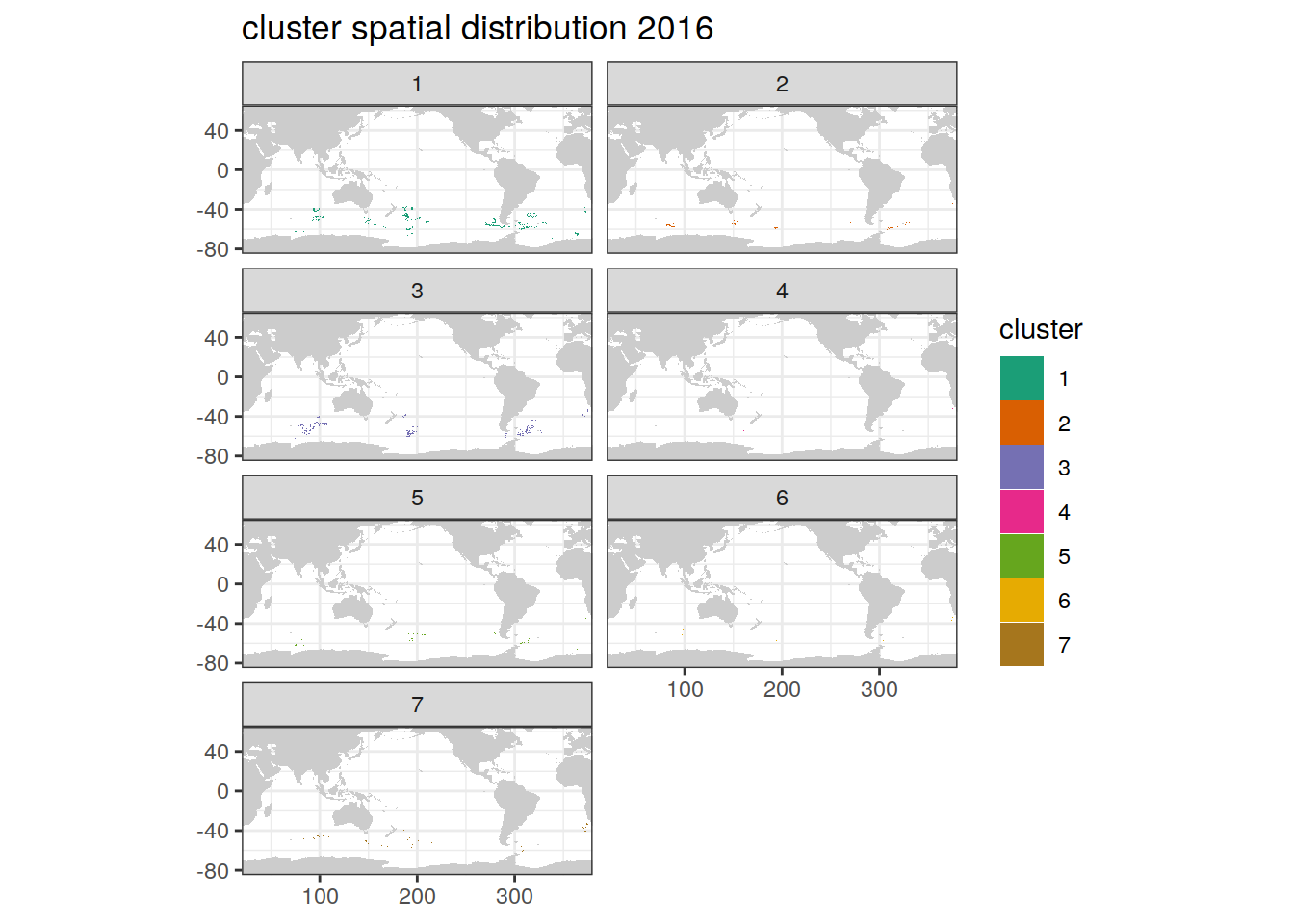
| Version | Author | Date |
|---|---|---|
| 80c16c2 | ds2n19 | 2023-11-15 |
[[4]]
| Version | Author | Date |
|---|---|---|
| 80c16c2 | ds2n19 | 2023-11-15 |
[[5]]
| Version | Author | Date |
|---|---|---|
| 80c16c2 | ds2n19 | 2023-11-15 |
[[6]]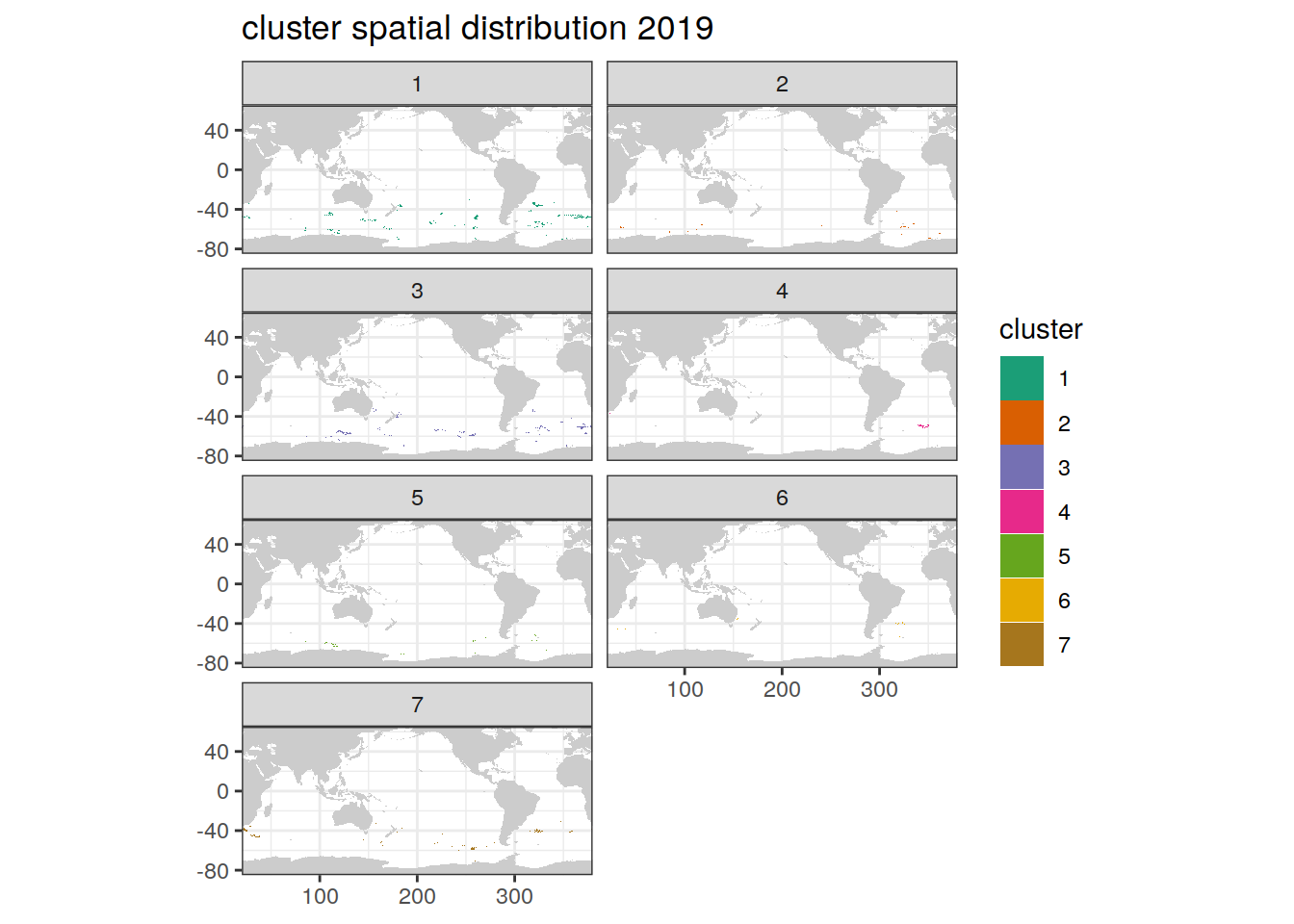
| Version | Author | Date |
|---|---|---|
| 80c16c2 | ds2n19 | 2023-11-15 |
[[7]]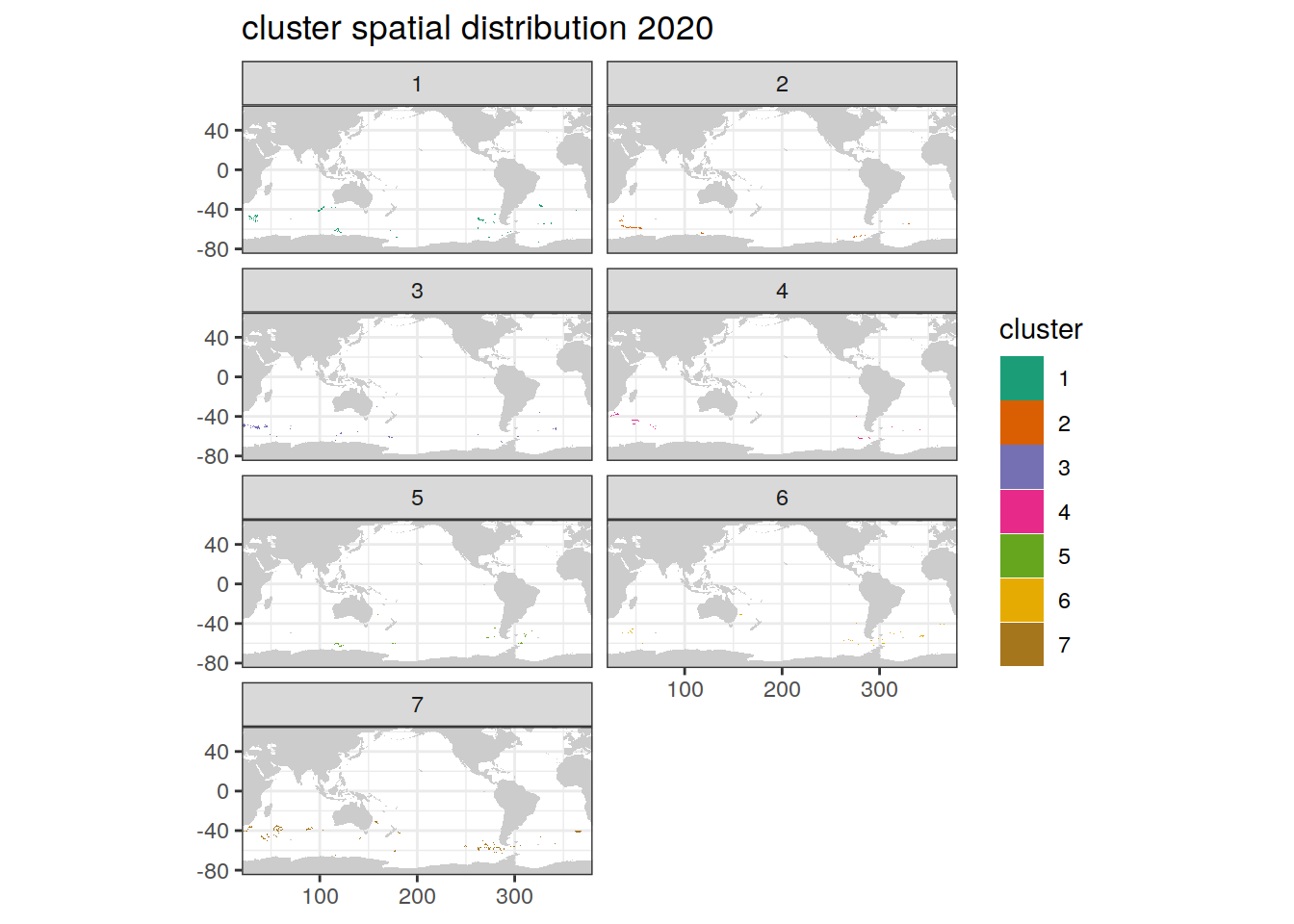
| Version | Author | Date |
|---|---|---|
| 80c16c2 | ds2n19 | 2023-11-15 |
[[8]]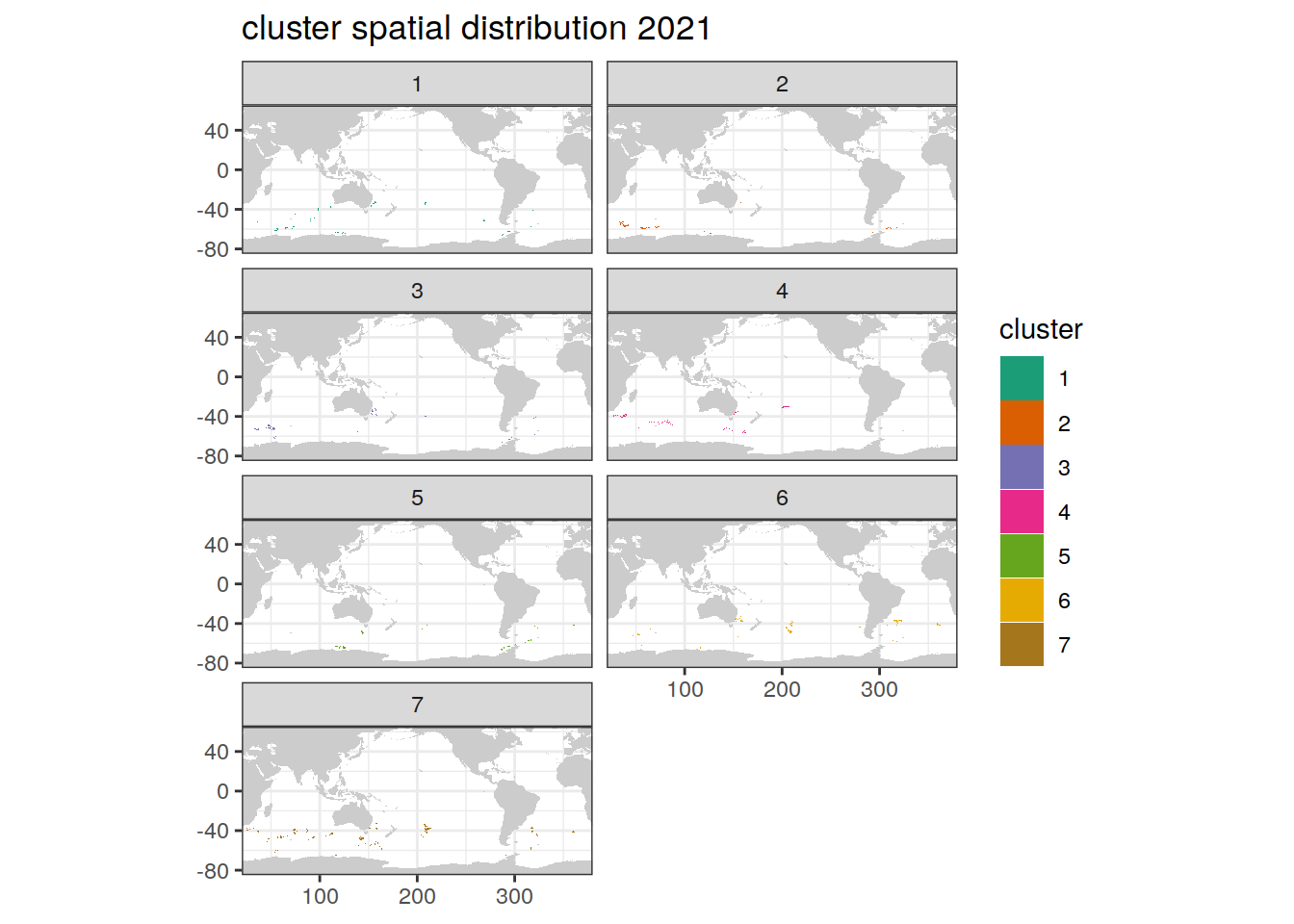
| Version | Author | Date |
|---|---|---|
| 80c16c2 | ds2n19 | 2023-11-15 |
[[9]]
| Version | Author | Date |
|---|---|---|
| 80c16c2 | ds2n19 | 2023-11-15 |
[[10]]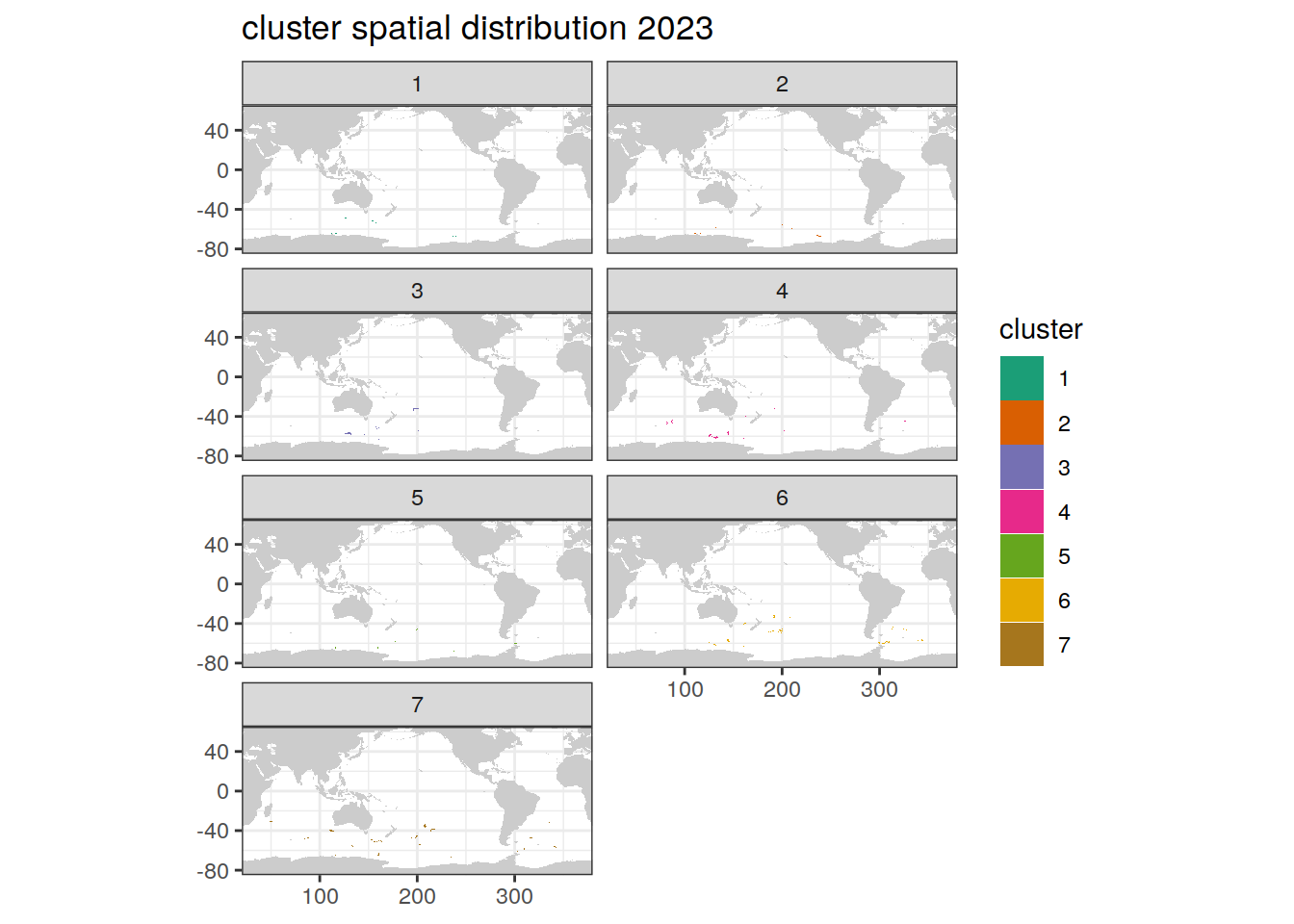
| Version | Author | Date |
|---|---|---|
| 80c16c2 | ds2n19 | 2023-11-15 |
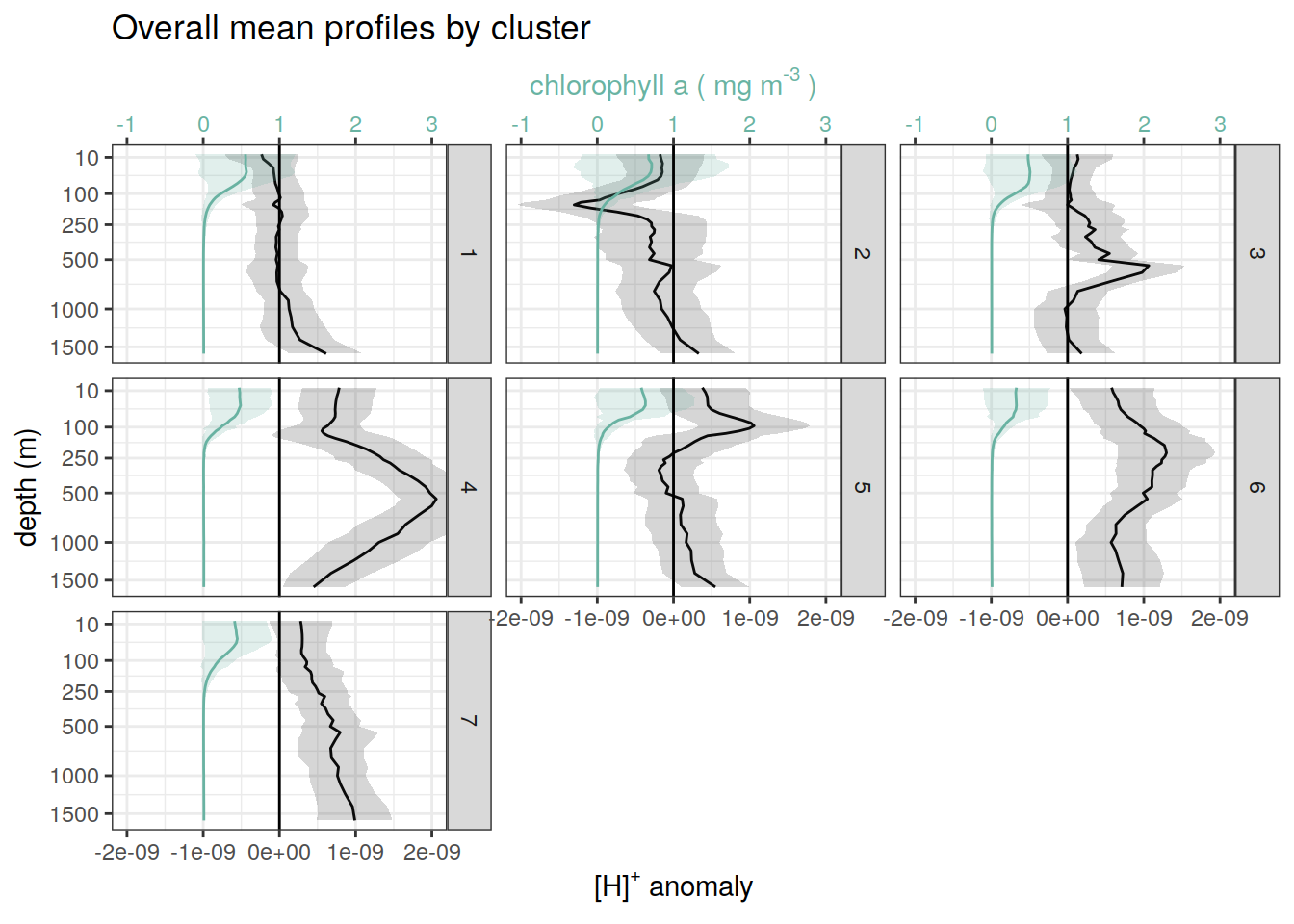
pH and Chl-a
for each cluster identified by pH cluster analysis show alongside chl-a
if (opt_analysis_type == 2) {
# Read chl-a data and link to cluster ID
chla_bgc_va <- read_rds(file = paste0(path_argo_preprocessed, "/chla_bgc_va.rds"))
chla_cluster <- right_join(chla_bgc_va, profile_id)
# summarise by cluster
chla_cluster_mean <- chla_cluster %>%
group_by(cluster, depth) %>%
summarise(chla_mean = mean(chla, na.rm = TRUE),
chla_sd = sd(chla, na.rm = TRUE)) %>%
ungroup()
# join pH anomaly with chl-a
cluster_ph_chla <- full_join(anomaly_cluster_mean, chla_cluster_mean)
# create figures
cluster_ph_chla %>%
ggplot()+
geom_path(aes(x = anomaly_mean,
y = depth))+
geom_ribbon(aes(xmax = anomaly_mean + anomaly_sd,
xmin = anomaly_mean - anomaly_sd,
y = depth),
alpha = 0.2)+
geom_vline(xintercept = 0)+
scale_y_continuous(trans = trans_reverser("sqrt"),
breaks = c(10, 100, 250, 500, seq(1000, 5000, 500)))+
facet_wrap(~cluster)+
coord_cartesian(xlim = opt_xlim)+
scale_x_continuous(breaks = opt_xbreaks)+
labs(title = paste0('Overall mean anomaly profiles by cluster'), x = opt_measure_label, y = 'depth (m)')
cluster_ph_chla %>%
ggplot()+
geom_path(aes(x = chla_mean,
y = depth))+
geom_ribbon(aes(xmax = chla_mean + chla_sd,
xmin = chla_mean - chla_sd,
y = depth),
alpha = 0.2)+
scale_y_continuous(trans = trans_reverser("sqrt"),
breaks = c(10, 100, 250, 500, seq(1000, 5000, 500)))+
facet_wrap(~cluster)+
coord_cartesian(xlim = opt_chla_xlim)+
scale_x_continuous(breaks = opt_chla_xbreaks)+
labs(title = paste0('Overall mean profiles by cluster'), x = opt_chla_measure_label, y = 'depth (m)')
