E. coli K12 analysis
Last updated: 2022-03-31
Checks: 7 0
Knit directory: rare-mutation-detection/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20210916) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 13af45c. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: scripts/
Untracked files:
Untracked: ._.DS_Store
Untracked: DOCNAME
Untracked: analysis/._.DS_Store
Untracked: analysis/cache/
Untracked: analysis/calc_nanoseq_metrics.Rmd
Untracked: data/
Untracked: prototype_code/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/ecoli_K12.Rmd) and HTML (docs/ecoli_K12.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | 05412f6 | Marek Cmero | 2022-03-28 | Build site. |
| Rmd | ea0ad82 | Marek Cmero | 2022-03-28 | Added singleton comparison + facet summary plots |
| html | 51aba0e | Marek Cmero | 2022-03-25 | Build site. |
| Rmd | a3895f7 | Marek Cmero | 2022-03-25 | Bug fix |
| html | ea4faf4 | Marek Cmero | 2022-03-25 | Build site. |
| Rmd | 5964f14 | Marek Cmero | 2022-03-25 | Added more comparison plots for ecoli K12 data |
| html | e5b39ad | Marek Cmero | 2022-03-25 | Build site. |
| Rmd | 1926d3d | Marek Cmero | 2022-03-25 | added K12 ecoli metrics |
Metrics for E. coli K12 data
MultiQC reports:
library(ggplot2)
library(data.table)
library(dplyr)
library(here)
library(tibble)
library(stringr)
library(Rsamtools)
library(GenomicRanges)
library(seqinr)
library(parallel)
library(readxl)source(here('code/load_data.R'))
source(here('code/plot.R'))
source(here('code/efficiency_nanoseq_functions.R'))# Ecoli genome max size
# genome_max <- 4528118
genome_max <- c('2e914854fabb46b9_1' = 4661751,
'2e914854fabb46b9_2' = 67365)
cores = 8genomeFile <- here('data/ref/Escherichia_coli_ATCC_10798.fasta')
rinfo_dir <- here('data/ecoli/AGRF_CAGRF22029764_HJK2GDSX3/QC/read_info')
markdup_dir <- here('data/ecoli/AGRF_CAGRF22029764_HJK2GDSX3/QC/mark_duplicates')
qualimap_dir <- here('data/ecoli/AGRF_CAGRF22029764_HJK2GDSX3/QC/qualimap')
qualimap_cons_dir <- here('data/ecoli/AGRF_CAGRF22029764_HJK2GDSX3/QC/consensus/qualimap')
