Human Chimp EB Differential Expression
karltayeb
2022-04-11
Last updated: 2022-04-12
Checks: 7 0
Knit directory: logistic-susie-gsea/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20220105) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 5e3a4e3. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .RData
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: library/
Ignored: renv/library/
Ignored: renv/staging/
Ignored: staging/
Untracked files:
Untracked: .ipynb_checkpoints/
Untracked: Untitled.ipynb
Untracked: _targets.R
Untracked: _targets.html
Untracked: _targets.md
Untracked: _targets/
Untracked: _targets_r/
Untracked: analysis/fetal_reference_cellid_gsea.Rmd
Untracked: analysis/fixed_intercept.Rmd
Untracked: analysis/iDEA_examples.Rmd
Untracked: analysis/latent_gene_list.Rmd
Untracked: analysis/libra_setup.Rmd
Untracked: analysis/linear_method_failure_modes.Rmd
Untracked: analysis/linear_regression_failure_regime.Rmd
Untracked: analysis/logistic_susie_veb_boost_vs_vb.Rmd
Untracked: analysis/logistic_susie_vis.Rmd
Untracked: analysis/references.bib
Untracked: analysis/simulations.Rmd
Untracked: analysis/test.Rmd
Untracked: baboon_diet_cache/
Untracked: build_site.R
Untracked: cache/
Untracked: code/enrichment_pipeline.R
Untracked: code/html_tables.R
Untracked: code/latent_logistic_susie.R
Untracked: code/logistic_susie_data_driver.R
Untracked: code/marginal_sumstat_gsea_collapsed.R
Untracked: code/sumstat_gsea.py
Untracked: code/susie_gsea_queries.R
Untracked: data/adipose_2yr_topsnp.txt
Untracked: data/de-droplet/
Untracked: data/deng/
Untracked: data/fetal_reference_cellid_gene_sets.RData
