Bio326_2023
Marie Saitou
2023-01-30
Last updated: 2023-02-28
Checks: 7 0
Knit directory: Bio326/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20210128) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 2016a67. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/Bio326_2023.Rmd) and HTML
(docs/Bio326_2023.html) files. If you’ve configured a
remote Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 2016a67 | mariesaitou | 2023-02-28 | wflow_publish(c("analysis/Bio326_2023.Rmd")) |
| html | 7c8df25 | mariesaitou | 2023-02-28 | Build site. |
| html | 8b730e0 | mariesaitou | 2023-02-27 | Build site. |
| html | 1d9c035 | mariesaitou | 2023-02-27 | Build site. |
| Rmd | f5fe8f7 | mariesaitou | 2023-02-27 | wflow_publish(c("analysis//Bio326_2023.Rmd")) |
| html | 942fc18 | mariesaitou | 2023-02-27 | Build site. |
| Rmd | 5ccee4b | mariesaitou | 2023-02-27 | wflow_publish(c("analysis//Bio326_2023.Rmd")) |
Overview of the tutorial
Last time, you started sequencing of cattle genome using Nanopore MinION. For the following three sessions we will learn:
How to use Orion and conduct genome analysis
Quality check, Read filtering, mapping to the reference genome and variant calling
How to interpret summary statistics of Nanopore sequence data
How to interpret variant data
In this tutorial, we will investigate a small subset of bull genome sequence.
You will have an access to the whole datasets later for your reports as soon as the computation is done.
Also, Matthew and I prepared some questions in each section.
Please discuss and try the quizzes to deepen your understandig on Nanopore data.
DAY1
Overview.
Quality check -> Trimming of low quality reads -> Quality check
Connect to Orion and the preparation
Go to https://orion.nmbu.no/ at NMBU or with VPN. 
In the Terminal/Command prompt, go to your directory. Review: the concept of current directry
cd your_directoryLet’s make a directory for analysis and enter in it.
mkdir bull_analysis # make directory "bull_analysis"
cd bull_analysis # set the current directory "bull_analysis"
Now, you will inspect the fastq file, which contains Nanopore read information.
Check the read quality by Nanoplot
Browse the inside of the read (fastq) file
Review: look into a file content in a command line
zcat /net/fs-2/scale/OrionStore/Courses/BIO326/EUK/bull_analysis/demo_data/bull_demodata_fastq.gz | more
How a fastq file looks.
Each entry in a FASTQ files consists of 4 lines:
- A sequence identifier with information about the sequencing run. (run time, run ID, cflow cell id … )
2.The sequence (the base calls; A, C, T, G and N).
A separator, which is simply a plus (+) sign.
The base call quality scores. These are Phred +33 encoded, using ASCII characters to represent the numerical quality scores.” quality score sheet

Get basic stats of the fastq file
“zcat”-> look inside
“wc” -> word count
“-l” -> line
zcat /net/fs-2/scale/OrionStore/Courses/BIO326/EUK/bull_analysis/demo_data/bull_demodata_fastq.gz | wc -l
Discussion Point
Now you got the number of lines in the fastq file.
How many sequence reads are in the fastq file?
Need Help?
We see that there are 96000 lines in the fastq file.
As we learned that “each entry in a FASTQ files consists of 4 lines”, one read is corresponding to four lines. So in this file we have 96000/4 = 24000 reads.
Run Nanoplot
The original fastq files may contain low quality reads. In this step, we will use “Nanoplot” to see the quality and lentgh of each read.
“Singularity” is a toolset on Orion to execute software. A variety of different bioinformatics tools are available in Singularity.
Make a slurm script like below and run it.
Review: make a slurm script and run it by sbatch
#!/bin/bash
#SBATCH --job-name=Nanoplot # sensible name for the job
#SBATCH --mail-user=yourname@nmbu.no # Email me when job is done.