if (opt_measure == 1){
chla_to_ph_factor <- 25
chla_to_ph_offset <- 1
} else {
chla_to_ph_factor <- 1e9
chla_to_ph_offset <- 1
}
chla_color <- "#69b3a2"
cluster_ph_chla %>%
ggplot() +
geom_path(aes(x = anomaly_mean,
y = depth)) +
geom_ribbon(aes(
xmax = anomaly_mean + anomaly_sd,
xmin = anomaly_mean - anomaly_sd,
y = depth
),
alpha = 0.2) +
geom_path(aes(
x = (chla_mean - chla_to_ph_offset) / chla_to_ph_factor,
y = depth
), color = chla_color) +
geom_ribbon(
aes(
xmax = (chla_mean + chla_sd - chla_to_ph_offset) / chla_to_ph_factor,
xmin = (chla_mean - chla_sd - chla_to_ph_offset) / chla_to_ph_factor,
y = depth
),
fill = chla_color,
alpha = 0.2
) +
geom_vline(xintercept = 0) +
scale_y_continuous(trans = trans_reverser("sqrt"),
breaks = c(10, 100, 250, 500, seq(1000, 5000, 500))) +
facet_wrap( ~ cluster,
strip.position = "right") +
coord_cartesian(xlim = opt_xlim) +
scale_x_continuous(
# First axis
name = opt_measure_label,
breaks = opt_xbreaks,
# Second axis
sec.axis = sec_axis(
trans = ~ . * chla_to_ph_factor + chla_to_ph_offset,
name = opt_chla_measure_label
)
) +
labs(
title = paste0('Overall mean profiles by cluster'),
x = opt_measure_label,
y = 'depth (m)'
) +
theme(axis.title.x.top = element_text(color = chla_color),
axis.text.x.top = element_text(color = chla_color))
}
| Version | Author | Date |
|---|---|---|
| 80c16c2 | ds2n19 | 2023-11-15 |
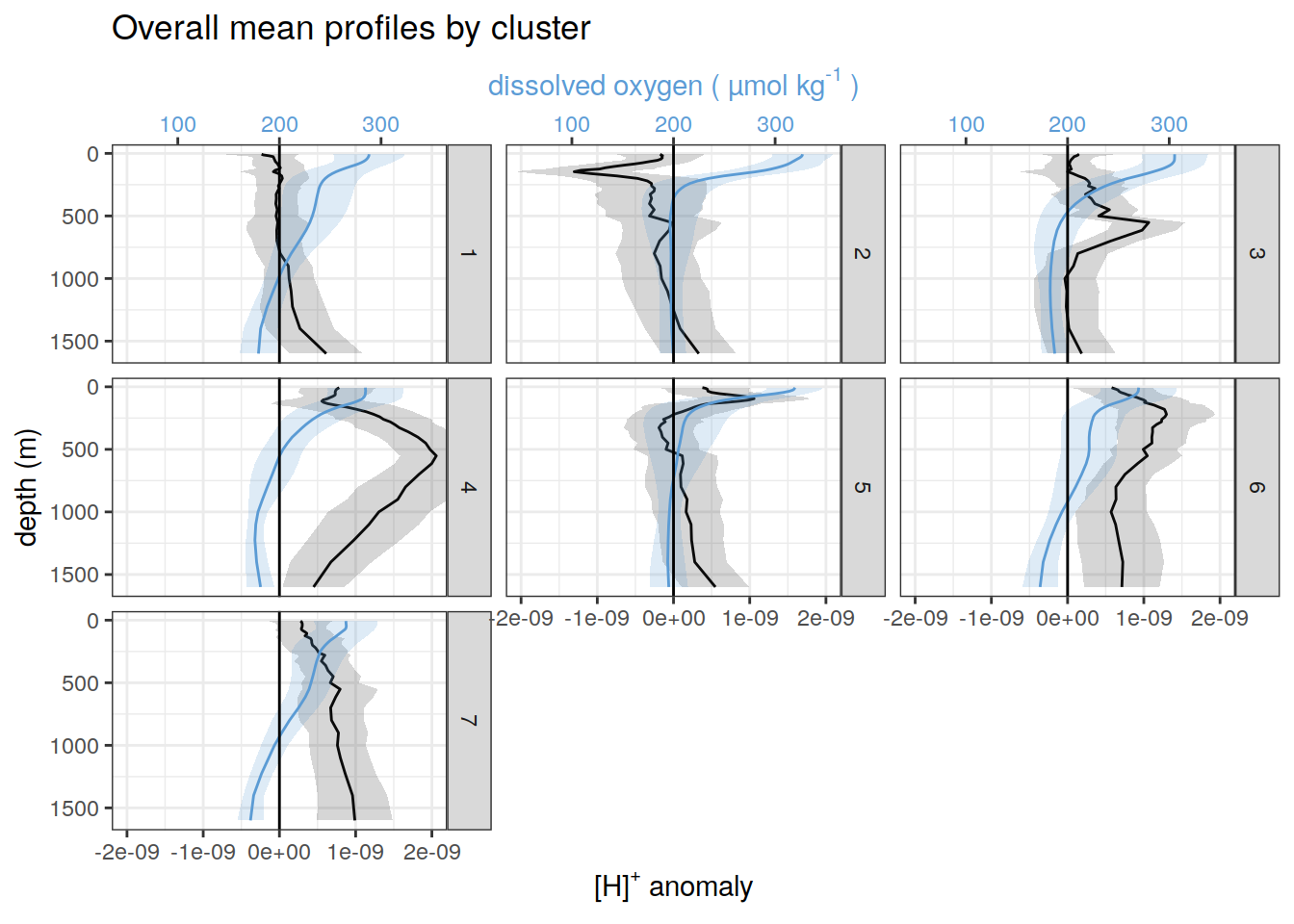
pH and oxygen
for each cluster identified by pH cluster analysis show alongside oxygen
if (opt_analysis_type == 2) {
# Read chl-a data and link to cluster ID
doxy_bgc_va <- read_rds(file = paste0(path_argo_preprocessed, "/doxy_bgc_va.rds"))
doxy_cluster <- right_join(doxy_bgc_va, profile_id)
# summarise by cluster
doxy_cluster_mean <- doxy_cluster %>%
group_by(cluster, depth) %>%
summarise(doxy_mean = mean(doxy, na.rm = TRUE),
doxy_sd = sd(doxy, na.rm = TRUE)) %>%
ungroup()
# join pH anomaly with chl-a
cluster_ph_doxy <- full_join(anomaly_cluster_mean, doxy_cluster_mean)
# create figure
cluster_ph_doxy %>%
ggplot()+
geom_path(aes(x = anomaly_mean,
y = depth))+
geom_ribbon(aes(xmax = anomaly_mean + anomaly_sd,
xmin = anomaly_mean - anomaly_sd,
y = depth),
alpha = 0.2)+
geom_vline(xintercept = 0)+
scale_y_reverse()+
facet_wrap(~cluster)+
coord_cartesian(xlim = opt_xlim)+
scale_x_continuous(breaks = opt_xbreaks)+
labs(title = paste0('Overall mean anomaly profiles by cluster'), x = opt_measure_label, y = 'depth (m)')