metadata_file <- here('data/metadata/NovaSeq data E coli.xlsx')sample_names <- list.files(rinfo_dir) %>%
str_split('\\.txt.gz') %>%
lapply(., dplyr::first) %>%
unlist() %>%
str_split('_') %>%
lapply(., head, 2) %>%
lapply(., paste, collapse='-') %>%
unlist()
# load and fetch duplicate rate from MarkDuplicates output
mdup <- load_markdup_data(markdup_dir, sample_names)
# get mean coverage for pre and post-consensus reads
qmap_cov <- get_qmap_coverage(qualimap_dir, sample_names)
qmap_cons_cov <- get_qmap_coverage(qualimap_cons_dir, sample_names)
# calculate metrics for nanoseq
rlen <- 151; skips <- 5
metrics_nano <- calc_metrics_new_rbs(rinfo_dir, pattern = 'Nano', cores = cores)
# calculate metrics for xGen
rlen <- 151; skips <- 8
metrics_xgen <- calc_metrics_new_rbs(rinfo_dir, pattern = 'xGEN', cores = cores)
metrics <- c(metrics_nano, metrics_xgen) %>% bind_rows()
metrics$duplicate_rate <- as.numeric(mdup)
metrics$duplex_coverage_ratio <- qmap_cov$coverage / qmap_cons_cov$coverage
metrics$duplex_coverage_ratio[qmap_cons_cov$coverage < 1] <- 0 # fix when < 1 duplex cov
metrics$sample <- sample_names
# load metadata
metadata <- read_excel(metadata_file)
metadata$`sample name` <- gsub('_', '-', metadata$`sample name`)
# prepare for plotting
mm <- data.frame(melt(metrics))
mm$protocol <- 'NanoSeq'
mm$protocol[grep('xGEN', mm$sample)] <- 'xGen'
mm$sample <- gsub('-HJK2GDSX3', '', mm$sample)
mm <- inner_join(mm, metadata, by=c('sample' = 'sample name'))
colnames(mm)[2] <- 'metric'
mm$nuclease <- paste(mm$`Mung bean unit`, mm$`S1 unit`, sep='+')Metric comparison plots
metrics_to_plot <- as.character(mm$metric) %>% unique()
for(metric in metrics_to_plot) {
p <- ggplot(mm[mm$metric == metric,], aes(sample, value)) +
geom_histogram(stat = 'identity', position = 'dodge') +
theme_bw() +
coord_flip() +
ggtitle(metric)
show(p)
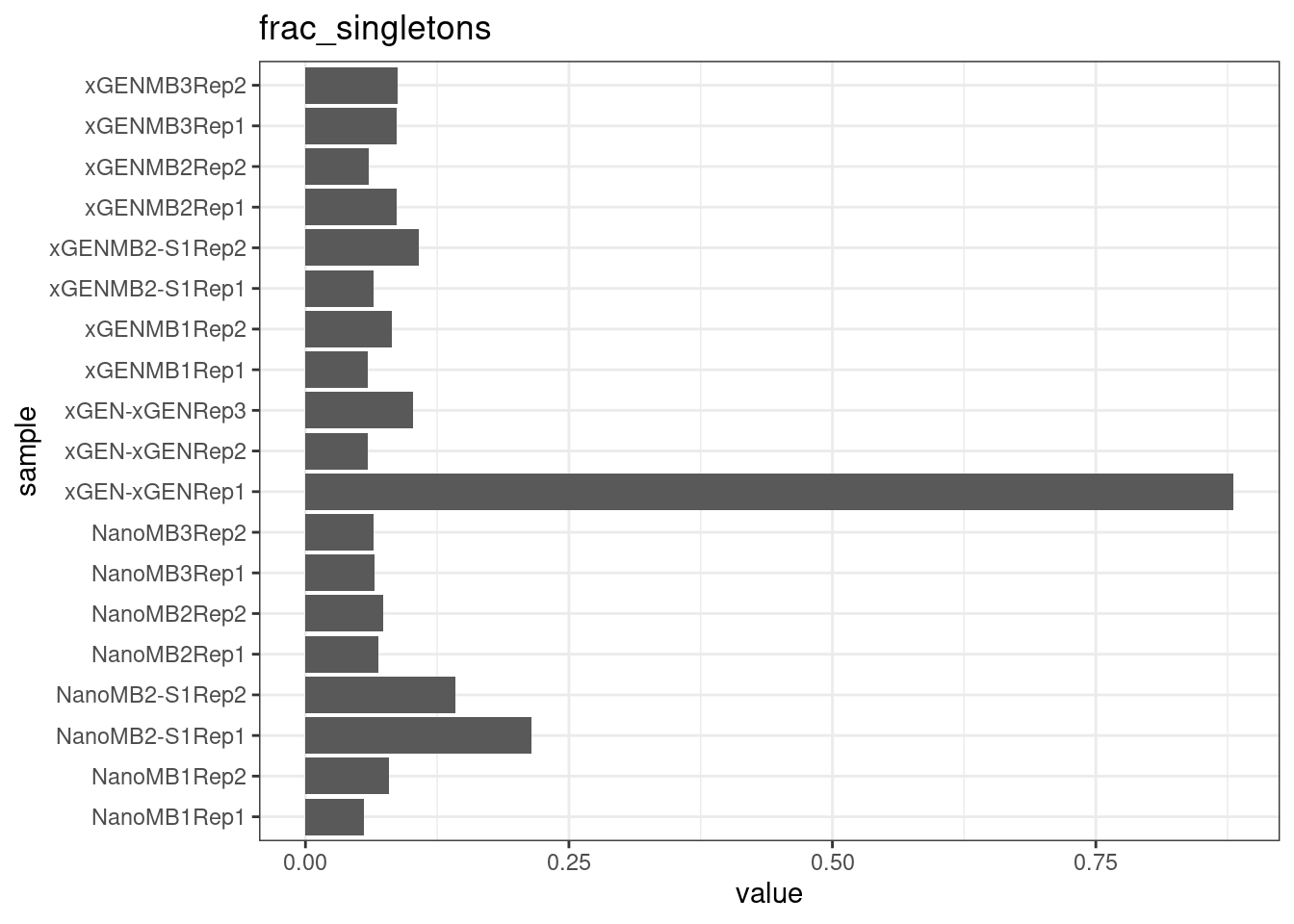
}
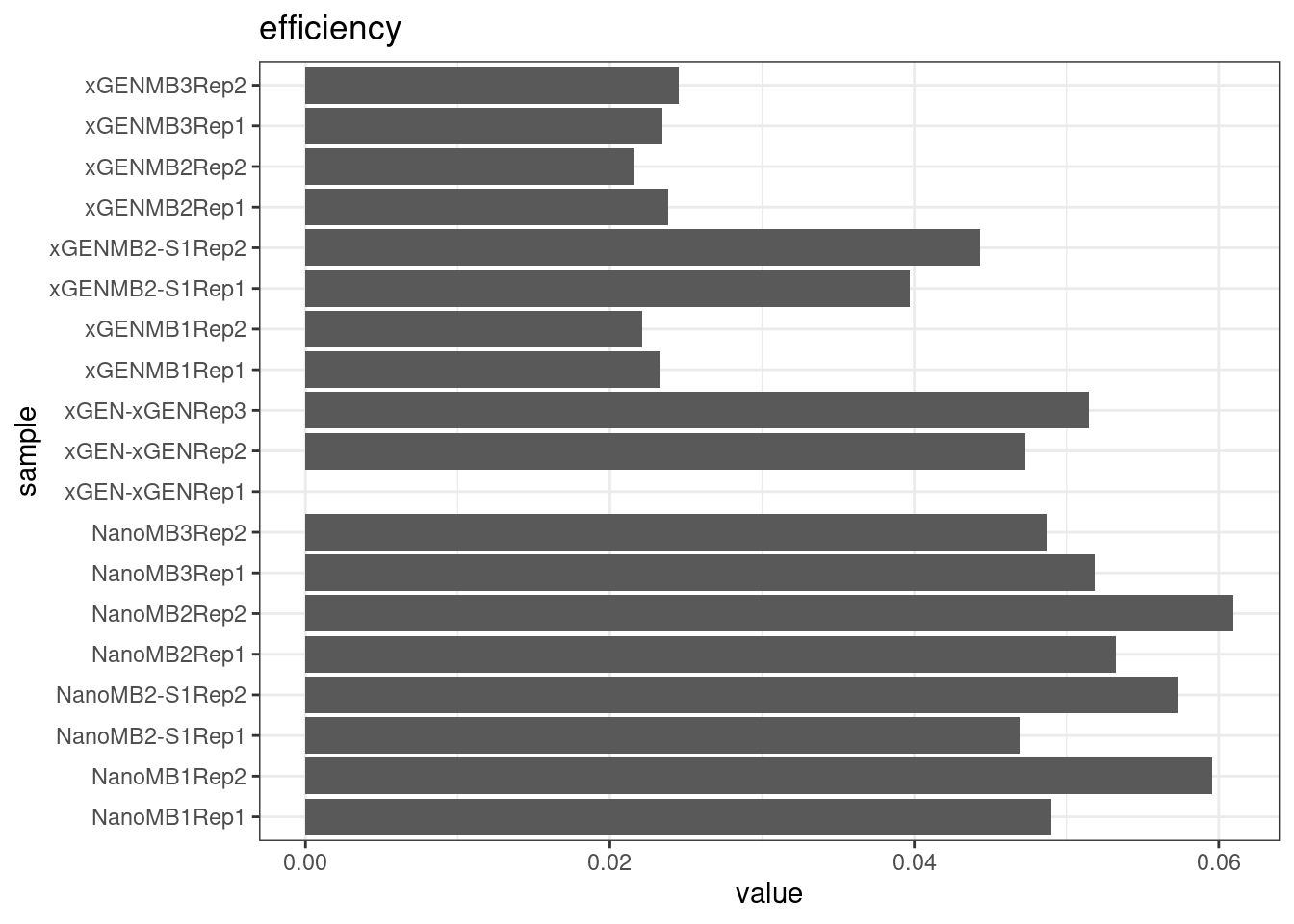
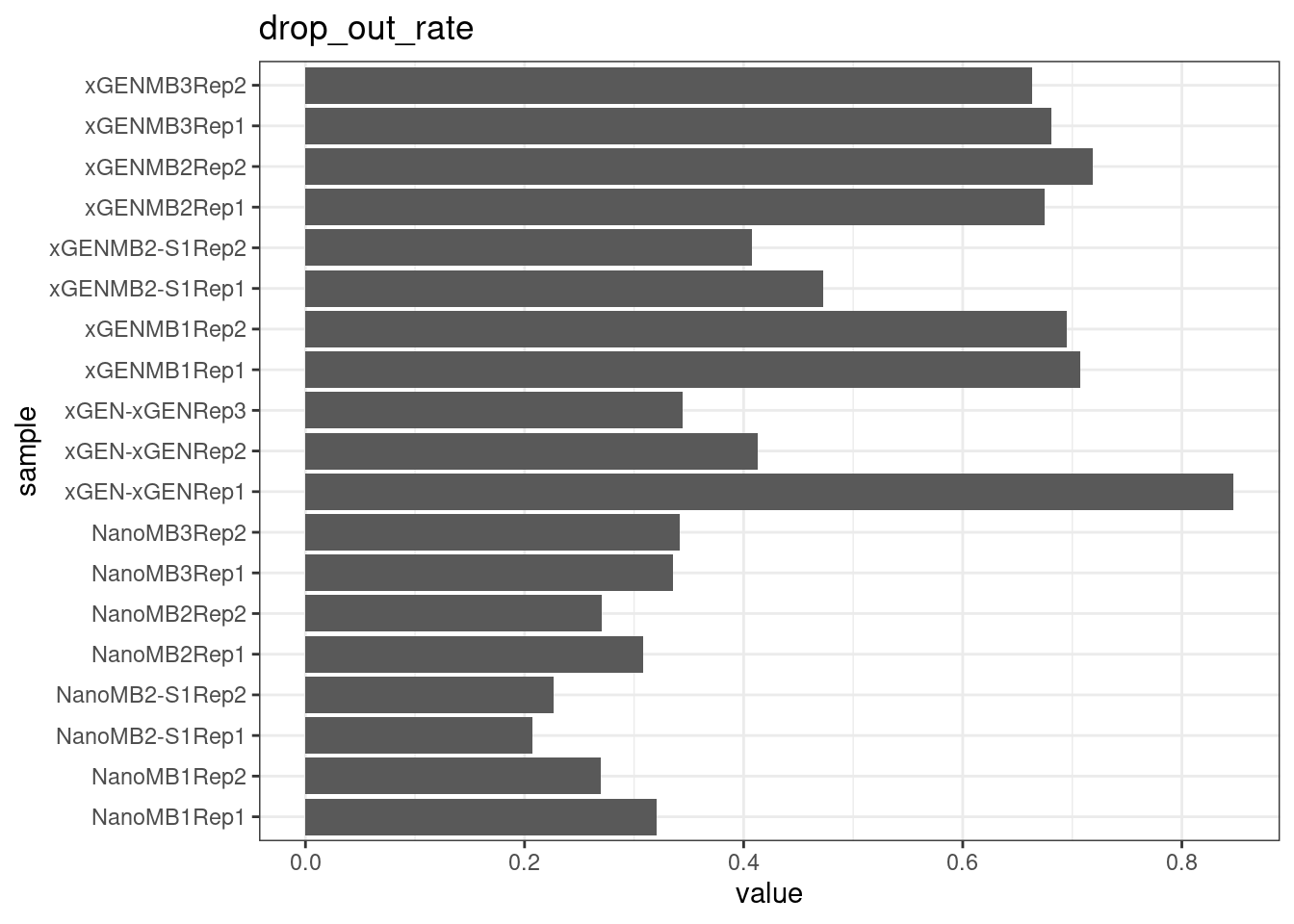
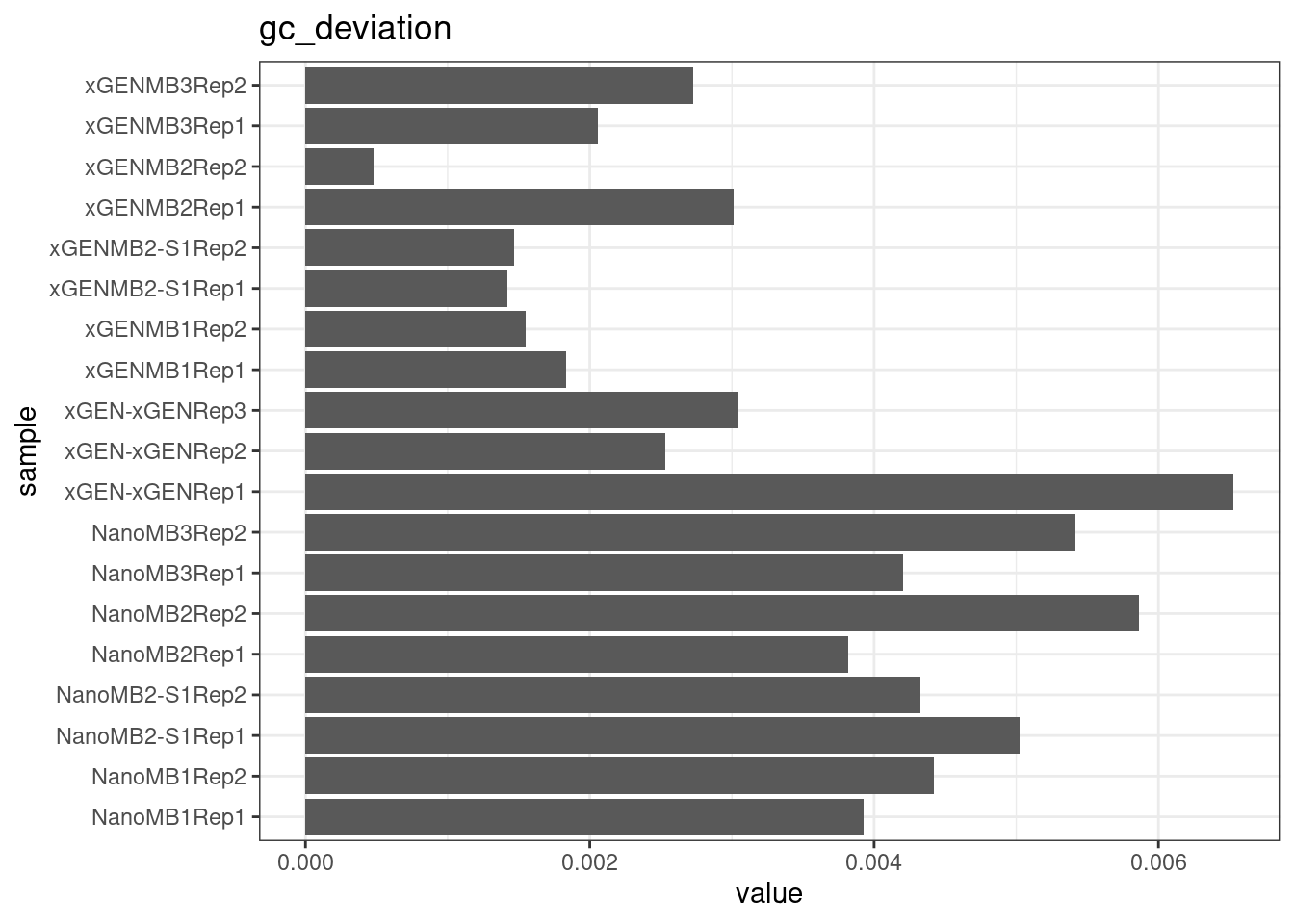

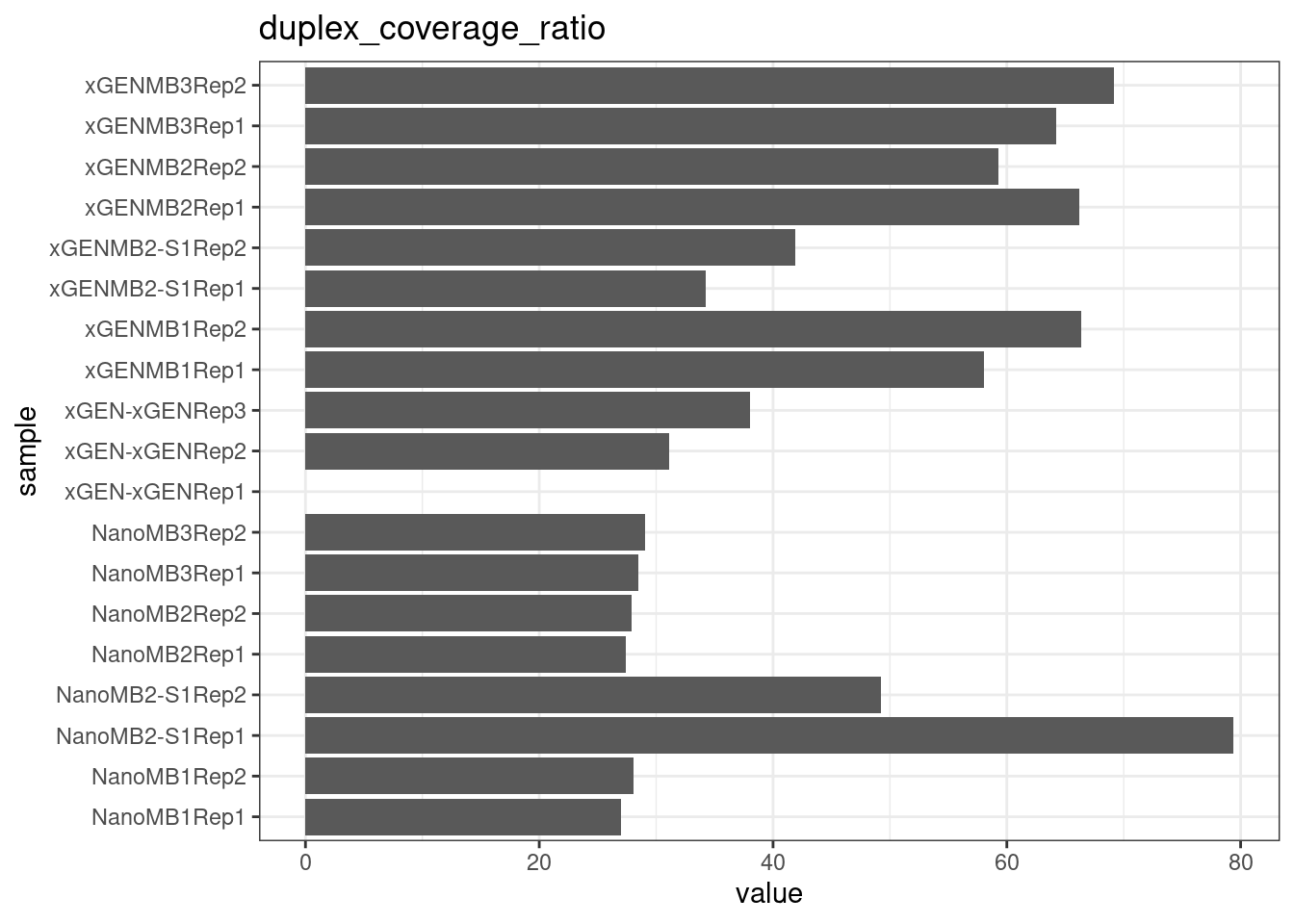
Compare protocols and nucleases directly.