#SBATCH --mem=12G
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=8
#SBATCH --mail-type=END
singularity exec /cvmfs/singularity.galaxyproject.org/all/nanoplot:1.41.0--pyhdfd78af_0 NanoPlot -t 8 --fastq /net/fs-2/scale/OrionStore/Courses/BIO326/EUK/bull_analysis/demo_data/bull_demodata_fastq.gz --plots dot --no_supplementary --no_static --N50 -p before
Nanoplot will generate the result files, named “before”xxx. Lets look into them…
# taking too long?
qlogin
cp /net/fs-2/scale/OrionStore/Courses/BIO326/EUK/bull_analysis/demo_data/beforeNanoPlot-report.html beforeNanoPlot-report.html
Open “beforeNanoPlot-report.html” on your local computer
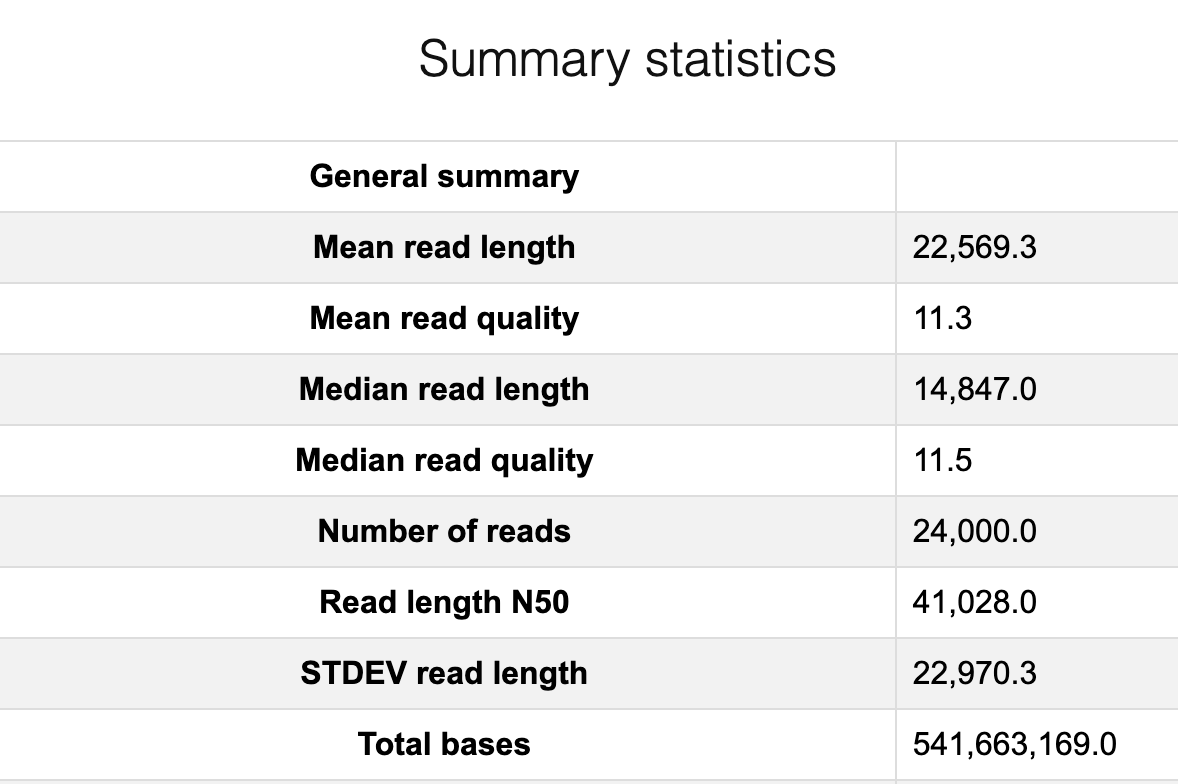
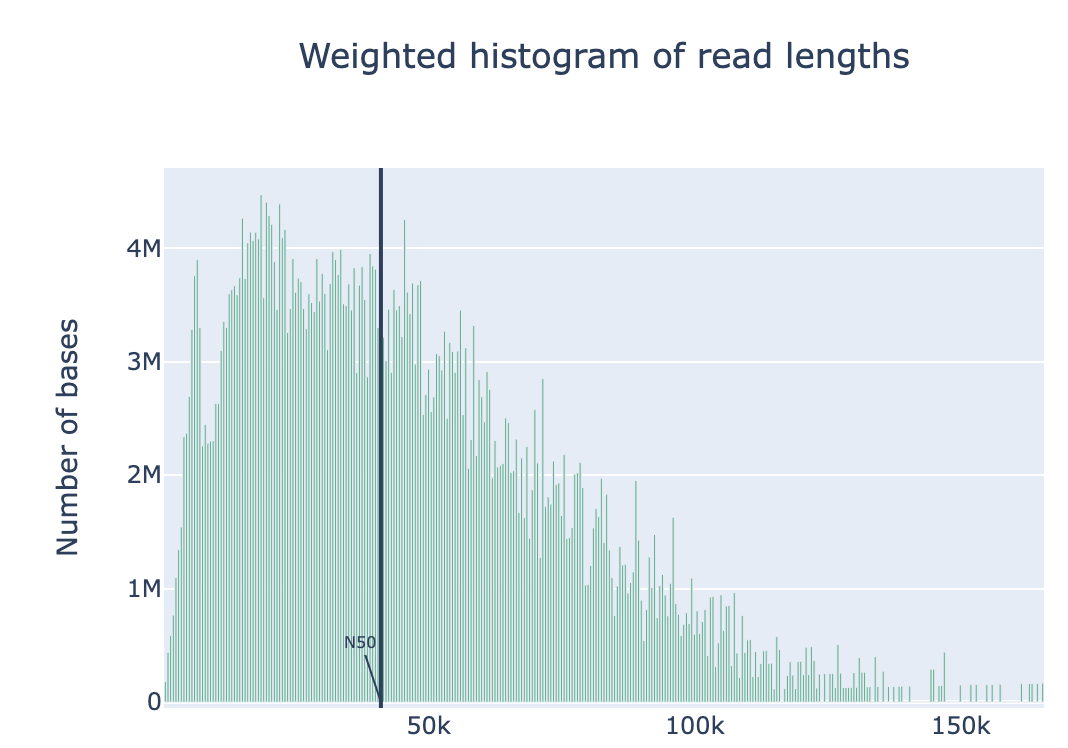
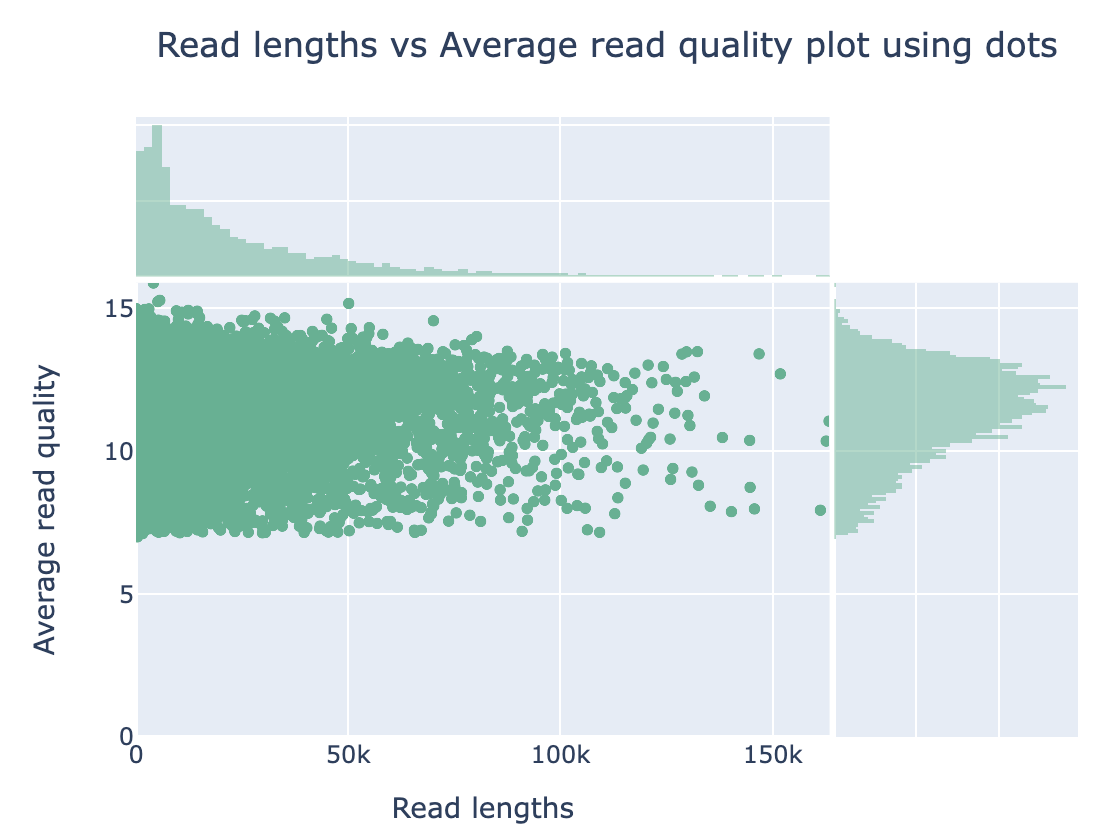
Filtering by Nanofilt
#!/bin/bash
#SBATCH --job-name=Nanoplot # sensible name for the job
#SBATCH --mail-user=yourname@nmbu.no # Email me when job is done.
#SBATCH --mem=12G
#SBATCH --ntasks=1
#SBATCH --mail-type=END
gunzip -c /net/fs-2/scale/OrionStore/Courses/BIO326/EUK/bull_analysis/demo_data/bull_demodata_fastq.gz | singularity exec /cvmfs/singularity.galaxyproject.org/all/nanofilt:2.8.0--py_0 NanoFilt -q 12 -l 500 | gzip > cleaned.bull.fastq.gz
-l, Filter on a minimum read length
-q, Filter on a minimum average read quality score
In this case, we are removing reads lower than quality score 12 and shorter than 500 bases.
Compare the before and after cleaning sequences
Run Nanoplot again on the cleaned sequences.
Need help?
#!/bin/bash
#SBATCH --job-name=Nanoplot # sensible name for the job
#SBATCH --mail-user=yourname@nmbu.no # Email me when job is done.
#SBATCH --mem=12G
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=8
#SBATCH --mail-type=END
singularity exec /cvmfs/singularity.galaxyproject.org/all/nanoplot:1.41.0--pyhdfd78af_0 NanoPlot -t 8 --fastq cleaned.bull.fastq.gz --N50 --no_supplementary --no_static --plots dot -p after
Open “afterNanoPlot-report.html” on your local computer.