cluster_ph_doxy %>%
ggplot()+
geom_path(aes(x = doxy_mean,
y = depth))+
geom_ribbon(aes(xmax = doxy_mean + doxy_sd,
xmin = doxy_mean - doxy_sd,
y = depth),
alpha = 0.2)+
scale_y_reverse()+
facet_wrap(~cluster)+
coord_cartesian(xlim = opt_doxy_xlim)+
scale_x_continuous(breaks = opt_doxy_xbreaks)+
labs(title = paste0('Overall mean profiles by cluster'), x = opt_doxy_measure_label, y = 'depth (m)')
if (opt_measure == 1){
doxy_to_ph_factor <- 1875
doxy_to_ph_offset <- 200
} else {
doxy_to_ph_factor <- 75e9
doxy_to_ph_offset <- 200
}
doxy_color <- "#5B9BD5"
cluster_ph_doxy %>%
ggplot() +
geom_path(aes(x = anomaly_mean,
y = depth)) +
geom_ribbon(aes(
xmax = anomaly_mean + anomaly_sd,
xmin = anomaly_mean - anomaly_sd,
y = depth
),
alpha = 0.2) +
geom_path(aes(
x = (doxy_mean - doxy_to_ph_offset) / doxy_to_ph_factor,
y = depth
), color = doxy_color) +
geom_ribbon(
aes(
xmax = (doxy_mean + doxy_sd - doxy_to_ph_offset) / doxy_to_ph_factor,
xmin = (doxy_mean - doxy_sd - doxy_to_ph_offset) / doxy_to_ph_factor,
y = depth
),
fill = doxy_color,
alpha = 0.2
) +
geom_vline(xintercept = 0) +
scale_y_reverse() +
facet_wrap( ~ cluster,
strip.position = "right") +
coord_cartesian(xlim = opt_xlim) +
scale_x_continuous(
# First axis
name = opt_measure_label,
breaks = opt_xbreaks,
# First axis
sec.axis = sec_axis(
trans = ~ . * doxy_to_ph_factor + doxy_to_ph_offset,
name = opt_doxy_measure_label
)
) +
labs(
title = paste0('Overall mean profiles by cluster'),
x = opt_measure_label,
y = 'depth (m)'
) +
theme(axis.title.x.top = element_text(color = doxy_color),
axis.text.x.top = element_text(color = doxy_color))
}
| Version | Author | Date |
|---|---|---|
| 80c16c2 | ds2n19 | 2023-11-15 |
Cluster by surface Extreme
# Carried out for 1600m profiles
opt_profile_range = 3
extreme_type <- c('L', 'N', 'H')
num_clusters <- c(5, 6, 5)
# ---------------------------------------------------------------------------------------------
# read data
# ---------------------------------------------------------------------------------------------
# load previously created OceanSODA extreme data. date, position and nature of extreme
pH_extreme <- read_rds(file = paste0(path_argo_preprocessed, "/OceanSODA_pH_anomaly_field.rds")) %>%
select(lon, lat, date, ph_extreme)
# read data
pH_anomaly_va <- read_rds(file = paste0(path_argo_preprocessed, "/pH_anomaly_va.rds")) %>%
mutate(date = ymd(format(date, "%Y-%m-15")))