# singletons
plot_metric_boxplot(mm, 'protocol', 'frac_singletons', 'Fraction of singleton reads') +
geom_jitter(width=0.1, aes(protocol, value, colour = nuclease))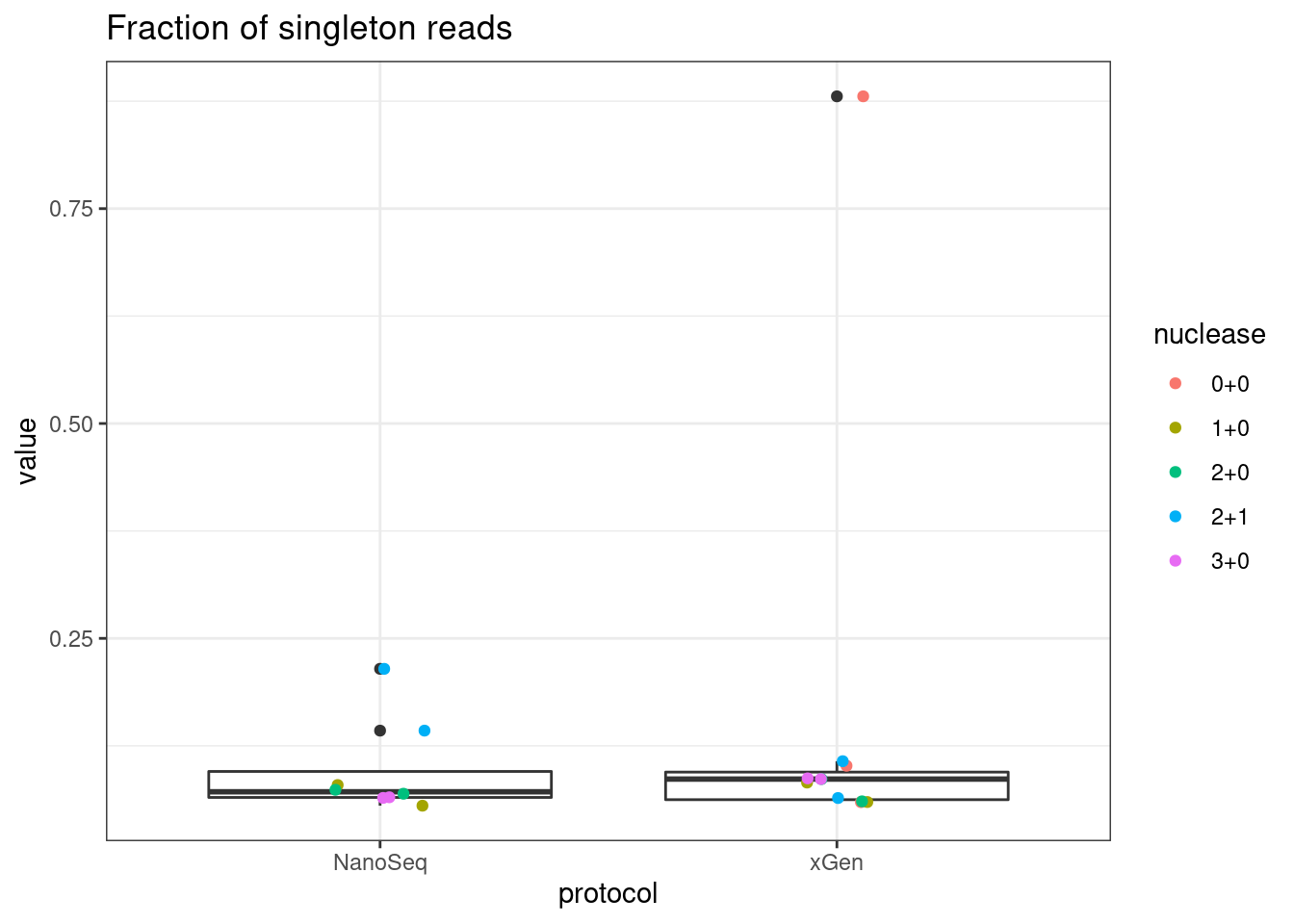
plot_metric_boxplot(mm, 'nuclease', 'frac_singletons', 'Fraction of singleton reads') +
geom_jitter(width=0.1, aes(nuclease, value, colour = protocol)) 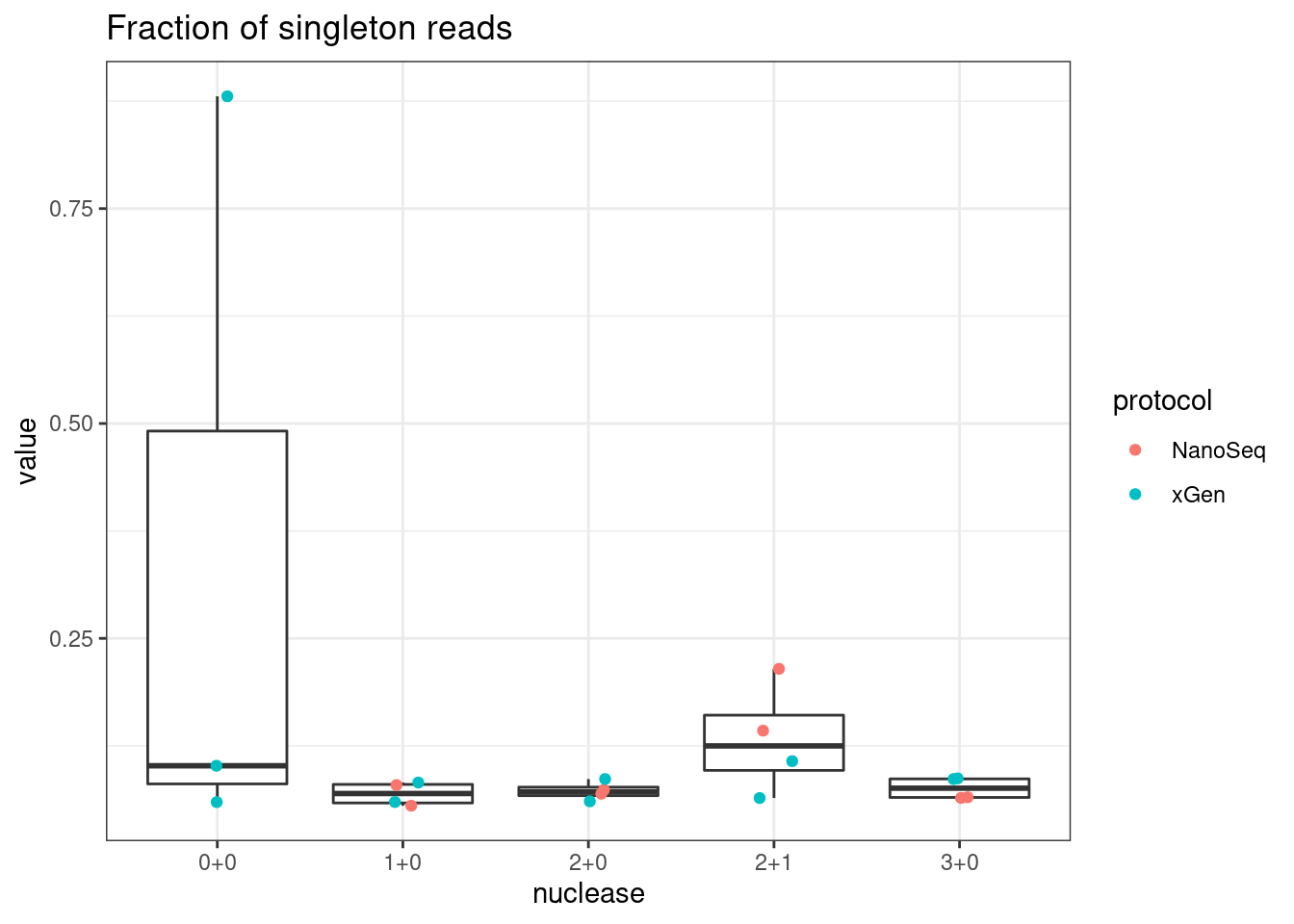
# duplicate rate
plot_metric_boxplot(mm, 'protocol', 'duplicate_rate', 'Duplicate rate (line = optimal)') +
geom_jitter(width=0.1, aes(protocol, value, colour = nuclease)) +