# taking too long?
qlogin
cp /net/fs-2/scale/OrionStore/Courses/BIO326/EUK/bull_analysis/demo_data/afterNanoPlot-report.html afterNanoPlot-report.html
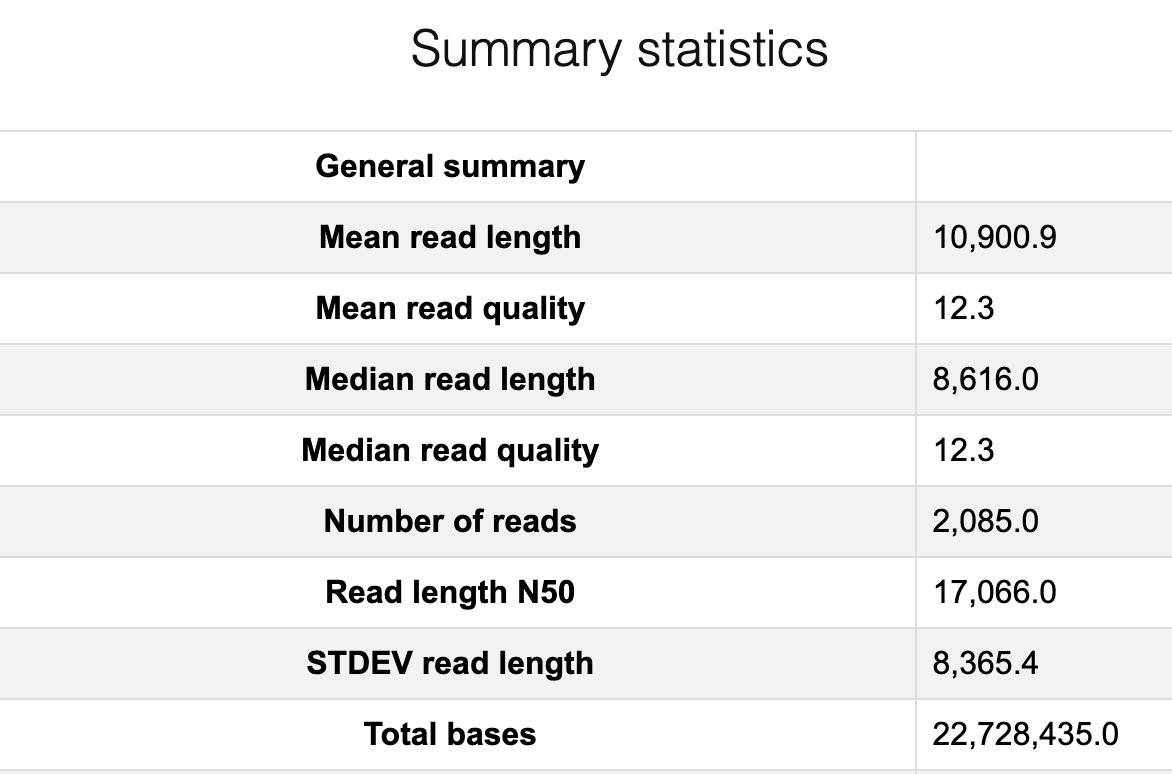