# Add the OceanSODA extreme condition
pH_anomaly_va <- left_join(pH_anomaly_va, pH_extreme)
# Select target variable
if (opt_measure == 1) {
pH_anomaly_va <- pH_anomaly_va %>%
rename(
anomaly = anomaly_pH
)
} else {
pH_anomaly_va <- pH_anomaly_va %>%
rename(
anomaly = anomaly_h_plus
)
}
for (i in 1:3) {
# ---------------------------------------------------------------------------------------------
# Preparation
# ---------------------------------------------------------------------------------------------
# select profile based on profile_range and he appropriate max depth
pH_anomaly_va_id <- pH_anomaly_va %>%
filter(profile_range == opt_profile_range & depth <= opt_max_depth[opt_profile_range] & ph_extreme == extreme_type[i])
# Simplified table ready to pivot
pH_anomaly_va_id <- pH_anomaly_va_id %>%
select(file_id,
depth,
anomaly,
year,
month,
lat,
lon)
# wide table with each depth becoming a column
pH_anomaly_va_wide <- pH_anomaly_va_id %>%
select(file_id, depth, anomaly) %>%
pivot_wider(names_from = depth, values_from = anomaly)
# Drop any rows with missing values N/A caused by gaps in climatology data
pH_anomaly_va_wide <- pH_anomaly_va_wide %>%
drop_na()
# Table for cluster analysis
points <- pH_anomaly_va_wide %>%
column_to_rownames(var = "file_id")
# ---------------------------------------------------------------------------------------------
# cluster analysis
# ---------------------------------------------------------------------------------------------
set.seed(1)
kclusts <-
tibble(k = num_clusters[i]) %>%
mutate(
kclust = map(k, ~ kmeans(points, .x, iter.max = opt_max_iterations, nstart = opt_n_start)),
tidied = map(kclust, tidy),
glanced = map(kclust, glance),
augmented = map(kclust, augment, points)
)
profile_id <-
kclusts %>%
unnest(cols = c(augmented)) %>%
select(file_id = .rownames,
cluster = .cluster) %>%
mutate(file_id = as.numeric(file_id),
cluster = as.character(cluster))
# Add cluster to pH_anomaly_va
pH_anomaly_cluster <- full_join(pH_anomaly_va, profile_id)
# Plot cluster mean
pH_anomaly_cluster <- pH_anomaly_cluster %>%
filter(!is.na(cluster))
# Plot cluster mean
anomaly_cluster_mean <- pH_anomaly_cluster %>%
group_by(cluster, depth) %>%
summarise(
count_cluster = n(),
anomaly_mean = mean(anomaly, na.rm = TRUE),
anomaly_sd = sd(anomaly, na.rm = TRUE)
) %>%
ungroup()
anomaly_cluster_mean_year <- pH_anomaly_cluster %>%
group_by(cluster, depth, year) %>%
summarise(
count_cluster = n(),
anomaly_mean = mean(anomaly, na.rm = TRUE),