geom_hline(yintercept = 0.81)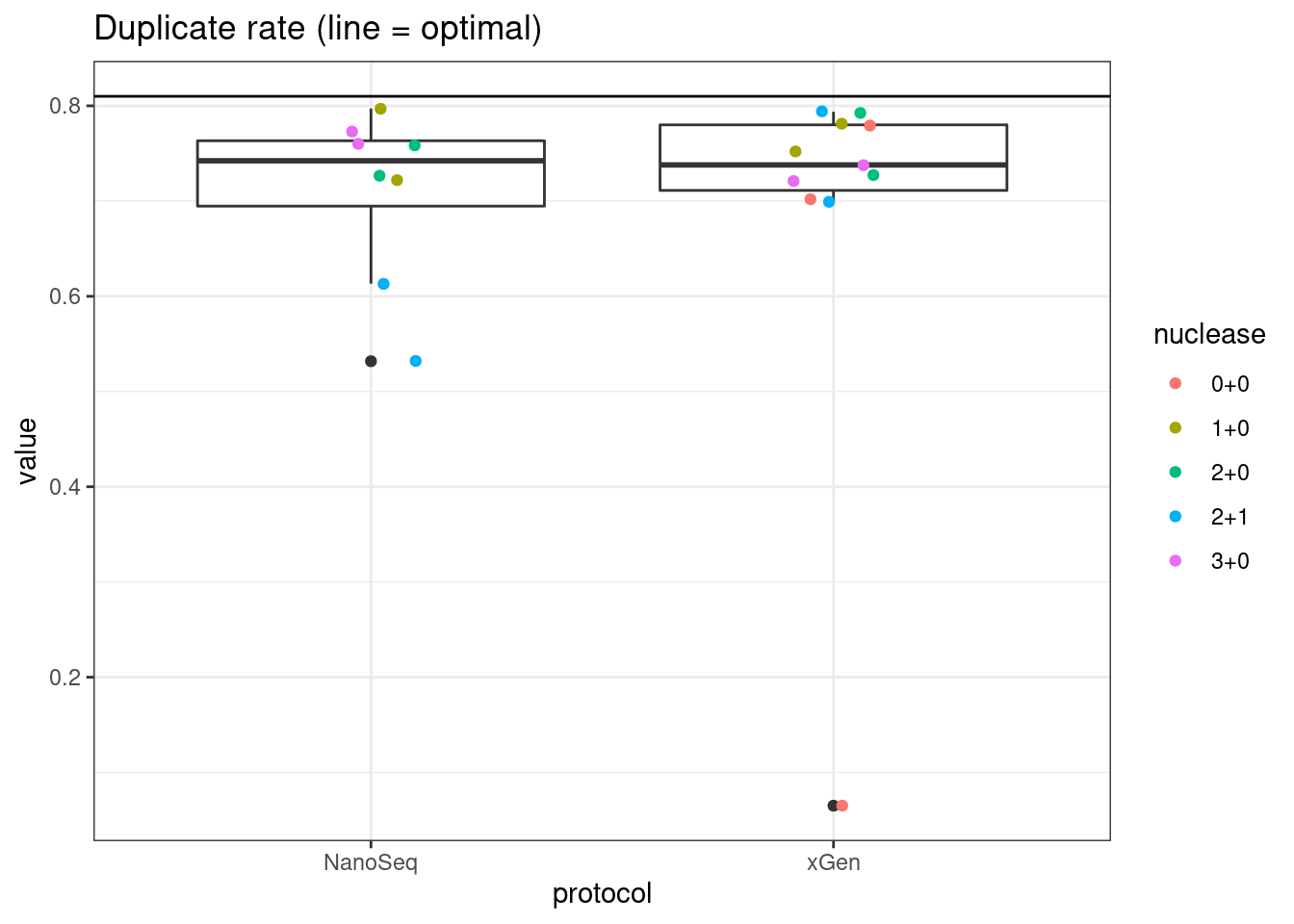
plot_metric_boxplot(mm, 'nuclease', 'duplicate_rate', 'Duplicate rate (line = optimal)') +
geom_jitter(width=0.1, aes(nuclease, value, colour = protocol)) +
geom_hline(yintercept = 0.81)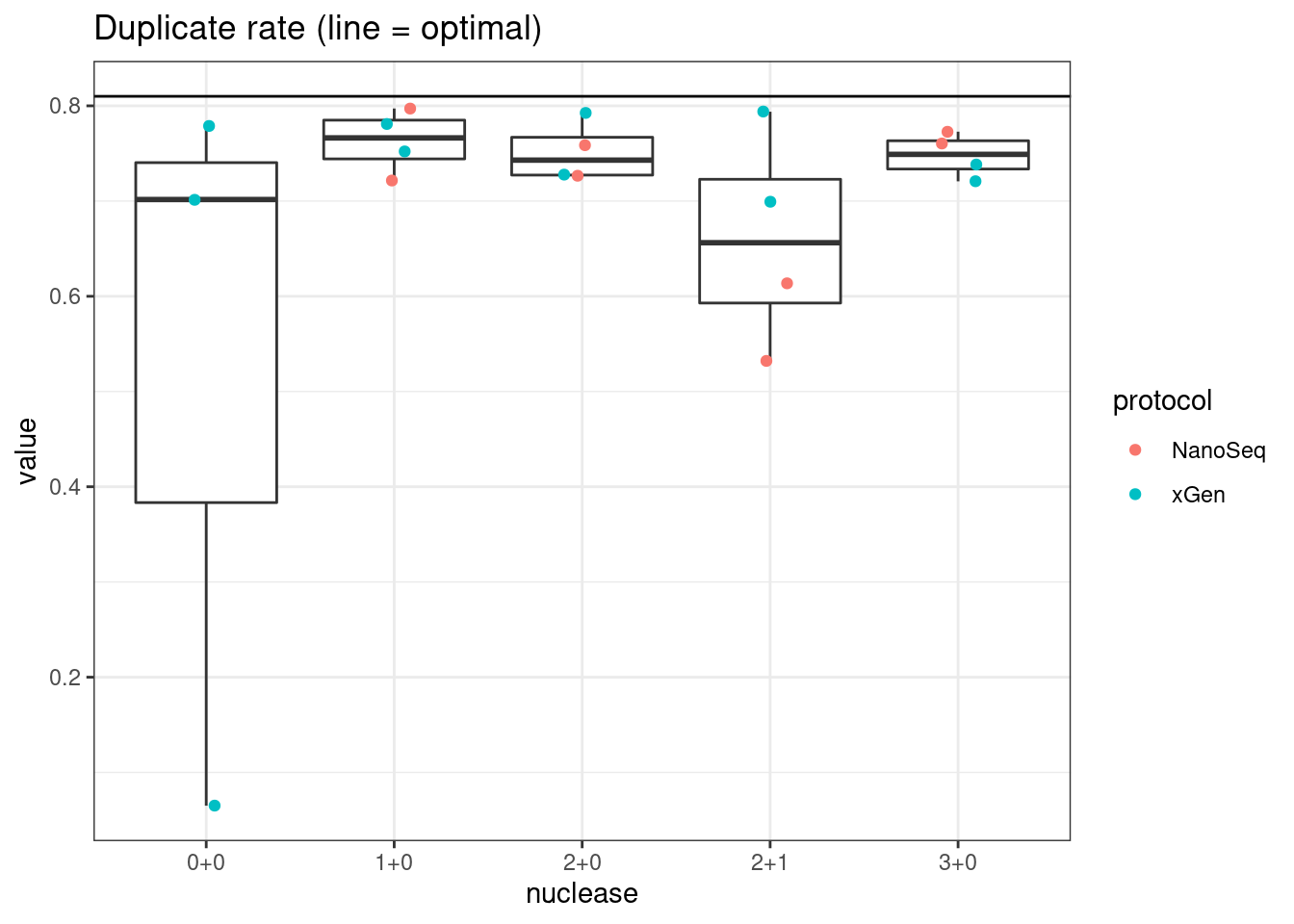
# efficiency
plot_metric_boxplot(mm, 'protocol', 'efficiency', 'Efficiency (line = optimal)') +
geom_jitter(width=0.1, aes(protocol, value, colour = nuclease)) +
geom_hline(yintercept = 0.07) 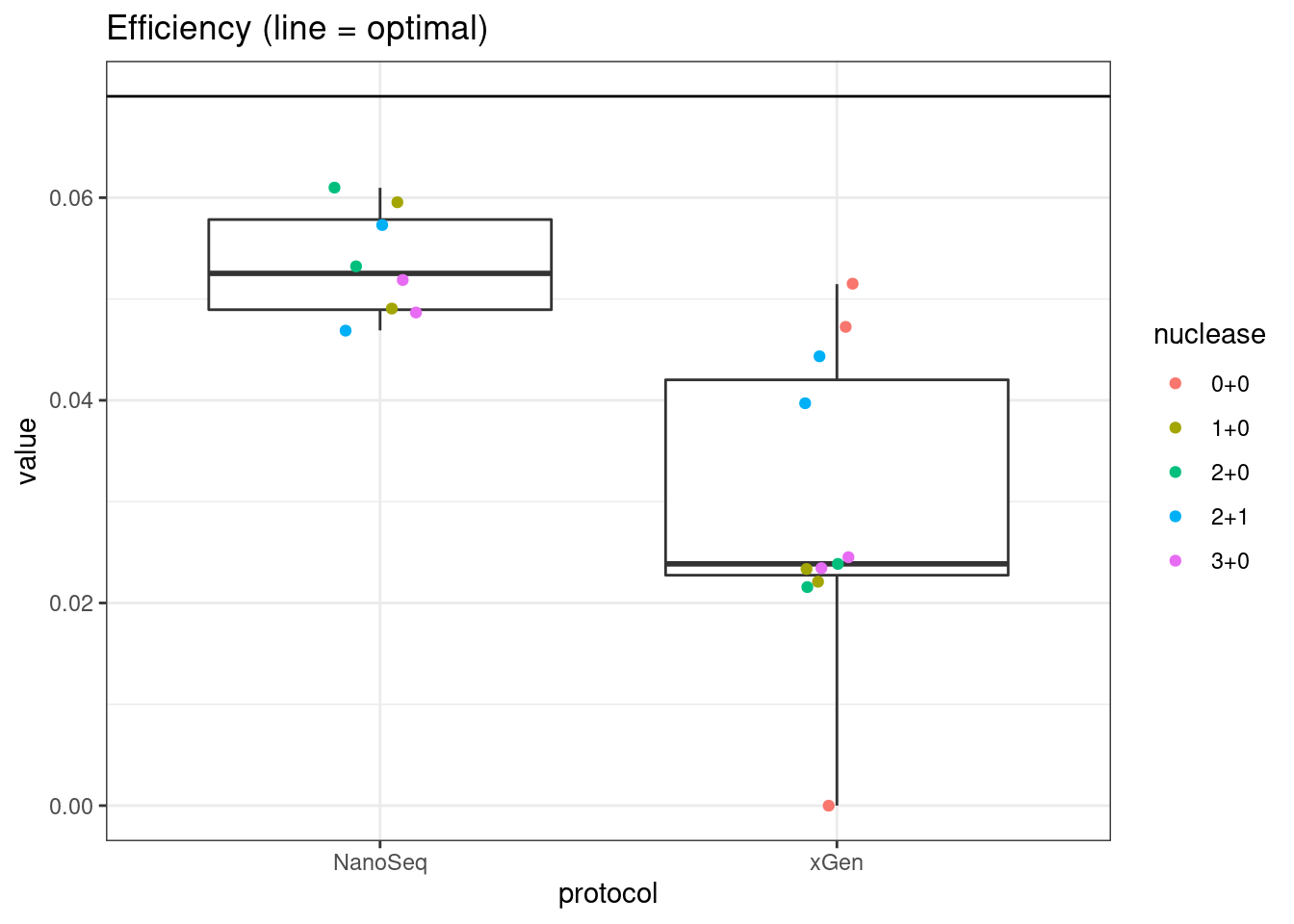
plot_metric_boxplot(mm, 'nuclease', 'efficiency', 'Efficiency (line = optimal)') +