Untracked: data/human_chimp_eb/
Untracked: data/pbmc-purified/
Untracked: data/wenhe_baboon_diet/
Untracked: deng_example_cache/
Untracked: docs.zip
Untracked: human_chimp_eb_de_example_cache/
Untracked: index.md
Untracked: latent_logistic_susie_cache/
Untracked: simulation_targets/
Untracked: single_cell_pbmc_cache/
Untracked: single_cell_pbmc_l1_cache/
Untracked: summary_stat_gsea_exploration_cache/
Untracked: summary_stat_gsea_sim_cache/
Unstaged changes:
Modified: _simulation_targets.R
Modified: _targets.Rmd
Modified: analysis/alpha_for_single_cell.Rmd
Modified: analysis/deng_example.Rmd
Modified: analysis/gseabenchmark_tcga.Rmd
Modified: analysis/single_cell_pbmc.Rmd
Modified: analysis/single_cell_pbmc_l1.Rmd
Deleted: analysis/summary_stat_gsea_univariate_simulations.Rmd
Modified: code/fit_baselines.R
Modified: code/fit_logistic_susie.R
Modified: code/fit_mr_ash.R
Modified: code/fit_susie.R
Modified: code/load_gene_sets.R
Modified: code/marginal_sumstat_gsea.R
Modified: code/simulate_gene_lists.R
Modified: target_components/factories.R
Modified: target_components/methods.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/human_chimp_eb_de_example.Rmd) and HTML (docs/human_chimp_eb_de_example.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 5e3a4e3 | karltayeb | 2022-04-12 | wflow_publish(c(“analysis/index.Rmd”, “analysis/baboon_diet.Rmd”, |
| Rmd | 8d47637 | karltayeb | 2022-04-11 | run human chimp EB with new standardized input/output |
Introduction
library(tidyverse)
library(htmltools)
source('code/utils.R')
source('code/logistic_susie_vb.R')
source('code/logistic_susie_veb_boost.R')