anomaly_sd = sd(anomaly, na.rm = TRUE)
) %>%
ungroup()
anomaly_year_mean <- pH_anomaly_cluster %>%
group_by(cluster, year) %>%
summarise(
count_cluster = n(),
anomaly_mean = mean(anomaly, na.rm = TRUE),
anomaly_sd = sd(anomaly, na.rm = TRUE)
) %>%
ungroup()
anomaly_year_mean <- anomaly_year_mean %>%
group_by(year) %>%
summarise(anomaly_mean = mean(anomaly_mean, na.rm = TRUE)) %>%
ungroup ()
if (i == 1) {
anomaly_cluster_mean_ext <- anomaly_cluster_mean %>% mutate(pH_extreme_order = i, pH_extreme = extreme_type[i])
anomaly_cluster_mean_year_ext <- anomaly_cluster_mean_year %>% mutate(pH_extreme_order = i, pH_extreme = extreme_type[i])
anomaly_year_mean_ext <- anomaly_year_mean %>% mutate(pH_extreme_order = i, pH_extreme = extreme_type[i])
pH_anomaly_cluster_ext <- pH_anomaly_cluster %>% mutate(pH_extreme_order = i, pH_extreme = extreme_type[i])
} else {
anomaly_cluster_mean_ext <- rbind(anomaly_cluster_mean_ext, anomaly_cluster_mean %>% mutate(pH_extreme_order = i, pH_extreme = extreme_type[i]))
anomaly_cluster_mean_year_ext <- rbind(anomaly_cluster_mean_year_ext, anomaly_cluster_mean_year %>% mutate(pH_extreme_order = i, pH_extreme = extreme_type[i]))
anomaly_year_mean_ext <- rbind(anomaly_year_mean_ext, anomaly_year_mean %>% mutate(pH_extreme_order = i, pH_extreme = extreme_type[i]))
pH_anomaly_cluster_ext <- rbind(pH_anomaly_cluster_ext, pH_anomaly_cluster_ext <- pH_anomaly_cluster %>% mutate(pH_extreme_order = i, pH_extreme = extreme_type[i]))
}
}Cluster mean
# create figure of mean clusters
anomaly_cluster_mean_ext %>%
group_split(pH_extreme_order) %>%
map(
~ ggplot(data = .x, ) +
geom_path(aes(x = anomaly_mean,
y = depth)) +
geom_ribbon(
aes(
xmax = anomaly_mean + anomaly_sd,
xmin = anomaly_mean - anomaly_sd,
y = depth
),
alpha = 0.2
) +
geom_vline(xintercept = 0) +
scale_y_reverse() +
facet_wrap(~ cluster) +
coord_cartesian(xlim = opt_xlim) +
scale_x_continuous(breaks = opt_xbreaks) +
labs(
title = paste0(
'Overall mean anomaly profiles by cluster \n',
'Surface extreme: ',
unique(.x$pH_extreme),
' pH'
),
x = opt_measure_label,
y = 'depth (m)'
)
)[[1]]
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
[[2]]
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
[[3]]
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
Clusters mean by year
# cluster means by year
anomaly_cluster_mean_year_ext %>%
mutate(year = as.factor(year)) %>%
group_split(pH_extreme_order) %>%
map(
~ ggplot(data = .x,) +
geom_path(aes(
x = anomaly_mean,