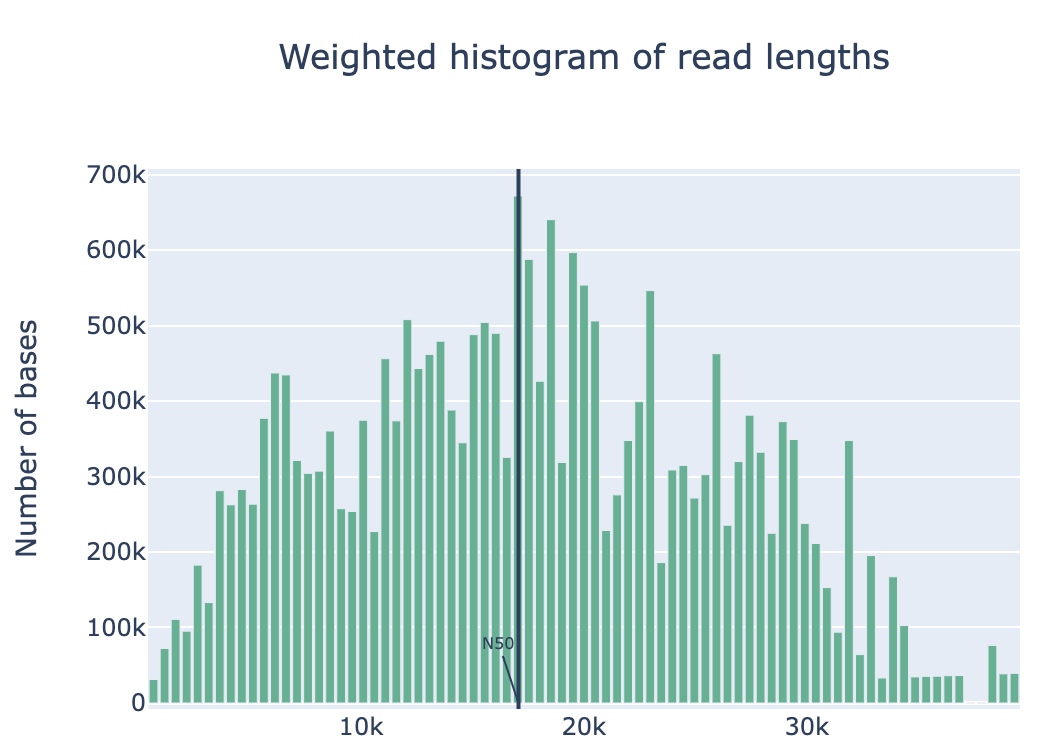
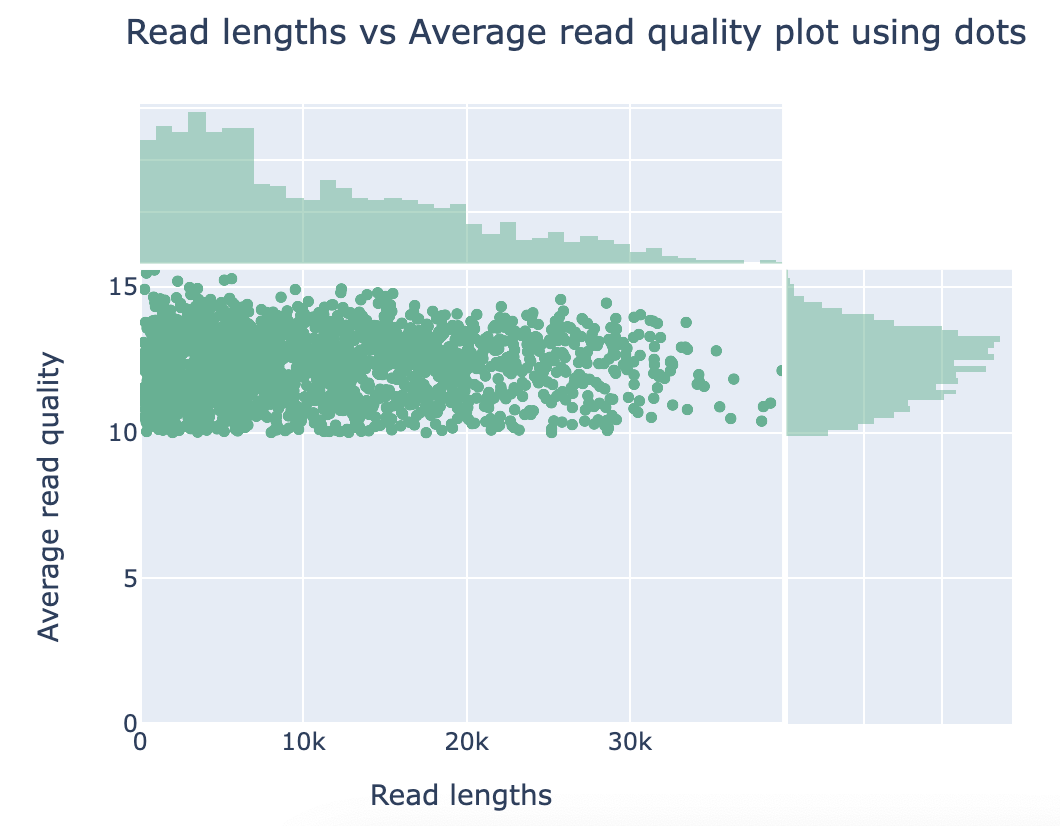
Discussion Point
Did you see the difference of read and quality distribution between before and after the filtering?
The end of Day1. Well done!