source('code/load_gene_sets.R')
source('code/enrichment_pipeline.R')Setup
Format
de <- readRDS('data/human_chimp_eb/big.df.rds')
genesets <- load_gene_sets()
hs <- org.Hs.eg.db::org.Hs.eg.db
gene_symbols <- unique(de$gene)
symbol2entrez <- AnnotationDbi::select(
hs, keys=gene_symbols,
columns=c('ENTREZID', 'SYMBOL'),
keytype = 'SYMBOL')
add_names = function(l, n){
names(l) <- n
return(l)
}
data <- de %>%
rename(SYMBOL=gene) %>%
left_join(symbol2entrez, by='SYMBOL') %>%
relocate(ENTREZID, .after=SYMBOL) %>%
mutate( # set default columns
beta = dream.logFC,
se = dream.SE,
threshold.on = dream.p.val
) %>%
group_by(celltype) %>%
group_map(~ .x, .keep = T) %>%
add_names(map_chr(., ~pluck(.x, 'celltype')[1]))Fit
# fit logistic susie
do_logistic_susie_cached = function(data,
db,
thresh,
prefix=''){
res <- xfun::cache_rds({
purrr::map_dfr(
names(data),
~do_logistic_susie(.x, db, thresh, genesets, data))
},
dir = params$cache_dir,
file=paste0(prefix, 'logistic_susie_', db, '_', thresh),
hash = list(data, db, thresh, prefix))
}
params.genesets <- eval(parse(text=params$genesets))
params.thresh <- eval(parse(text=params$thresh))
fits <- map_dfr(params.genesets, ~do_logistic_susie_cached(data, .x, params.thresh))
# fit ora
do_ora_cached = function(data, db, thresh, prefix='', ...){
res <- xfun::cache_rds({
purrr::map_dfr(names(data), ~do_ora(.x, db, thresh, genesets, data))
}, dir = params$cache_dir, file=paste0(prefix, 'ora_', db, '_', thresh), ...)
}
ora <- map_dfr(params.genesets, ~do_ora_cached(data, .x, params.thresh))Overview
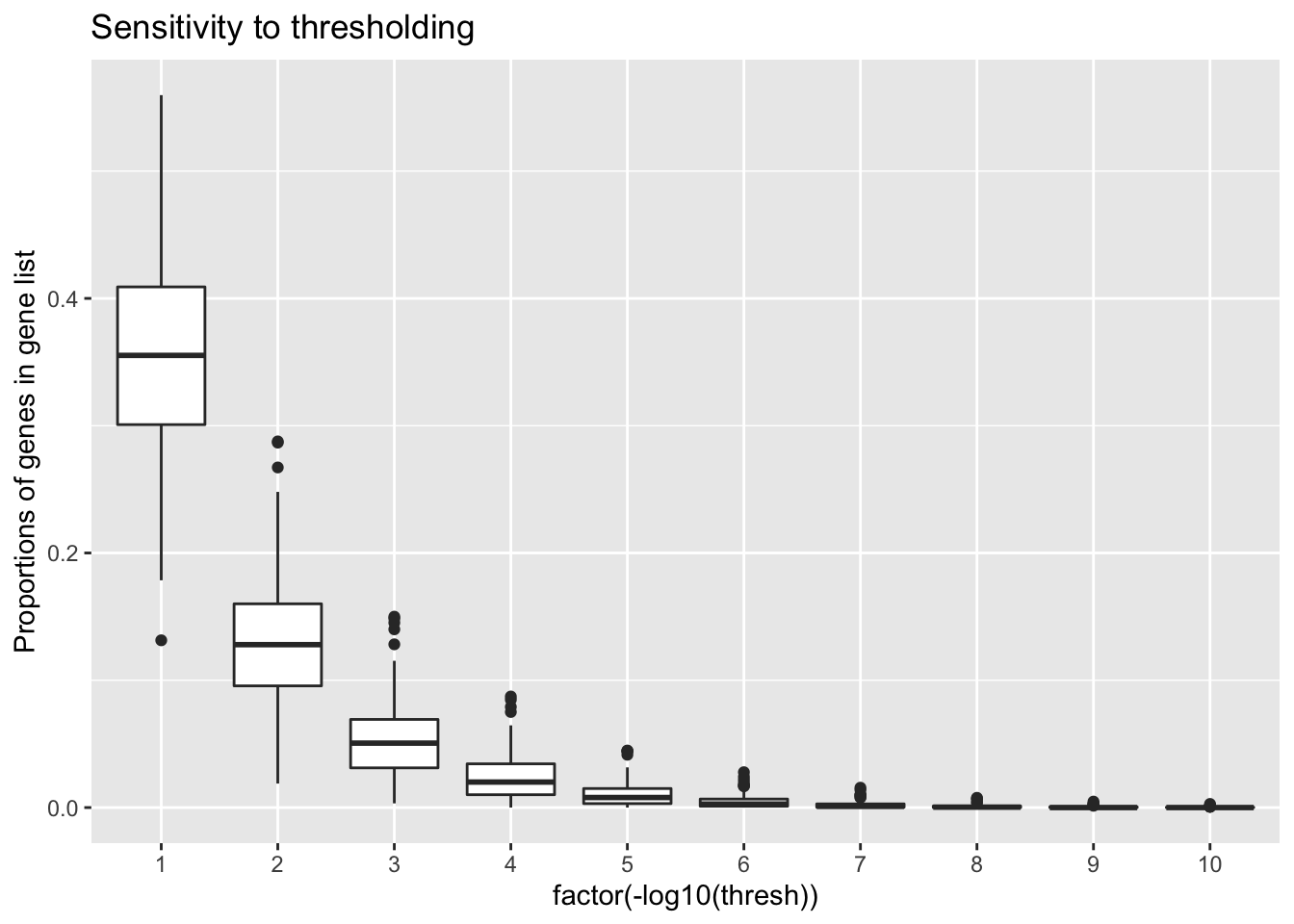
Threshold sensitivity
mean.gene.prop = function(l){