y = depth,
col = year
)) +
geom_vline(xintercept = 0) +
scale_y_reverse() +
facet_wrap(~ cluster) +
coord_cartesian(xlim = opt_xlim) +
scale_x_continuous(breaks = opt_xbreaks) +
scale_color_viridis_d() +
labs(
title = paste0(
'Overall mean anomaly profiles by cluster by year \n',
'Surface extreme: ',
unique(.x$pH_extreme),
' pH'
),
x = opt_measure_label,
y = 'depth (m)'
)
)[[1]]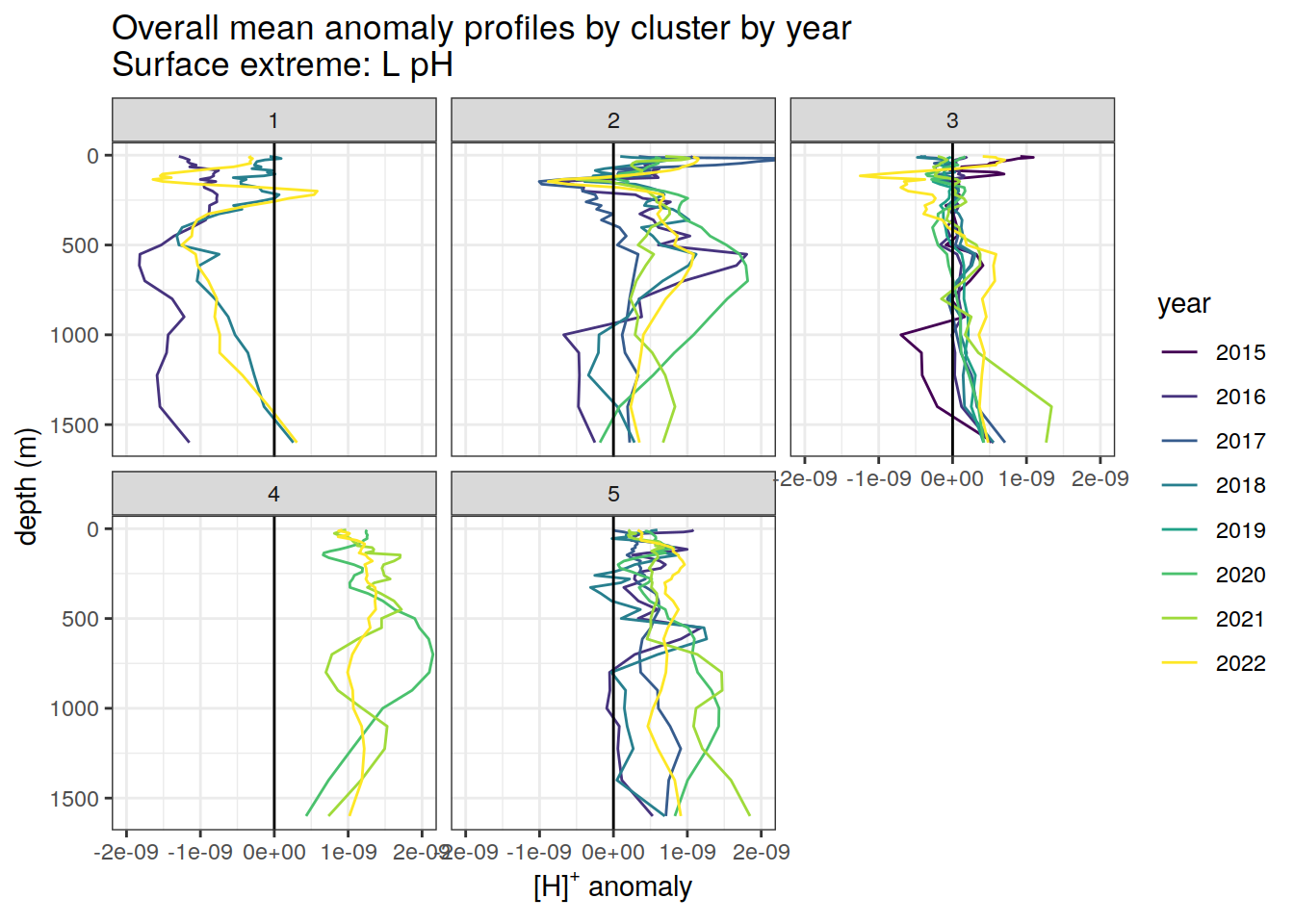
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
[[2]]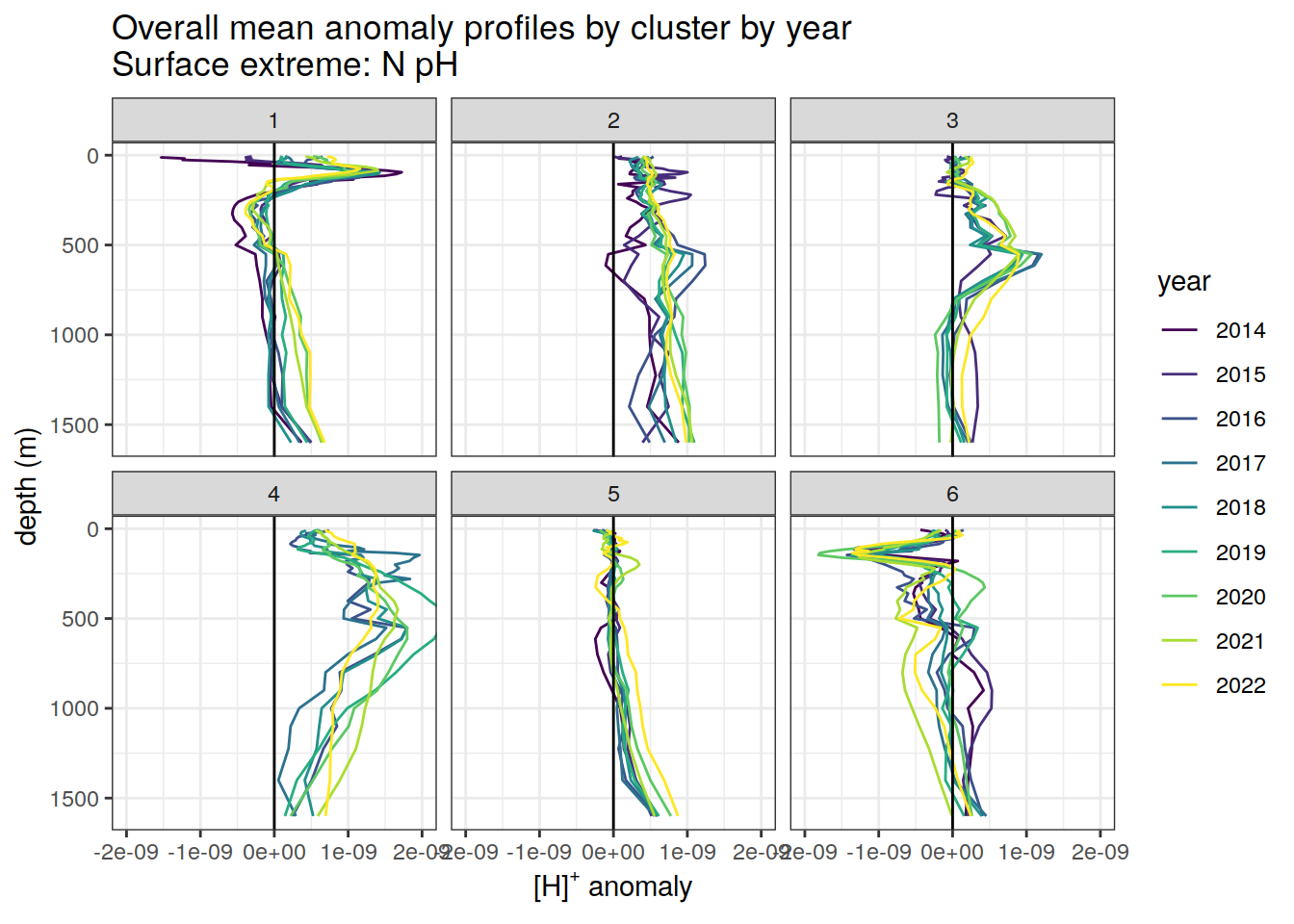
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
[[3]]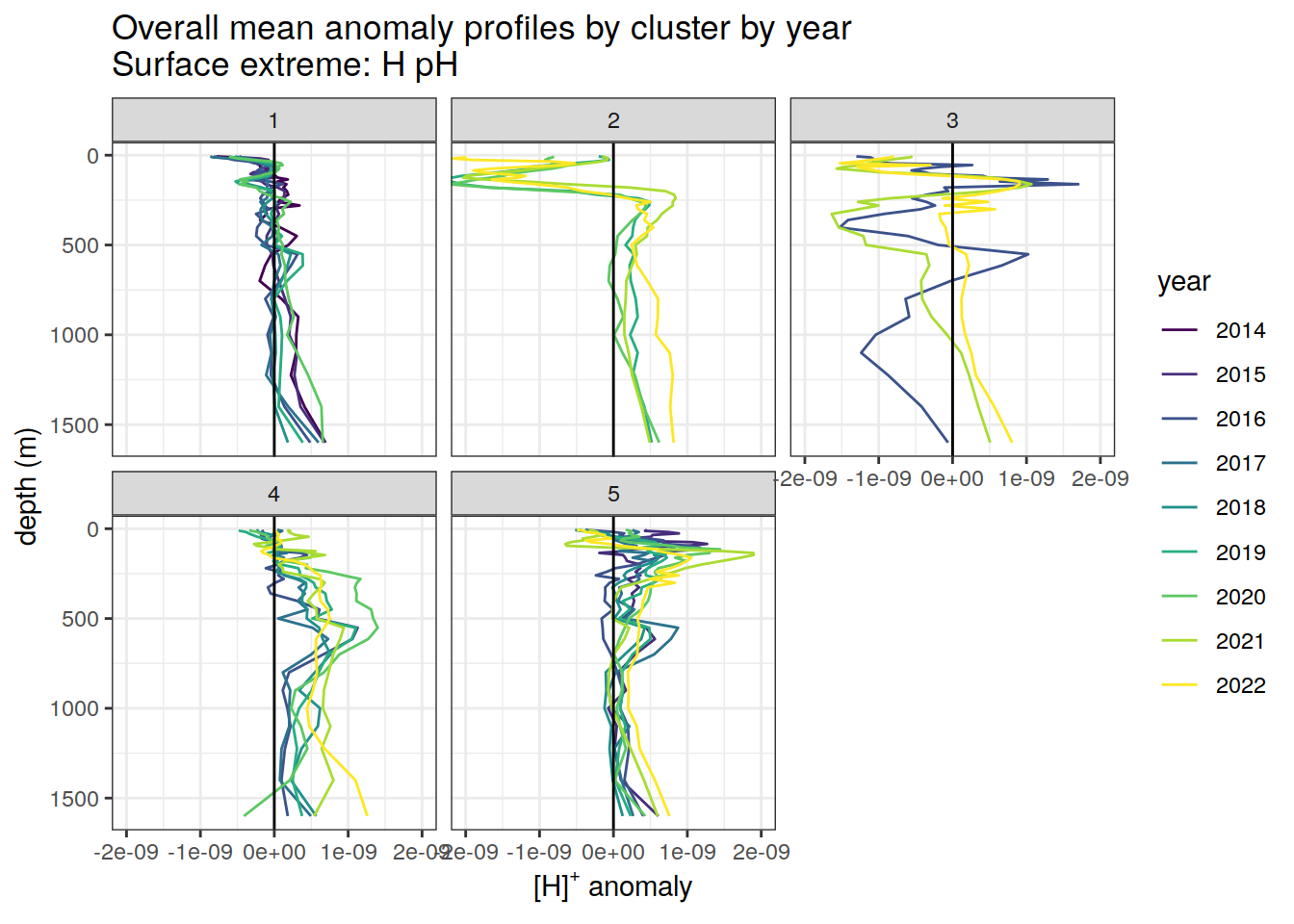
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
Cluster by year
count of each cluster by year
# Determine profile count by extreme and cluster and year
# Count the measurements
cluster_by_year <- pH_anomaly_cluster_ext %>%
count(file_id, pH_extreme_order, pH_extreme, cluster, year,
name = "count_cluster")
# Convert to profiles
cluster_by_year <- cluster_by_year %>%
count(pH_extreme_order, pH_extreme, cluster, year,
name = "count_cluster")
year_min <- min(cluster_by_year$year)
year_max <- max(cluster_by_year$year)
# create figure
cluster_by_year %>%
group_split(pH_extreme_order) %>%
map(
~ ggplot(
data = .x,
aes(
x = year,
y = count_cluster,
col = cluster,
group = cluster
)
) +
geom_point() +
geom_line() +
scale_x_continuous(breaks = seq(year_min, year_max, 2)) +
scale_color_brewer(palette = 'Dark2') +
labs(
x = 'year',
y = 'number of profiles',
col = 'cluster',
title = paste0(
'Count of profiles by year and cluster \n',
'Surface extreme: ',
unique(.x$pH_extreme),
' pH'
)
)
)[[1]]
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
[[2]]
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
[[3]]
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
Cluster by month
count of each cluster by month of year
# Determine profile count by cluster and year
# Count the measurements
cluster_by_year <- pH_anomaly_cluster_ext %>%
count(file_id, pH_extreme_order, pH_extreme, cluster, month,
name = "count_cluster")
# Convert to profiles
cluster_by_year <- cluster_by_year %>%
count(pH_extreme_order, pH_extreme, cluster, month,
name = "count_cluster")
# create figure
cluster_by_year %>%
group_split(pH_extreme_order) %>%
map(
~ ggplot(
data = .x,
aes(
x = month,
y = count_cluster,
col = cluster,
group = cluster
)
) +
geom_point() +
geom_line() +
scale_x_continuous(breaks = seq(1, 12, 2)) +
scale_color_brewer(palette = 'Dark2') +