geom_jitter(width=0.1, aes(nuclease, value, colour = protocol)) +
geom_hline(yintercept = 0.07) 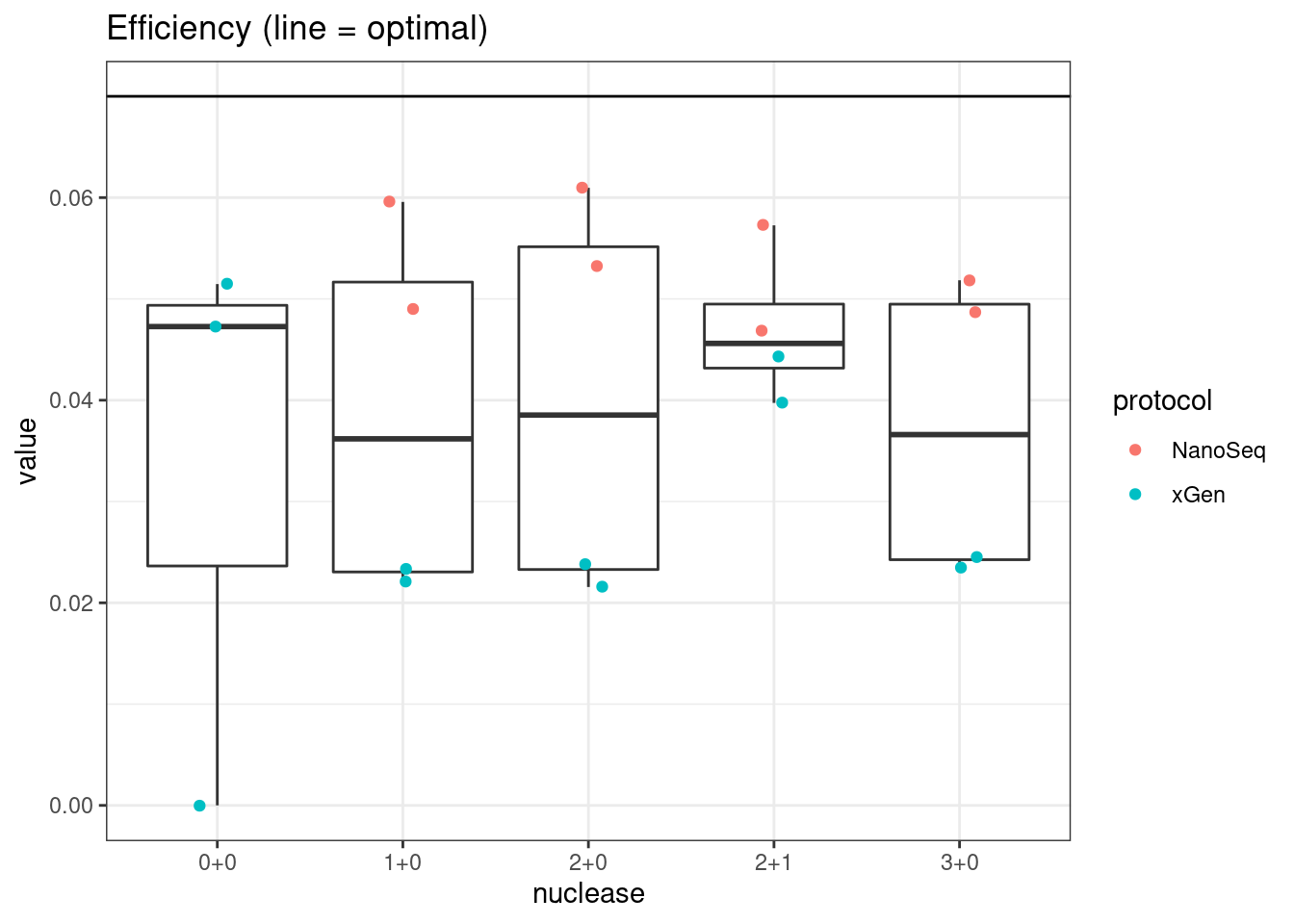
# drop out rate
plot_metric_boxplot(mm, 'protocol', 'drop_out_rate', 'Strand drop-out fraction (lines = optimal range)') +
geom_jitter(width=0.1, aes(protocol, value, colour = nuclease)) +
geom_hline(yintercept = c(0.1, 0.3))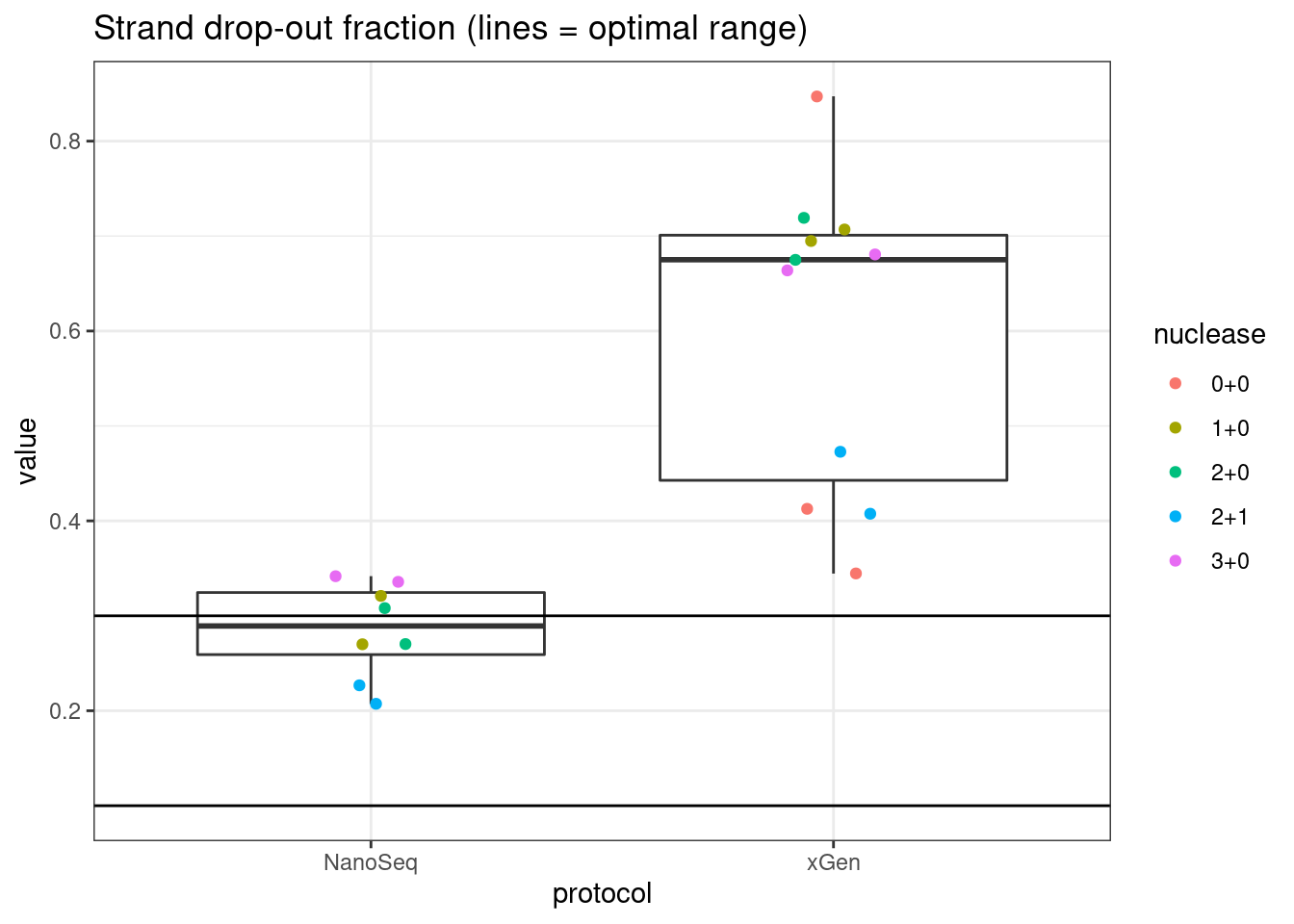
plot_metric_boxplot(mm, 'nuclease', 'drop_out_rate', 'Strand drop-out fraction (lines = optimal range)') +
geom_jitter(width=0.1, aes(nuclease, value, colour = protocol)) +
geom_hline(yintercept = c(0.1, 0.3))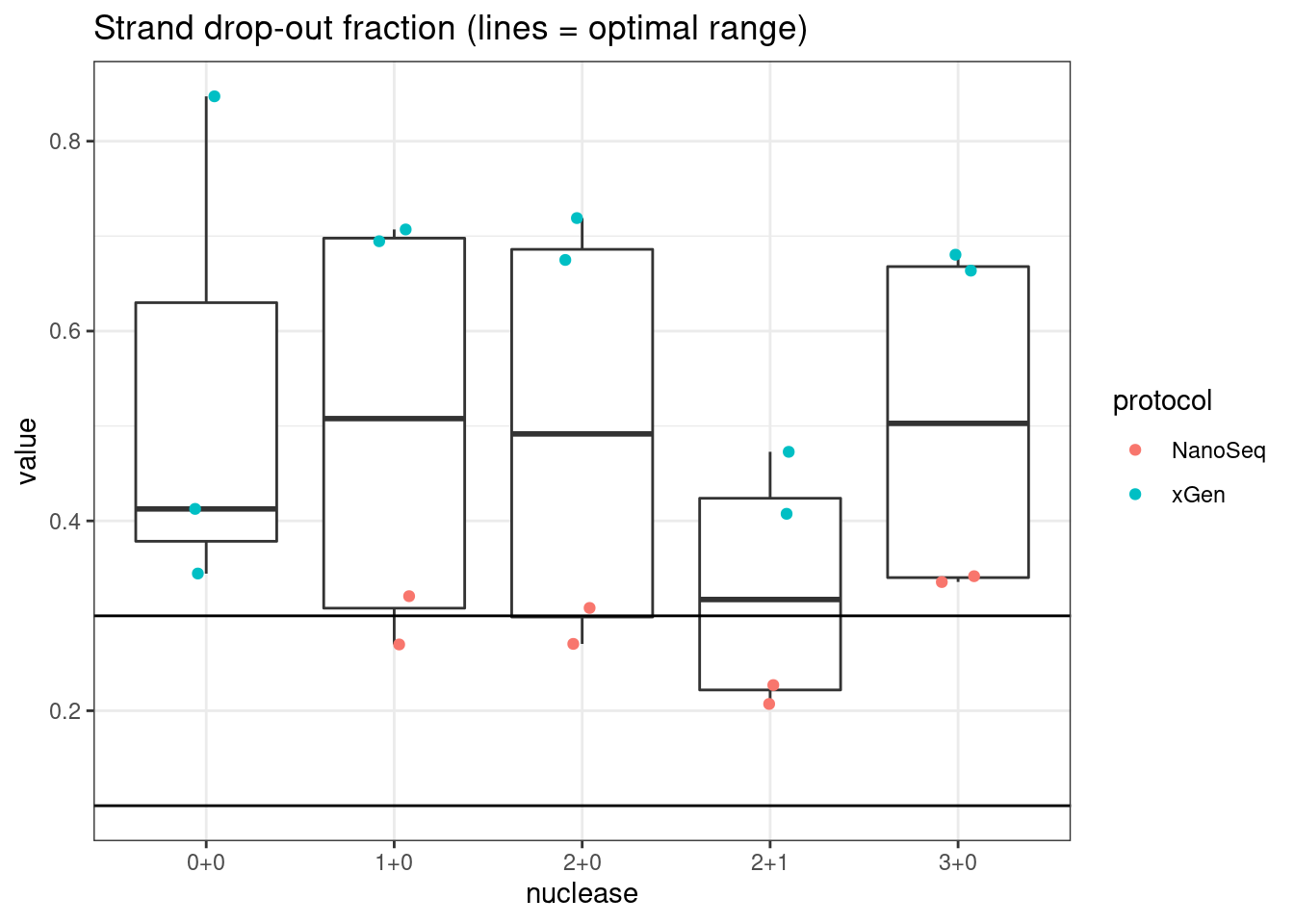