purrr::map_dbl(3:10, ~get_y(l, 10^(-.x)) %>% mean())
}
thresh <- map_dbl(1:10, ~10**-.x)
.prop.ones = function(experiment){
map_dbl(thresh, ~ prep_binary_data(
genesets[['gobp']], data[[experiment]], thresh=.x)$y %>% mean())
}
prop.ones <- xfun::cache_rds({map_dfc(names(data), ~.prop.ones(.x))},
dir=params$cache_dir,
file='threshold_sensitivity')
colnames(prop.ones) <- names(data)
prop.ones <- prop.ones %>% mutate(thresh = thresh)
prop.ones %>%
pivot_longer(one_of(names(data))) %>%
group_by(name) %>%
mutate(value = value) %>%
ggplot(aes(x=factor(-log10(thresh)), y=value)) +
geom_boxplot() +
labs(
y = 'Proportions of genes in gene list',
title = 'Sensitivity to thresholding'
)
Big volcano plot
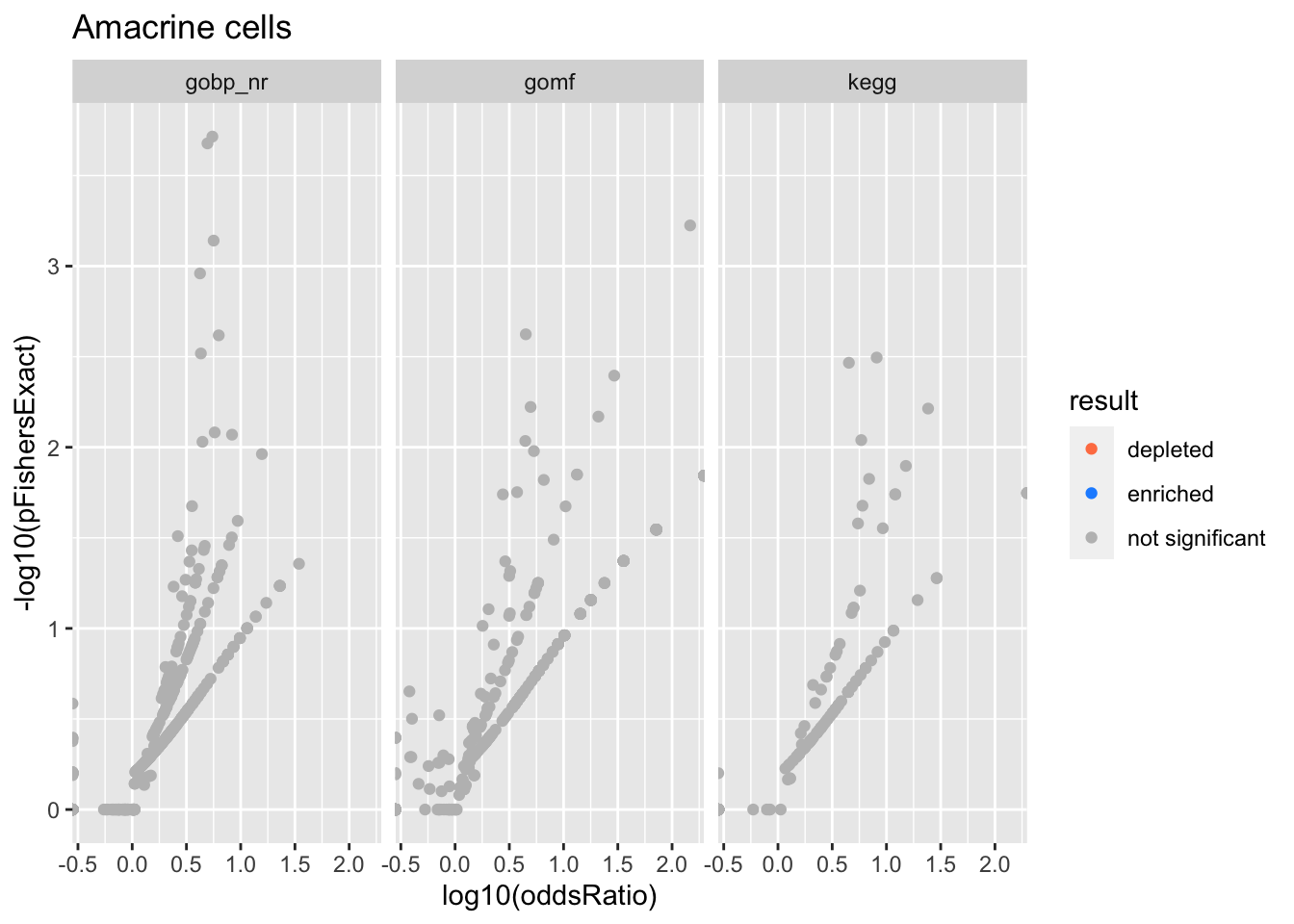
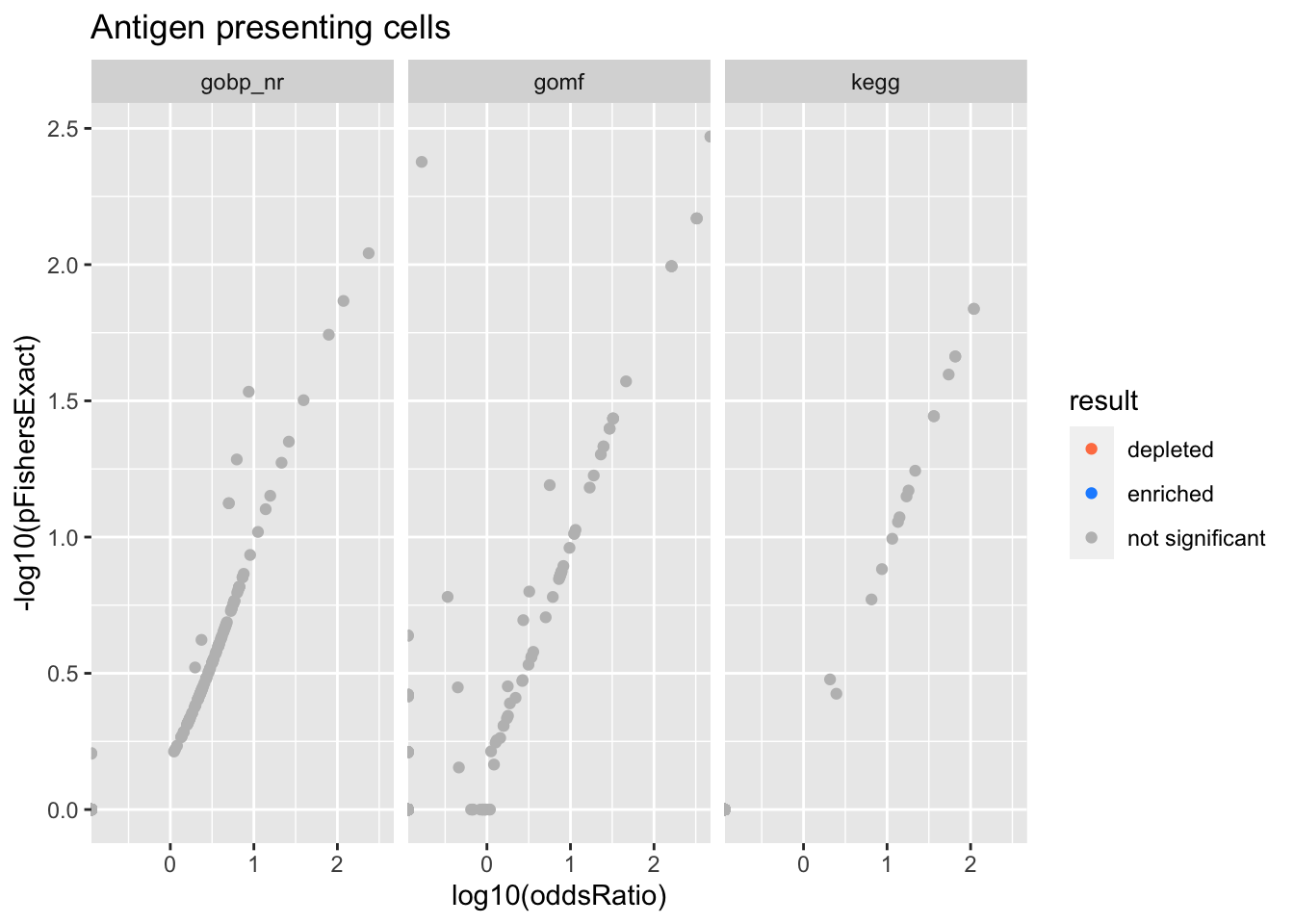
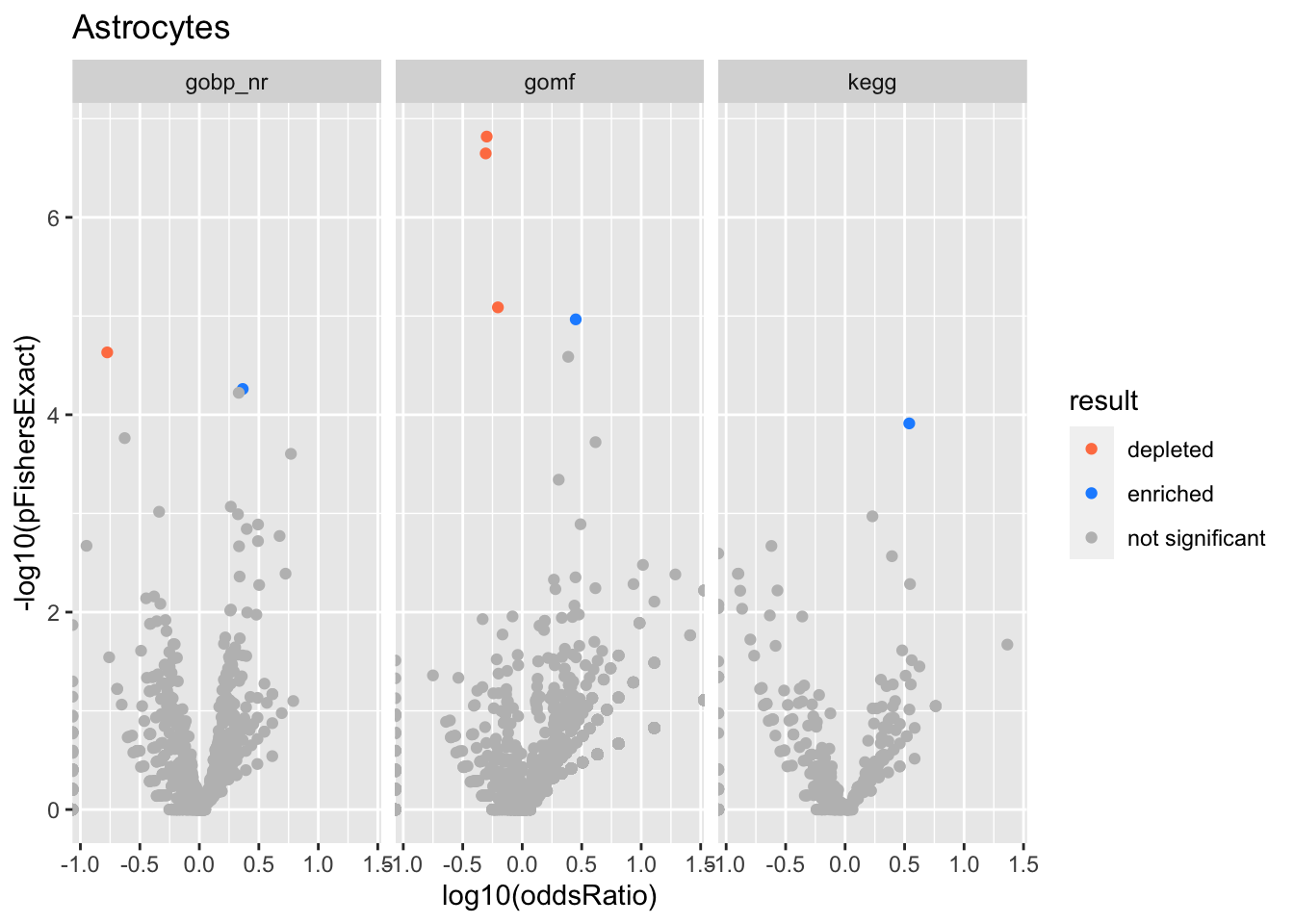
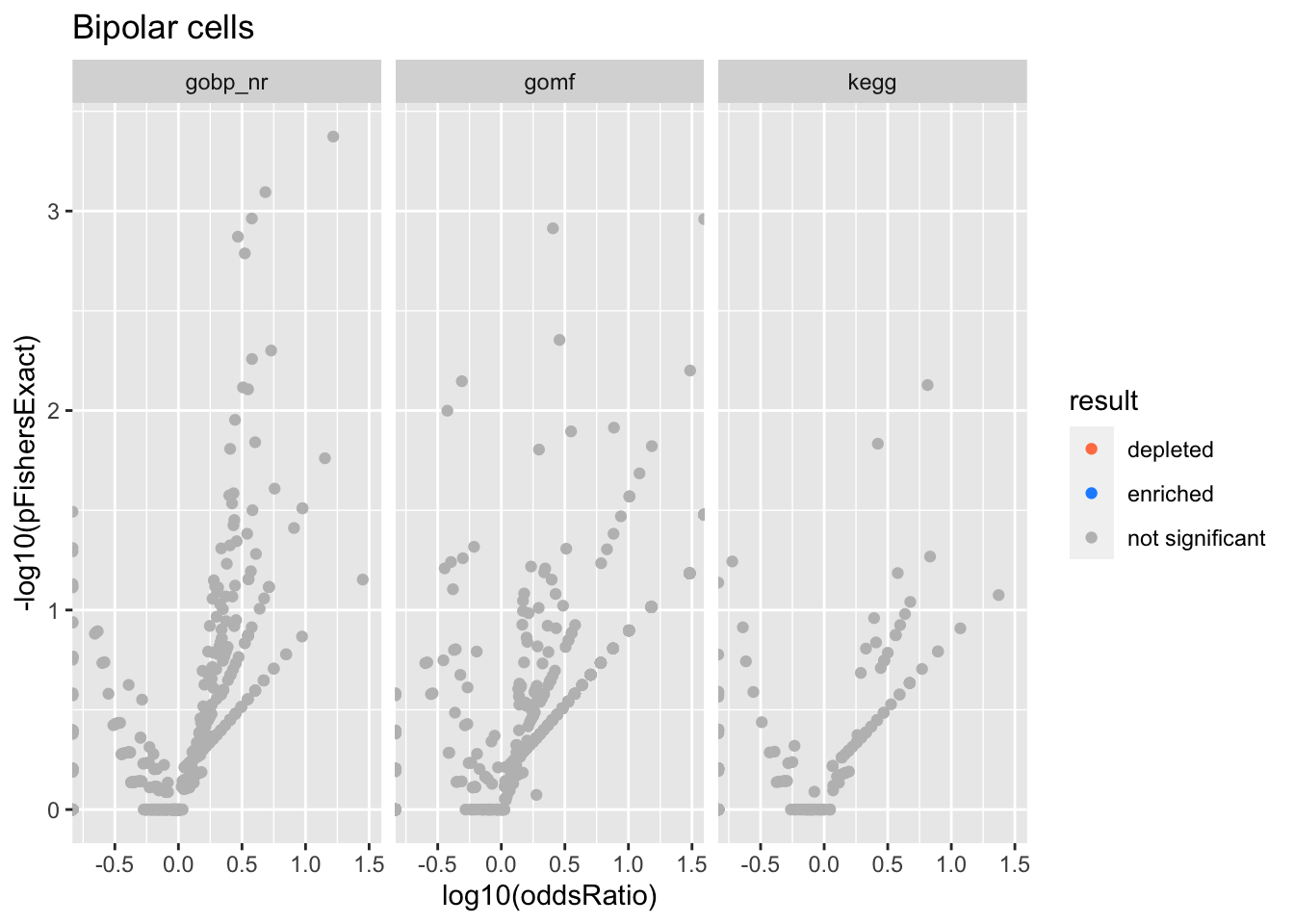
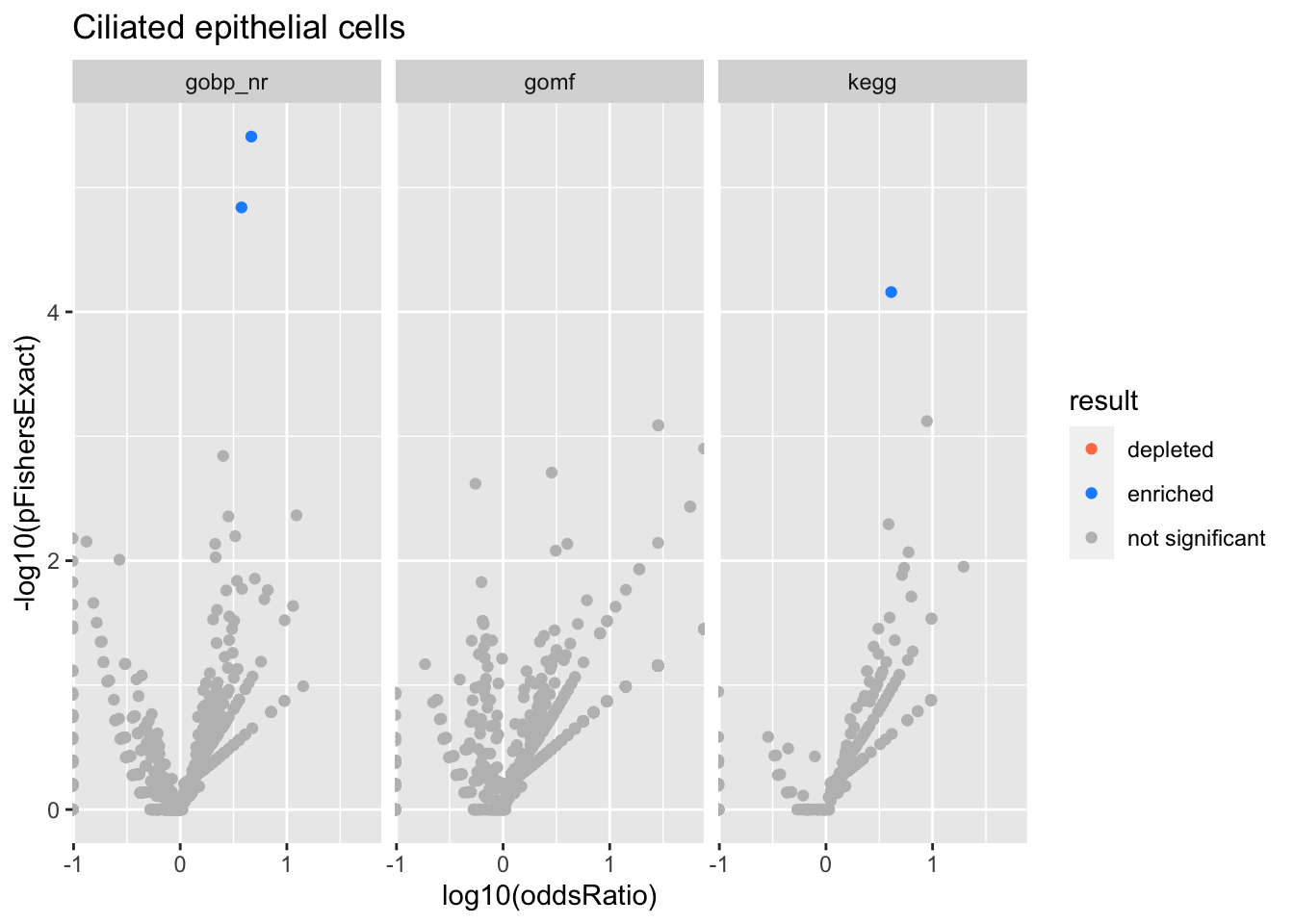
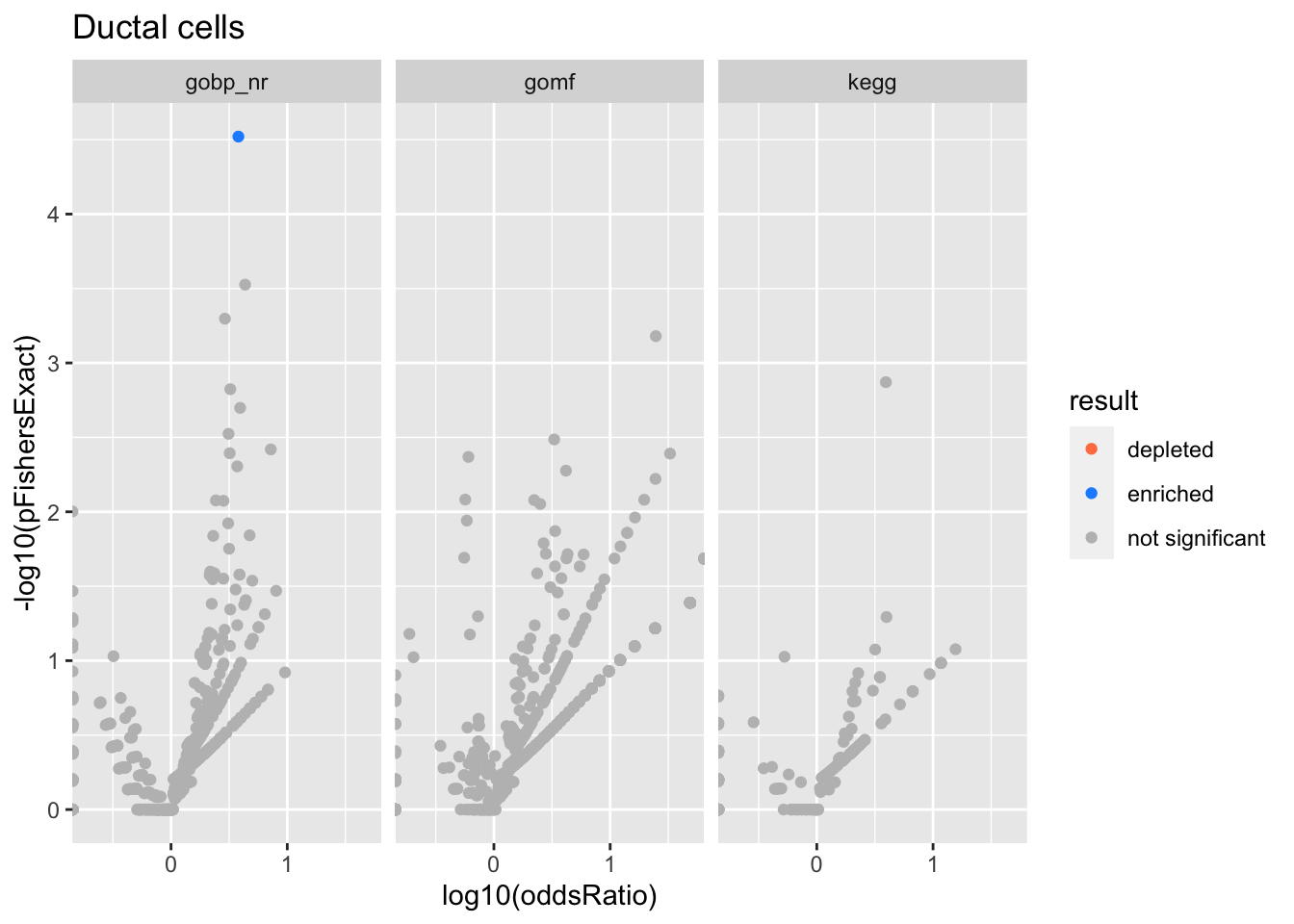
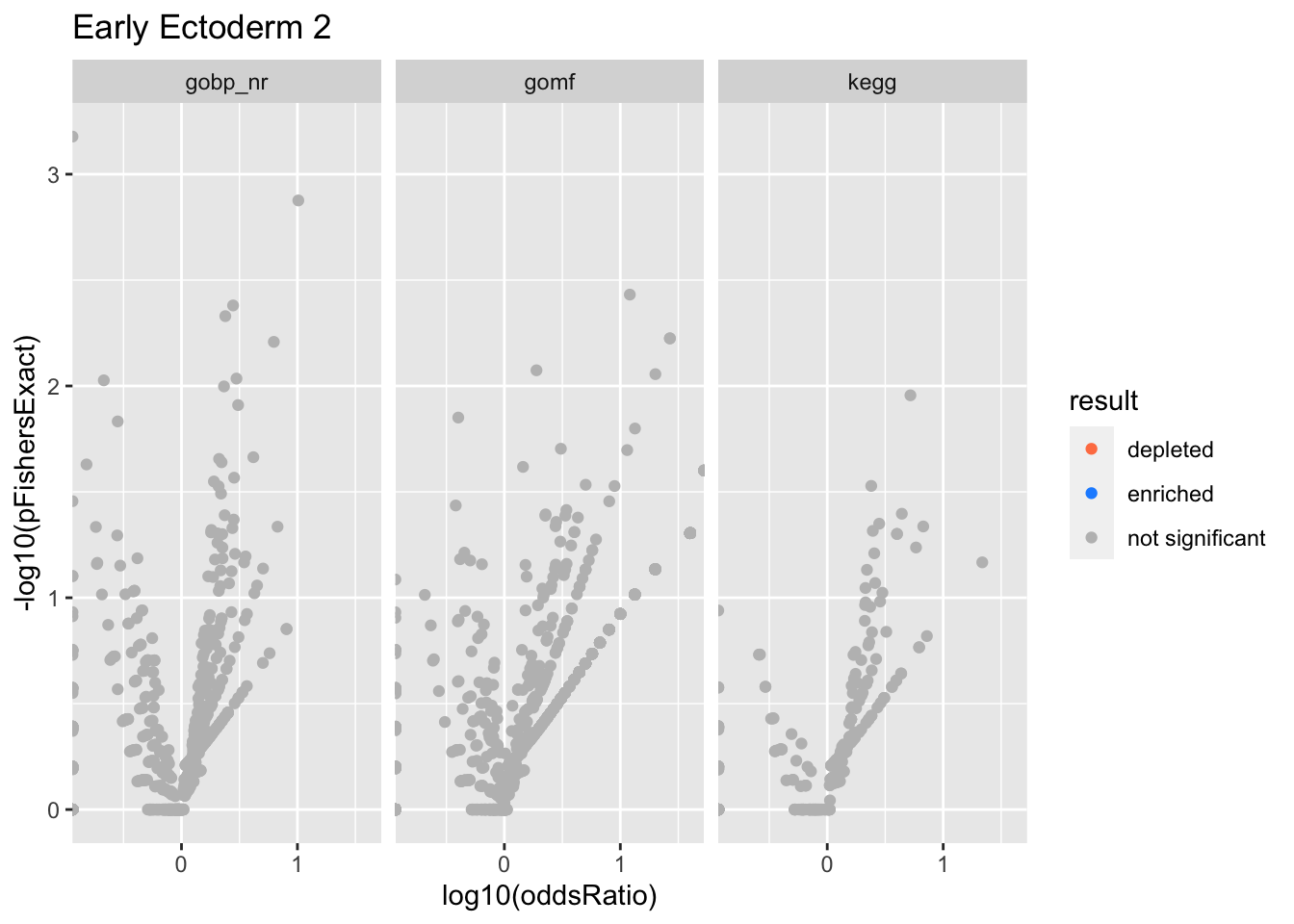
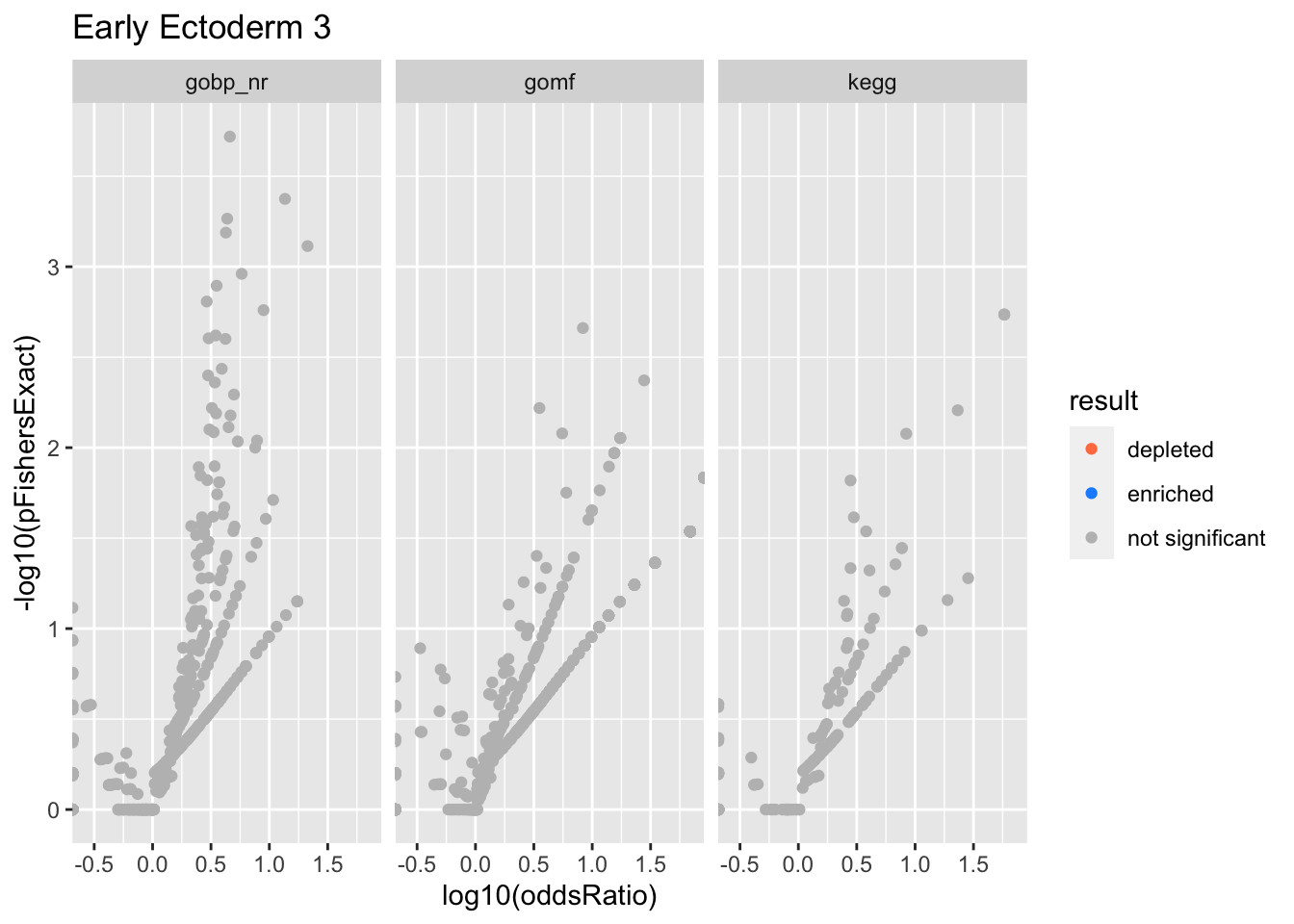
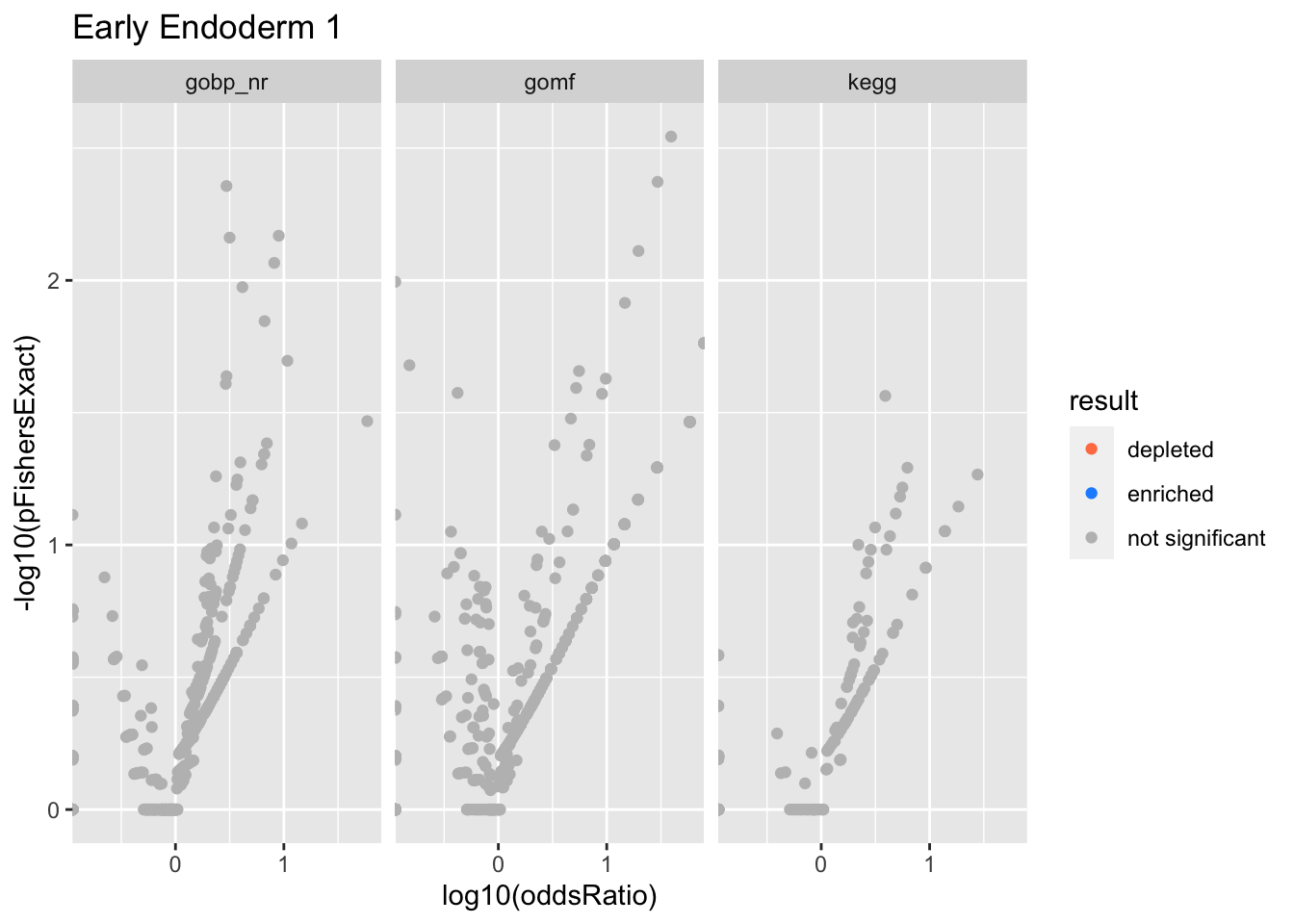
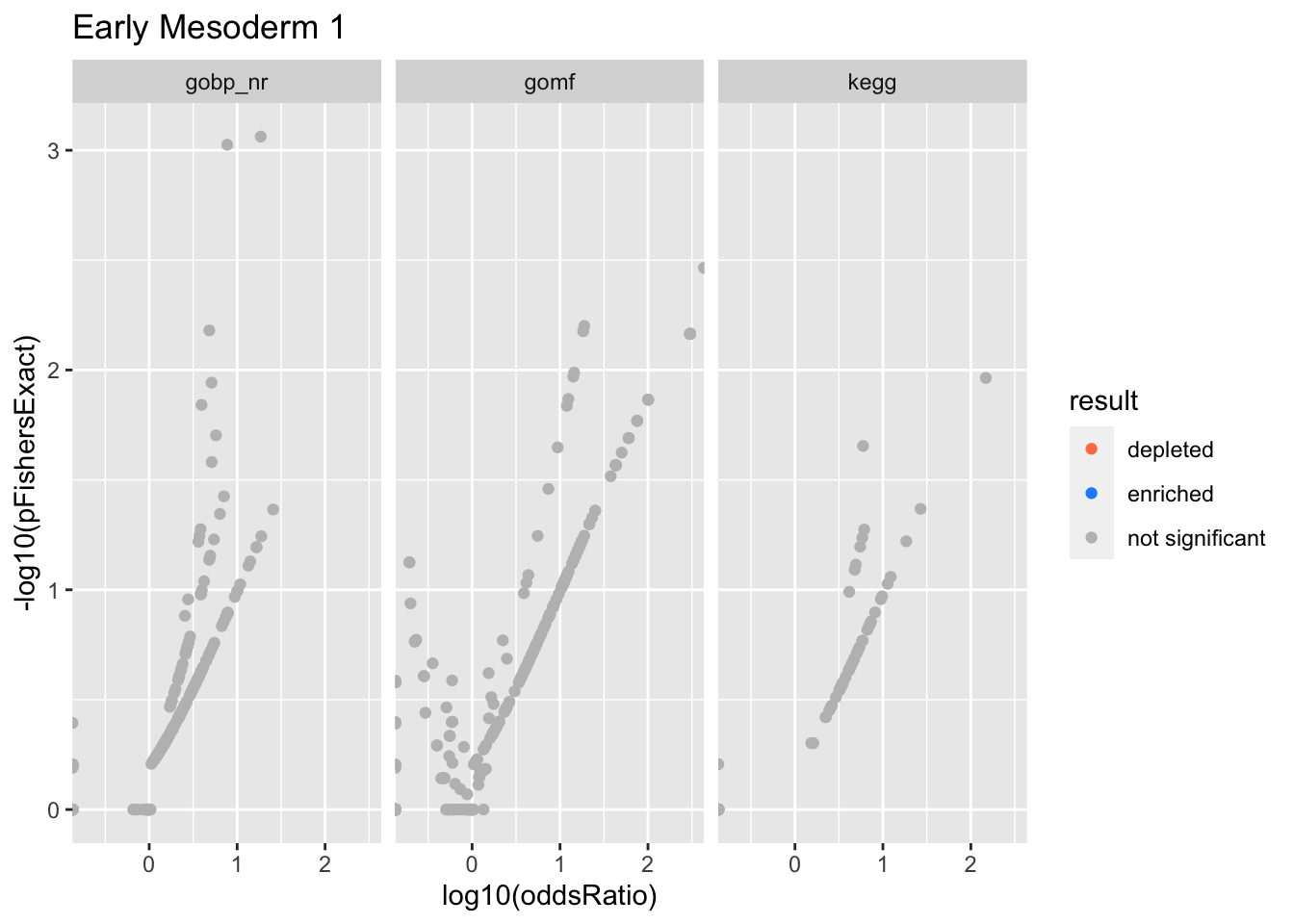
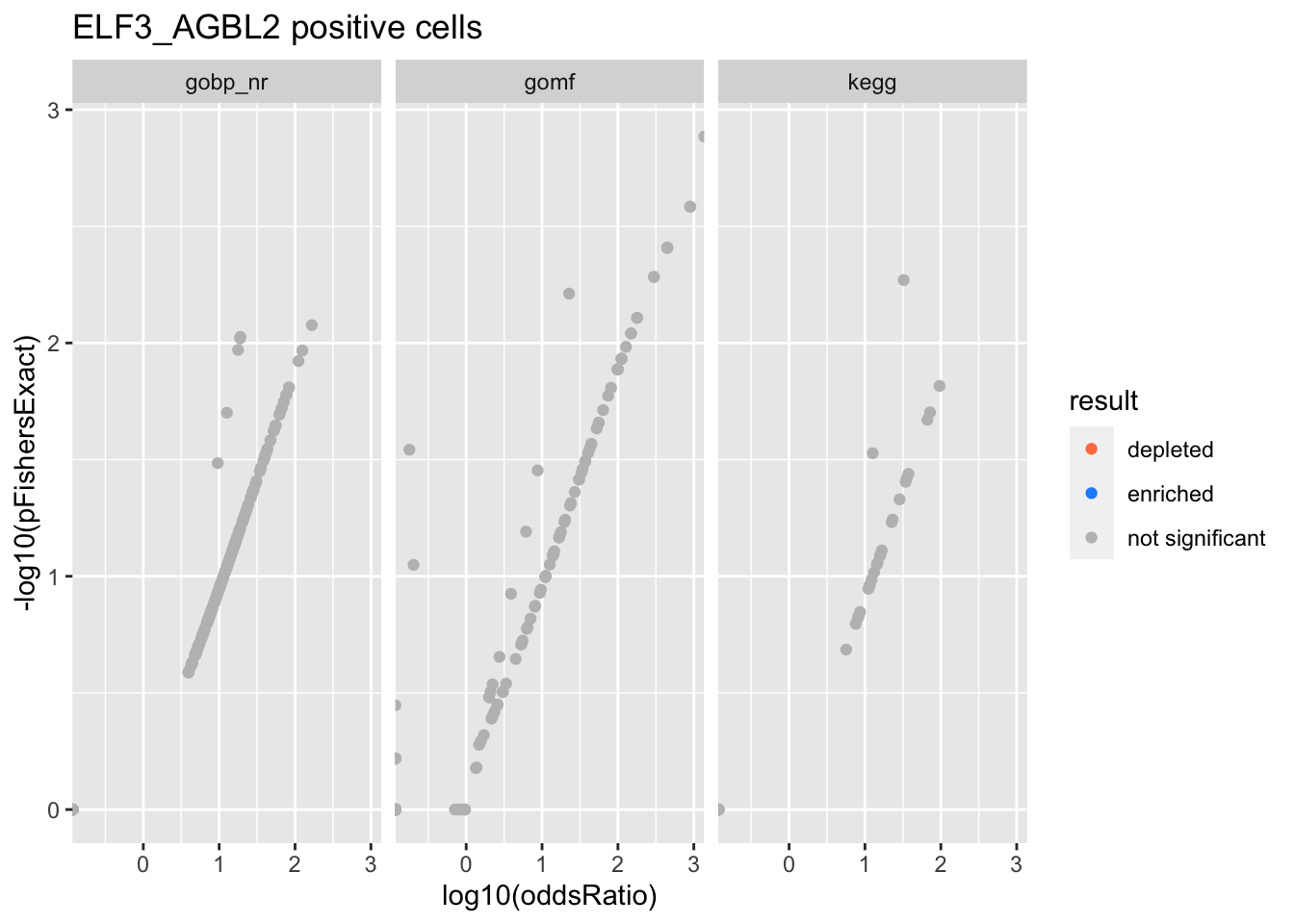
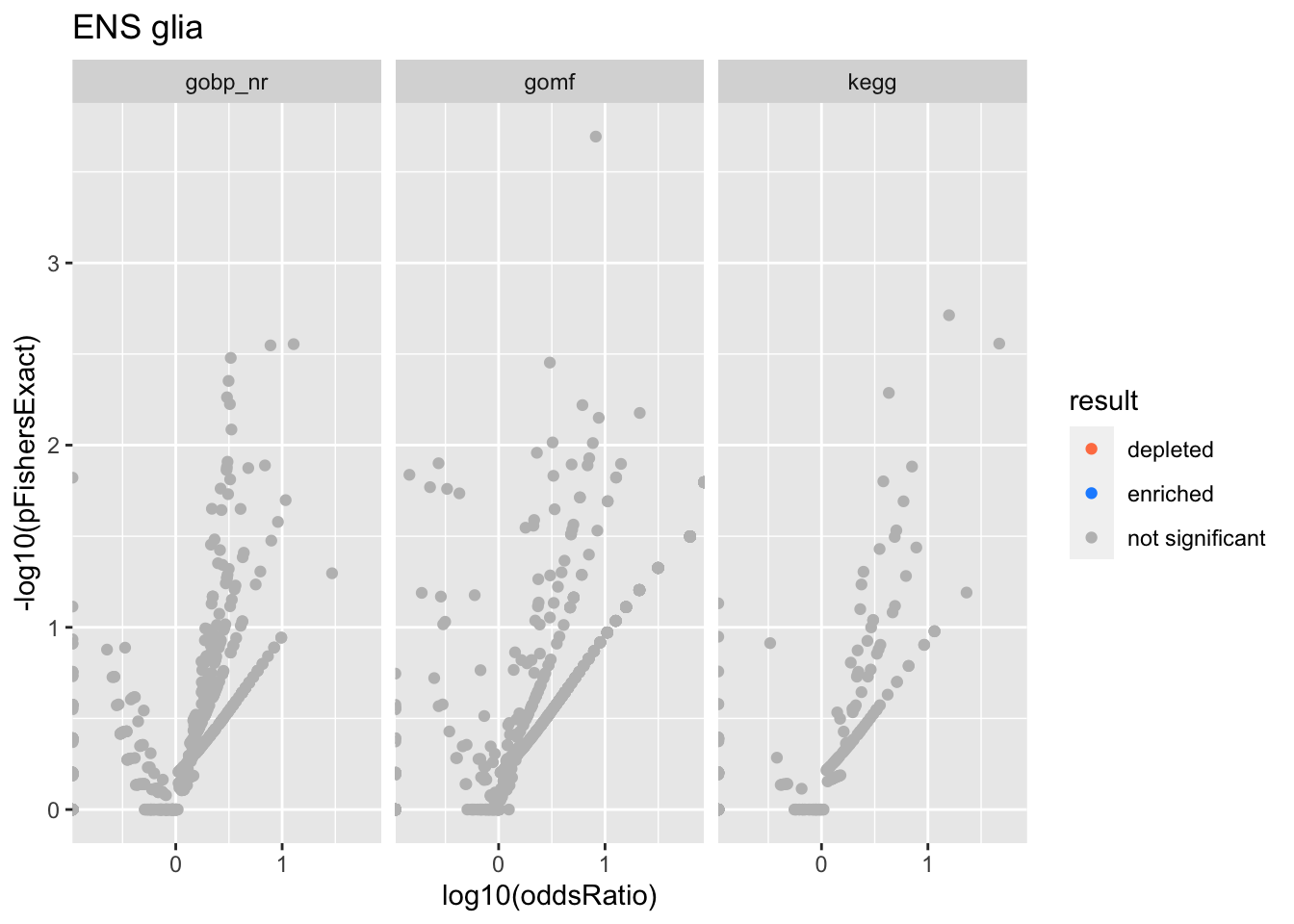
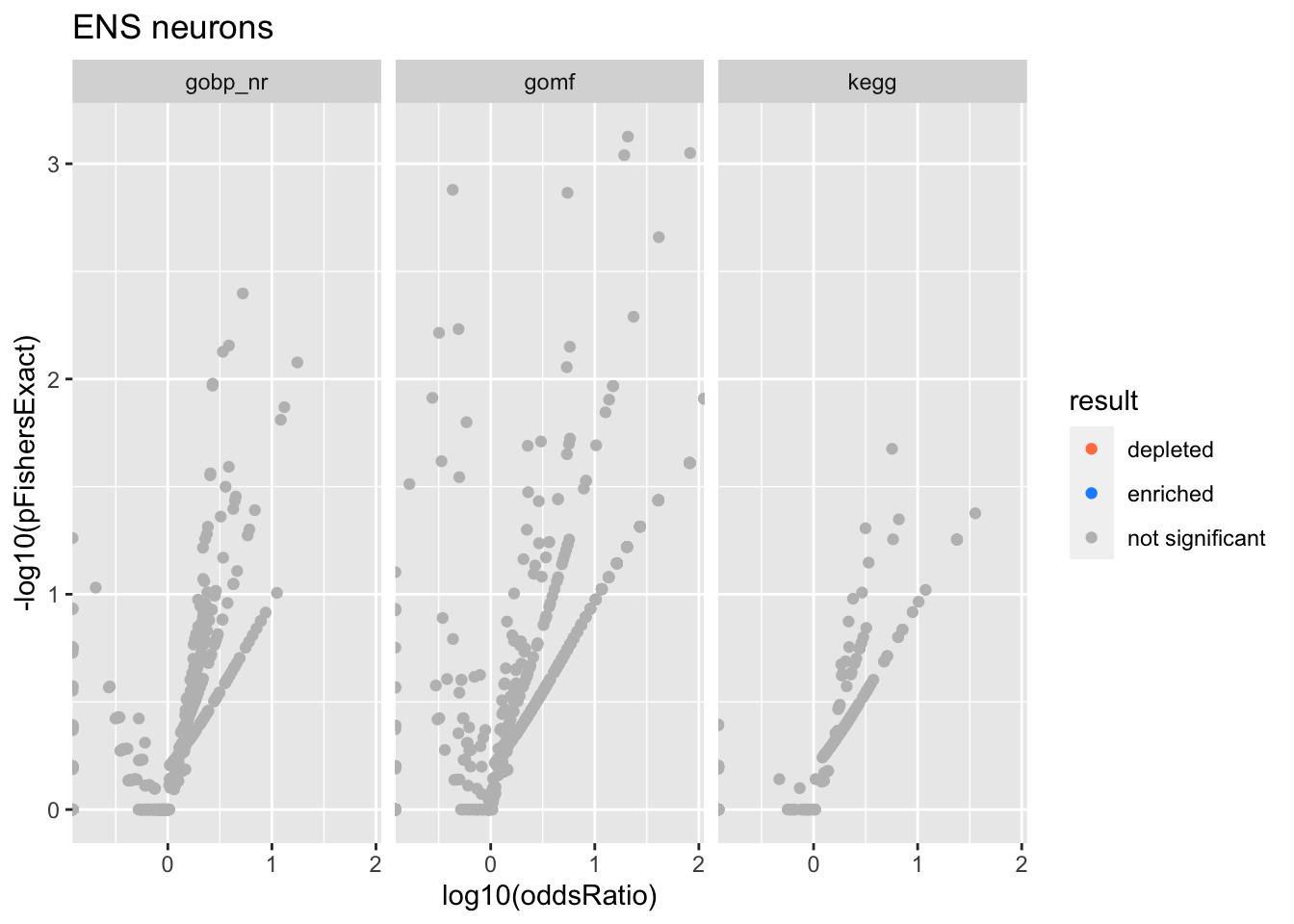
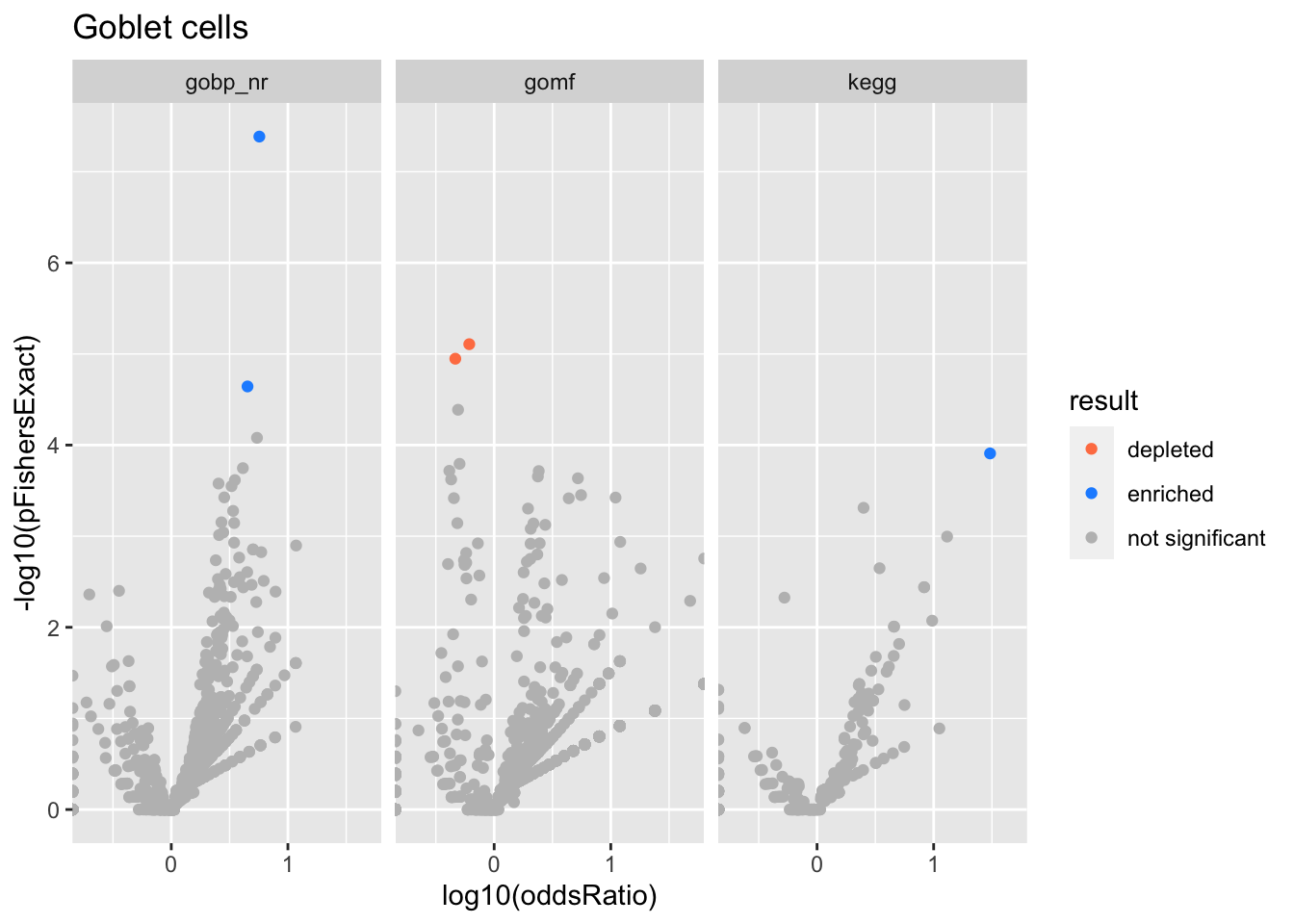
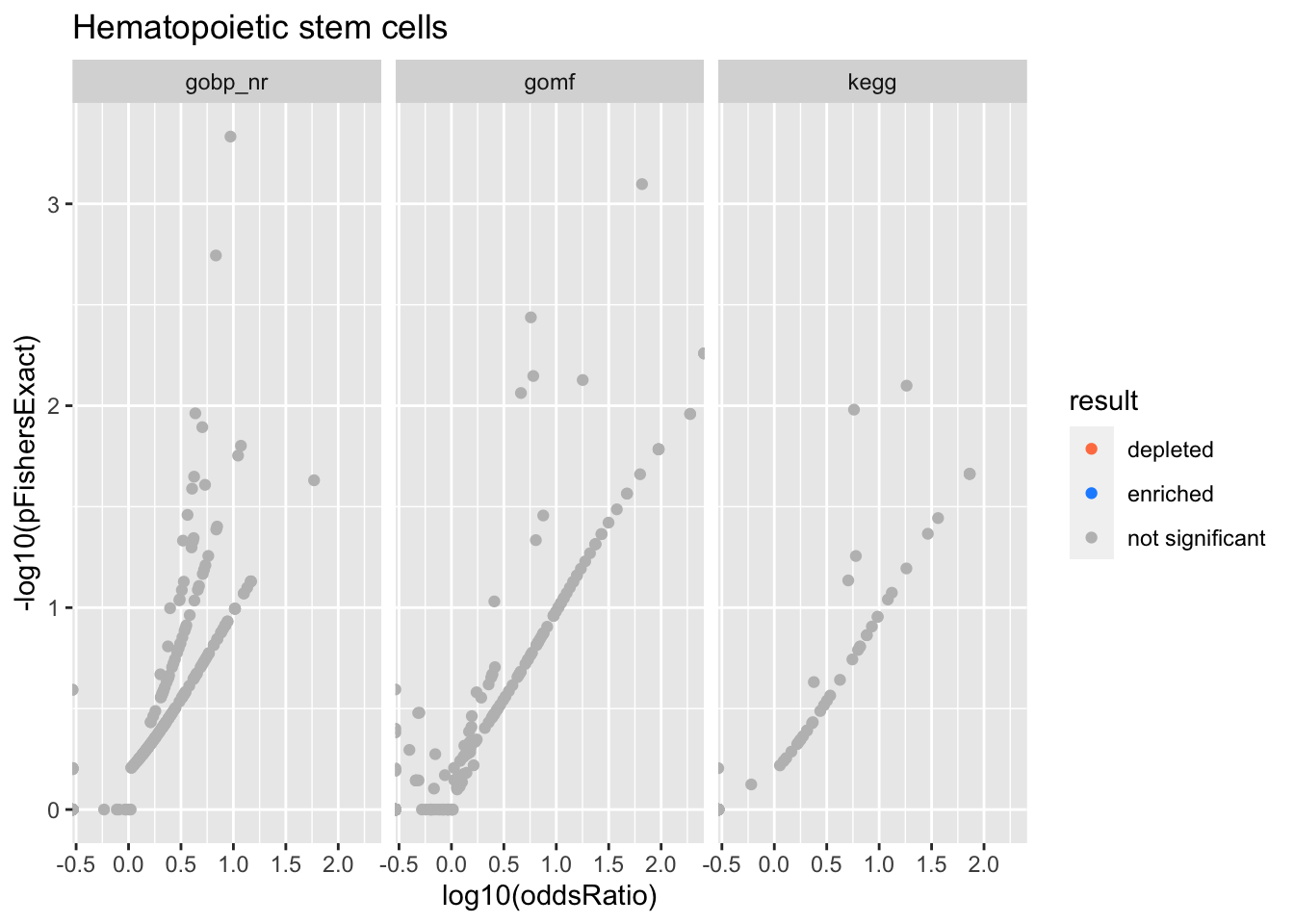
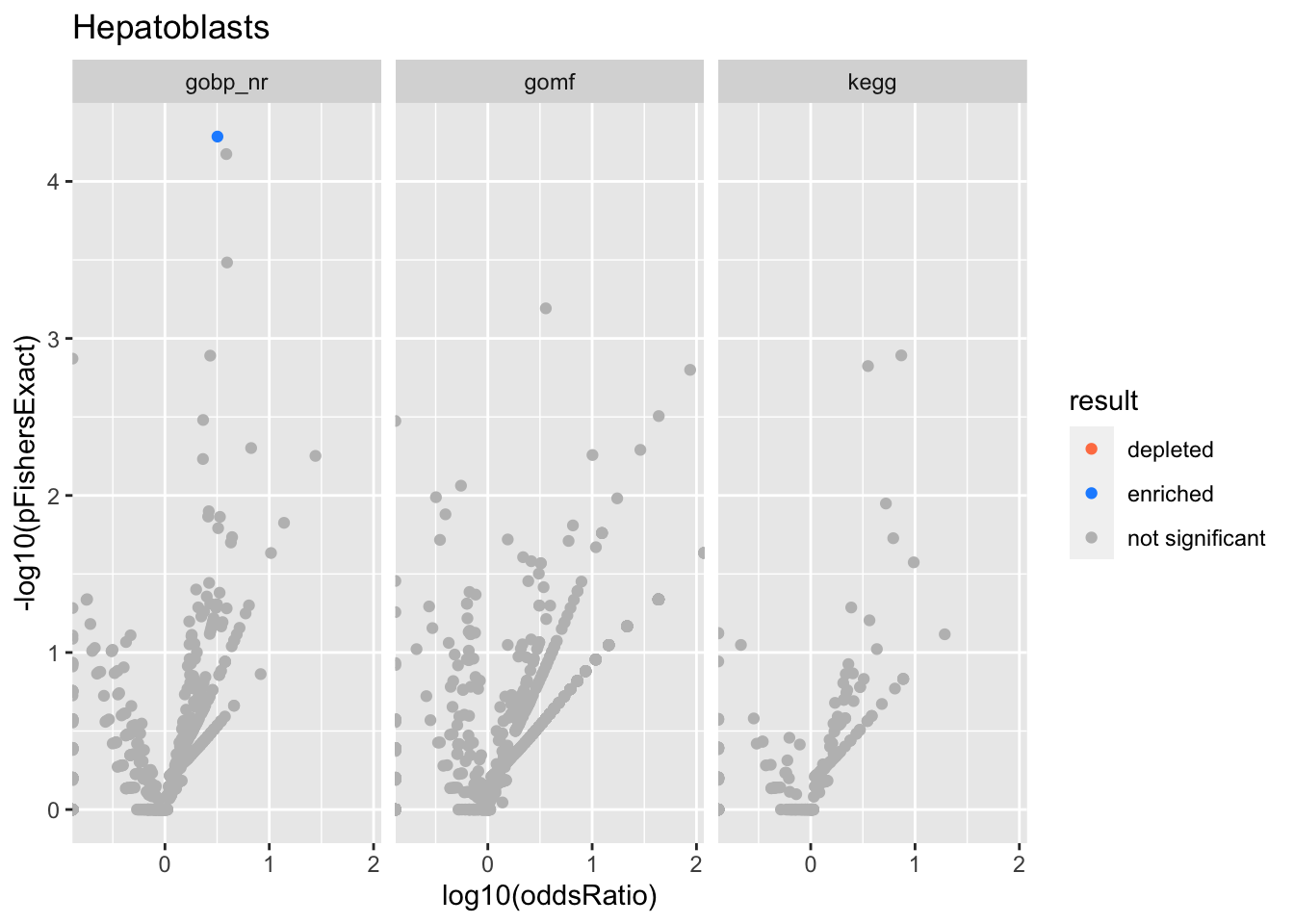
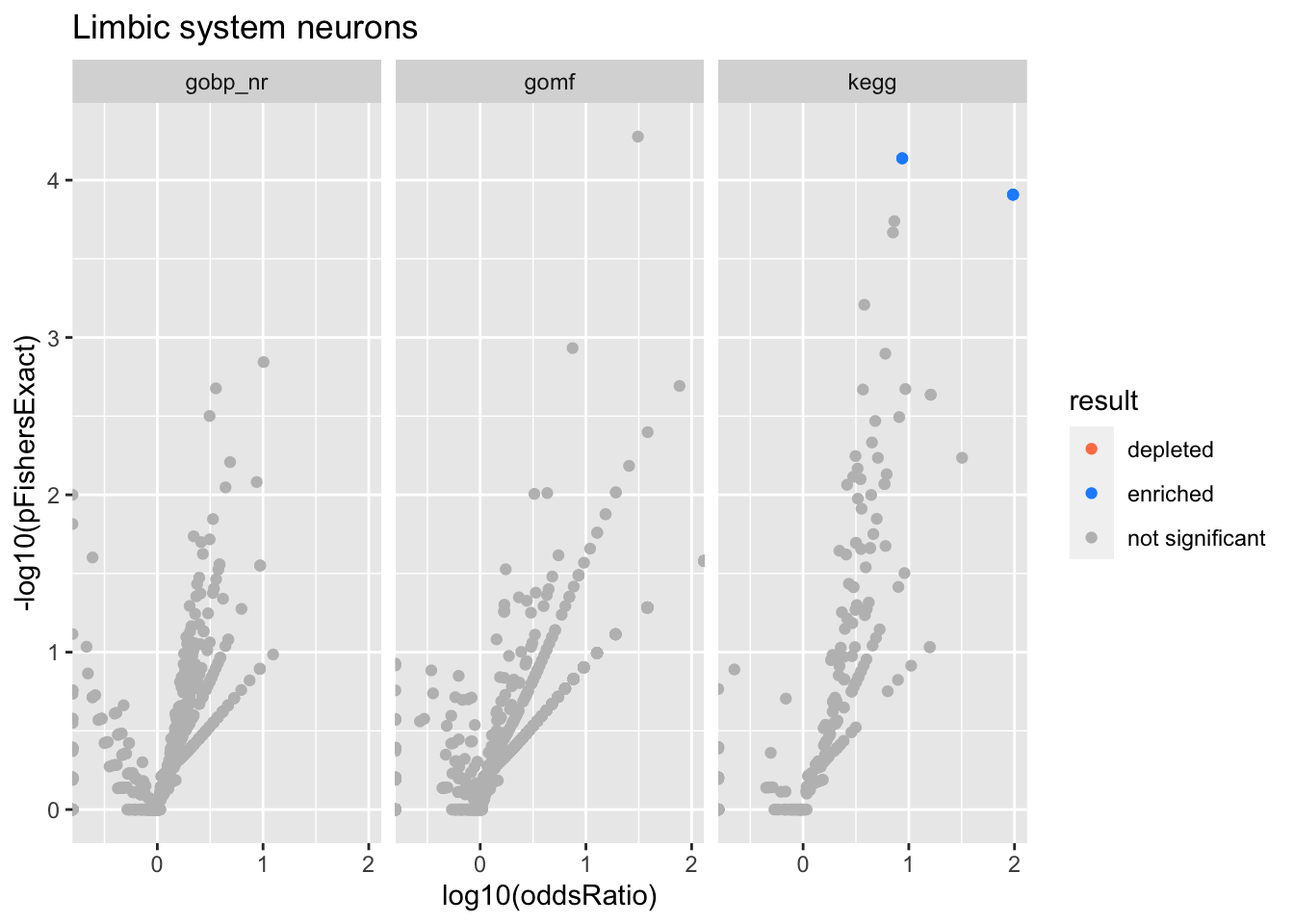
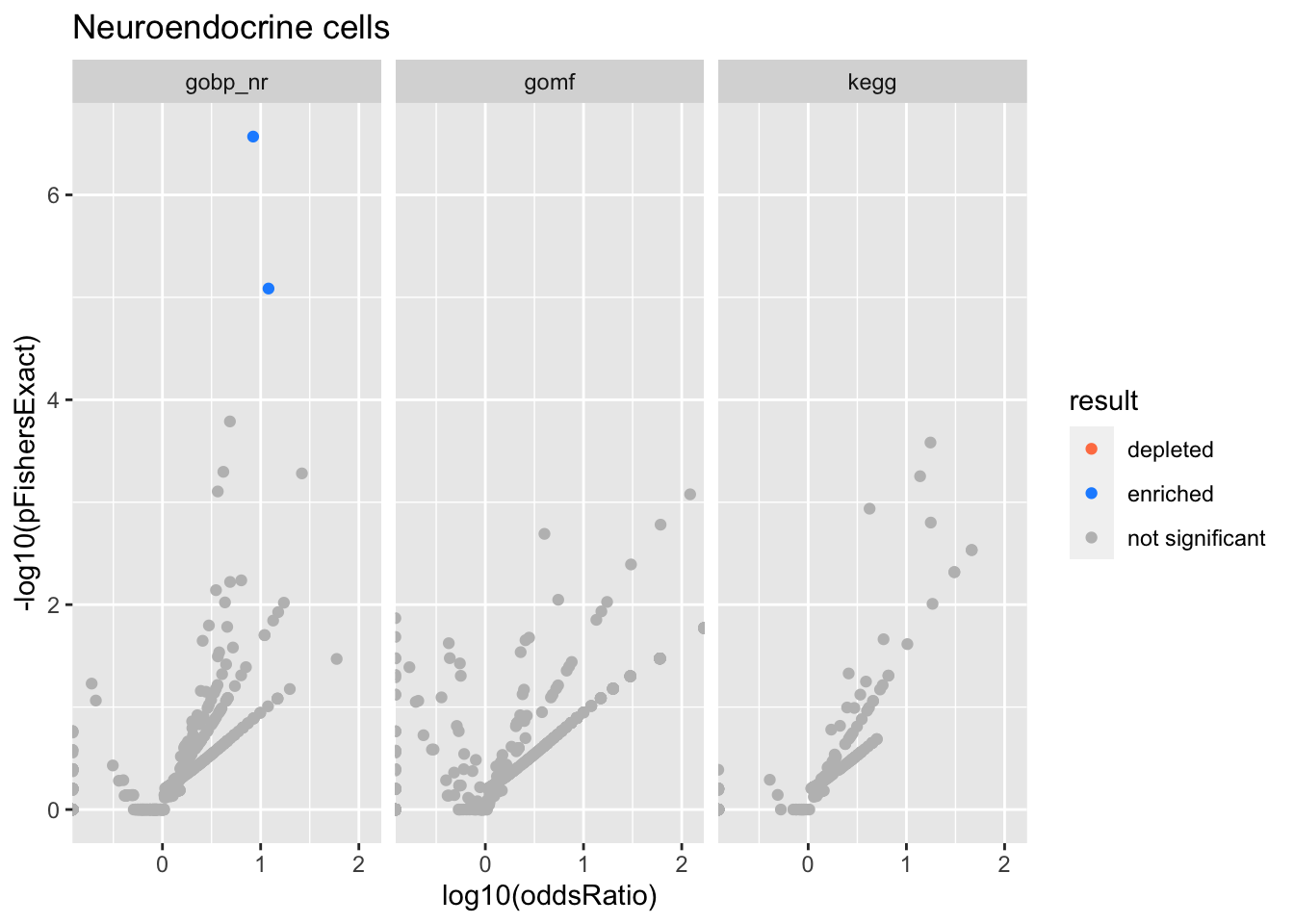
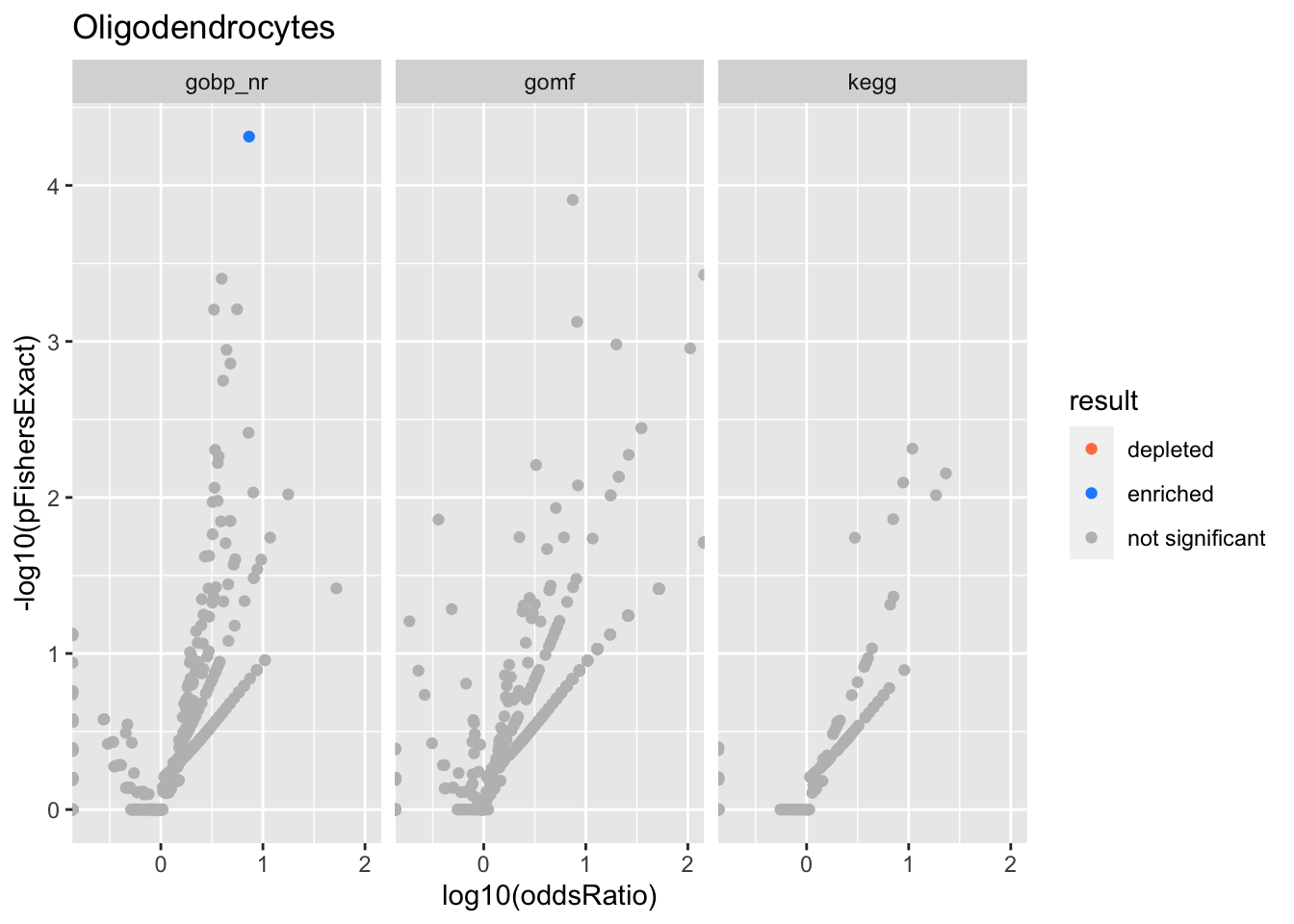
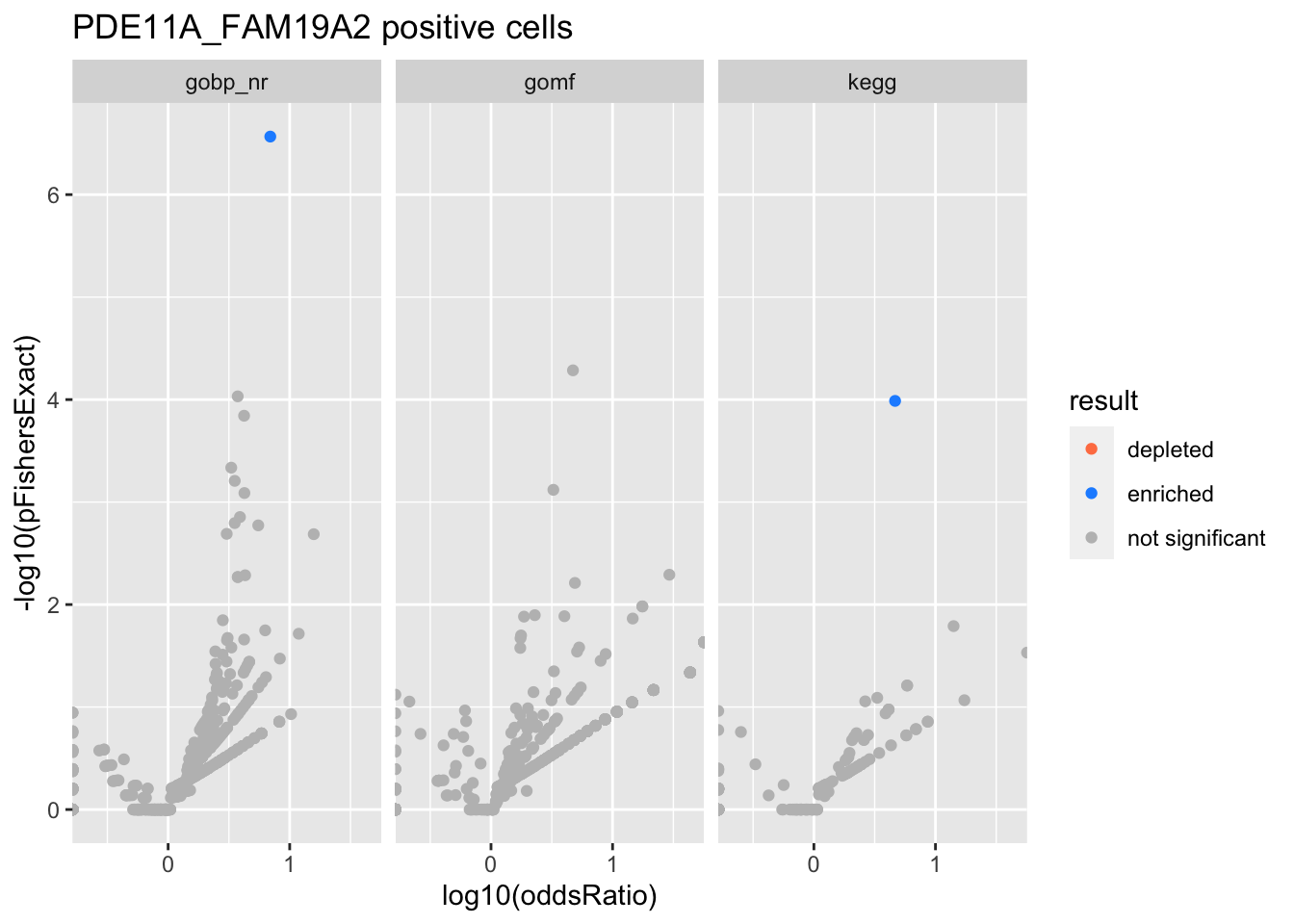
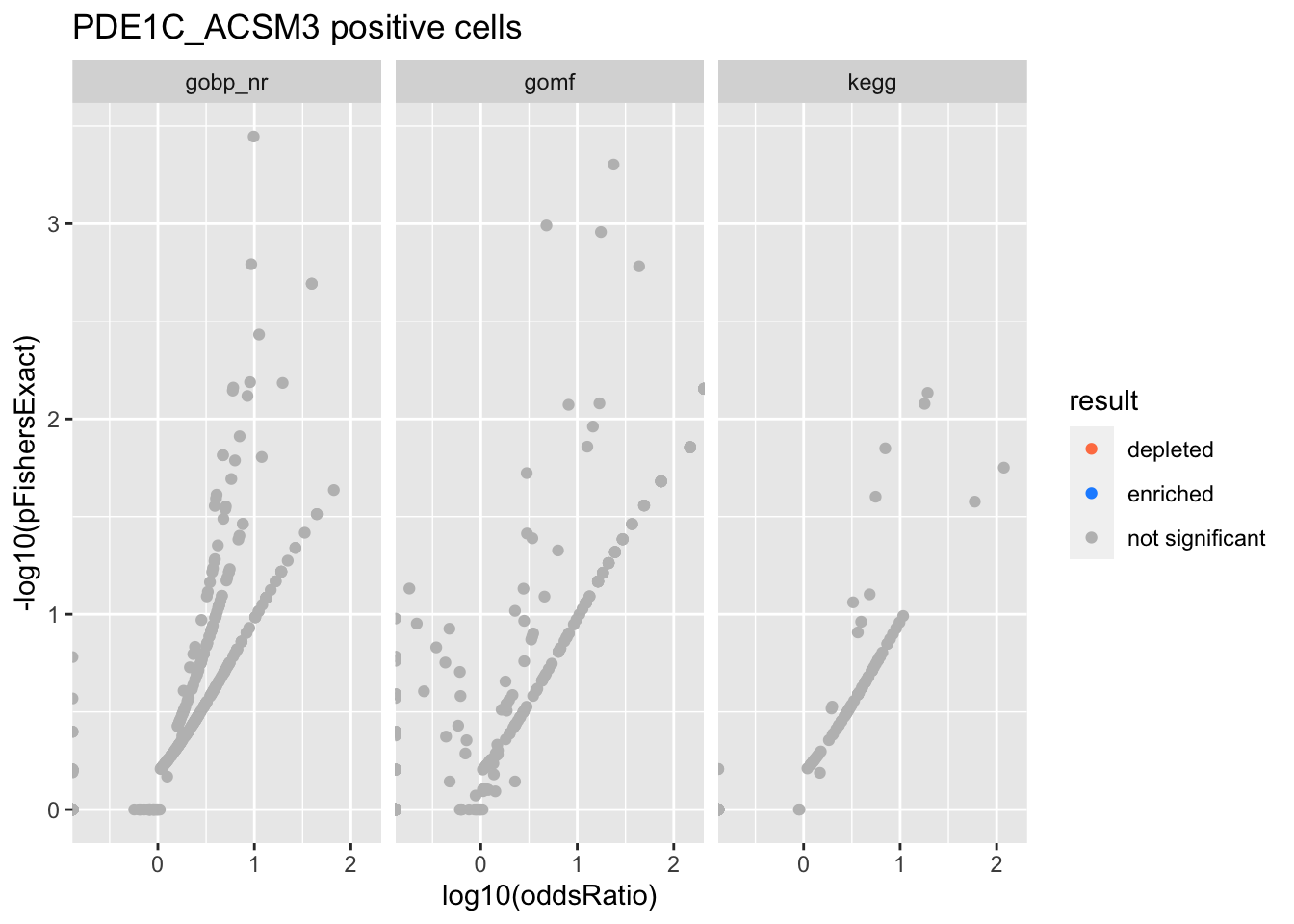
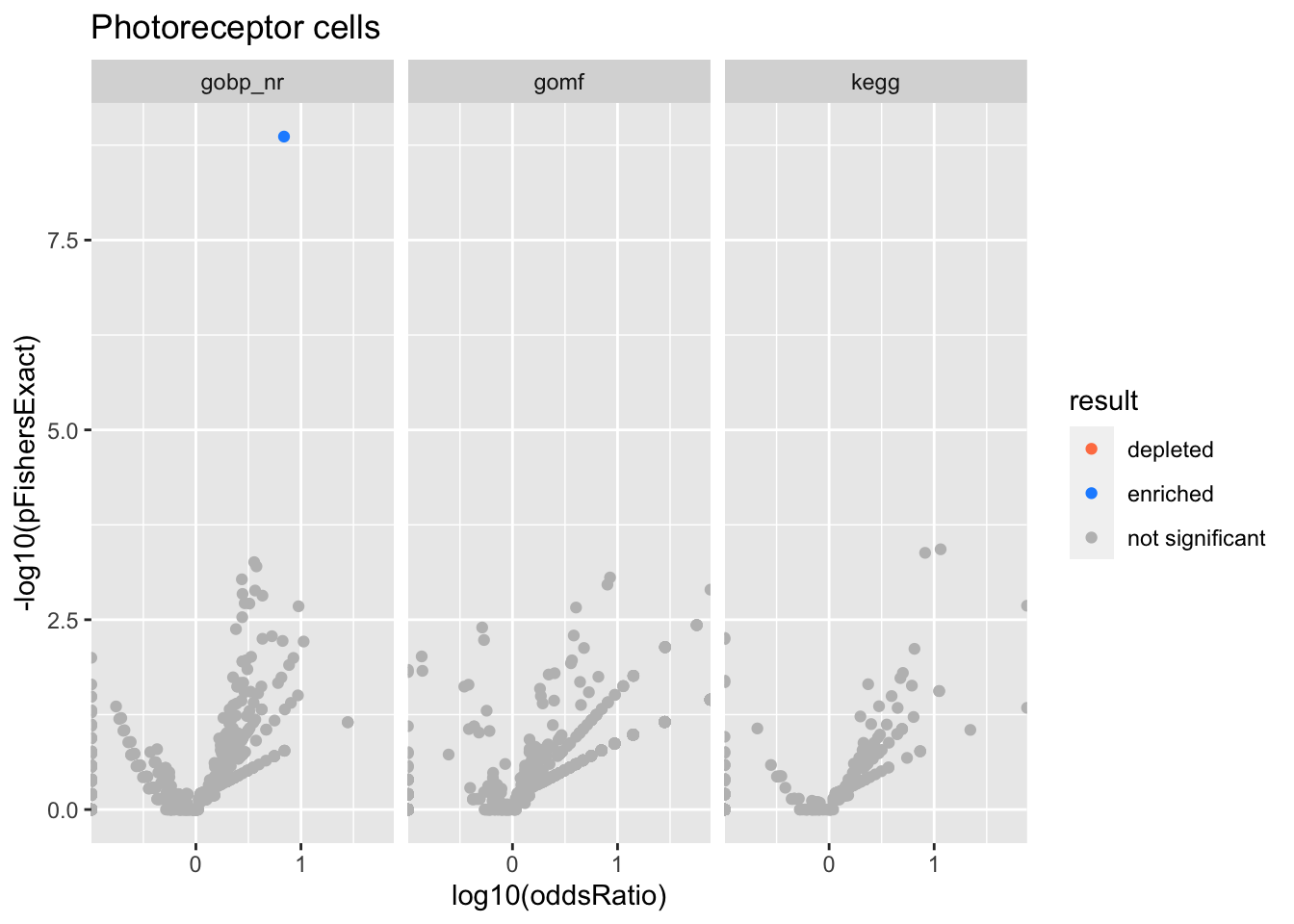