labs(
x = 'month',
y = 'number of profiles',
col = 'cluster',
title = paste0(
'Count of profiles by month and cluster \n',
'Surface extreme: ',
unique(.x$pH_extreme),
' pH'
)
)
)[[1]]
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
[[2]]
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
[[3]]
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
Cluster spatial
location of each cluster on map, spatial analysis
# create figure combined
pH_anomaly_cluster_ext %>%
group_split(pH_extreme_order) %>%
map(
~ map +
geom_tile(data = .x,
aes(
x = lon,
y = lat,
fill = cluster
)) +
lims(y = c(-85,-30)) +
scale_fill_brewer(palette = 'Dark2') +
labs(
title = paste0(
'cluster spatial distribution \n',
'Surface extreme: ',
unique(.x$pH_extreme),
' pH'
)
)
)[[1]]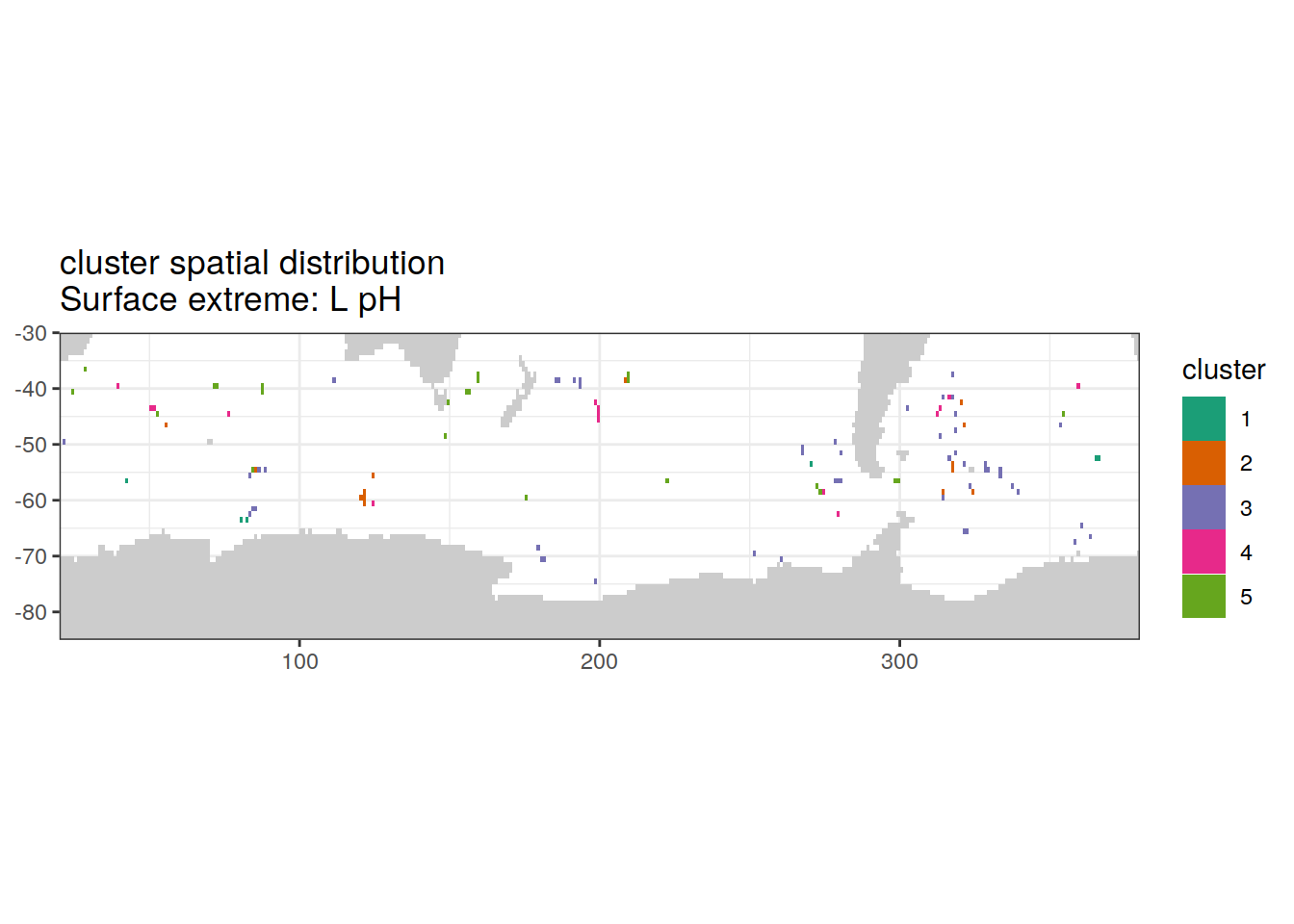
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
[[2]]
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
[[3]]
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
Spatial by cluster
location of each cluster on map, spatial analysis
# create figure by cluster
pH_anomaly_cluster_ext %>%
group_split(pH_extreme_order) %>%
map(
~ map +
geom_tile(data = .x,
aes(
x = lon,
y = lat,
fill = cluster
)) +
lims(y = c(-85,-30)) +
scale_fill_brewer(palette = 'Dark2') +
facet_wrap( ~ cluster, ncol = 2) +
labs(
title = paste0(
'cluster spatial distribution \n',
'Surface extreme: ',
unique(.x$pH_extreme),
' pH'
)
)
)[[1]]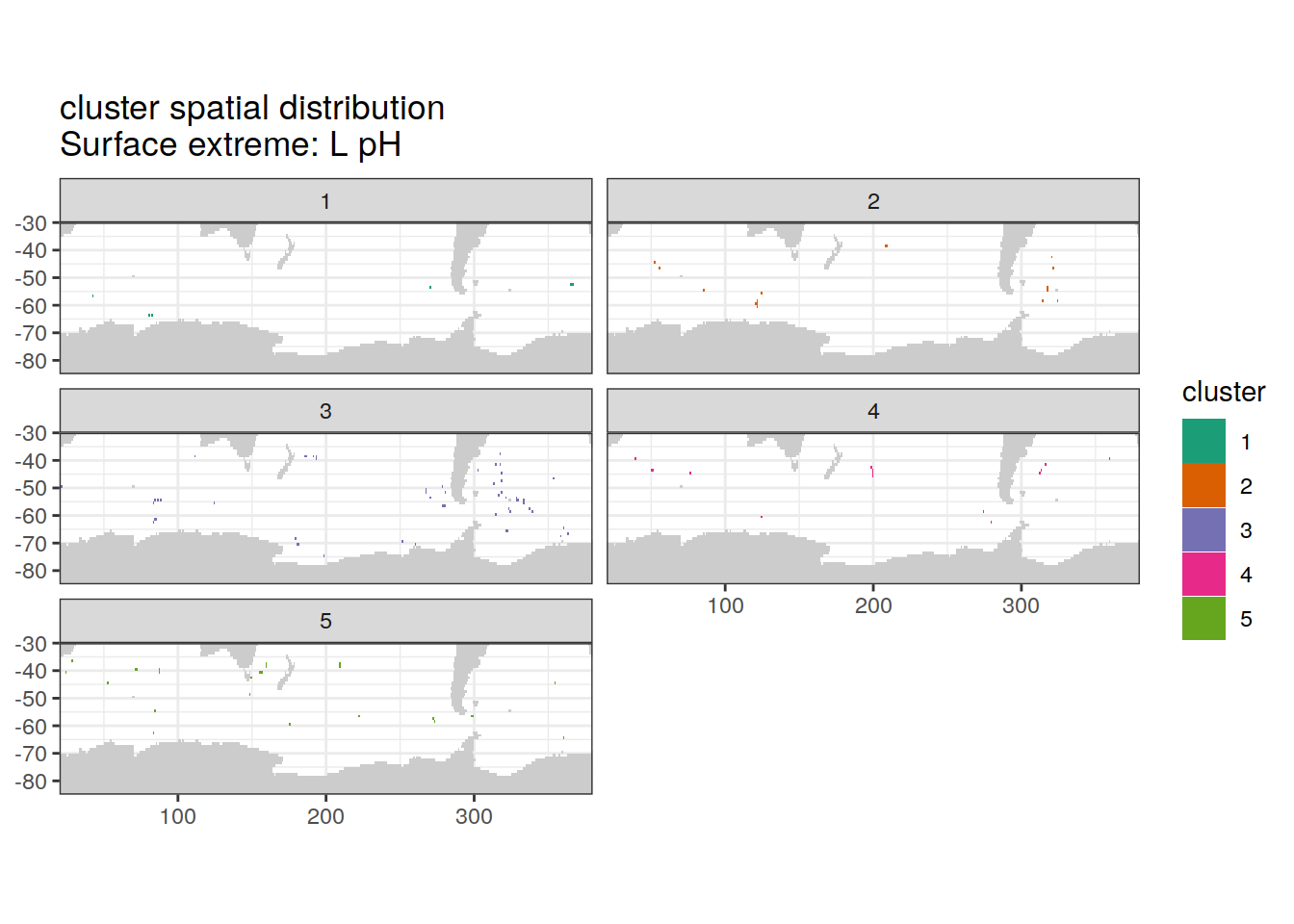
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
[[2]]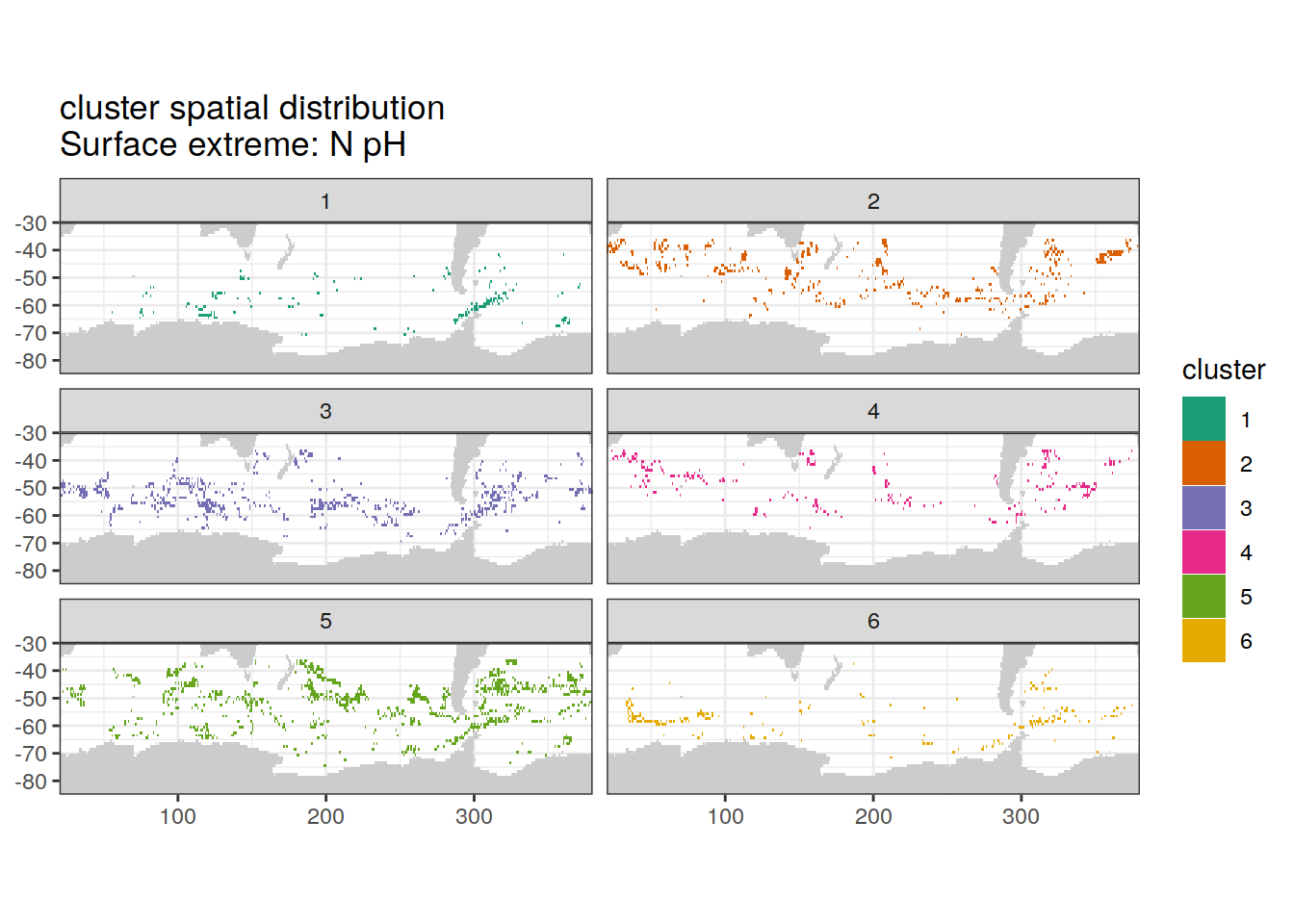
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
[[3]]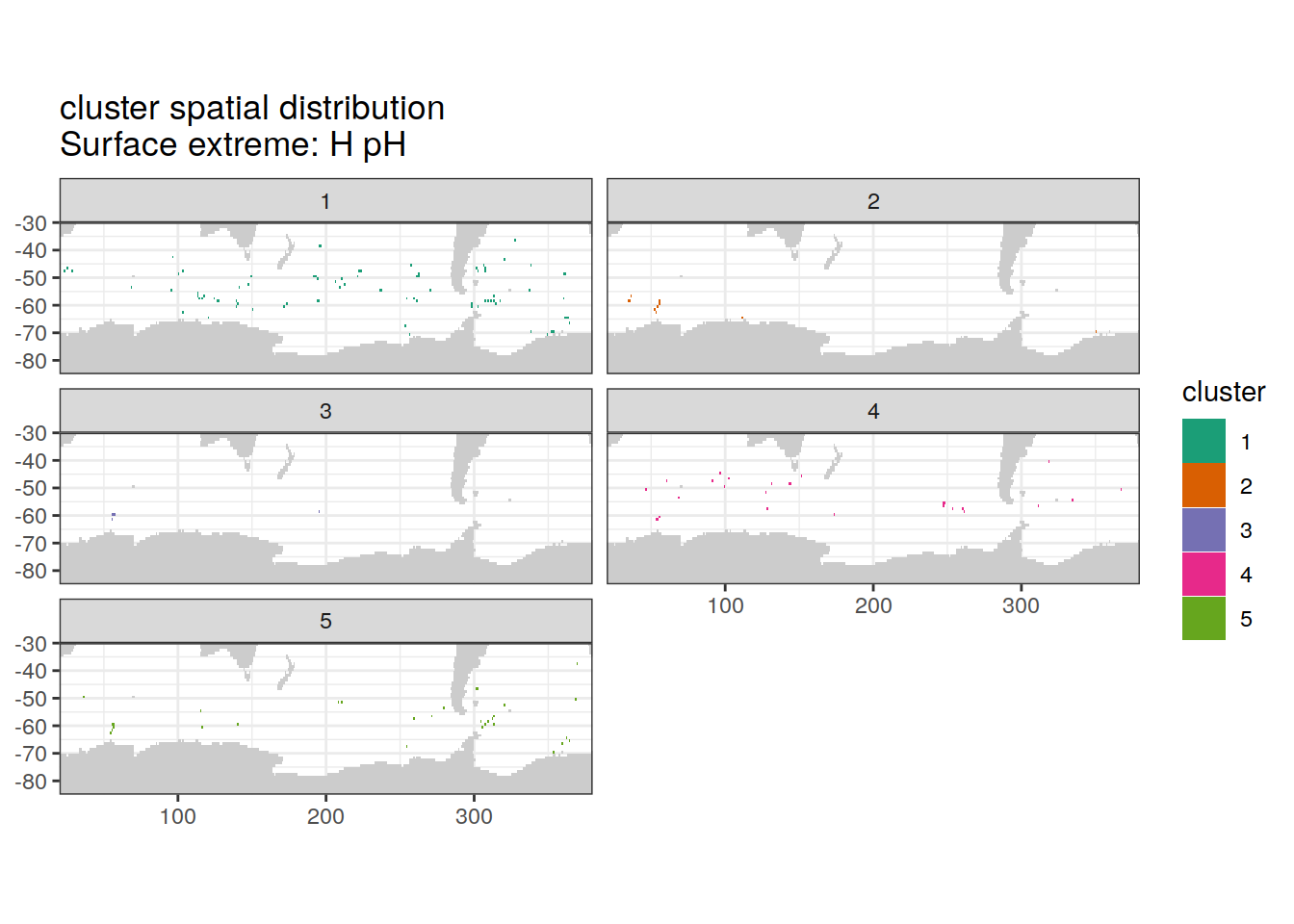
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
Spatial count
location of each cluster on map, spatial analysis
# create figure
pH_anomaly_cluster_ext %>%
group_split(pH_extreme_order) %>%
map(
~ map +
geom_tile(data = .x %>%
count(lat, lon, cluster),
aes(
x = lon,
y = lat,
fill = n
)) +
lims(y = c(-85,-30)) +
scale_fill_viridis_c(
option = "cividis",
direction = -1,
trans = "log10"
) +
facet_wrap(~ cluster, ncol = 2) +
labs(
title = paste0(
'cluster spatial distribution \n',
'Surface extreme: ',
unique(.x$pH_extreme),
' pH'
)
)
)[[1]]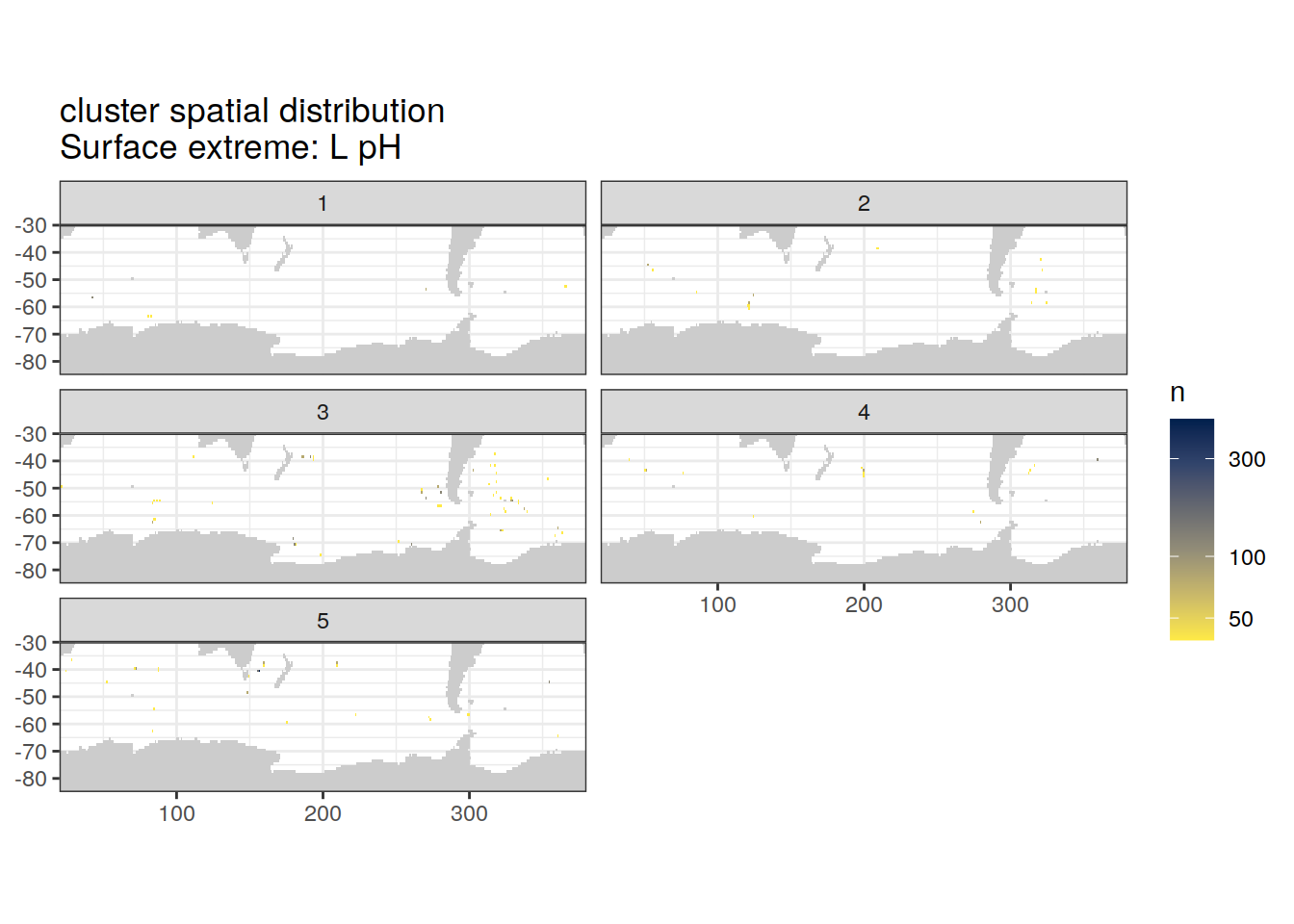
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
[[2]]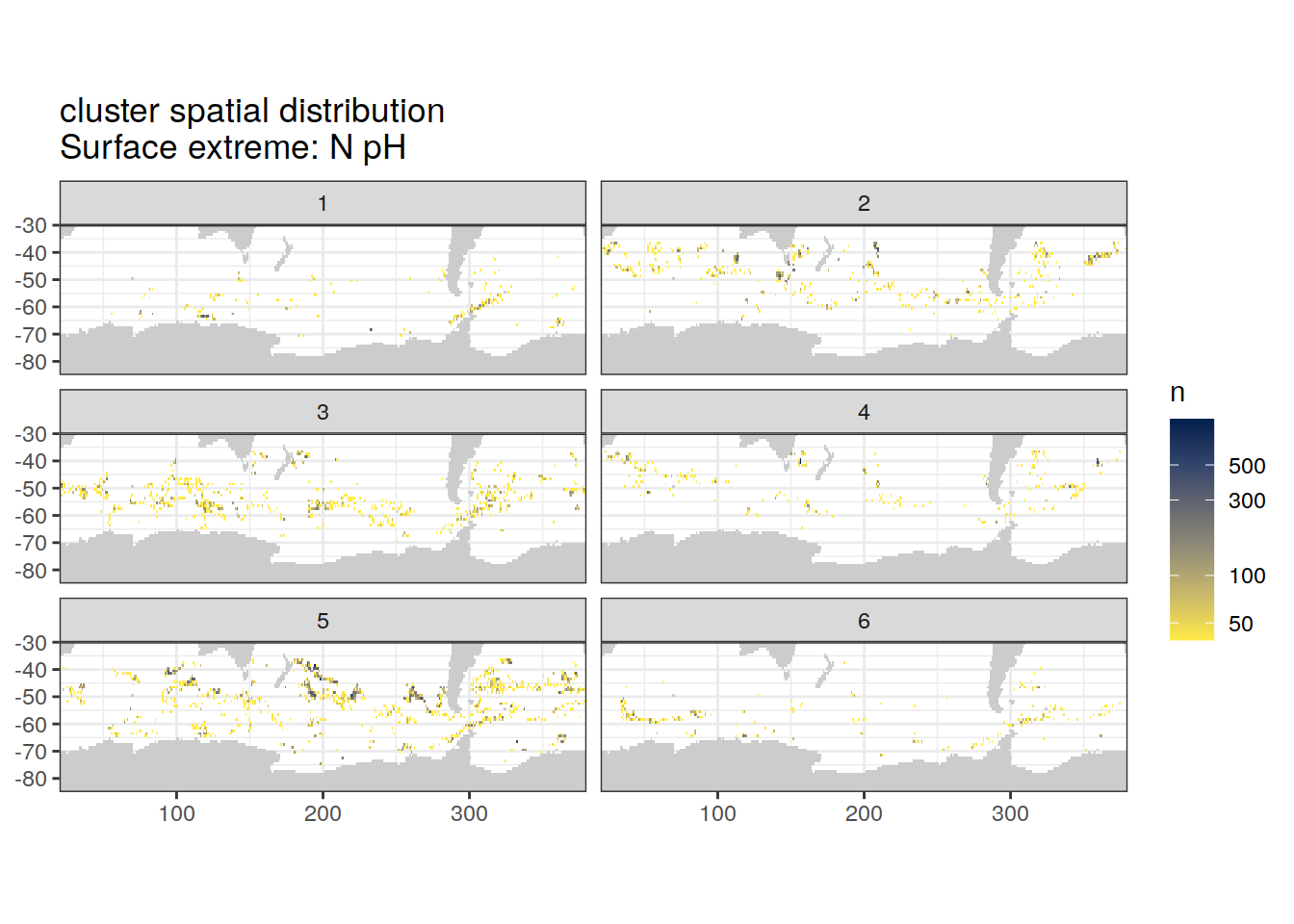
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
[[3]]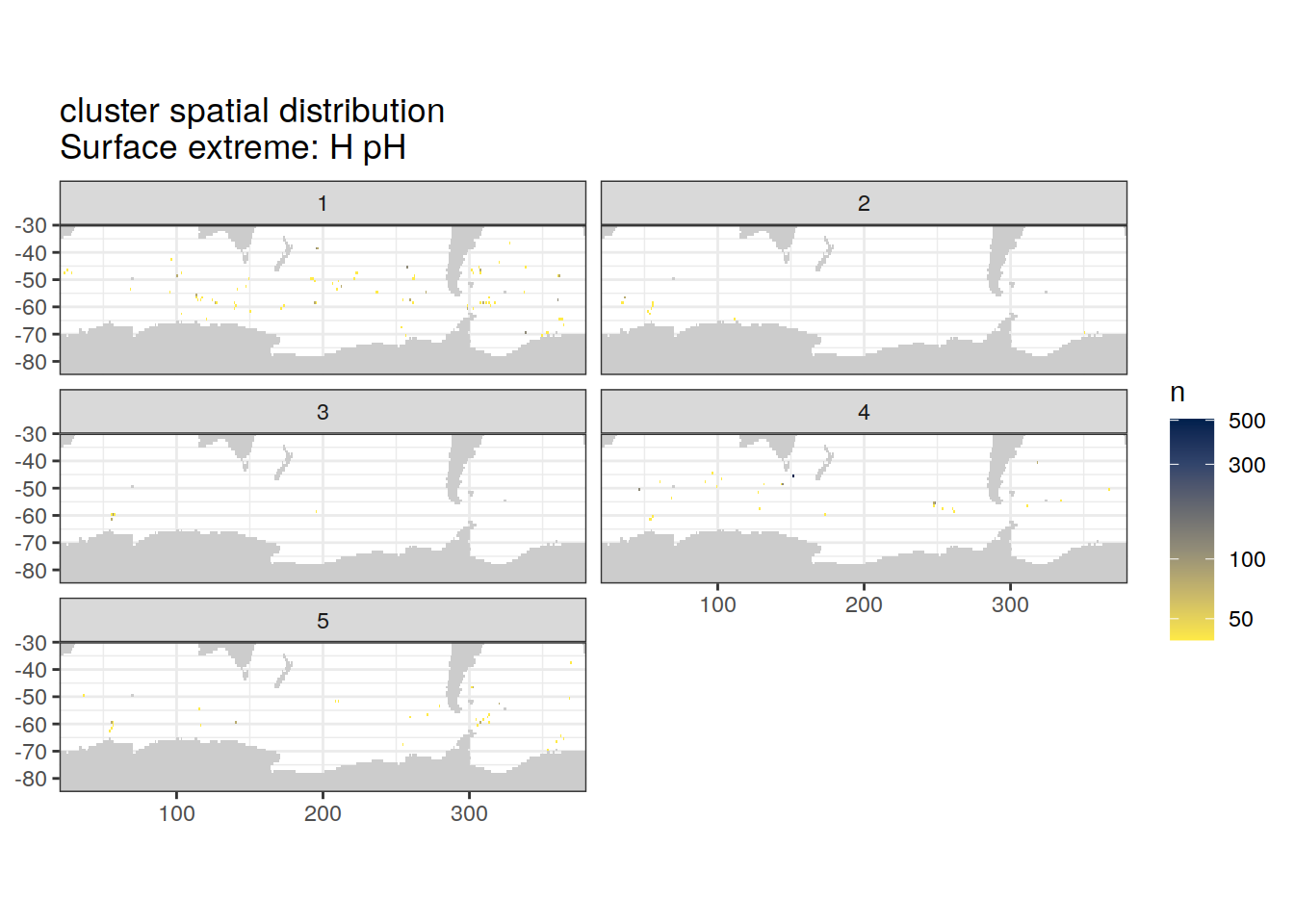