# GC deviation between strands
plot_metric_boxplot(mm, 'protocol', 'gc_deviation', 'GC deviation (both strands vs. one)') +
geom_jitter(width=0.1, aes(protocol, value, colour = nuclease))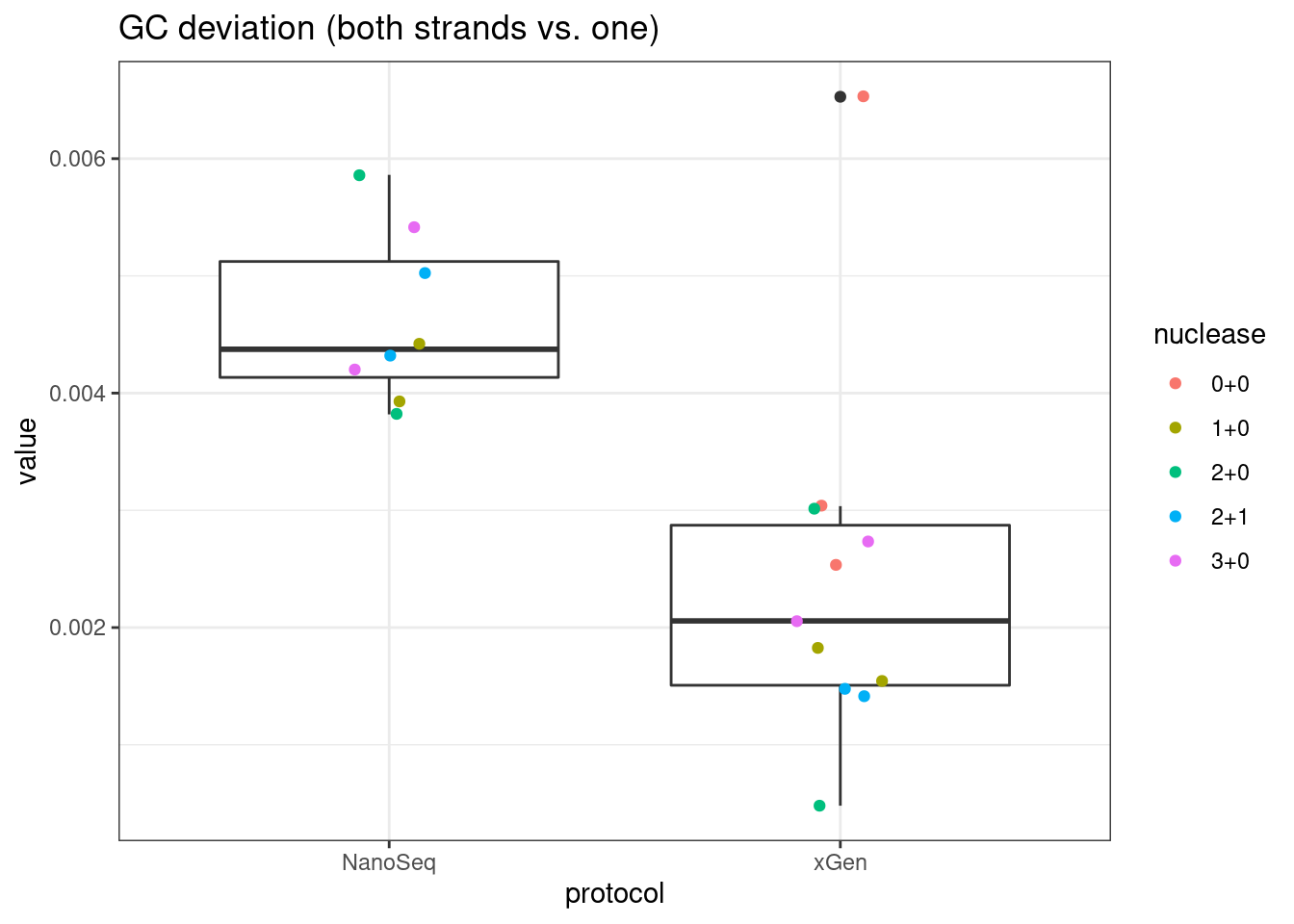
plot_metric_boxplot(mm, 'nuclease', 'gc_deviation', 'GC deviation (both strands vs. one)') +
geom_jitter(width=0.1, aes(nuclease, value, colour = protocol))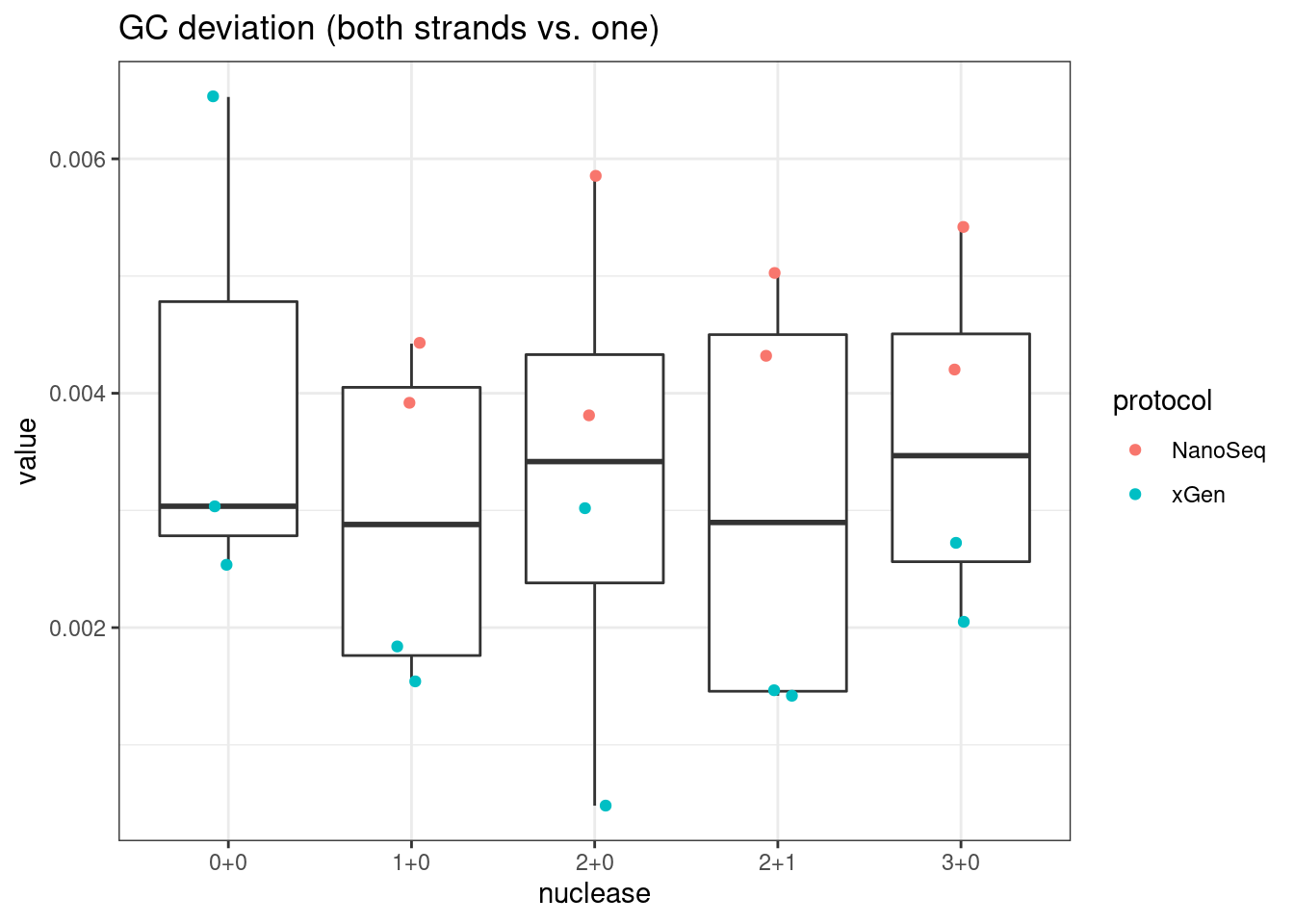
# duplex coverage ratio
plot_metric_boxplot(mm, 'protocol', 'duplex_coverage_ratio', 'Duplex coverage ratio (total cov / duplex cov)') +