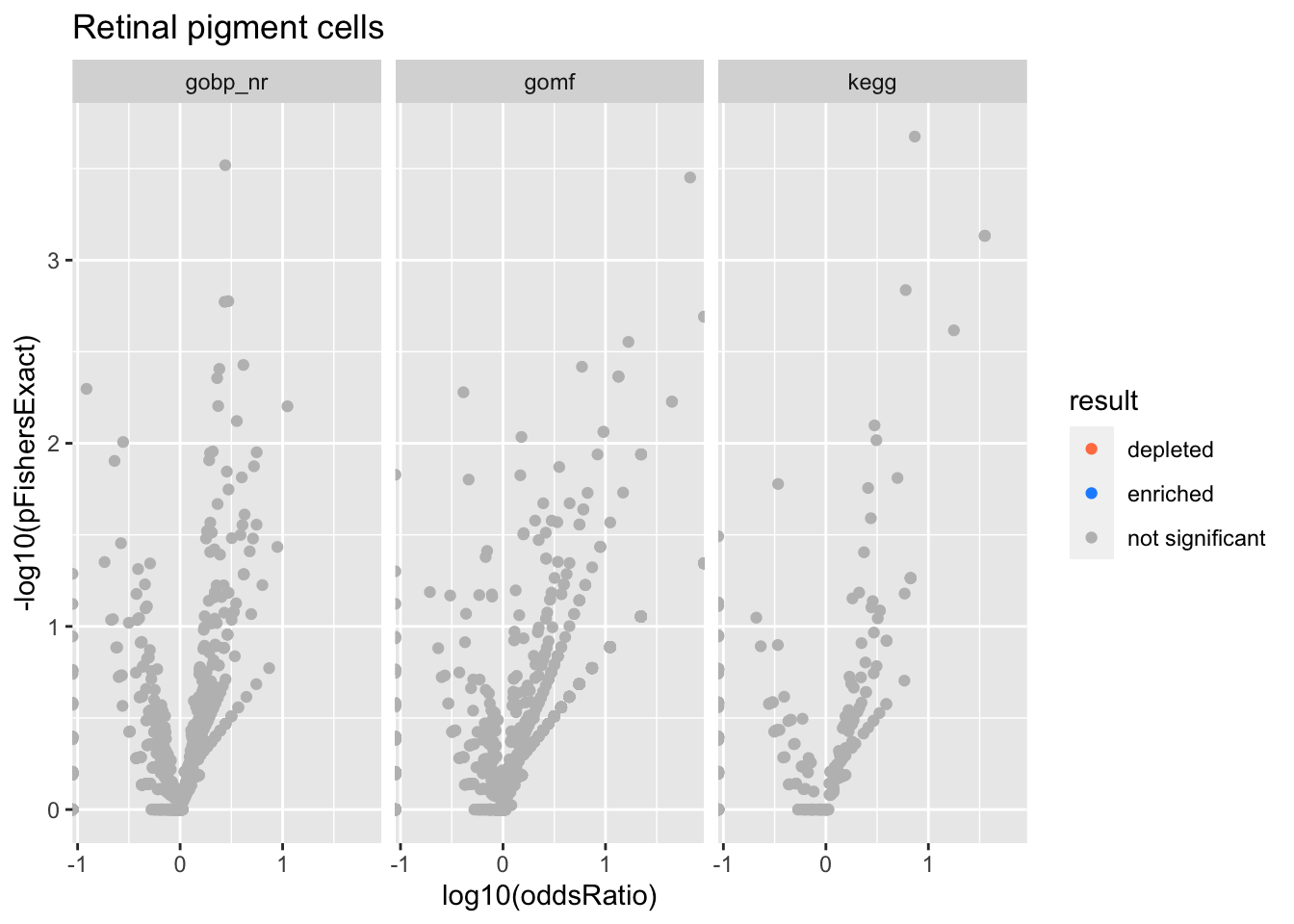
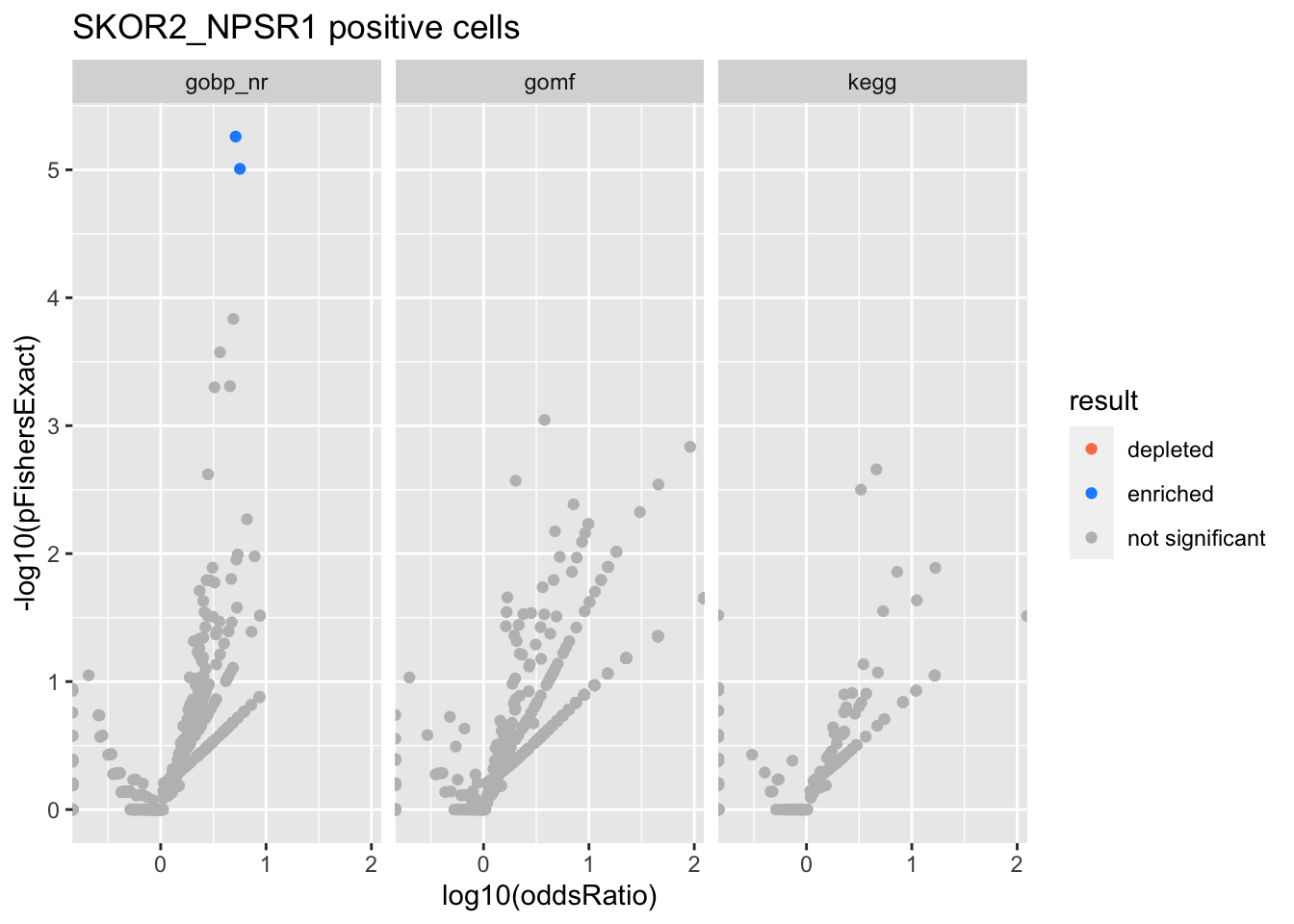
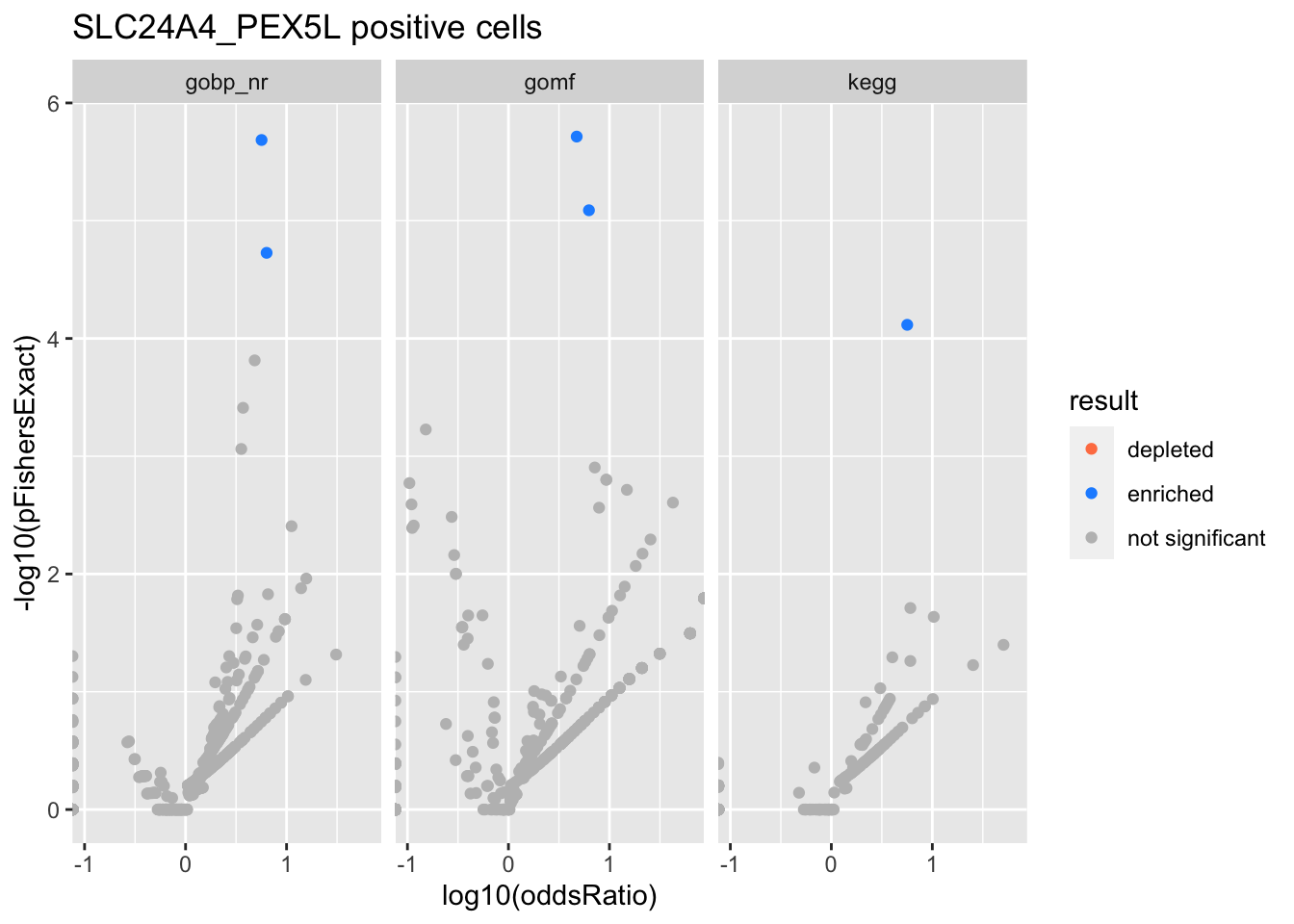
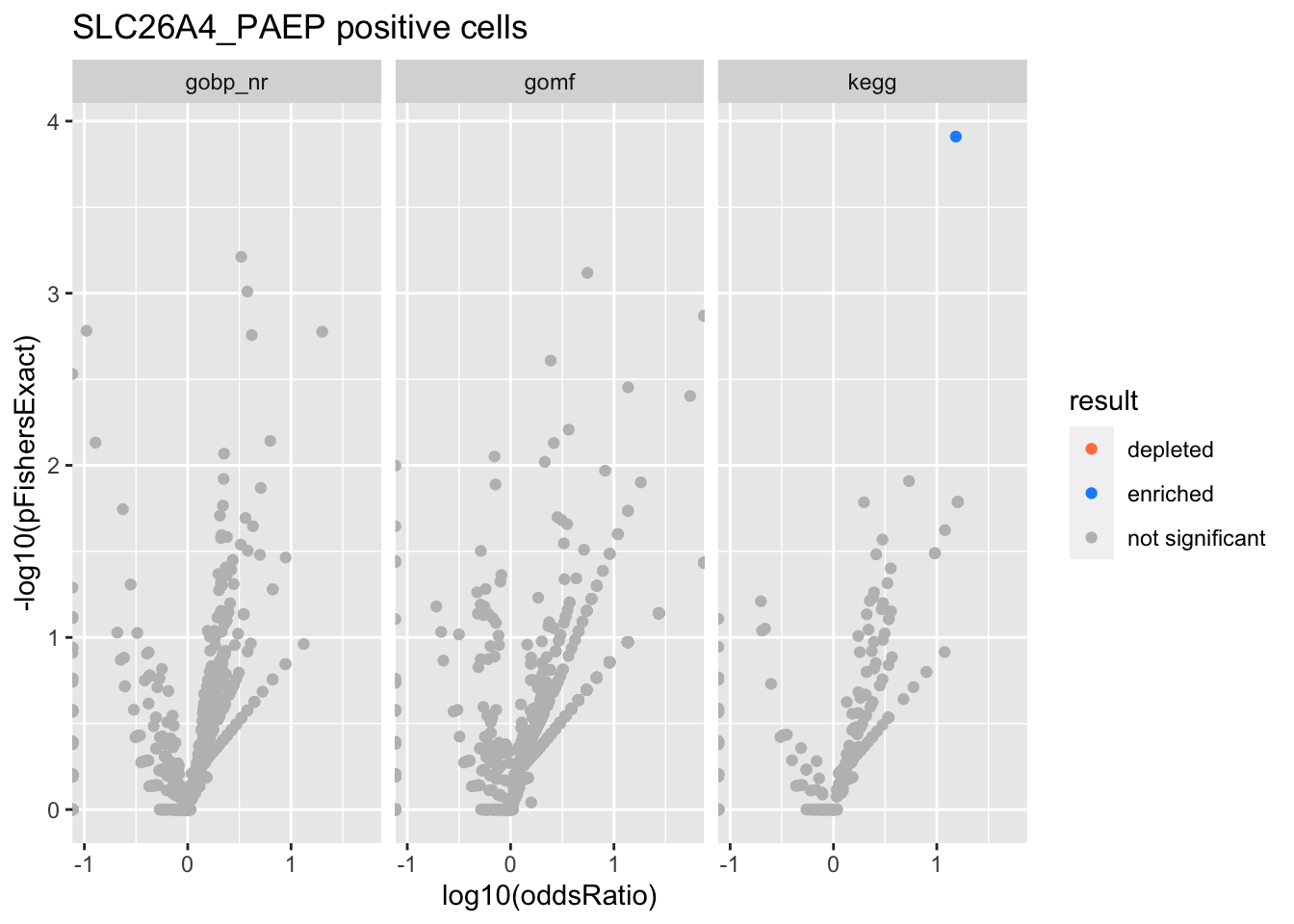
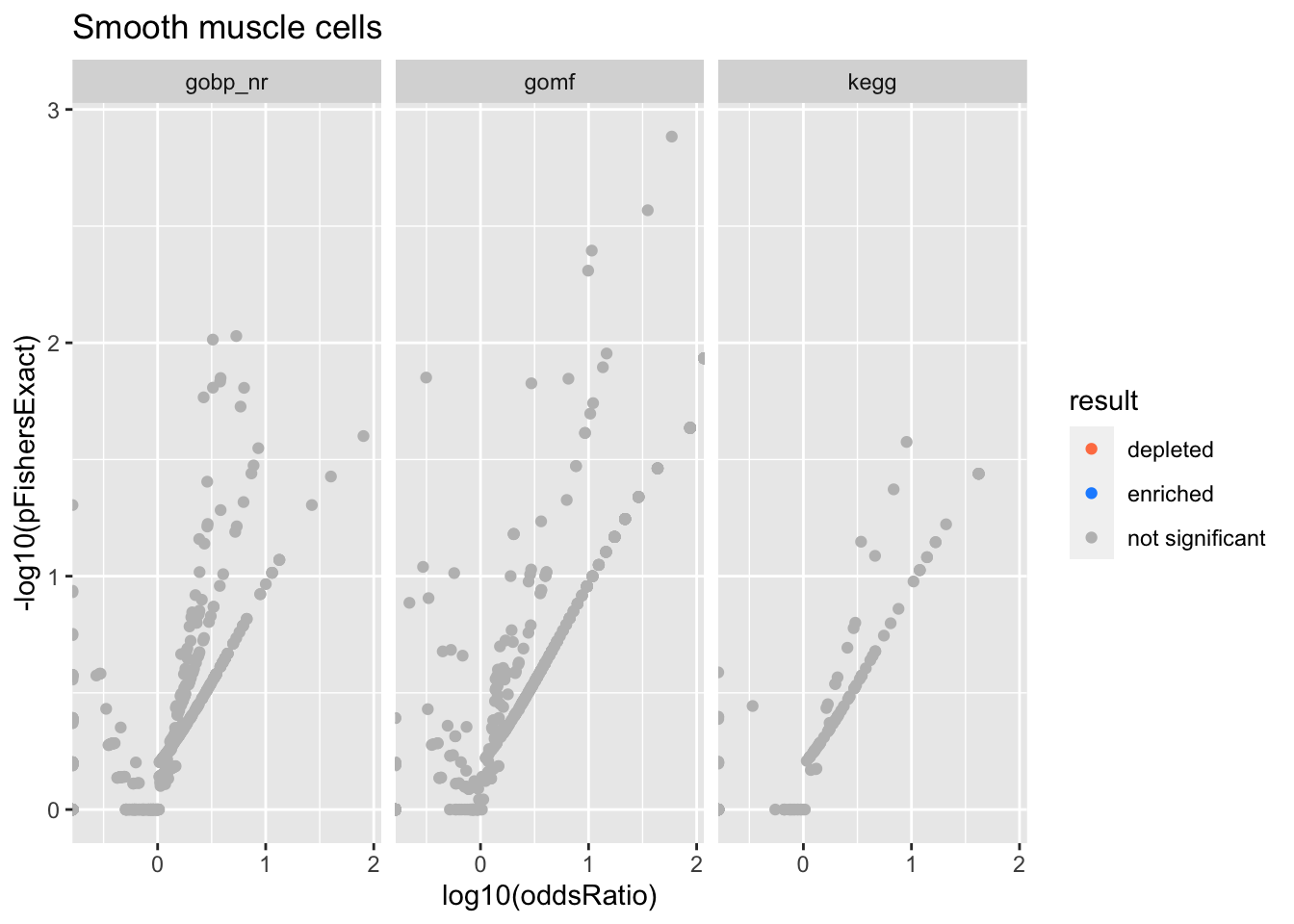
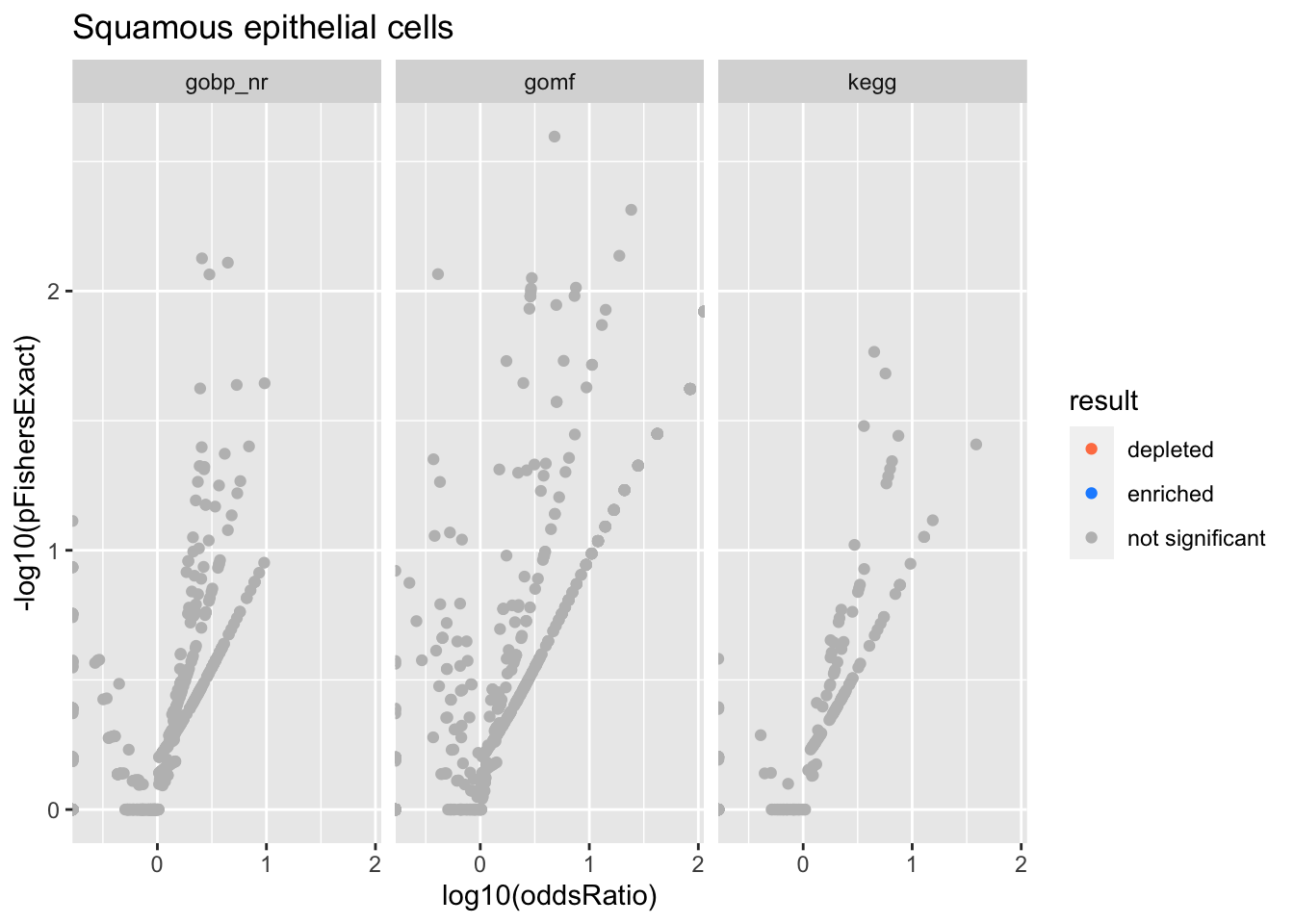
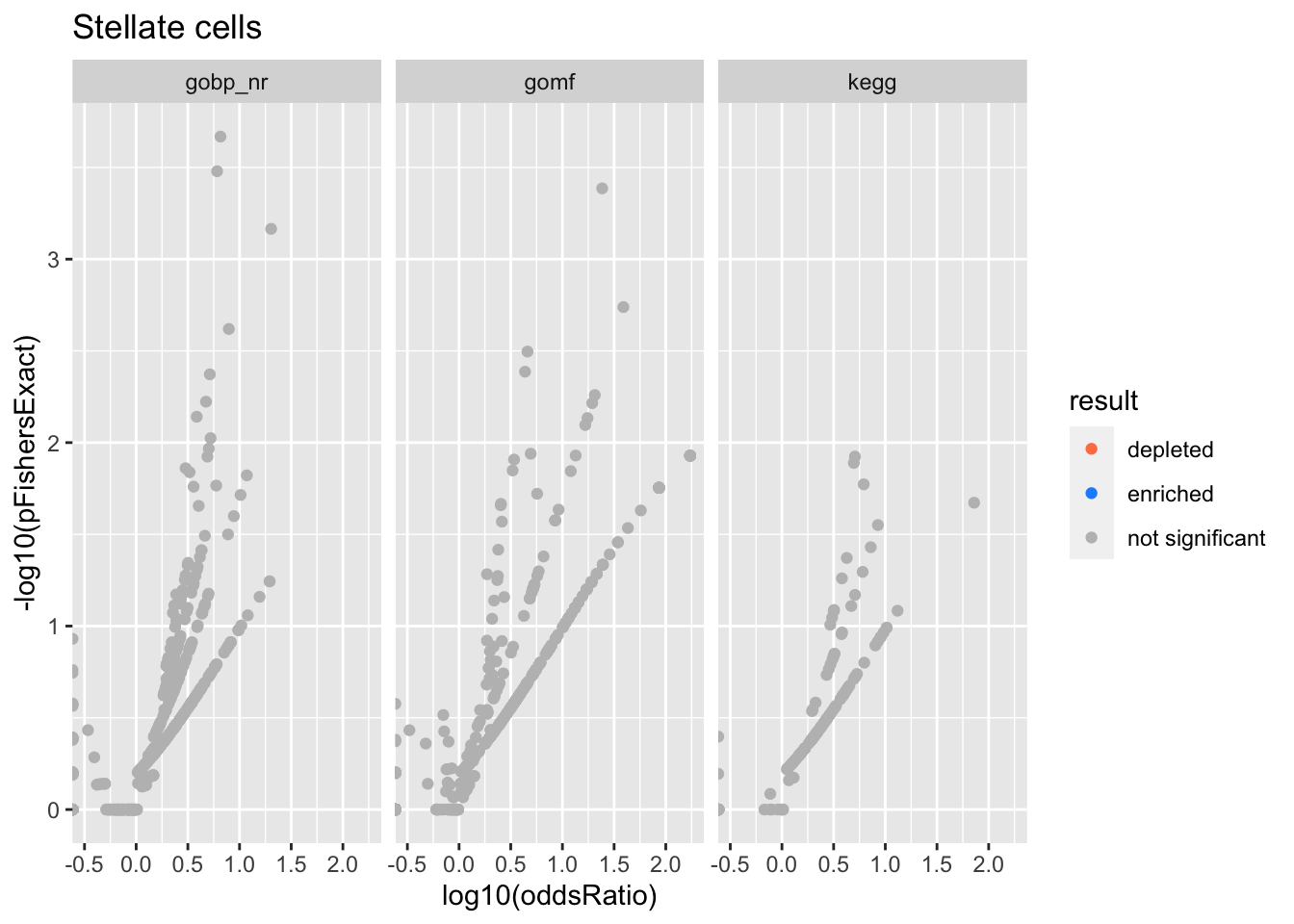
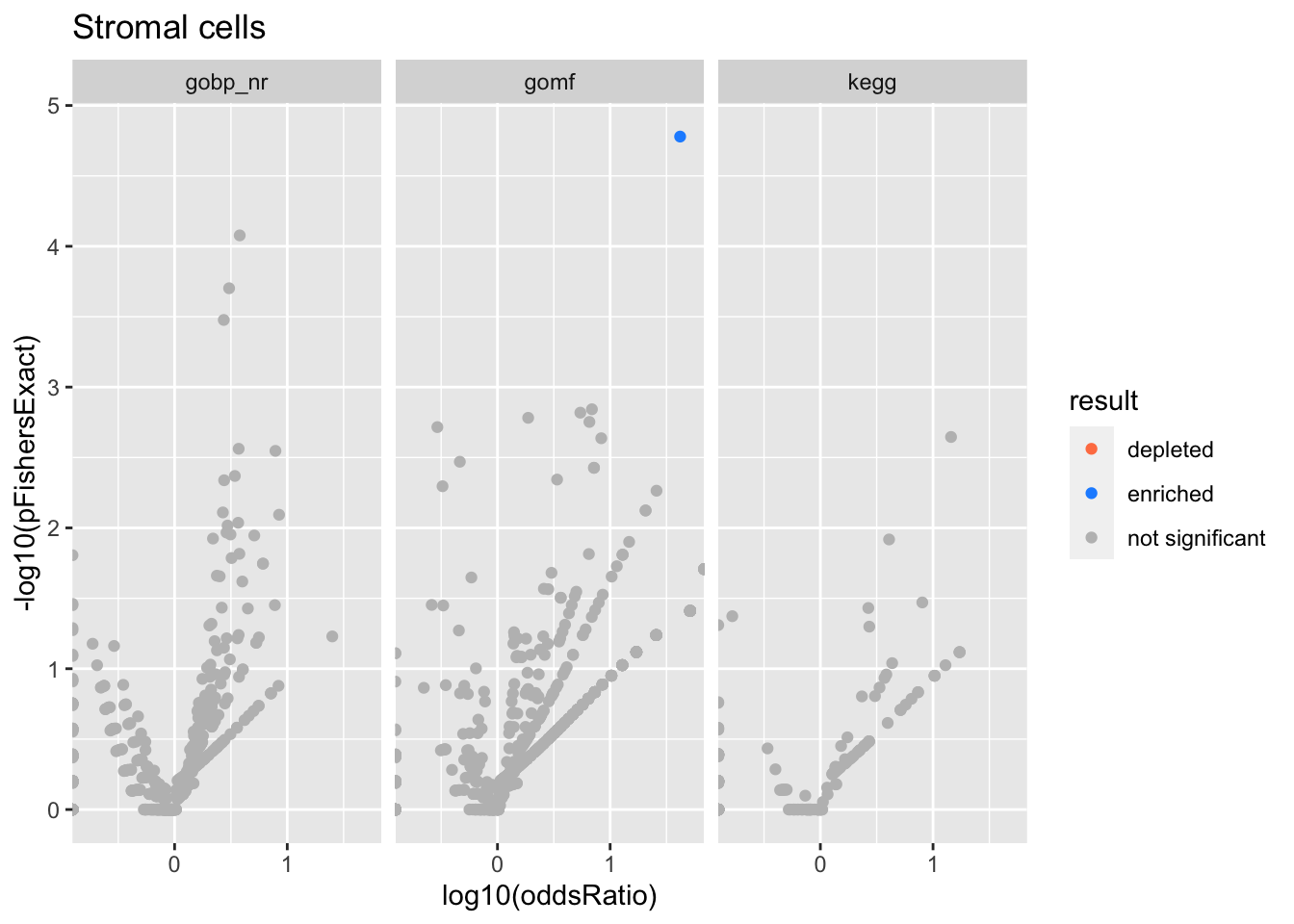
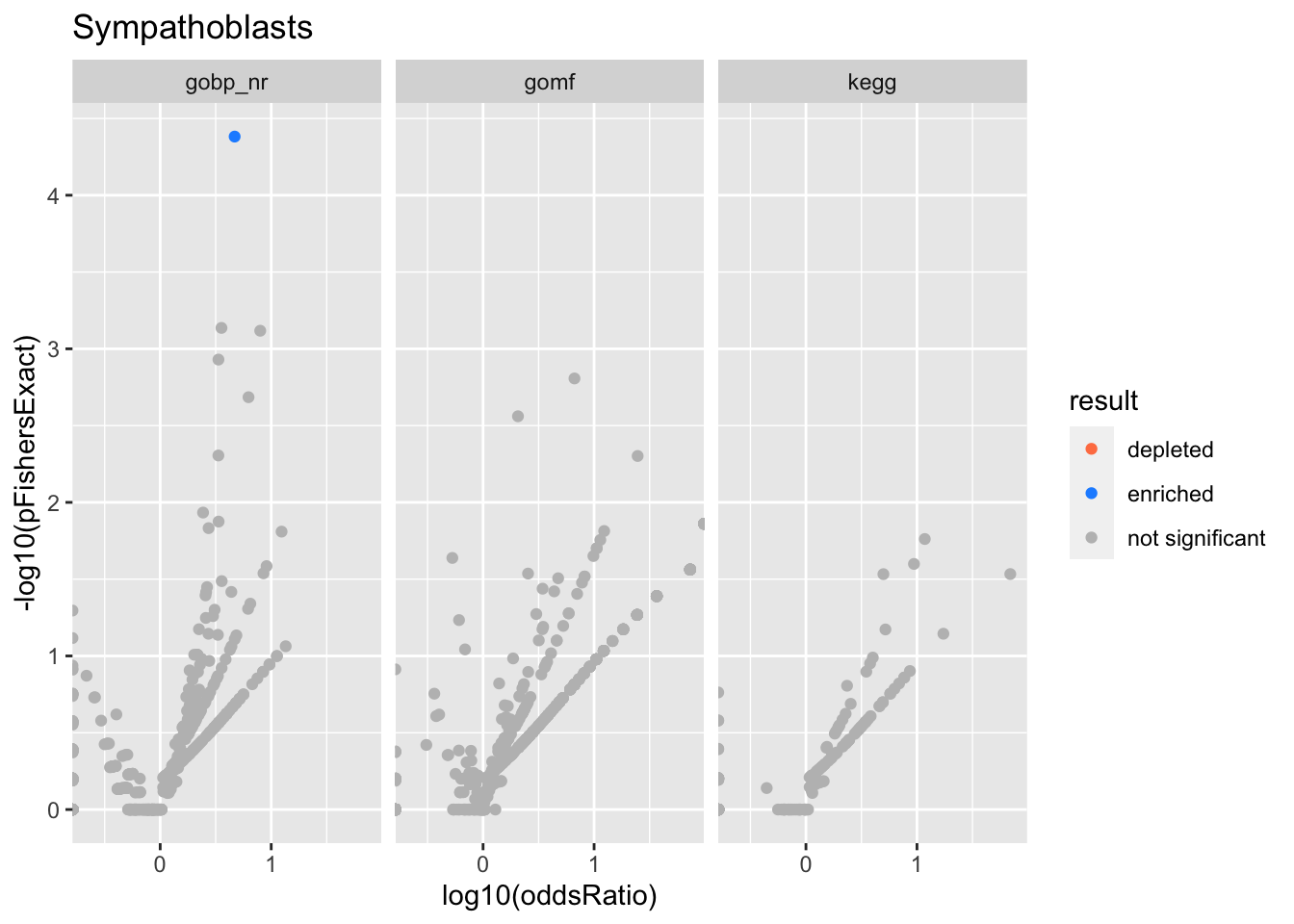
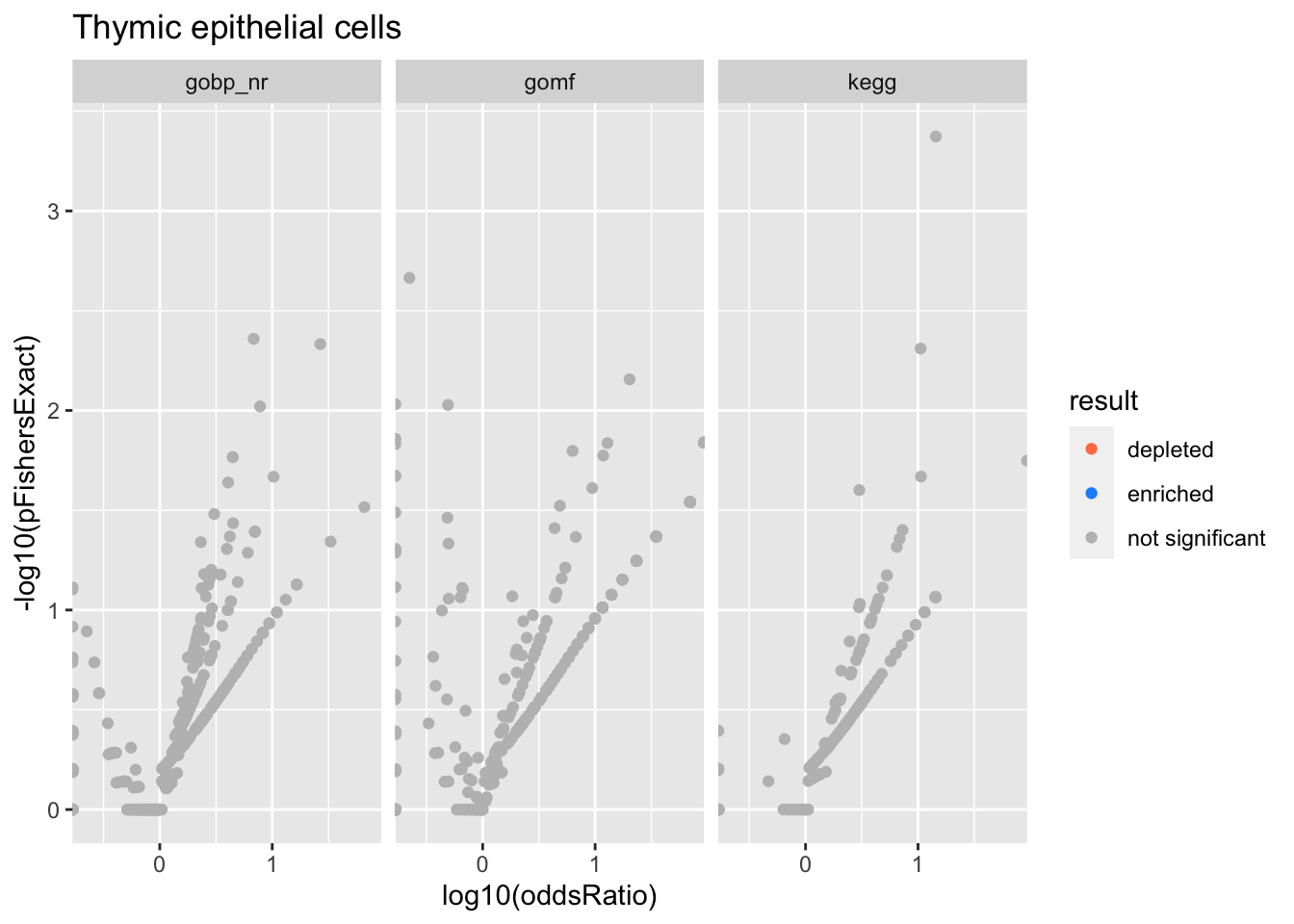
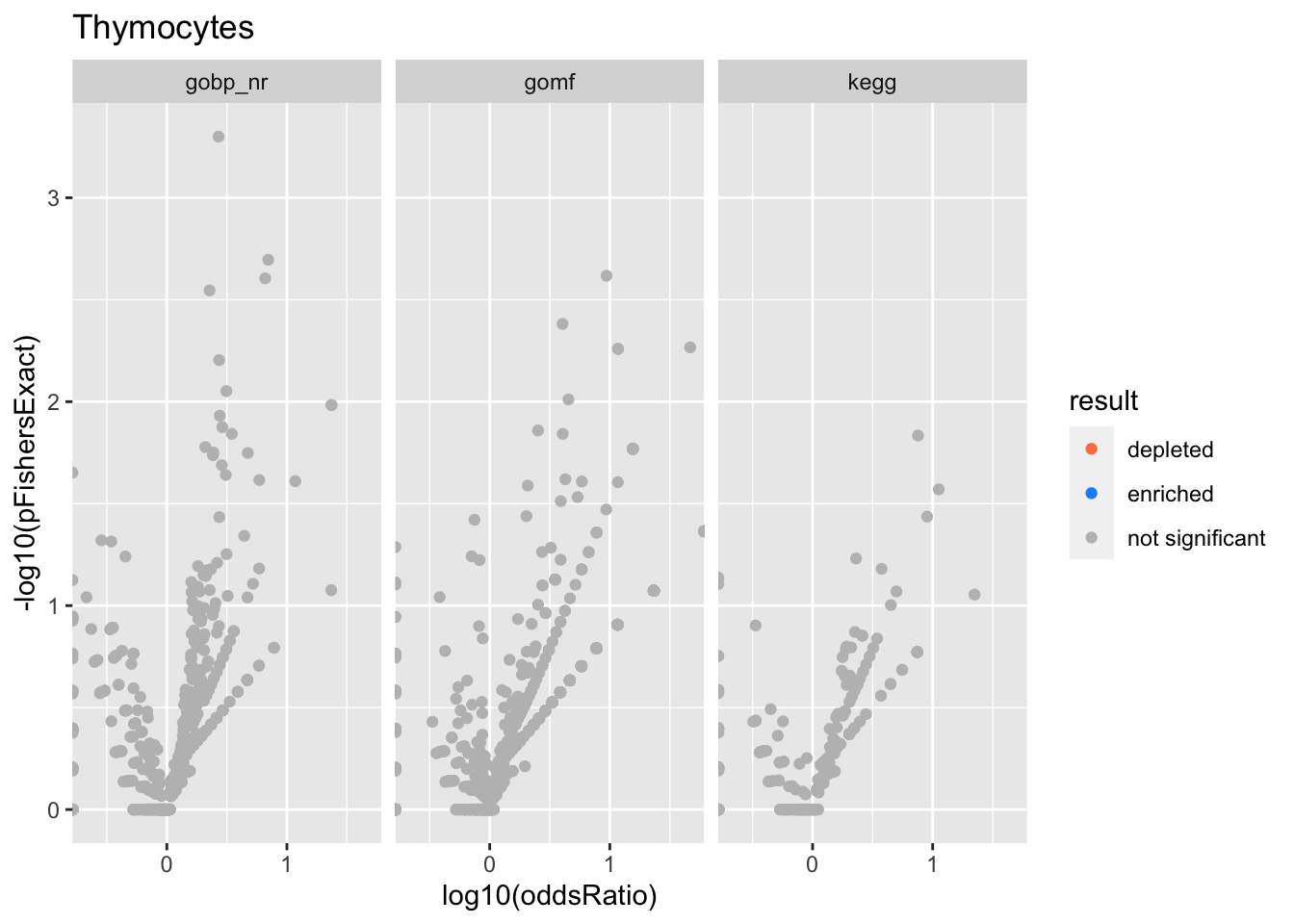
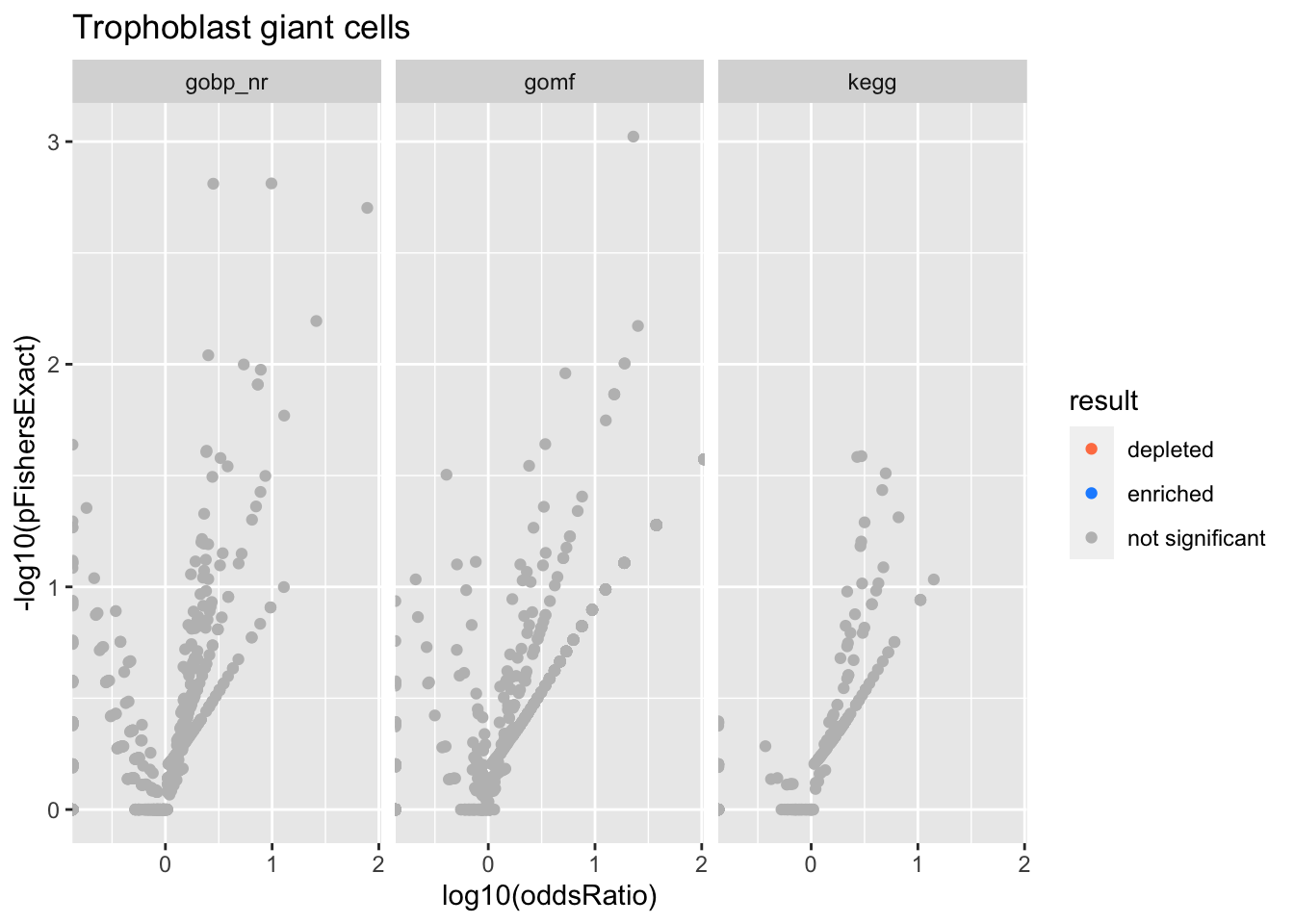
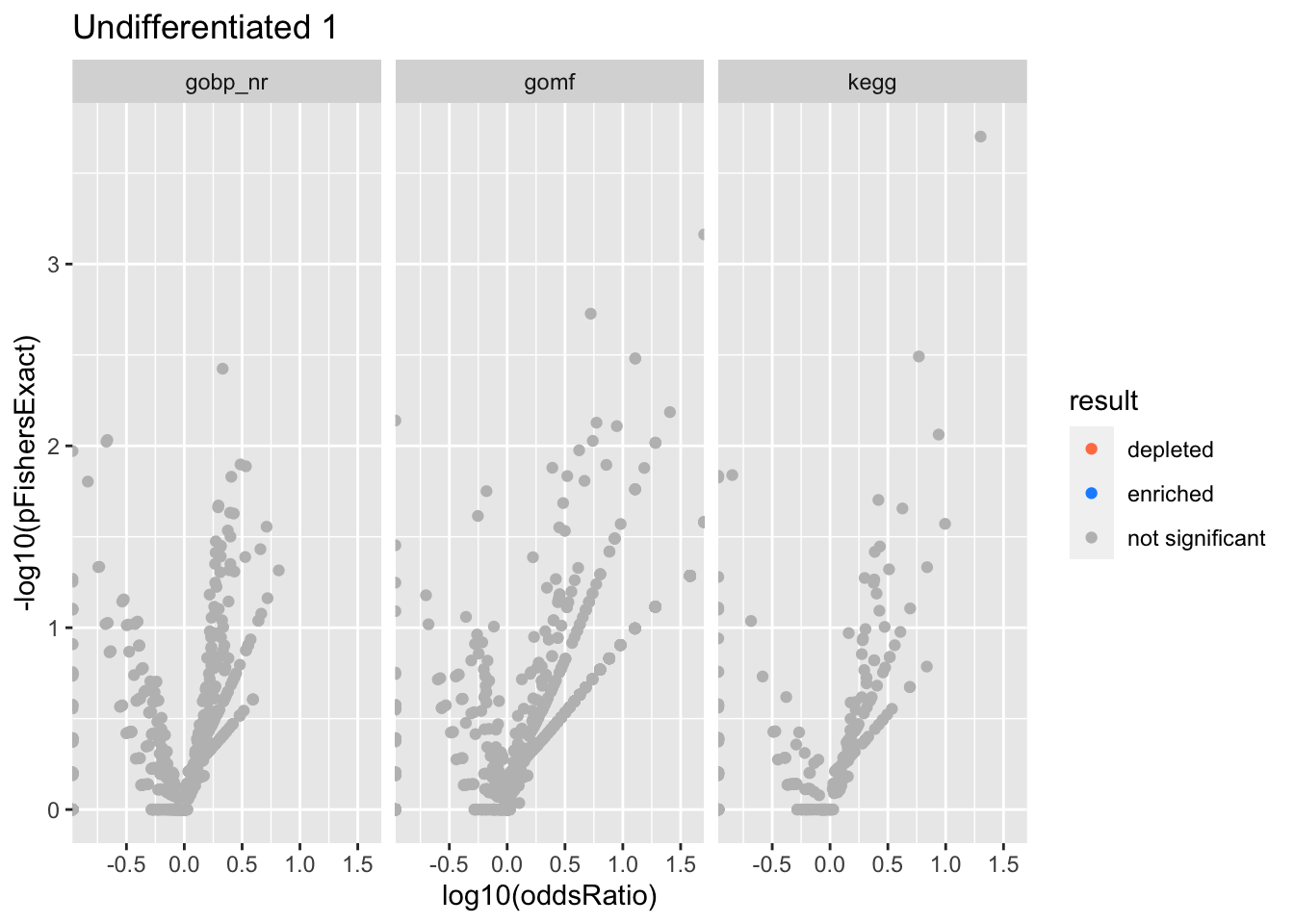
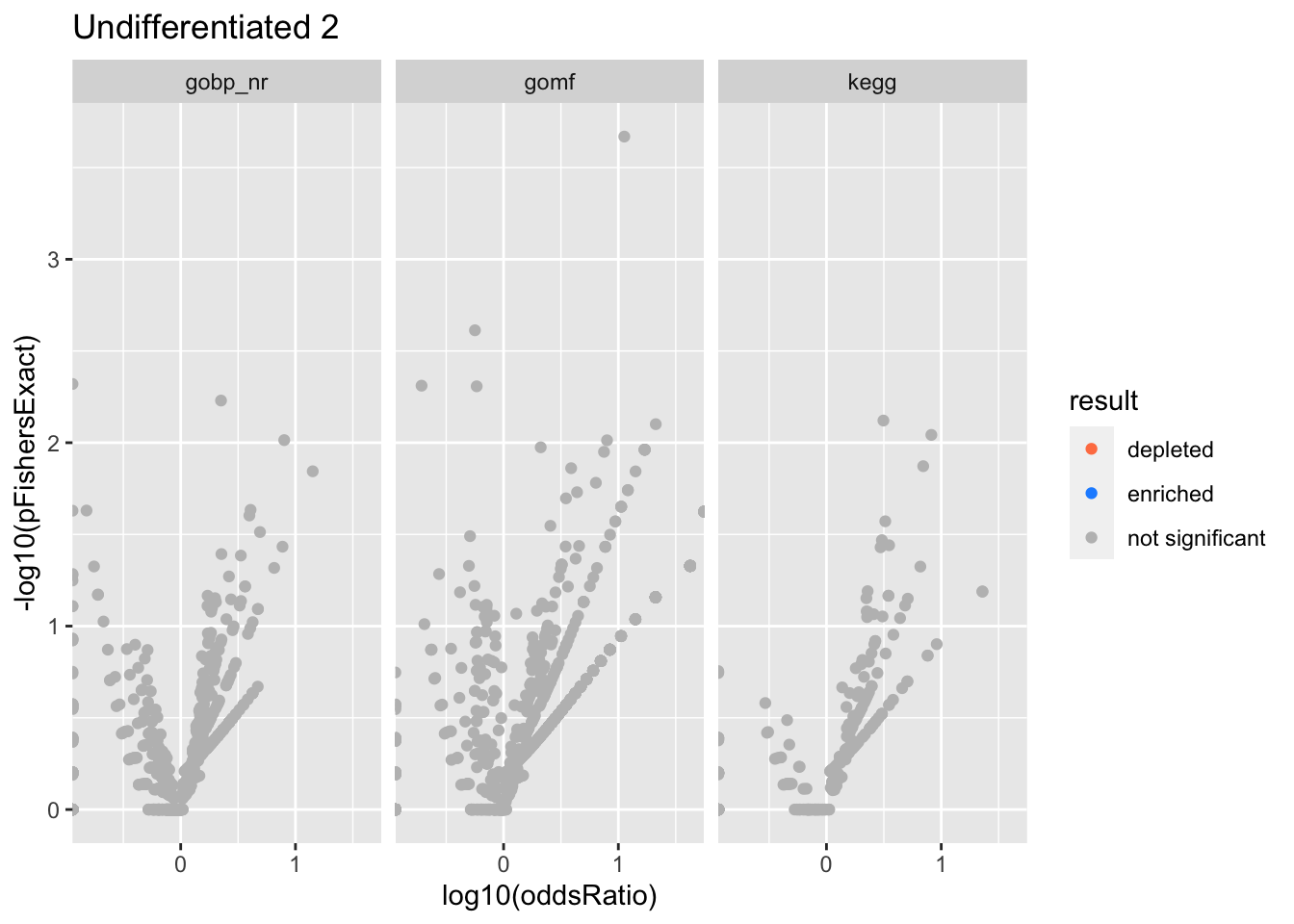
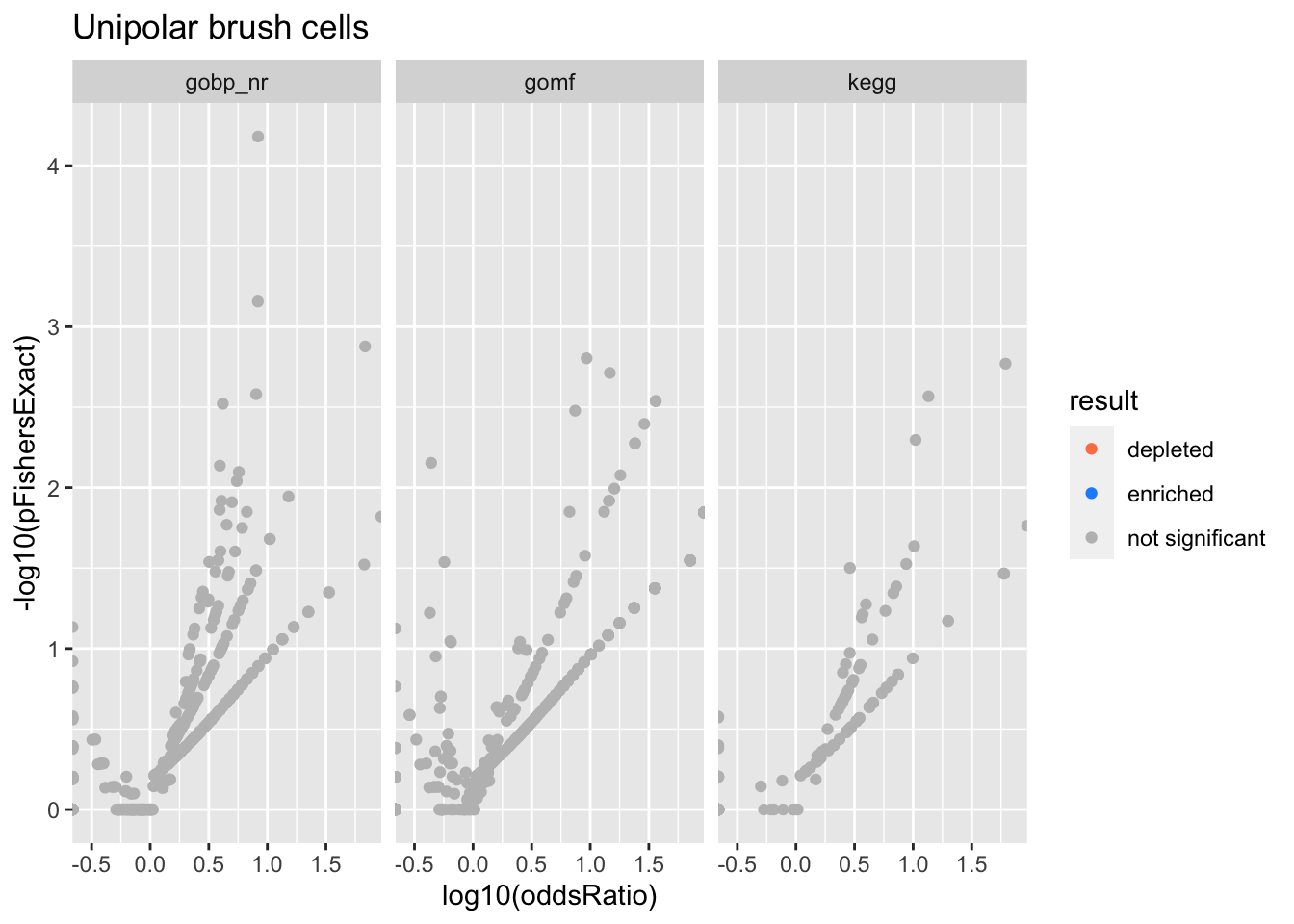
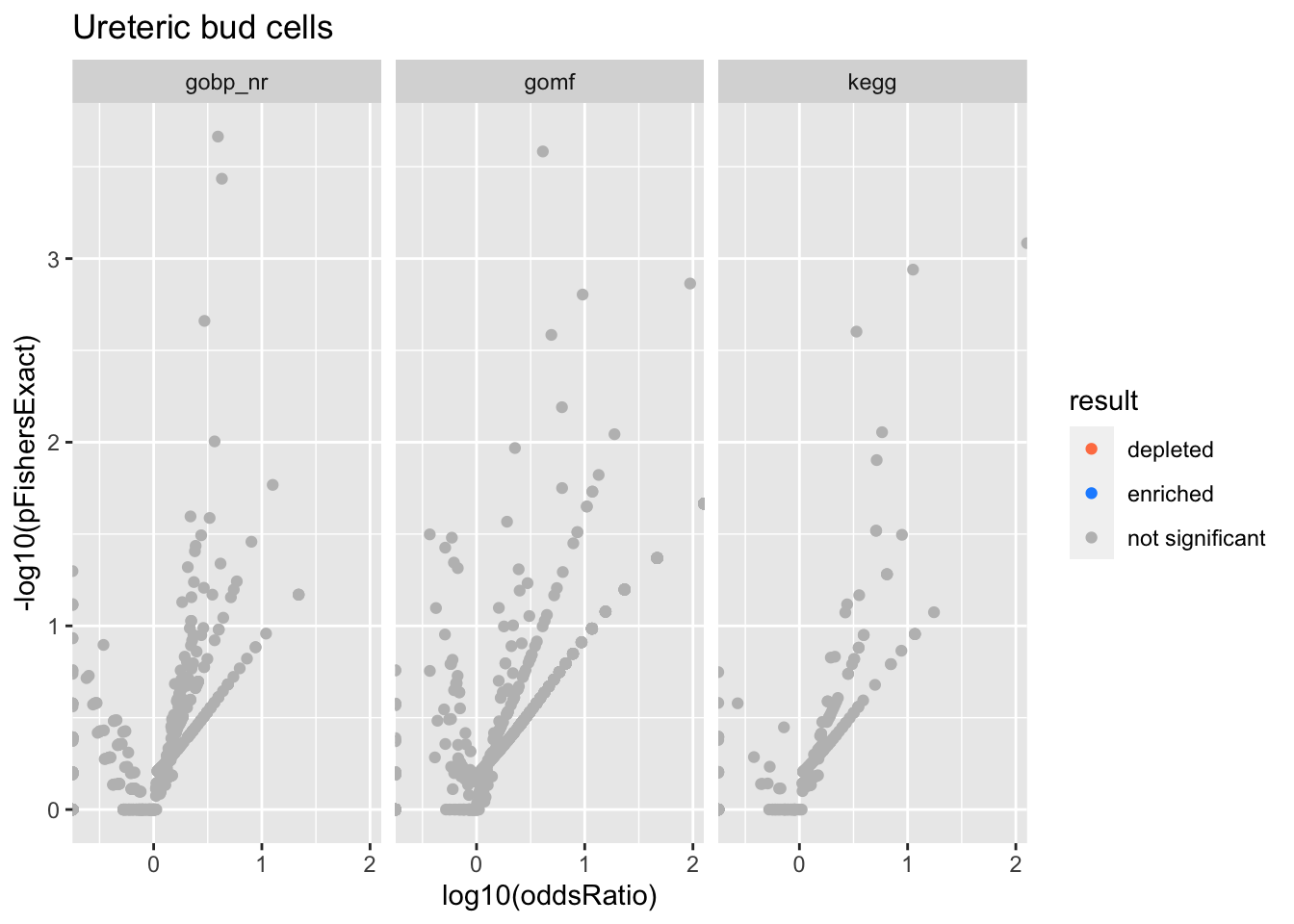
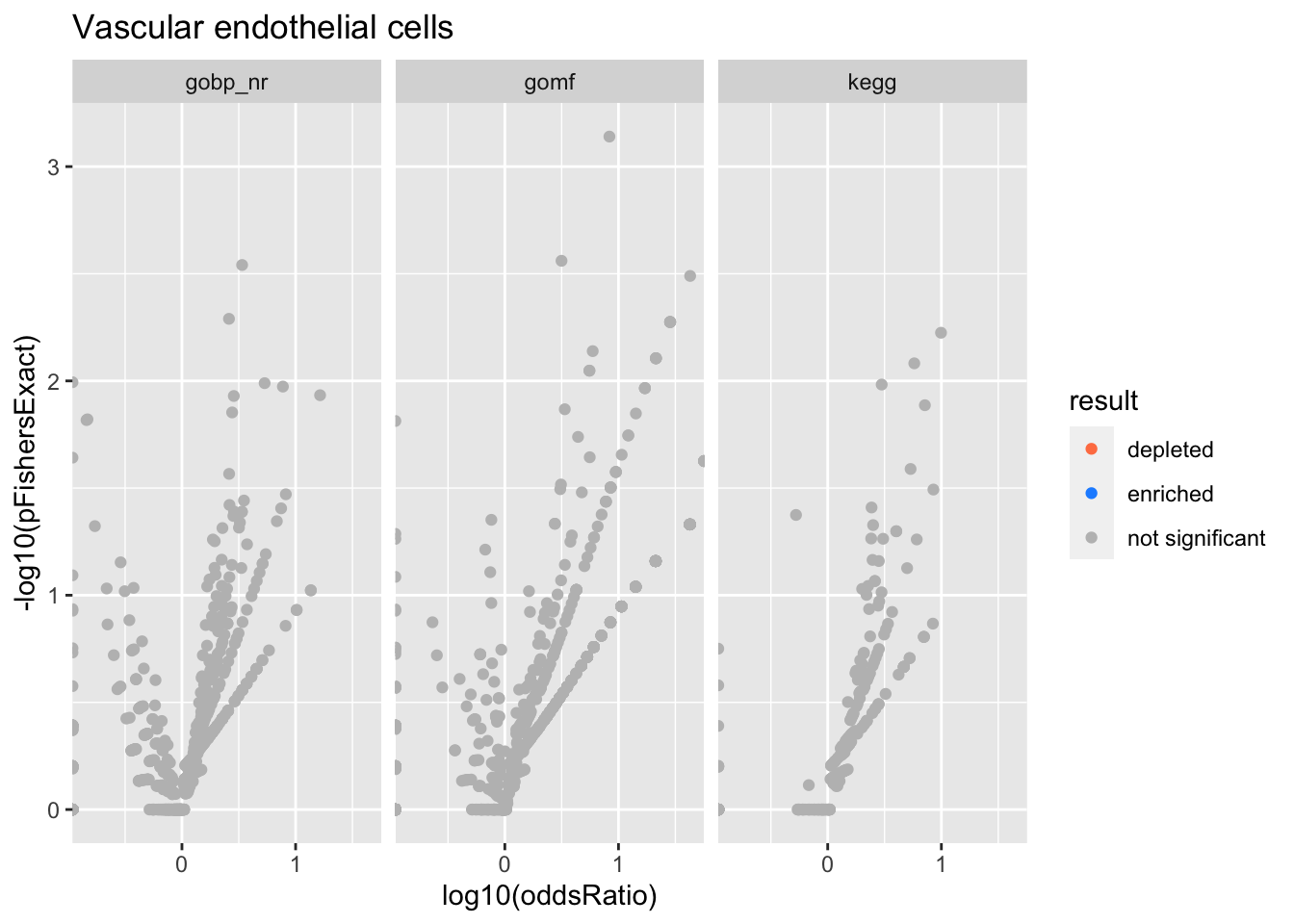
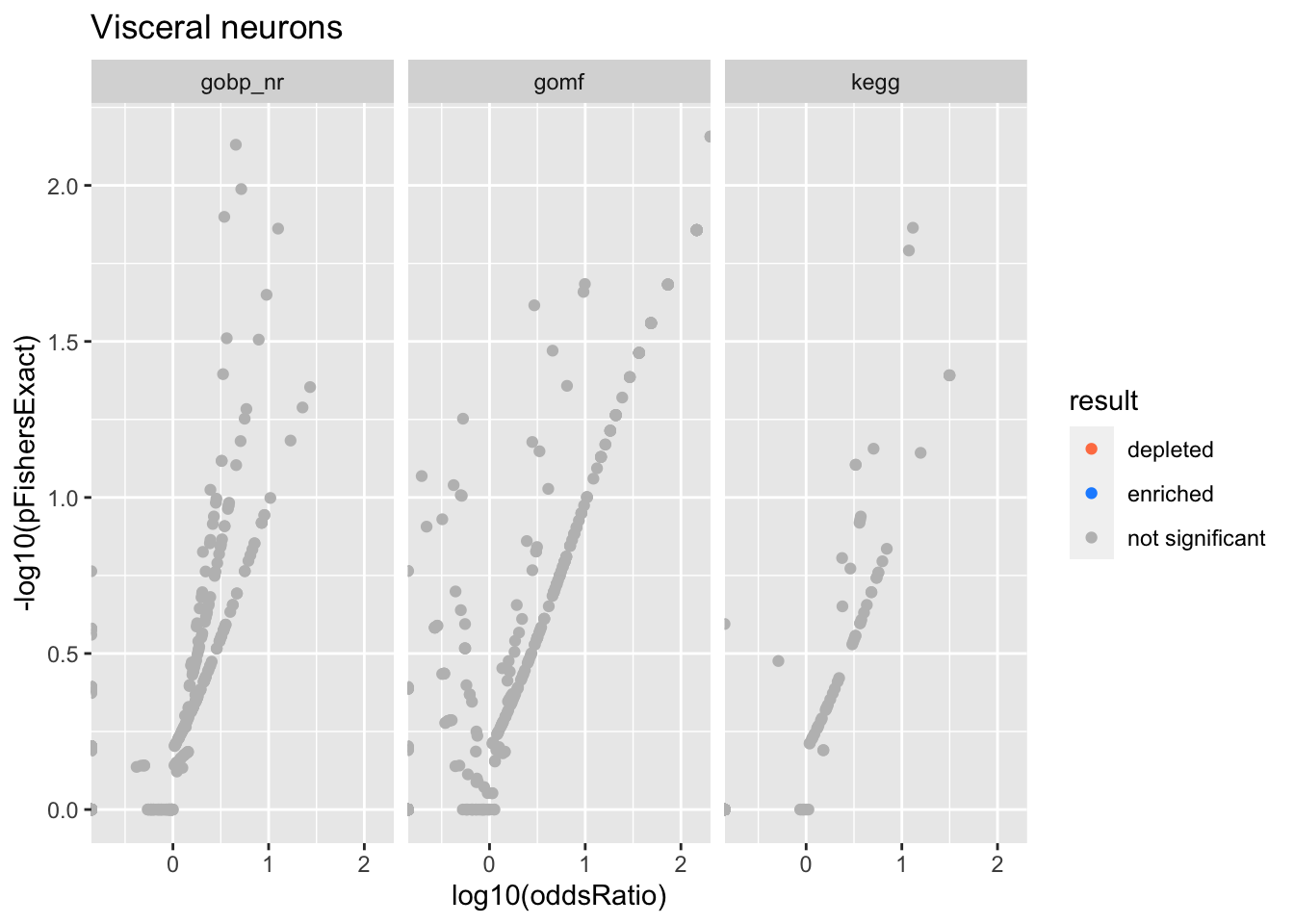
Colors represent enrichment/depletion detected by Fishers exact test (Benjamini Hochberg corrected p-values < \(0.05\)). Gene sets that belong to a SuSiE credible set are circled.
get_ora_enrichments = function(tbl){
tbl %>% mutate(
padj = p.adjust(pFishersExact),
result = case_when(
padj < 0.05 & oddsRatio < 1 ~ 'depleted',
padj < 0.05 & oddsRatio > 1 ~ 'enriched',
TRUE ~ 'not significant'
)
)
}
# plot all enrichments, highlight gene sets in credible set
csdat <- res2 %>%
filter(in_cs, active_cs)
res %>%
group_by(experiment, db) %>%
get_ora_enrichments %>%
ggplot(aes(x=log10(oddsRatio), y=-log10(pFishersExact), color=result)) +
geom_point() +
geom_point(
csdat,
mapping=aes(x=log10(oddsRatio), y=-log10(pFishersExact)),
color='black', pch=21, size=5) +
scale_color_manual(values = c('depleted' = 'coral',
'enriched' = 'dodgerblue',
'not significant' = 'grey')) +
facet_wrap(vars(db))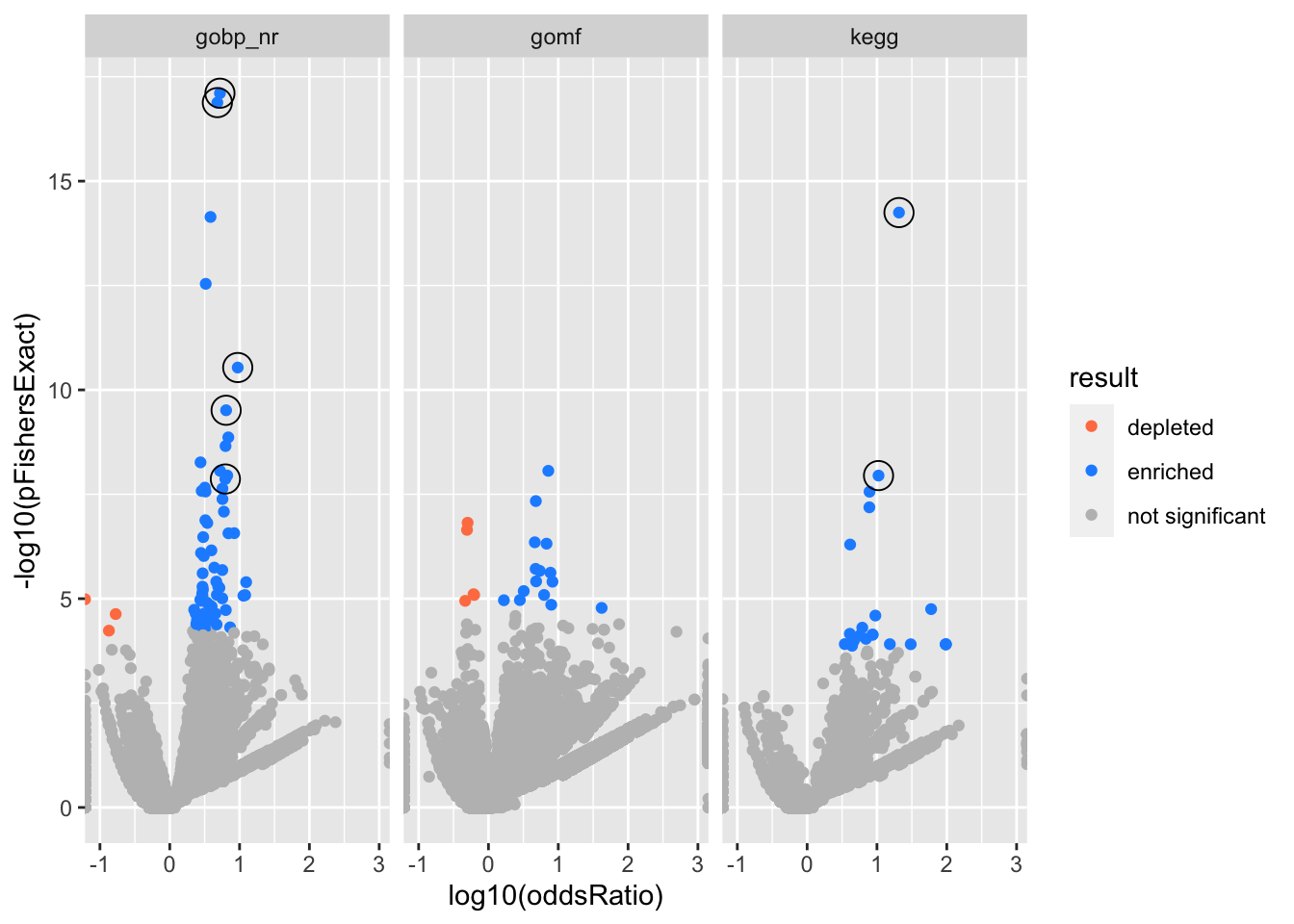