DAY2
Overview
Map the reads to the reference genome -> Detect variants (difference from the reference genome)
Find out where “minimap2” is in Singularity
minimap2
find /cvmfs/singularity.galaxyproject.org/all/ -name minimap2*
/cvmfs/singularity.galaxyproject.org/all/minimap2:2.24--h7132678_1
run Minimap and map the reads to the reference genome
#!/bin/bash
#SBATCH --job-name=Nanoplot # sensible name for the job
#SBATCH --mail-user=yourname@nmbu.no # Email me when job is done.
#SBATCH --mem=12G
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=8
#SBATCH --mail-type=END
singularity exec /cvmfs/singularity.galaxyproject.org/all/minimap2:2.24--h7132678_1 minimap2 -t 8 -a Bos_taurus.fa.gz cleaned.bull.fastq.gz > bull.sam
Browse the mapped file
cat bull.sam | head bull.sam
# taking too long?
qlogin
cp /net/fs-2/scale/OrionStore/Courses/BIO326/EUK/bull_analysis/demo_data/bull.sam bull.sam
File format conversion
#!/bin/bash
#SBATCH --job-name=Nanoplot # sensible name for the job
#SBATCH --mail-user=yourname@nmbu.no # Email me when job is done.
#SBATCH --mem=12G
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=8
#SBATCH --mail-type=END
# convert the sam file to bam format
singularity exec /cvmfs/singularity.galaxyproject.org/all/samtools:1.16.1--h6899075_1 samtools view -S -b bull.sam > bull0.bam
## sort the bam file
singularity exec /cvmfs/singularity.galaxyproject.org/all/samtools:1.16.1--h6899075_1 samtools sort bull0.bam -o bull.bam
# index the bam file
singularity exec /cvmfs/singularity.galaxyproject.org/all/samtools:1.16.1--h6899075_1 samtools index -M bull.bam
Error correction with Pilon/Medaka
(Skip this time as it takes time… You will learn the error correction in the prokaryotic part. )
Variant Calling using Sniffles
#!/bin/bash
#SBATCH --job-name=Nanoplot # sensible name for the job
#SBATCH --mail-user=yourname@nmbu.no # Email me when job is done.
#SBATCH --mem=12G
#SBATCH --ntasks=1
#SBATCH --mail-type=END
singularity exec /cvmfs/singularity.galaxyproject.org/all/sniffles:2.0.7--pyhdfd78af_0 sniffles --input bull.bam --vcf bull.vcf
Now you got the variant file!
Inspect variants
more -s 2255 bull.vcf
grep -v '^##' bull.vcf | head
DAY3
Now you have variants! Lets see what genes are affected by the variants.
sessionInfo()R version 4.2.2 (2022-10-31)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur ... 10.16
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] workflowr_1.7.0
loaded via a namespace (and not attached):
[1] Rcpp_1.0.10 compiler_4.2.2 pillar_1.8.1 bslib_0.4.2
[5] later_1.3.0 git2r_0.31.0 jquerylib_0.1.4 tools_4.2.2
[9] getPass_0.2-2 digest_0.6.31 jsonlite_1.8.4 evaluate_0.20
[13] lifecycle_1.0.3 tibble_3.1.8 pkgconfig_2.0.3 rlang_1.0.6
[17] cli_3.6.0 rstudioapi_0.14 yaml_2.3.7 xfun_0.37
[21] fastmap_1.1.0 httr_1.4.4 stringr_1.5.0 knitr_1.42
[25] fs_1.6.1 vctrs_0.5.2 sass_0.4.5 rprojroot_2.0.3
[29] glue_1.6.2 R6_2.5.1 processx_3.8.0 fansi_1.0.4
[33] rmarkdown_2.20 callr_3.7.3 magrittr_2.0.3 whisker_0.4.1
[37] ps_1.7.2 promises_1.2.0.1 htmltools_0.5.4 httpuv_1.6.8
[41] utf8_1.2.3 stringi_1.7.12 cachem_1.0.6