| Version | Author | Date |
|---|---|---|
| efa9686 | ds2n19 | 2023-11-24 |
sessionInfo()R version 4.2.2 (2022-10-31)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: openSUSE Leap 15.5
Matrix products: default
BLAS: /usr/local/R-4.2.2/lib64/R/lib/libRblas.so
LAPACK: /usr/local/R-4.2.2/lib64/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] yardstick_1.2.0 workflowsets_1.0.1 workflows_1.1.3 tune_1.1.2
[5] rsample_1.2.0 recipes_1.0.8 parsnip_1.1.1 modeldata_1.2.0
[9] infer_1.0.5 dials_1.2.0 scales_1.2.1 broom_1.0.5
[13] tidymodels_1.1.1 ggpmisc_0.5.4-1 ggpp_0.5.5 ggforce_0.4.1
[17] gsw_1.1-1 gridExtra_2.3 lubridate_1.9.0 timechange_0.1.1
[21] argodata_0.1.0 forcats_0.5.2 stringr_1.5.0 dplyr_1.1.3
[25] purrr_1.0.2 readr_2.1.3 tidyr_1.3.0 tibble_3.2.1
[29] ggplot2_3.4.4 tidyverse_1.3.2
loaded via a namespace (and not attached):
[1] googledrive_2.0.0 colorspace_2.0-3 ellipsis_0.3.2
[4] class_7.3-20 rprojroot_2.0.3 fs_1.5.2
[7] rstudioapi_0.15.0 furrr_0.3.1 listenv_0.8.0
[10] farver_2.1.1 MatrixModels_0.5-1 prodlim_2019.11.13
[13] fansi_1.0.3 xml2_1.3.3 codetools_0.2-18
[16] splines_4.2.2 cachem_1.0.6 confintr_1.0.2
[19] knitr_1.41 polyclip_1.10-4 polynom_1.4-1
[22] jsonlite_1.8.3 workflowr_1.7.0 dbplyr_2.2.1
[25] compiler_4.2.2 httr_1.4.4 backports_1.4.1
[28] assertthat_0.2.1 Matrix_1.5-3 fastmap_1.1.0
[31] gargle_1.2.1 cli_3.6.1 later_1.3.0
[34] tweenr_2.0.2 htmltools_0.5.3 quantreg_5.94
[37] tools_4.2.2 gtable_0.3.1 glue_1.6.2
[40] Rcpp_1.0.10 cellranger_1.1.0 jquerylib_0.1.4
[43] RNetCDF_2.6-1 DiceDesign_1.9 vctrs_0.6.4
[46] iterators_1.0.14 timeDate_4021.106 xfun_0.35
[49] gower_1.0.0 globals_0.16.2 rvest_1.0.3
[52] lifecycle_1.0.3 googlesheets4_1.0.1 future_1.29.0
[55] MASS_7.3-58.1 ipred_0.9-13 hms_1.1.2
[58] promises_1.2.0.1 parallel_4.2.2 SparseM_1.81
[61] RColorBrewer_1.1-3 yaml_2.3.6 sass_0.4.4
[64] rpart_4.1.19 stringi_1.7.8 highr_0.9
[67] foreach_1.5.2 lhs_1.1.6 hardhat_1.3.0
[70] lava_1.7.0 rlang_1.1.1 pkgconfig_2.0.3
[73] evaluate_0.18 lattice_0.20-45 labeling_0.4.2
[76] tidyselect_1.2.0 parallelly_1.32.1 magrittr_2.0.3
[79] R6_2.5.1 generics_0.1.3 DBI_1.1.3
[82] pillar_1.9.0 haven_2.5.1 whisker_0.4
[85] withr_2.5.0 survival_3.4-0 nnet_7.3-18
[88] future.apply_1.10.0 modelr_0.1.10 crayon_1.5.2
[91] utf8_1.2.2 tzdb_0.3.0 rmarkdown_2.18
[94] grid_4.2.2 readxl_1.4.1 git2r_0.30.1
[97] reprex_2.0.2 digest_0.6.30 httpuv_1.6.6
[100] GPfit_1.0-8 munsell_0.5.0 viridisLite_0.4.1
[103] bslib_0.4.1