Enrichment results
Glossary
alphais the posterior probability of SuSiE including this gene set in this component which is different from PIP (probability of SuSiE including this gene set in ANY component)betaposterior mean/standard error of posterior mean for effect size. Standard errors are likely too small.oddsRatio, pHypergeometric, pFishersExactconstruct a contingency table (gene list membersip) x (gene set membership), estimate theoddsRatiogives the odds of being in the gene list conditional on being in the gene set / odds of being in the gene list conditional on NOT being in the gene set.pHypergeometricandpFishersExactare pvalues from 1 and 2 sided test respectively.
experiments <- unique(res$experiment)
do.experiment.volcano = function(this_experiment){
res %>%
filter(experiment == this_experiment) %>%
group_by(db) %>%
get_ora_enrichments %>%
ggplot(aes(x=log10(oddsRatio), y=-log10(pFishersExact), color=result)) +
geom_point() +
geom_point(
csdat %>% filter(experiment == this_experiment),
mapping=aes(x=log10(oddsRatio), y=-log10(pFishersExact)),
color='black', pch=21, size=5) +
scale_color_manual(values = c('depleted' = 'coral',
'enriched' = 'dodgerblue',
'not significant' = 'grey')) +
facet_wrap(vars(db)) +
labs(title = this_experiment)
}
for (i in 1:length(experiments)){
this_experiment <- experiments[i]
cat("\n")
cat("###", this_experiment, "\n") # Create second level headings with the names.
do.experiment.volcano(this_experiment) %>% print()
cat("\n\n")
for(db in names(html_tables[[this_experiment]])){
cat("####", db, "\n") # Create second level headings with the names.