geom_jitter(width=0.1, aes(protocol, value, colour = nuclease)) +
geom_hline(yintercept = 30)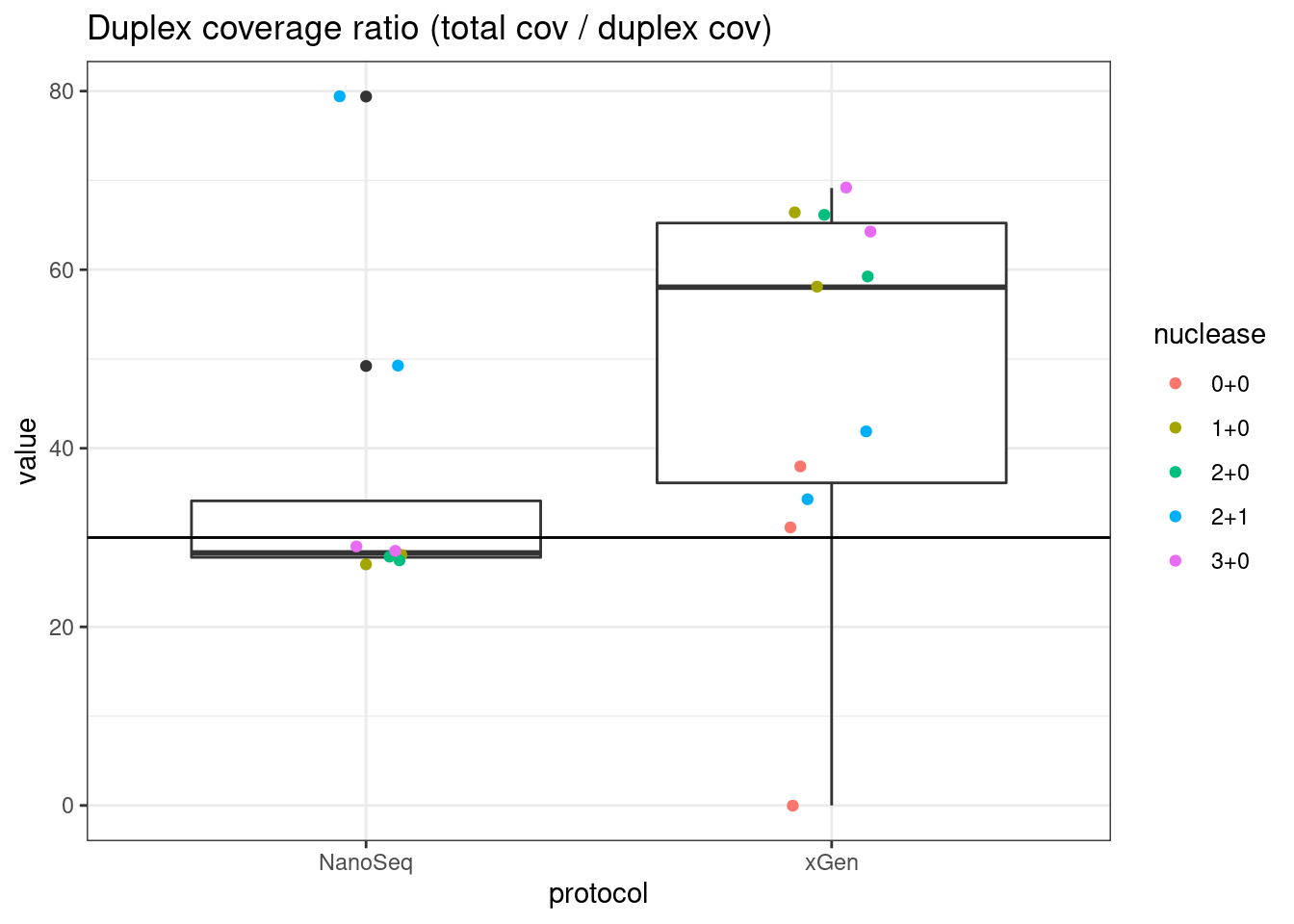
| Version | Author | Date |
|---|---|---|
| 05412f6 | Marek Cmero | 2022-03-28 |
plot_metric_boxplot(mm, 'nuclease', 'duplex_coverage_ratio', 'Duplex coverage ratio (total cov / duplex cov)') +
geom_jitter(width=0.1, aes(nuclease, value, colour = protocol)) +
geom_hline(yintercept = 30)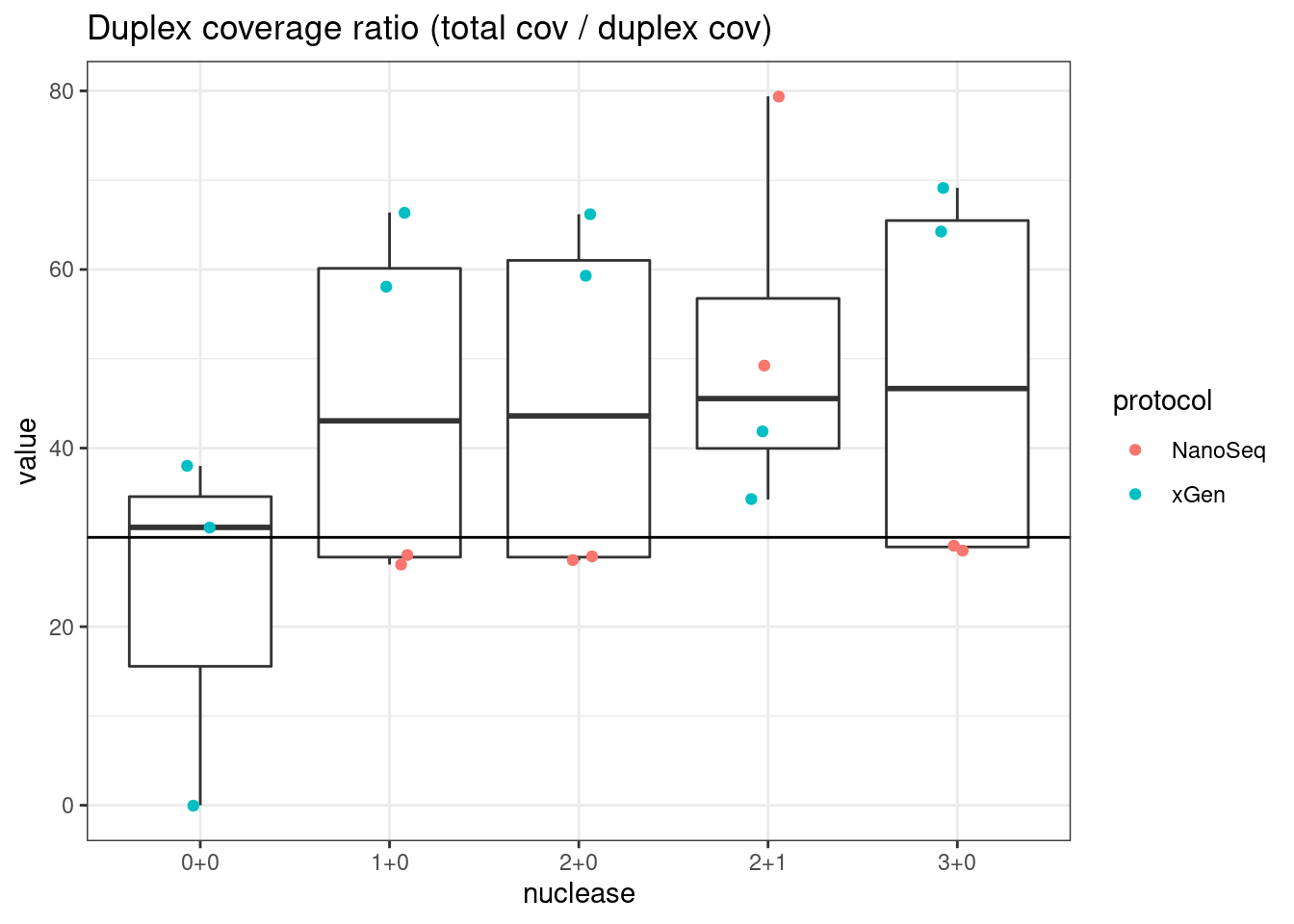
| Version | Author | Date |
|---|---|---|
| 05412f6 | Marek Cmero | 2022-03-28 |
Facet boxplots by nuclease and protocol to show overall results.