to_print <- html_tables[[this_experiment]][[db]] %>% distinct()
to_print %>% report_susie_credible_sets() %>% htmltools::HTML() %>% print()
cat("\n")
}}Acinar cells
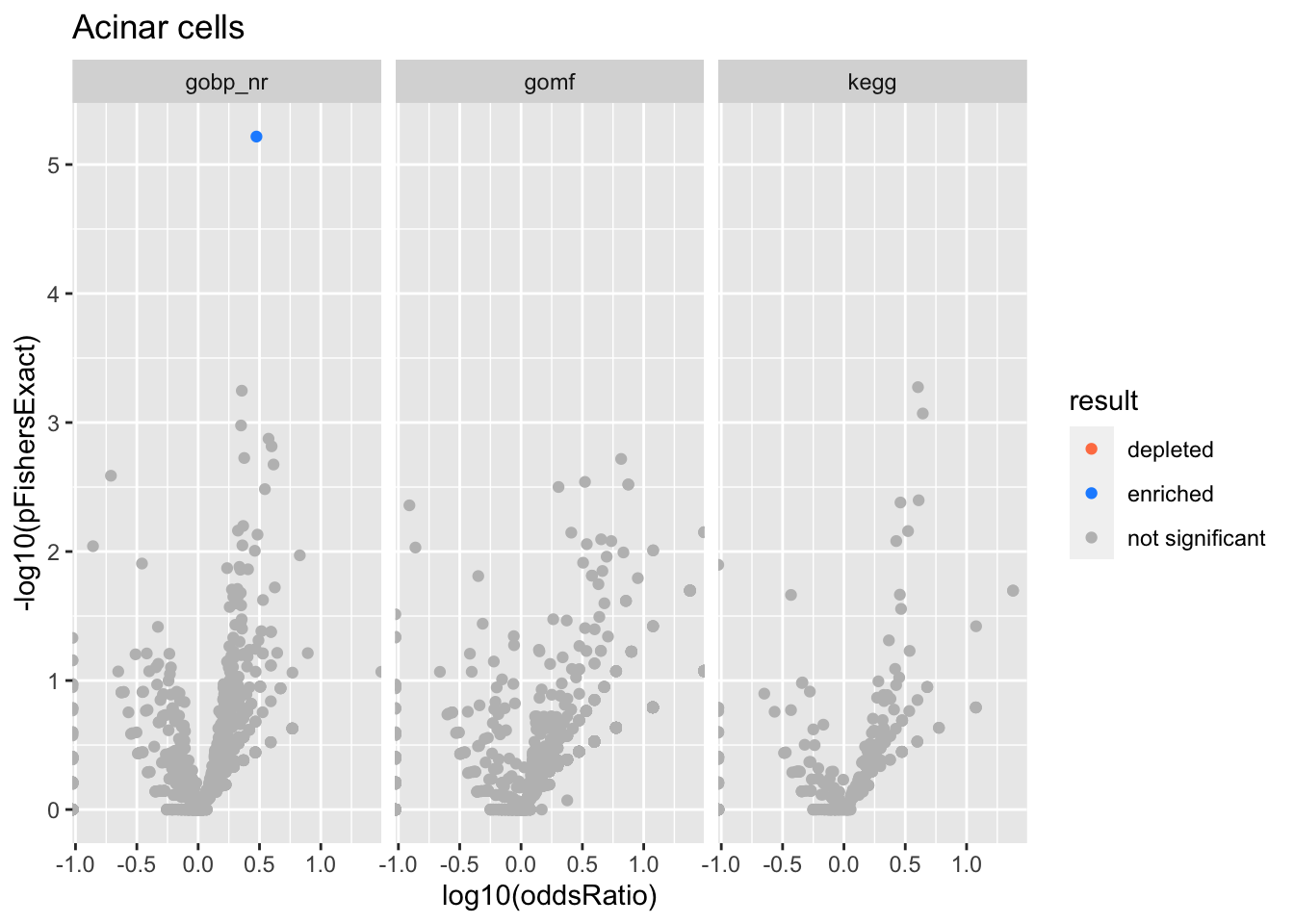
Adrenocortical cells
Amacrine cells
Antigen presenting cells
Astrocytes
Bipolar cells
Bronchiolar and alveolar epithelial cells
Cardiomyocytes
Chromaffin cells
Ciliated epithelial cells
CLC_IL5RA positive cells
Corneal and conjunctival epithelial cells
Ductal cells
Early Ectoderm 1
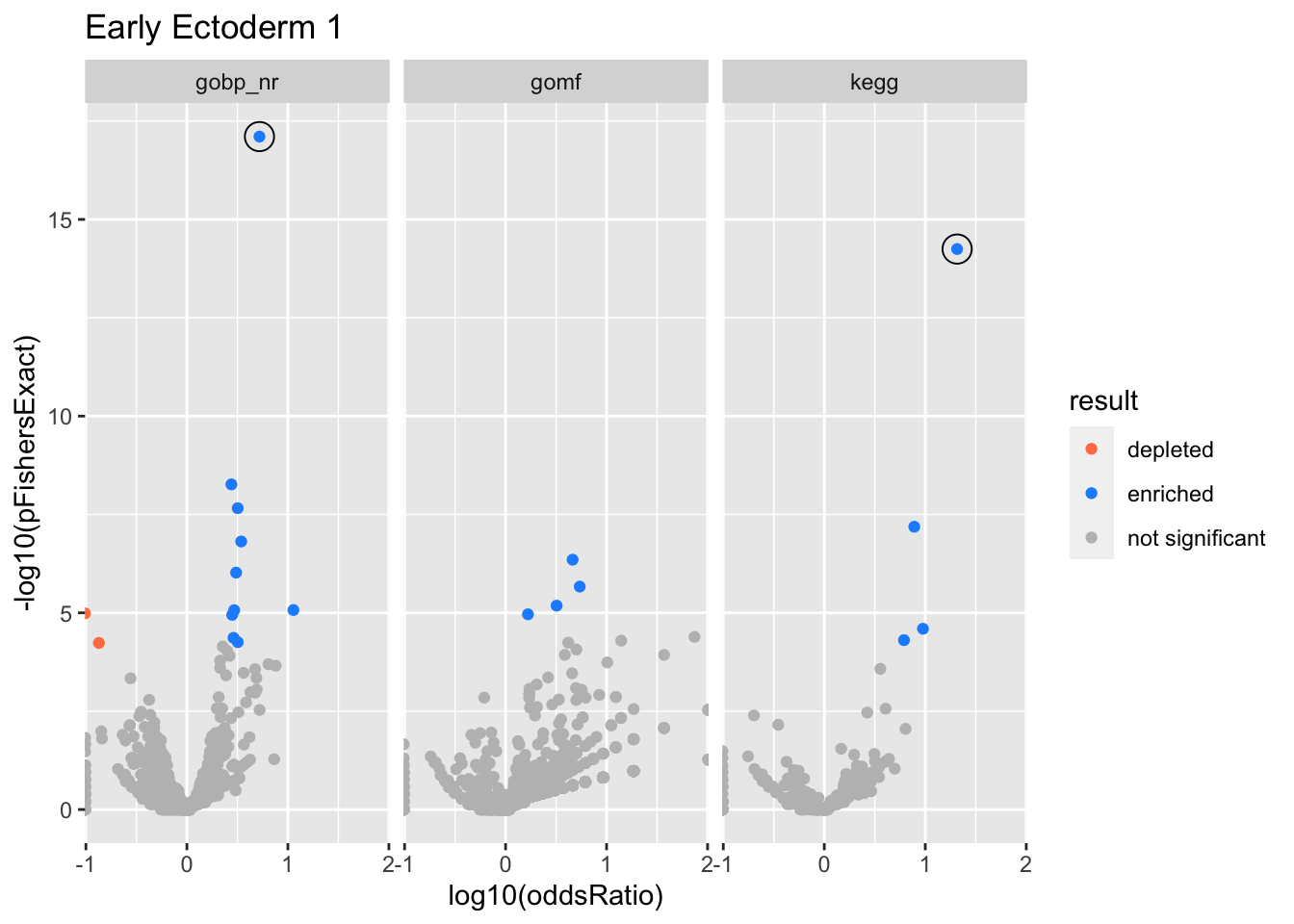
gobp_nr
| geneSet | description | alpha | beta | beta.se | pHypergeometric | pFishersExact | overlap | geneSetSize | oddsRatio |
|---|---|---|---|---|---|---|---|---|---|
| L1 | |||||||||
| GO:0006260 | DNA replication | 1 | 1.58 | 0.134 | 7.85e-18 | 7.85e-18 | 53 | 252 | 5.23 |
kegg
| geneSet | description | alpha | beta | beta.se | pHypergeometric | pFishersExact | overlap | geneSetSize | oddsRatio |
|---|---|---|---|---|---|---|---|---|---|
| L1 | |||||||||
| hsa03030 | DNA replication | 1 | 2.93 | 0.333 | 5.67e-15 | 5.67e-15 | 19 | 36 | 20.7 |
Early Ectoderm 2
Early Ectoderm 3
Early Ectoderm 4
Early Endoderm 1
Early Mesoderm 1
ELF3_AGBL2 positive cells
ENS glia
ENS neurons
Epicardial fat cells
Erythroblasts
Extravillous trophoblasts
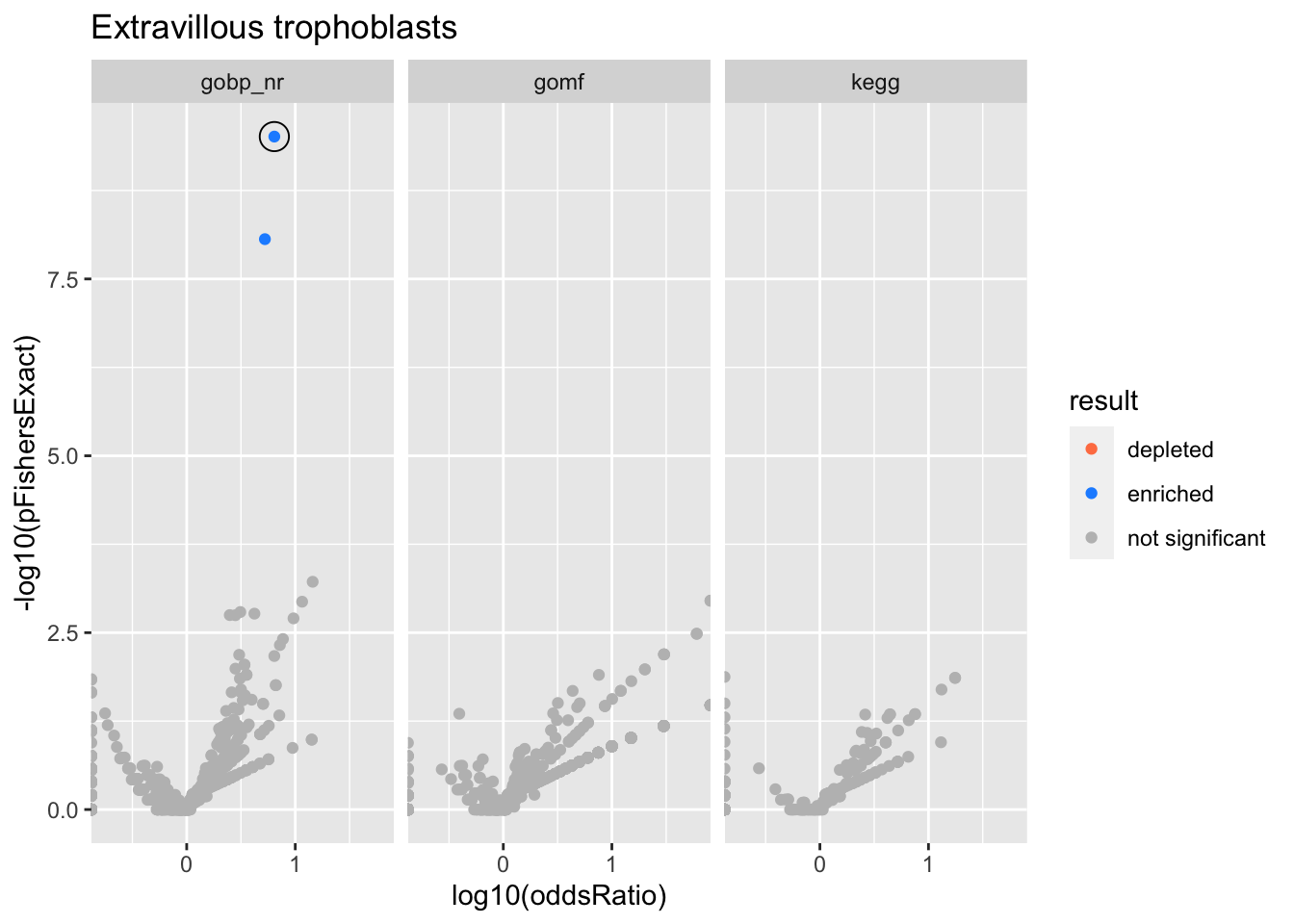
gobp_nr
| geneSet | description | alpha | beta | beta.se | pHypergeometric | pFishersExact | overlap | geneSetSize | oddsRatio |
|---|---|---|---|---|---|---|---|---|---|
| L1 | |||||||||
| GO:0007059 | chromosome segregation | 1 | 1.78 | 0.186 | 3.07e-10 | 3.07e-10 | 23 | 140 | 6.41 |
Ganglion cells
Goblet cells
Granule neurons
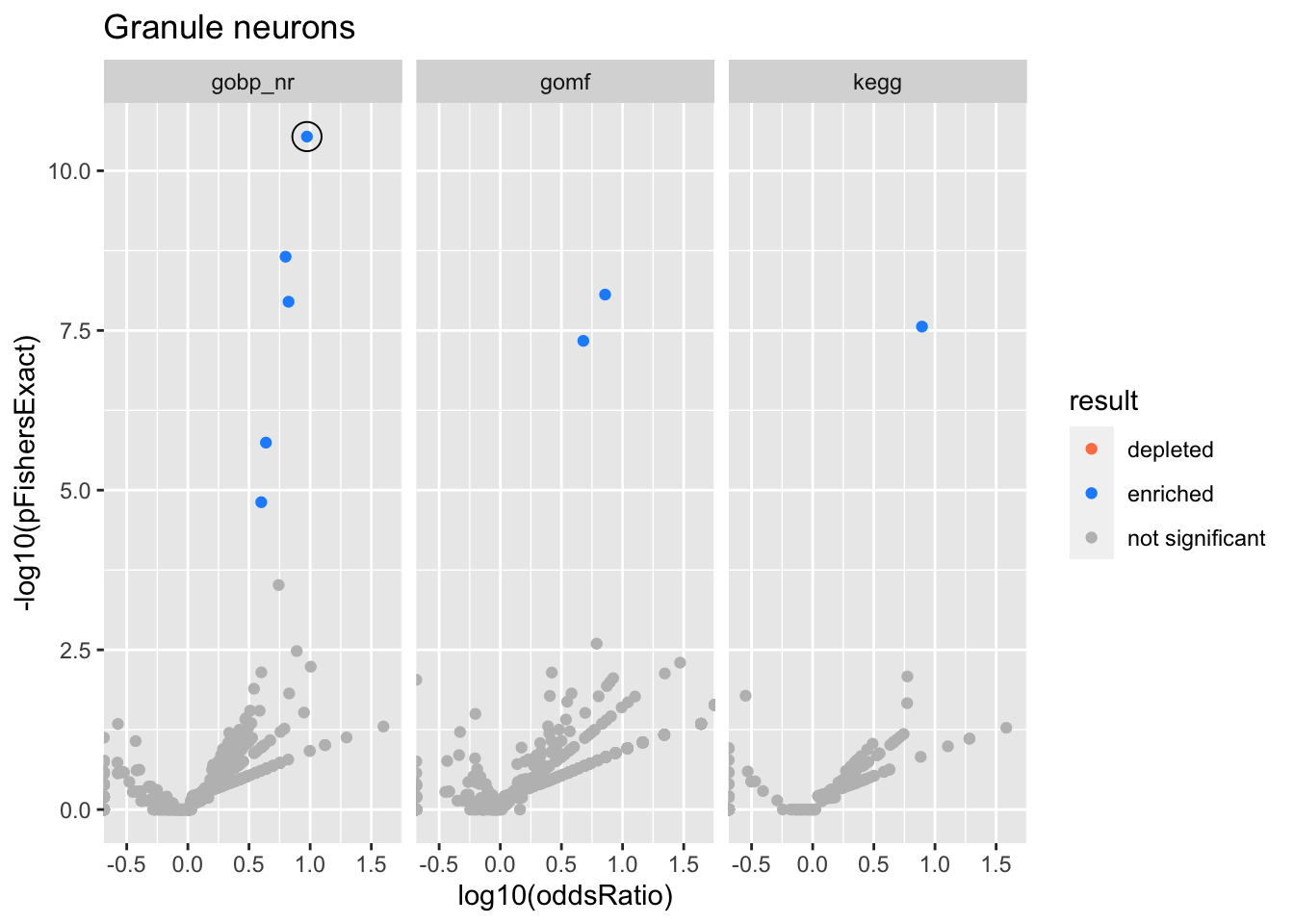
gobp_nr
| geneSet | description | alpha | beta | beta.se | pHypergeometric | pFishersExact | overlap | geneSetSize | oddsRatio |
|---|---|---|---|---|---|---|---|---|---|
| L1 | |||||||||
| GO:0070972 | protein localization to endoplasmic reticulum | 1 | 2.17 | 0.205 | 2.91e-11 | 2.91e-11 | 19 | 115 | 9.41 |
Hematopoietic stem cells
Hepatoblasts
Horizontal cells
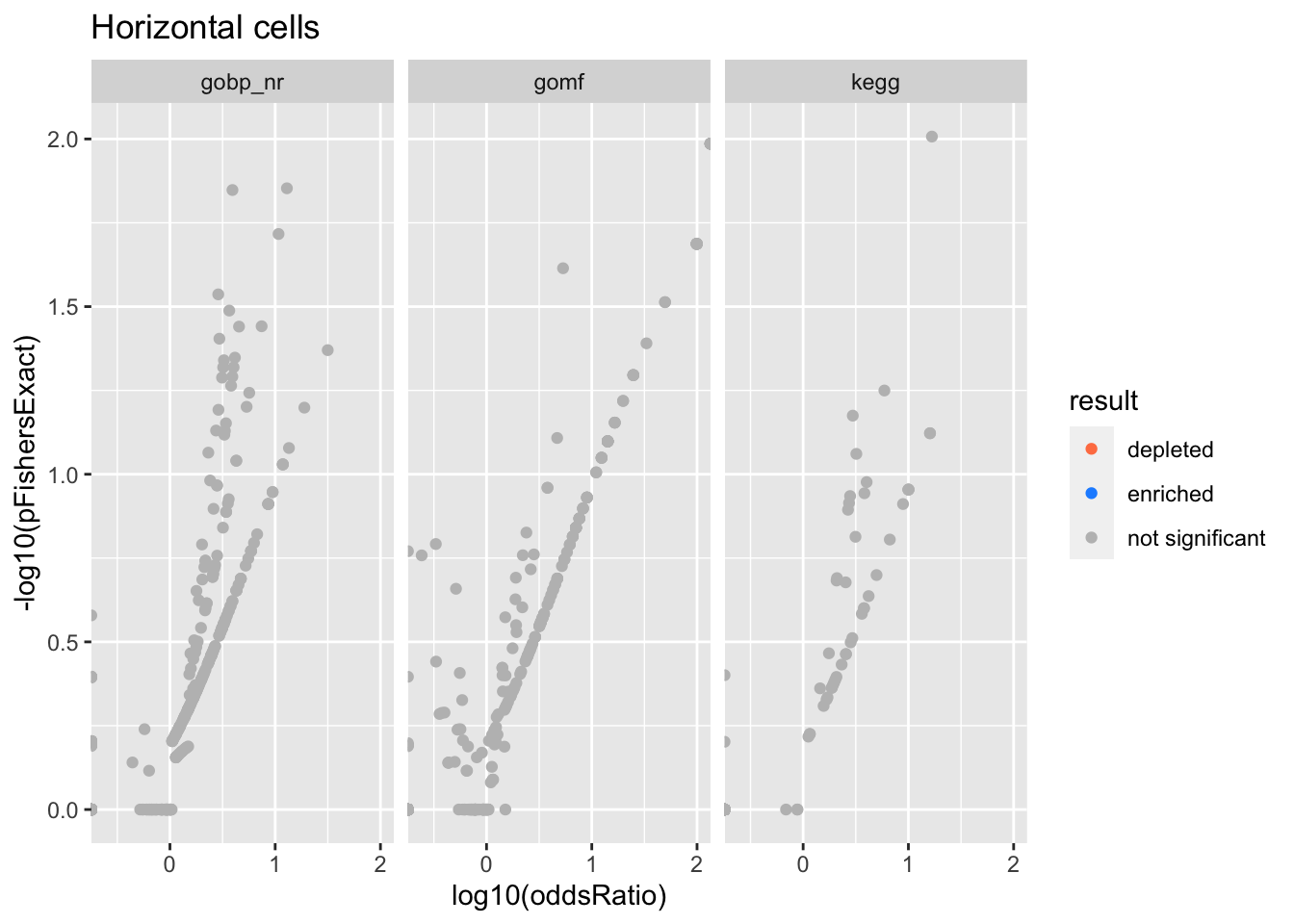
IGFBP1_DKK1 positive cells
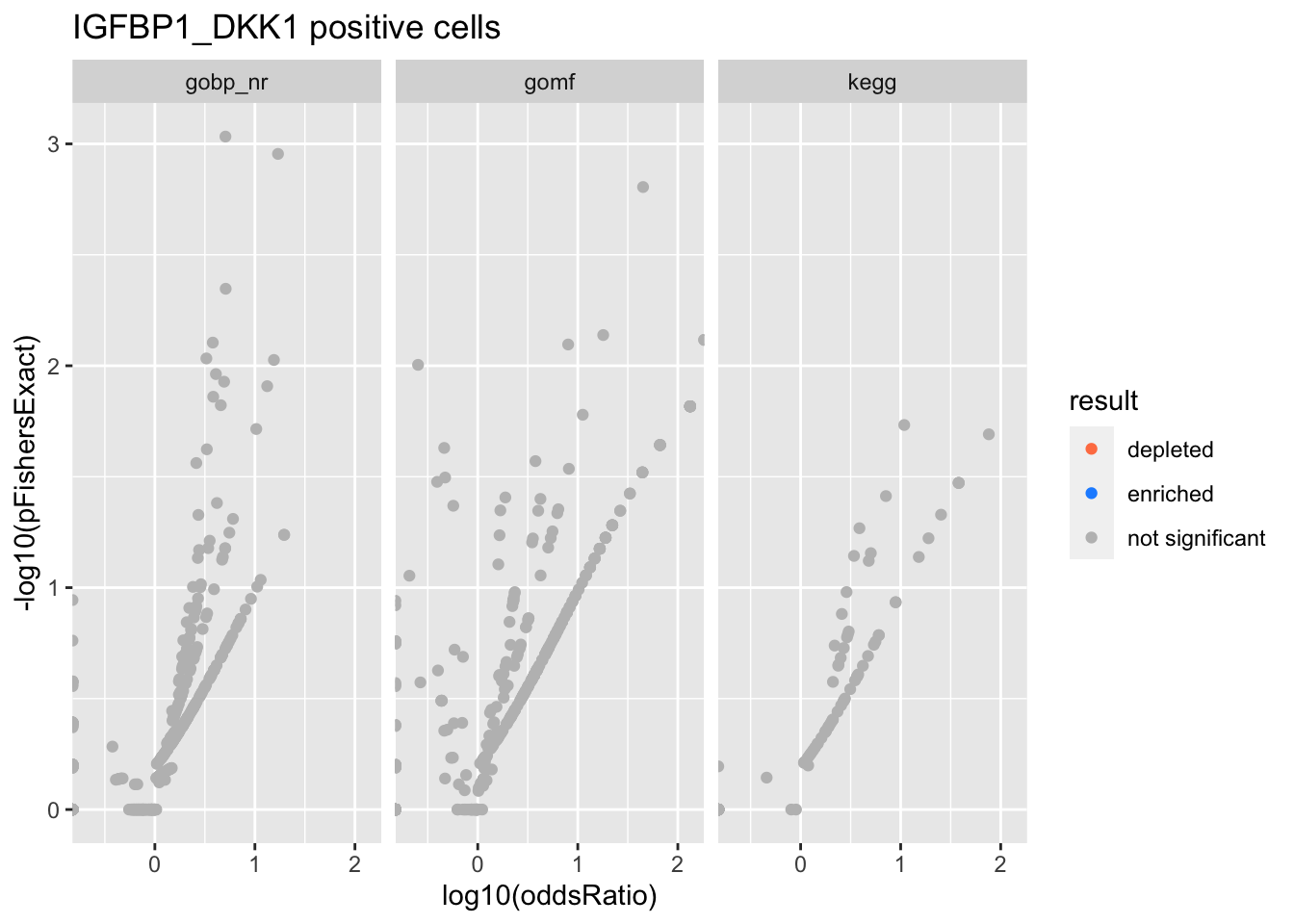
Inhibitory interneurons
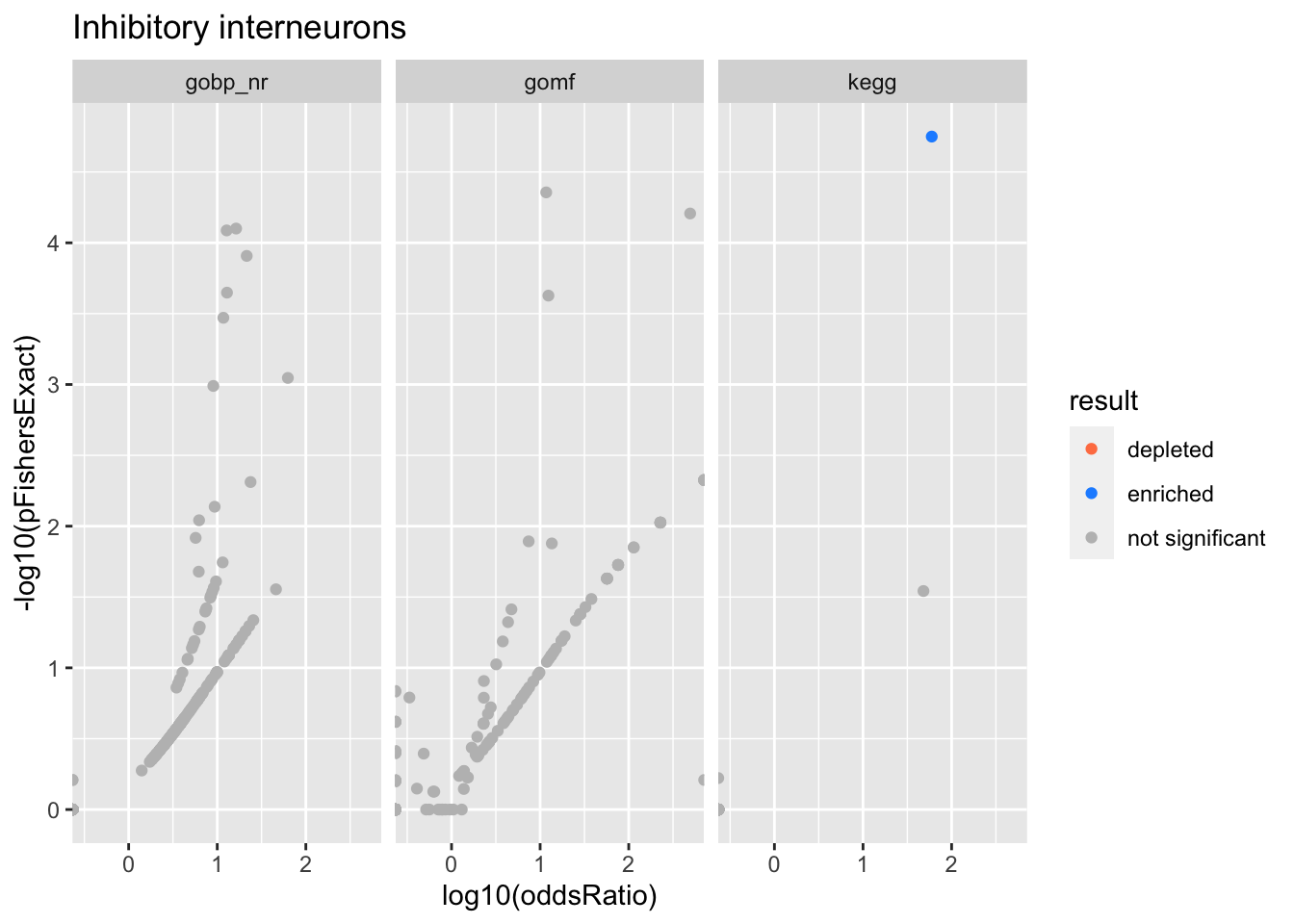
Islet endocrine cells
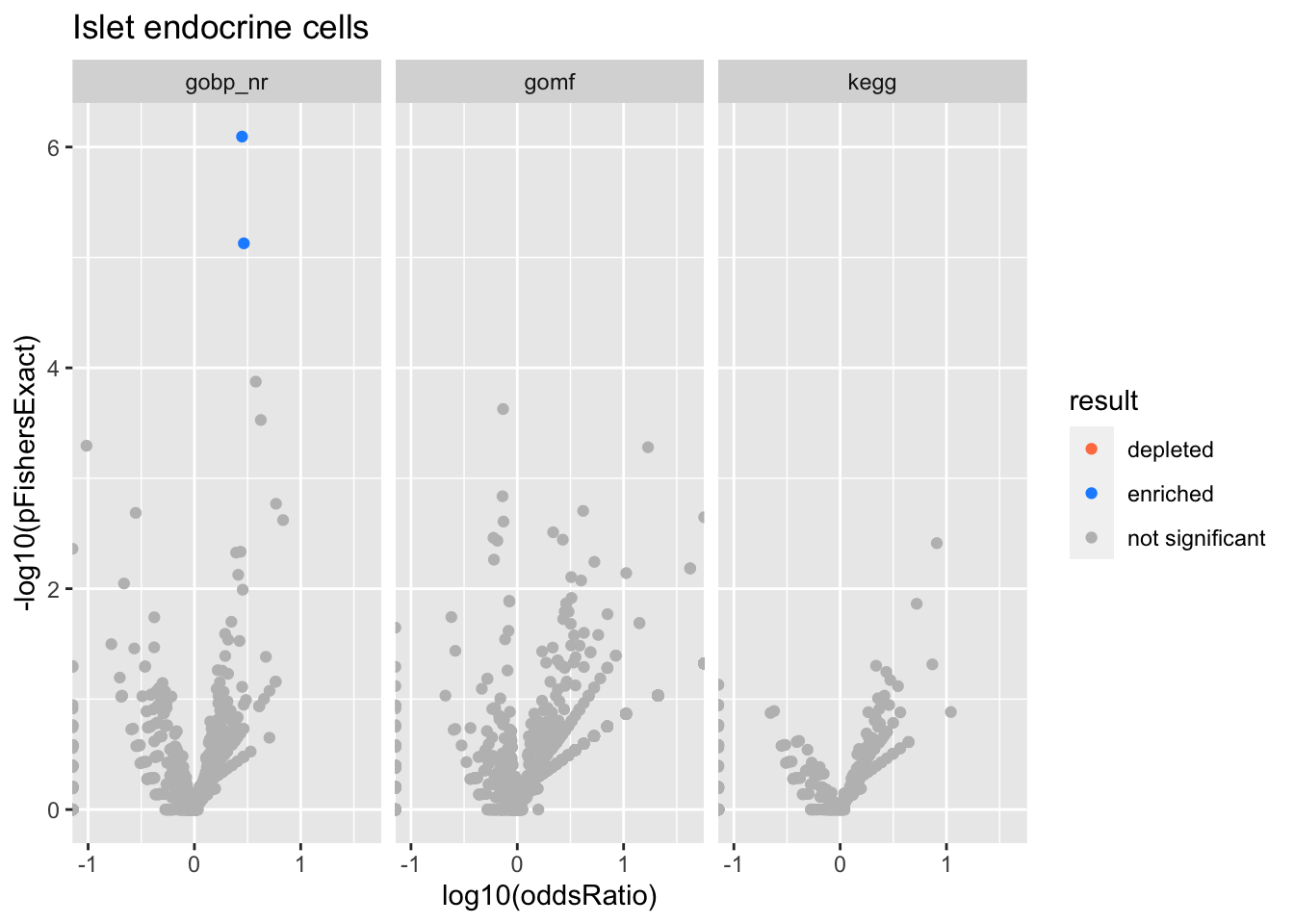
Lens fibre cells
Limbic system neurons
Megakaryocytes
Mesangial cells
Mesothelial cells
Metanephric cells
Microglia
MUC13_DMBT1 positive cells
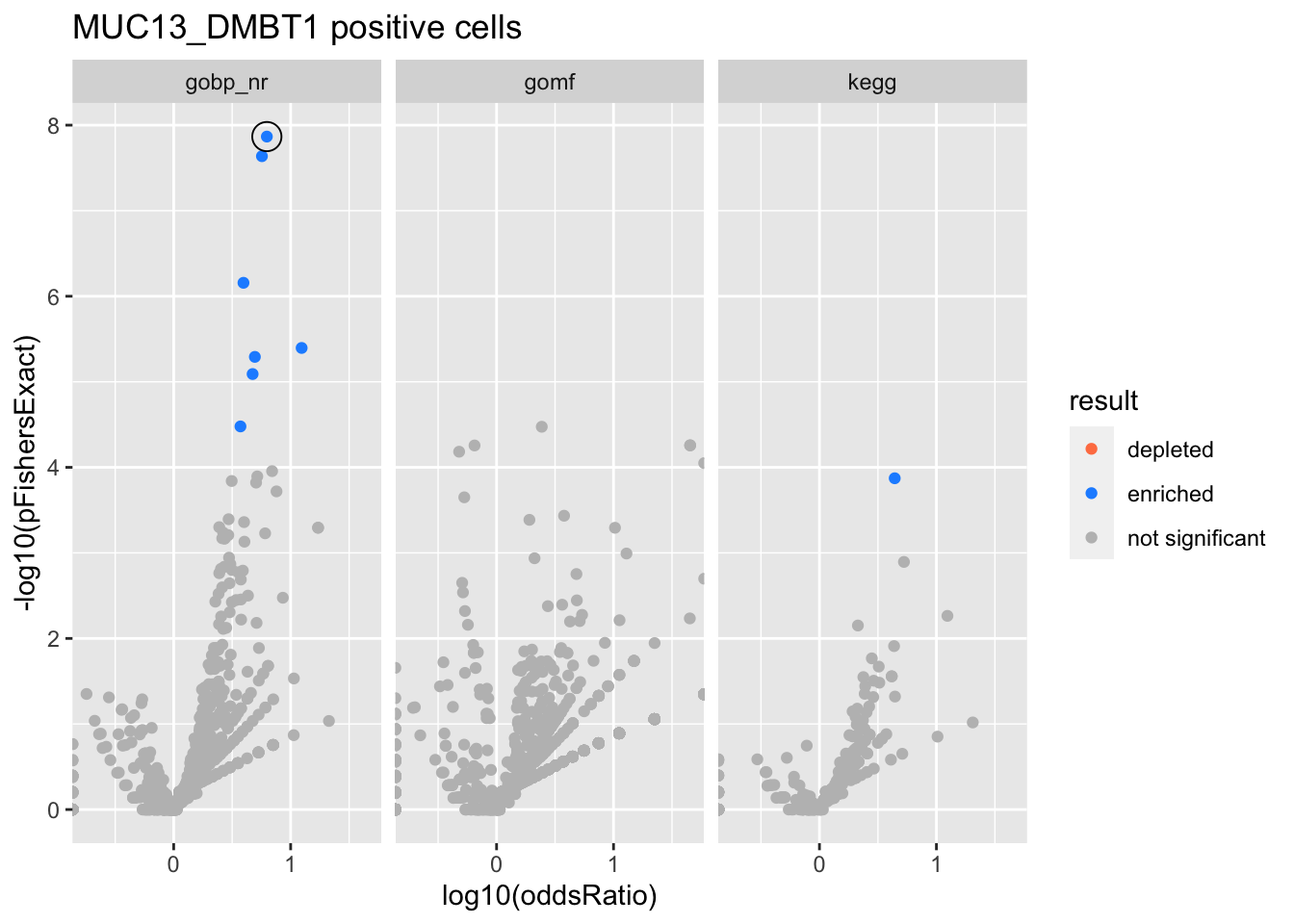
gobp_nr
| geneSet | description | alpha | beta | beta.se | pHypergeometric | pFishersExact | overlap | geneSetSize | oddsRatio |
|---|---|---|---|---|---|---|---|---|---|
| L1 | |||||||||
| GO:0045165 | cell fate commitment | 1 | 1.74 | 0.228 | 1.36e-08 | 1.36e-08 | 19 | 87 | 6.26 |
Neuroendocrine cells
Oligodendrocytes
PAEP_MECOM positive cells
Parietal and chief cells
PDE11A_FAM19A2 positive cells
PDE1C_ACSM3 positive cells
Photoreceptor cells
Retinal pigment cells
Retinal progenitors and Muller glia
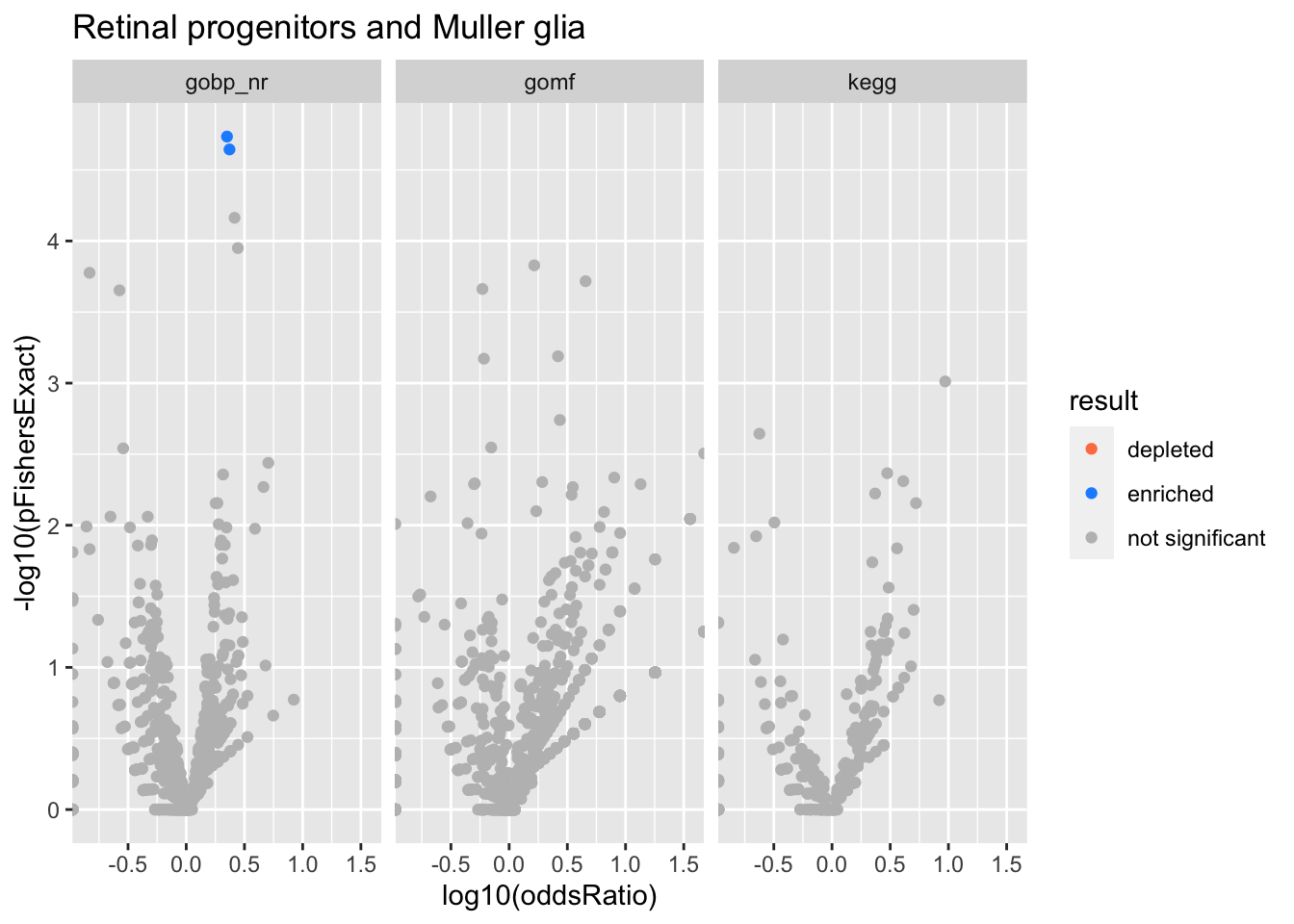
SATB2_LRRC7 positive cells
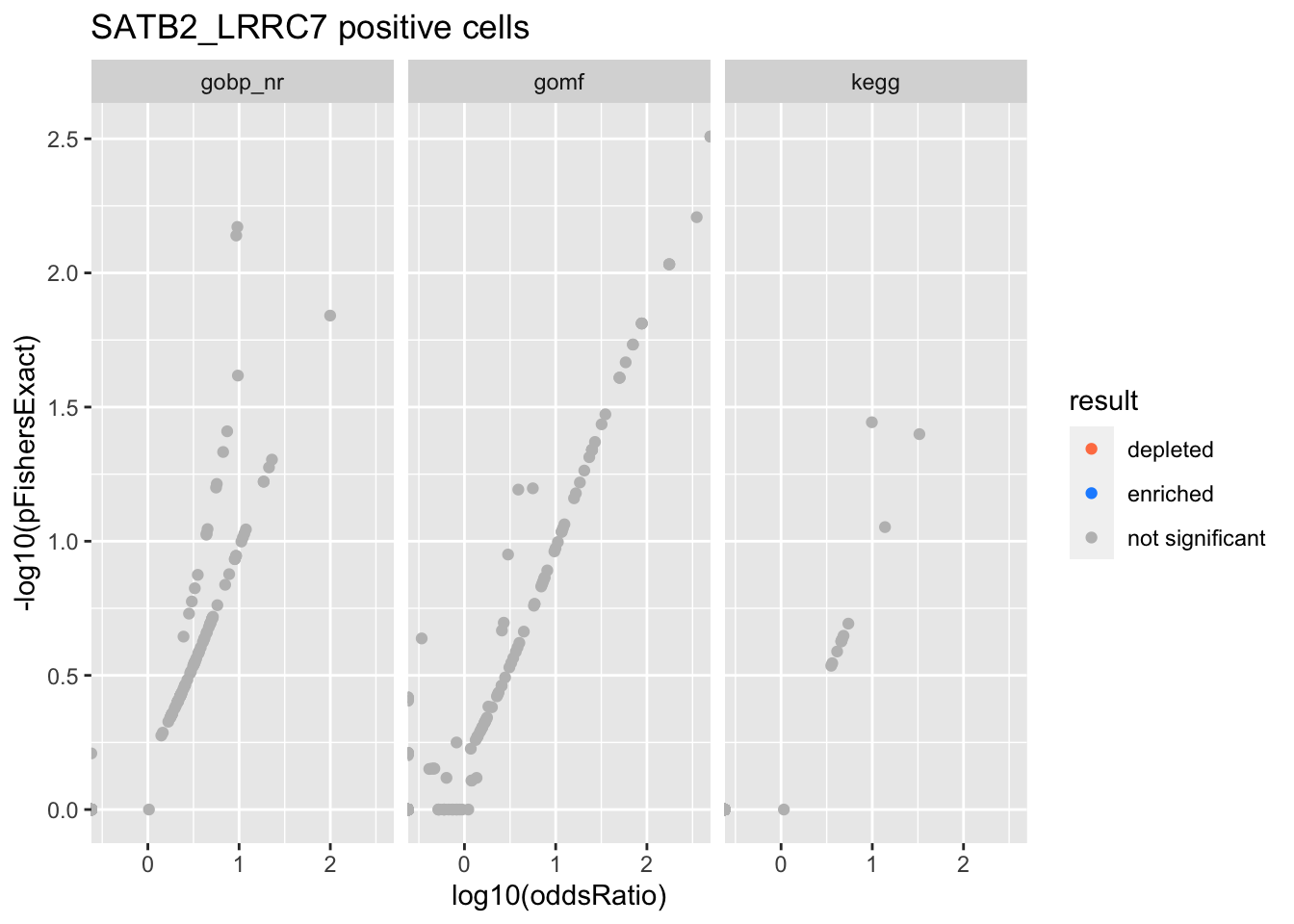
Schwann cells
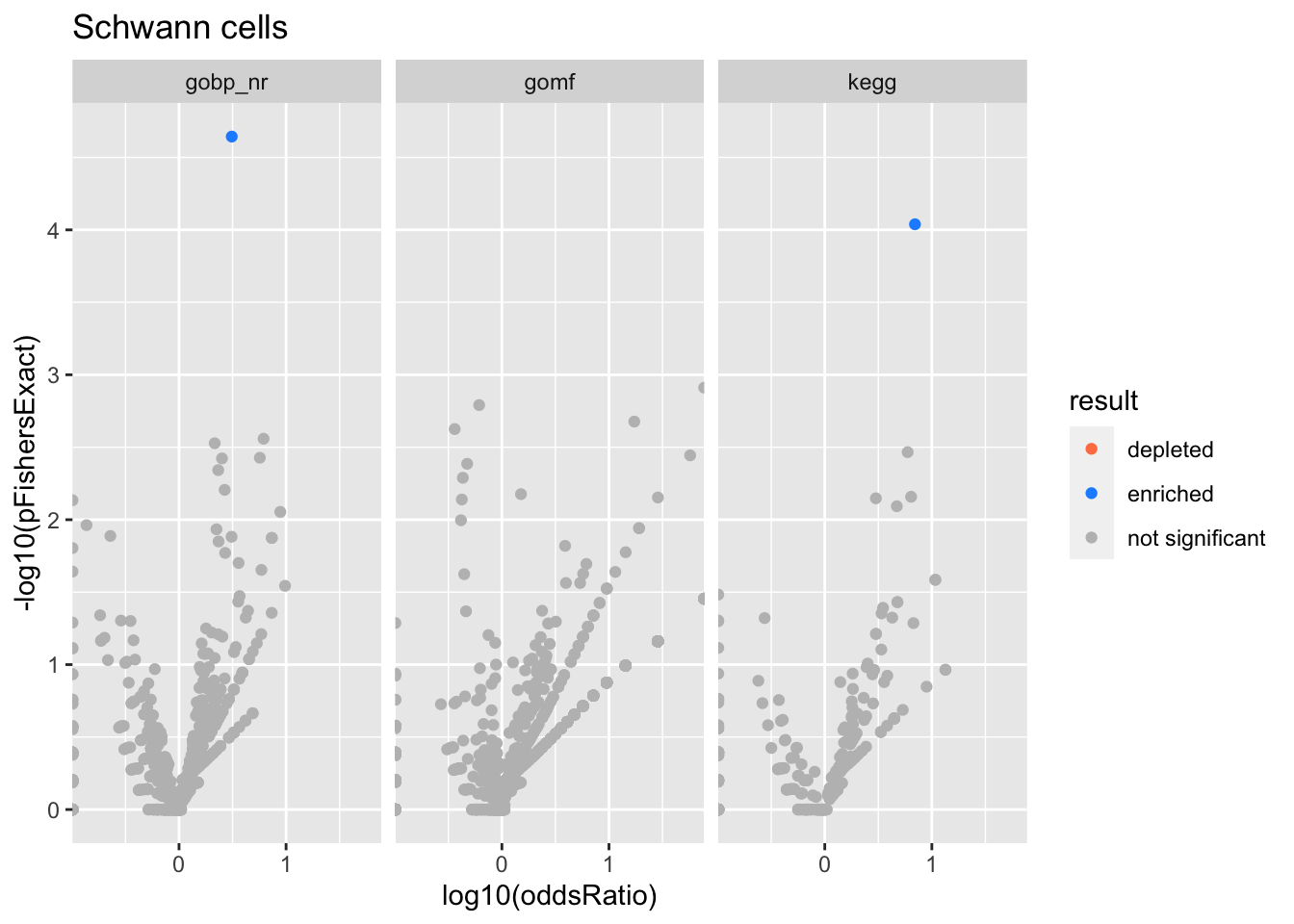
Skeletal muscle cells
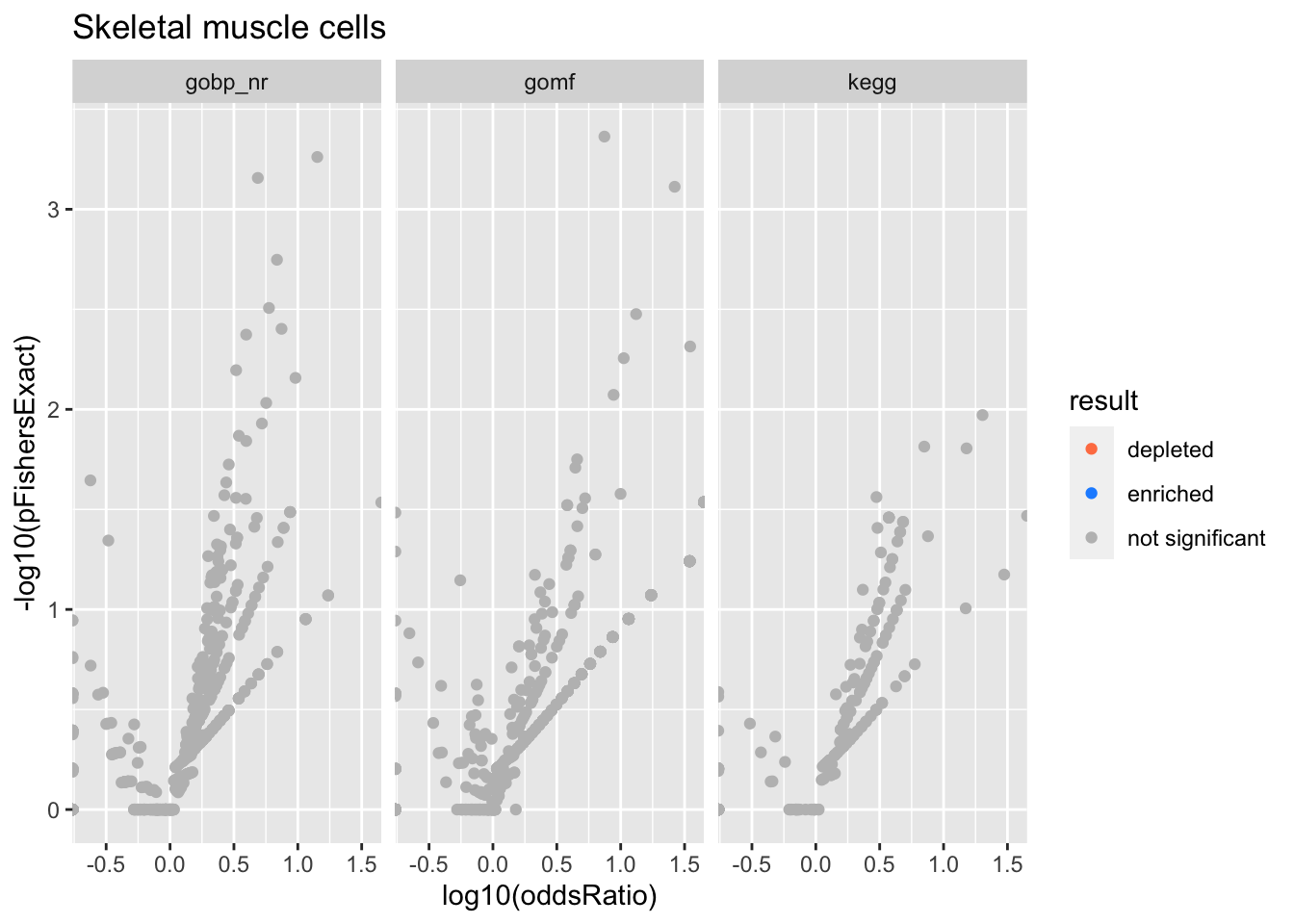
SKOR2_NPSR1 positive cells
SLC24A4_PEX5L positive cells
SLC26A4_PAEP positive cells
Smooth muscle cells
Squamous epithelial cells
Stellate cells
Stromal cells
Sympathoblasts
Syncytiotrophoblasts and villous cytotrophoblasts
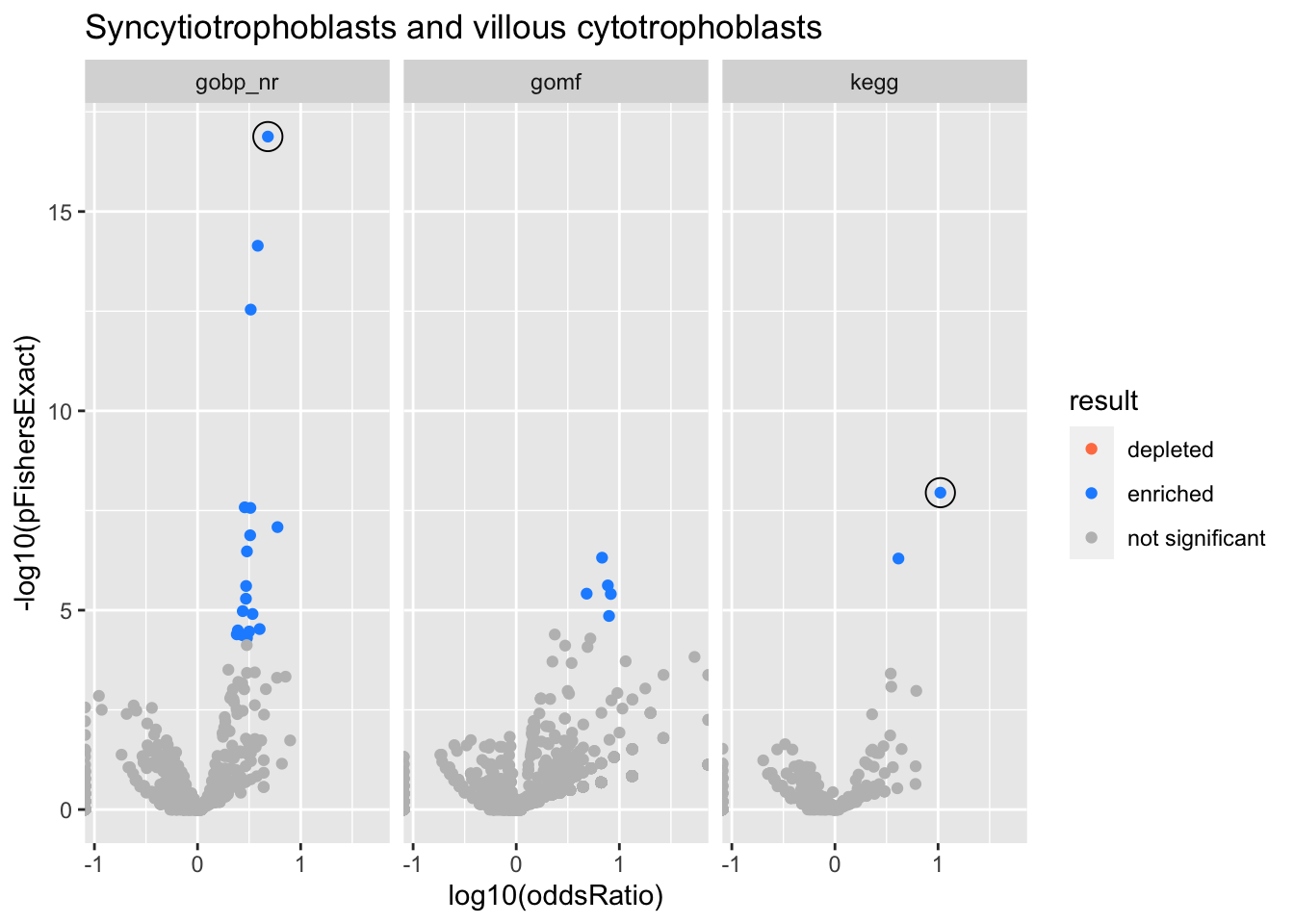
gobp_nr
| geneSet | description | alpha | beta | beta.se | pHypergeometric | pFishersExact | overlap | geneSetSize | oddsRatio |
|---|---|---|---|---|---|---|---|---|---|
| L1 | |||||||||
| GO:0007059 | chromosome segregation | 1 | 1.43 | 0.134 | 1.32e-17 | 1.32e-17 | 61 | 243 | 4.81 |
kegg
| geneSet | description | alpha | beta | beta.se | pHypergeometric | pFishersExact | overlap | geneSetSize | oddsRatio |
|---|---|---|---|---|---|---|---|---|---|
| L1 | |||||||||
| hsa03030 | DNA replication | 1 | 2.21 | 0.346 | 1.13e-08 | 1.13e-08 | 15 | 33 | 10.5 |
Thymic epithelial cells
Thymocytes
Trophoblast giant cells
Undifferentiated 1
Undifferentiated 2
Unipolar brush cells
Ureteric bud cells
Vascular endothelial cells
Visceral neurons
sessionInfo()#> R version 4.1.2 (2021-11-01)
#> Platform: x86_64-apple-darwin17.0 (64-bit)
#> Running under: macOS Big Sur 10.16
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRlapack.dylib
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> attached base packages:
#> [1] stats graphics grDevices datasets utils methods base
#>
#> other attached packages:
#> [1] kableExtra_1.3.4 BiocGenerics_0.40.0 htmltools_0.5.2
#> [4] forcats_0.5.1 stringr_1.4.0 dplyr_1.0.8
#> [7] purrr_0.3.4 readr_2.1.2 tidyr_1.2.0
#> [10] tibble_3.1.6 ggplot2_3.3.5 tidyverse_1.3.1
#>
#> loaded via a namespace (and not attached):
#> [1] colorspace_2.0-3 ellipsis_0.3.2 rprojroot_2.0.2
#> [4] XVector_0.34.0 fs_1.5.2 rstudioapi_0.13
#> [7] farver_2.1.0 bit64_4.0.5 AnnotationDbi_1.56.2
#> [10] fansi_1.0.2 lubridate_1.8.0 xml2_1.3.3
#> [13] codetools_0.2-18 doParallel_1.0.17 cachem_1.0.6
#> [16] knitr_1.38 jsonlite_1.8.0 workflowr_1.7.0
#> [19] apcluster_1.4.9 WebGestaltR_0.4.4 broom_0.7.12
#> [22] dbplyr_2.1.1 png_0.1-7 BiocManager_1.30.16
#> [25] compiler_4.1.2 httr_1.4.2 backports_1.4.1
#> [28] assertthat_0.2.1 Matrix_1.4-0 fastmap_1.1.0
#> [31] cli_3.2.0 later_1.3.0 tools_4.1.2
#> [34] igraph_1.2.11 gtable_0.3.0 glue_1.6.2
#> [37] GenomeInfoDbData_1.2.7 doRNG_1.8.2 Rcpp_1.0.8.2
#> [40] Biobase_2.54.0 cellranger_1.1.0 jquerylib_0.1.4
#> [43] vctrs_0.3.8 Biostrings_2.62.0 svglite_2.1.0
#> [46] iterators_1.0.14 xfun_0.30 rvest_1.0.2
#> [49] lifecycle_1.0.1 renv_0.15.4 rngtools_1.5.2
#> [52] org.Hs.eg.db_3.14.0 zlibbioc_1.40.0 scales_1.1.1
#> [55] vroom_1.5.7 hms_1.1.1 promises_1.2.0.1
#> [58] parallel_4.1.2 yaml_2.3.5 curl_4.3.2
#> [61] memoise_2.0.1 sass_0.4.0 stringi_1.7.6
#> [64] RSQLite_2.2.10 highr_0.9 S4Vectors_0.32.3
#> [67] foreach_1.5.2 GenomeInfoDb_1.30.1 rlang_1.0.2
#> [70] pkgconfig_2.0.3 systemfonts_1.0.4 bitops_1.0-7
#> [73] evaluate_0.15 lattice_0.20-45 labeling_0.4.2
#> [76] bit_4.0.4 tidyselect_1.1.2 magrittr_2.0.2
#> [79] R6_2.5.1 IRanges_2.28.0 generics_0.1.2
#> [82] DBI_1.1.2 pillar_1.7.0 haven_2.4.3
#> [85] whisker_0.4 withr_2.5.0 KEGGREST_1.34.0
#> [88] RCurl_1.98-1.6 modelr_0.1.8 crayon_1.5.0
#> [91] utf8_1.2.2 tzdb_0.2.0 rmarkdown_2.13
#> [94] grid_4.1.2 readxl_1.3.1 blob_1.2.2
#> [97] git2r_0.29.0 webshot_0.5.2 reprex_2.0.1
#> [100] digest_0.6.29 httpuv_1.6.5 stats4_4.1.2
#> [103] munsell_0.5.0 viridisLite_0.4.0 bslib_0.3.1