ggplot(mm, aes(nuclease, value)) +
geom_boxplot() +
theme_bw() +
facet_wrap(~metric, scales = 'free')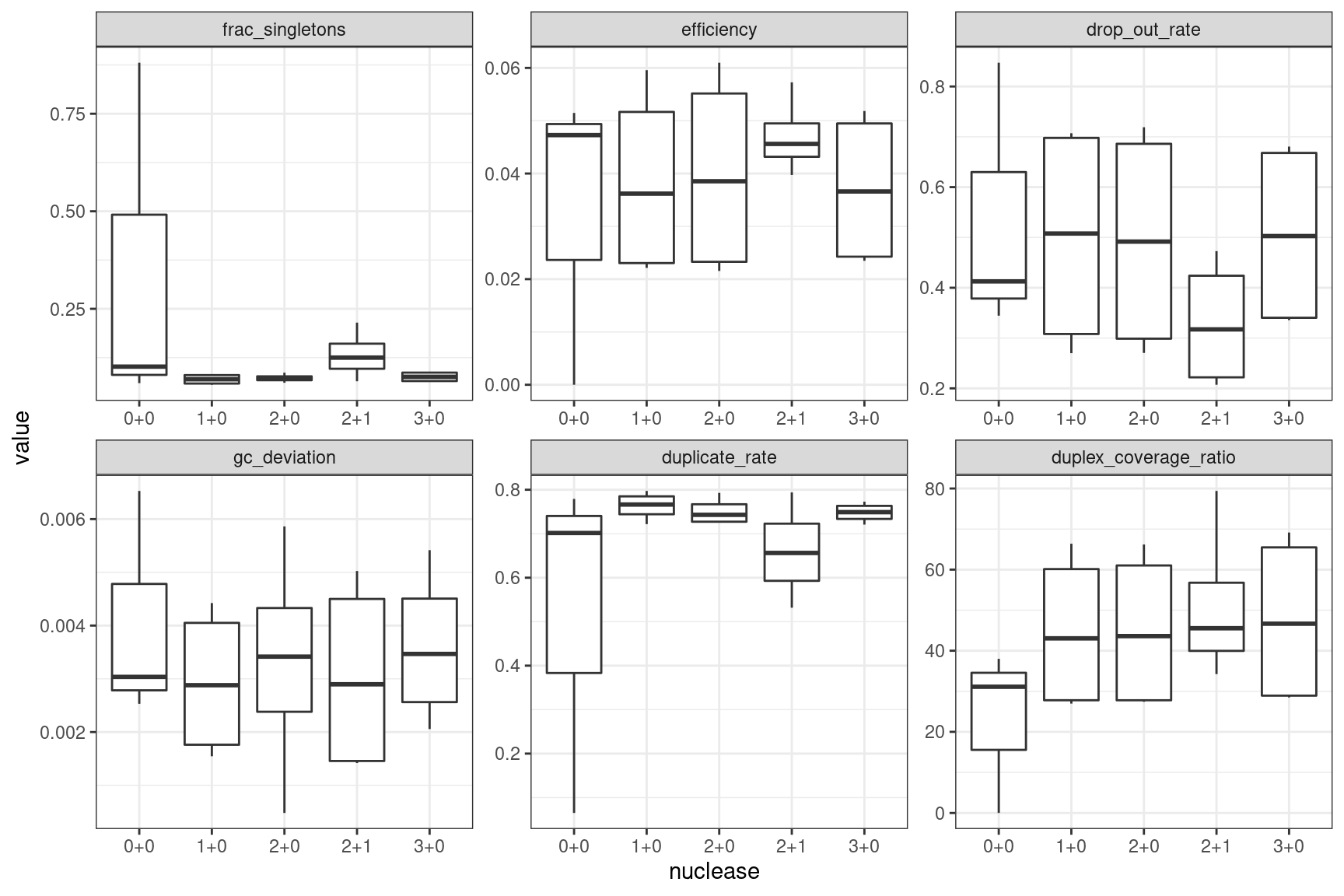
| Version | Author | Date |
|---|---|---|
| 05412f6 | Marek Cmero | 2022-03-28 |
ggplot(mm, aes(protocol, value)) +
geom_boxplot() +
theme_bw() +
facet_wrap(~metric, scales = 'free')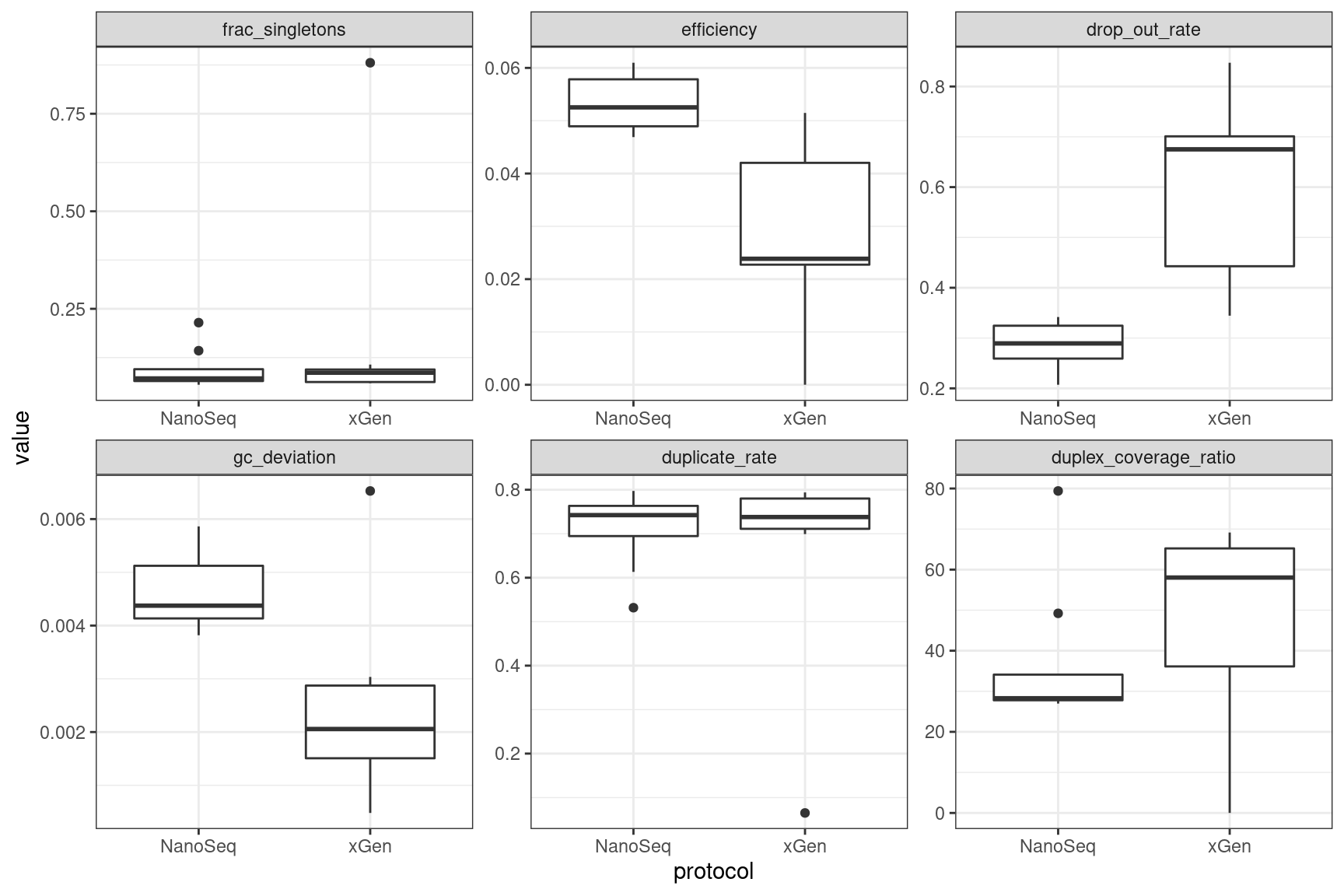
| Version | Author | Date |
|---|---|---|
| 05412f6 | Marek Cmero | 2022-03-28 |
sessionInfo()R version 4.0.5 (2021-03-31)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS: /stornext/System/data/apps/R/R-4.0.5/lib64/R/lib/libRblas.so
LAPACK: /stornext/System/data/apps/R/R-4.0.5/lib64/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 parallel stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] readxl_1.3.1 seqinr_4.2-8 Rsamtools_2.6.0
[4] Biostrings_2.58.0 XVector_0.30.0 GenomicRanges_1.42.0
[7] GenomeInfoDb_1.26.7 IRanges_2.24.1 S4Vectors_0.28.1
[10] BiocGenerics_0.36.1 stringr_1.4.0 tibble_3.1.5
[13] here_1.0.1 dplyr_1.0.7 data.table_1.14.0
[16] ggplot2_3.3.5 workflowr_1.6.2
loaded via a namespace (and not attached):
[1] Rcpp_1.0.7 assertthat_0.2.1 rprojroot_2.0.2
[4] digest_0.6.27 utf8_1.2.2 plyr_1.8.6
[7] cellranger_1.1.0 R6_2.5.1 evaluate_0.14
[10] highr_0.9 pillar_1.6.4 zlibbioc_1.36.0
[13] rlang_0.4.12 whisker_0.4 jquerylib_0.1.4
[16] rmarkdown_2.11 labeling_0.4.2 BiocParallel_1.24.1
[19] RCurl_1.98-1.3 munsell_0.5.0 compiler_4.0.5
[22] httpuv_1.6.3 xfun_0.22 pkgconfig_2.0.3
[25] htmltools_0.5.2 tidyselect_1.1.1 GenomeInfoDbData_1.2.4
[28] fansi_0.5.0 crayon_1.4.2 withr_2.4.2
[31] later_1.3.0 MASS_7.3-53.1 bitops_1.0-7
[34] grid_4.0.5 jsonlite_1.7.2 gtable_0.3.0
[37] lifecycle_1.0.1 DBI_1.1.1 git2r_0.28.0
[40] magrittr_2.0.1 scales_1.1.1 stringi_1.7.5
[43] farver_2.1.0 reshape2_1.4.4 fs_1.5.0
[46] promises_1.2.0.1 bslib_0.3.0 ellipsis_0.3.2
[49] generics_0.1.1 vctrs_0.3.8 tools_4.0.5
[52] ade4_1.7-18 glue_1.4.2 purrr_0.3.4
[55] fastmap_1.1.0 yaml_2.2.1 colorspace_2.0-0
[58] knitr_1.33 sass_0.4.0